

Abstract
Background and purpose:
As a calcium channel blocker, diltiazem acts mainly on the voltage-gated calcium channels, Cav1.2, for its beneficial effects in cardiovascular diseases such as hypertension, angina and/or supraventricular arrhythmias. However, the effects of diltiazem on different isoforms of Cav1.2 channels expressed in heart and vascular smooth muscles remain to be investigated. Here, we characterized the effects of diltiazem on the splice variants of Cav1.2 channels, predominant in cardiac and vascular smooth muscles.
Experimental approach:
Cardiac and smooth muscle isoforms of Cav1.2 channels were expressed in human embryonic kidney cells and their electrophysiological properties were characterized using whole-cell patch-clamp techniques.
Key results:
Under closed-channel and use-dependent block (0.03 Hz), cardiac splice variant Cav1.2CM was less sensitive to diltiazem than two major smooth muscle splice variants, Cav1.2SM and Cav1.2b. Cav1.2CM has a more positive half-inactivation potential than the smooth muscle channels, and diltiazem shifted it less to negative potential. Additionally, the current decay was slower in Cav1.2CM channels. When we modified alternatively spliced exons of cardiac Cav1.2CM channels into smooth muscle exons, we found that all three loci contribute to the different diltiazem sensitivity between cardiac and smooth muscle splice isoforms.
Conclusions and implications:
Alternative splicing of Cav1.2 channels modifies diltiazem sensitivity in the heart and blood vessels. Gating properties altered by diltiazem are different in the three channels.
Keywords: alternative splicing, calcium channel, diltiazem, smooth muscle, heart
Introduction
Rapid entry of Ca2+ through the voltage-gated L-type calcium channels Cav1.2 (channel nomenclature follows Alexander et al., 2009) is a critical step in initiating exciting-contraction coupling of cardiac and vascular smooth muscles. Drugs that inhibit Ca2+ influx via Cav1.2 channels are therefore useful in the treatment of a variety of cardiovascular disorders. Three distinct chemical classes of small-molecule calcium channel blockers are currently in clinical use: 1,4-dihydropyridines (DHPs), phenylalkylamines (PAAs) and benzothiazepines (BTZs). Calcium channels comprise a pore-forming α1-subunit and auxiliary β-, α2δ- and/or γ-subunit. The α1-subunit consists of four homologous domains (I–IV), each having six transmembrane segments (S1–S6). Calcium channel blockers bind to the pore-forming α1-subunit to inhibit Ca2+ influx. The structure of Cav1.2 calcium channels can be diversified by alternative splicing, and these are categorized as cardiac and arterial smooth muscle isoforms (Soldatov, 1994; Welling et al., 1997; Liao et al., 2004; 2005; 2009b;). These splice variants have been shown to exhibit various sensitivities to blocking by DHPs. This correlates with different DHP sensitivities of Cav1.2 calcium channels expressed in the heart and blood vessels (Triggle, 2003).
Diltiazem belongs to the family of BTZs and is widely used in treating cardiovascular diseases. It has mild negative inotropic effects on cardiac muscles and can reduce coronary resistance. Despite a lower vascular selectivity than the DHPs, diltiazem is considered a potent vasodilator and is used to treat mild to moderate hypertension and angina pectoris. Its ability to suppress sino-atrial node activity makes it an ideal candidate to treat supraventricular arrhythmias (Budriesi et al., 2007). Block of Cav1.2 calcium channel activity by diltiazem is preferentially to the inactivated states of the channel (Lee and Tsien, 1983). Diltiazem acts selectively to inhibit more effectively calcium channels expressed on smooth muscles of blood vessels than on heart muscles. The IC50 for cardiac calcium channels is almost twice to that for arterial calcium channels (Sun and Triggle, 1995). It is well known that diltiazem binds to the IIIS6 and IVS6 of Cav1.2 calcium channels to exhibit its actions (Hering et al., 1996; Hockerman et al., 2000). The IIIS6 and IVS6 segments belong to constitutive exons. Thus, no difference exists at diltiazem-binding regions between cardiac and vascular Cav1.2 calcium channels. In this report, we sought to understand whether the alternative splicing of Cav1.2 calcium channels could differentially modulate diltiazem blockade of smooth muscle and cardiac muscle isoforms of Cav1.2 channels.
The genotypes of the predominant isoforms of cardiac and arterial smooth muscle Cav1.2 channels are mainly different in three alternatively spliced loci (Figure 1): the mutually exclusive exons 1/1a at the N-terminus and exons 8/8a at the IS6 segment and/or cassette exon 9* in the I–II loop. Previous reports indicated that mutually exclusive exons 31/32 at IVS3 segment are expressed differently in cardiac and smooth muscles. However, we and others have shown that exon 32 is predominantly utilized in the Cav1.2 channels of both cardiac and smooth muscles (Diebold et al., 1992; Gidh-Jain et al., 1995; Liao et al., 2007). There is an additional alternatively spliced exon 33 located at the IVS3-4 linker that is also expressed in different ratios in the heart and arteries. But the inclusion of exon 33 is predominant in both tissues. In rat heart, over 95% of Cav1.2 channels contain exon 33, while in the aorta, the expression is around 30% (Liao et al., 2007). As a result, two subpopulations of smooth muscle channels, Cav1.2b and Cav1.2SM, can be distinguished by the inclusion or exclusion of exon 33 (Figure 1). In this study, we investigated the molecular component(s) that may determine diltiazem sensitivity of Cav1.2 channels expressed in the heart and blood vessels by examining the diltiazem blockade of the two predominant vascular smooth muscle isoforms and one cardiac isoform in a human embryonic kidney (HEK) 293 heterologous expression system.
Figure 1.
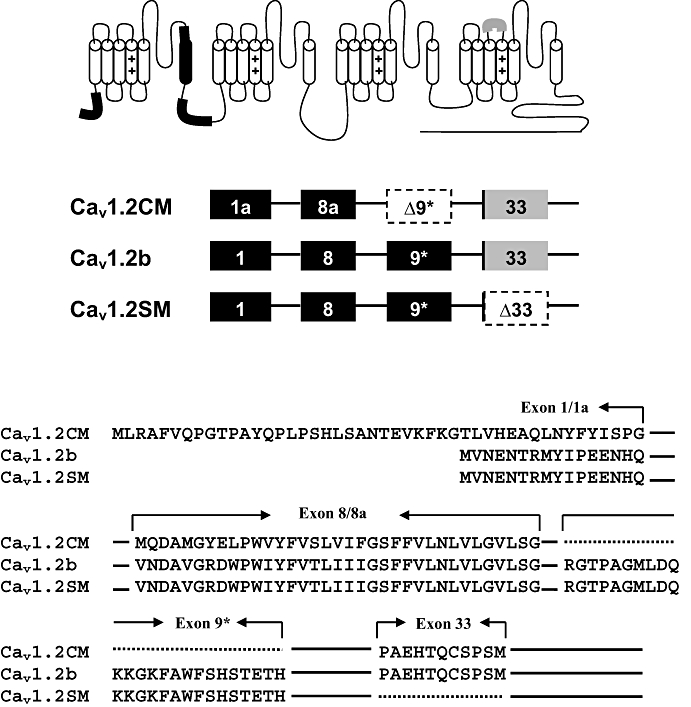
Schematic representation of Cav1.2 channels between cardiac (Cav1.2CM) and smooth muscle (Cav1.2b and Cav1.2SM) isoforms. Exons 1/1a and 8/8a are mutually exclusive exons. Exon 9* is excluded in cardiac Cav1.2CM. They are labelled in black. Exon 33 is partially deleted in the smooth muscles. Around 30% of smooth muscle calcium channels do not contain exon 33 (Liao et al., 2007). Cav1.2CM and Cav1.2b contain exon 33, while Cav1.2SM does not. Exon 33 is labelled in grey.
Methods
Construction of chimeras and cell culture
The construction of Cav1.2b, Cav1.2SM and Cav1.2CM channels has been described previously (Liao et al., 2007). To generate Cav1.2CM-1, a fragment containing exon 1 was cloned into Cav1.2CM with HindIII and AfeI sites. To generate Cav1.2CM-8, a fragment containing exon 8 was cloned into Cav1.2CM using BstApI and ClaI sites. To generate Cav1.2CM-1-8, a fragment with exon 9* deletion was cloned into Cav1.2b using ClaI and SgrAI sites. The identities of all constructs were confirmed by DNA sequencing.
HEK 293 cells were grown to around 20% confluence before being transiently transfected with α1- (1.25 µg), β2a- (1.25 µg) and α2δ- (1.25 µg) subunits using the calcium phosphate transfection method. They were maintained at 37°C for 48–72 h in Dulbecco's modified Eagle medium without serum before electrophysiological recordings.
Electrophysiology and data analysis
Whole-cell patch clamp techniques were used to characterize the channel properties as described before (Shen et al., 2006; Liao et al., 2009a). In brief, calcium currents (ICa) or barium currents (IBa) were recorded at room temperature (∼25°C). Patch electrodes were pulled from a flaming/brown micropipette puller (Sutter Instrument, Novato, CA, USA) and polished with a microforge (NARISHIGE Scientific Instrument Laboratory, Tokyo, Japan). The external solution contained (in millimolar) 140 tetraethylammonium methanesulfonate, 10 HEPES, 1.8 CaCl2 or 5 BaCl2 (pH was adjusted to 7.4 with CsOH and osmolarity to 300–310 mOsm with glucose). The internal solution (pipette solution) contained (in millimolar) 138 Cs-MeSO3, 5 CsCl2, 0.5 EGTA, 10 HEPES, 1 MgCl2, 2 mg·mL−1 MgATP, pH 7.3 (adjusted with CsOH). The osmolarity was adjusted to 290–300 mOsm with glucose. Under voltage clamp protocol and using an Axopatch 200B amplifier (MDS Analytical Technologies, Union City, CA, USA), whole-cell currents were recorded and filtered at 1–5 kHz and sampled at 5–50 kHz. The series resistance was normally <5 MΩ after compensation. The capacity transient was compensated using an online P/4 protocol. The voltages are uncorrected for a −10 mV junction potential, and actual voltage can be obtained by subtracting 10 mV from the reported values.
Data analyis
Values are expressed as mean ± SEM. GraphPad Prism (GraphPad Software, Inc., La Jolla, CA, USA) software was used for data plotting and statistical analysis. Statistical significance of differences between means was calculated with the Student's t-test or one-way anova.
Materials
(+)-cis-Diltiazem hydrochloride (Sigma, St Louis, MO, USA) was dissolved in saline water to make a stock solution of 100 mM and stored at −20°C. Diltiazem at 0.01–1 mM concentrations was freshly prepared in bath solution from stock and then applied to the HEK 293 cells.
Results
Closed-channel block by diltiazem
Closed-channel block was measured by stepping to 10 mV from a holding potential of −90 mV every 30 s till saturation. Ca2+ (1.8 mM) was used as the charge carrier. Diltiazem at five concentrations (10, 50, 100, 500 and 1000 µM) were applied to the cells. At 10 µM, Cav1.2b, Cav1.2SM and Cav1.2CM showed very minor responses to diltiazem. Only around 10% of the currents were inhibited (Figure 2A). We also observed that at concentrations lower than 10 µM, diltiazem can exert agonist effects in some cells (data not shown). At 1000 µM, diltiazem blocked 80–90% of the currents. Among the three isoforms, Cav1.2b and Cav1.2SM exhibited similar inhibition by diltiazem. Their sensitivities to diltiazem were almost identical (see table in Figure 2C). However, the cardiac Cav1.2CM channel isoform showed a lower sensitivity to diltiazem at all concentrations except 10 µM. The IC50 of diltiazem for Cav1.2CM was about twice that for the two vascular smooth muscle isoforms (Figure 2C, P < 0.01, one-way anova and Newman–Keuls test).
Figure 2.
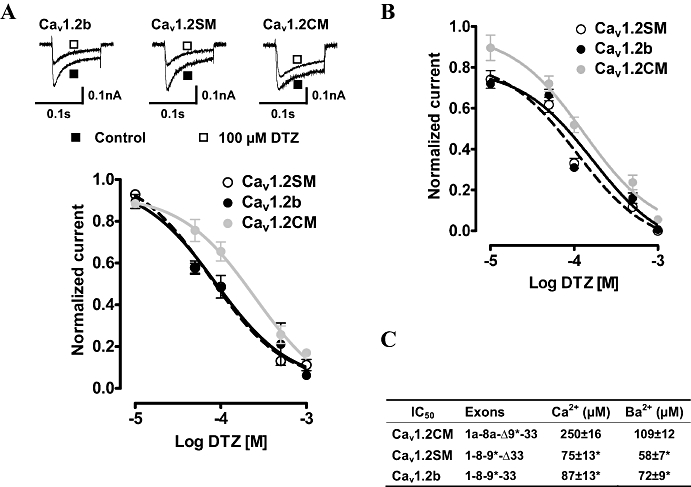
Closed-channel block of Cav1.2 channels by diltiazem (DTZ) at 0.03 Hz in Ca2+ and Ba2+. (A) DTZs (10, 50, 100, 500 and 1000 µM) were added to the bath solution containing 1.8 mM Ca2+ until equilibrium. Sample currents in the absence and presence of 100 µM DTZ are illustrated. (B) Dose–response curves of three Cav1.2 channels in 5 mM Ba2+. IC50 was calculated from fits of individual dose–response curves to the Hill equation and summarized in (C). Δ indicates the exclusion of an exon. *P < 0.01, versus Cav1.2CM (one-way anova).
We further compared the response of the three channels to diltiazem block in the absence of Ca2+. Five millimolar Ba2+ was used as the charge carrier, and similar experiments were carried out. The IC50 of all three channels was lower in solutions containing Ba2+ than in solutions containing Ca2+ (Figure 2B,C). Interestingly, the change of IC50 for Cav1.2CM was greater than the other two channels. Thus, the IC50 of diltiazem for Cav1.2CM was decreased using Ba2+ to less than 50% of that using Ca2+, while the other two channels showed around 15 µM decrease (Figure 2C). Again, Cav1.2CM exhibits less sensitivity to diltiazem block in Ba2+ external solution than the other two vascular channels (Figure 2C, P < 0.01 as compared with Cav1.2CM, one-way anova and Newman–Keuls test).
Diltiazem alters steady-state inactivation (SSI) properties
Our results so far showed that Cav1.2SM and Cav1.2b channels were more sensitive to diltiazem than Cav1.2CM channels in closed-state block. Diltiazem was known to affect the kinetics of Cav1.2 channels. Thus, we characterized the effect of diltiazem on the SSI properties of the three channels. A concentration (100 µM) close to the IC50 of diltiazem was applied to the cells. The SSI protocols were carried out before and after diltiazem block. To examine the effect of diltiazem on the SSI properties of the three Cav1.2 channels, we applied a three-pulse protocol. A normalization pulse to 10 mV was added before a classical two-pulse protocol to eliminate the rundown effect during recording. The 5 s prepulse was applied to fully inactivate the channels. As shown in Figure 3A–C, diltiazem significantly shifted the inactivation curves to the negative potential in all three channels. Application of 100 µM diltiazem shifted the voltage for half-maximal inactivation (V0.5) of Cav1.2CM channels (Figure 3D), and these shifts were significant for each channel (P < 0.001, Student's t-test). In both absence and presence of diltiazem, Cav1.2CM channels had the most positive V0.5 when compared with the two smooth muscle channels (Figure 3, P < 0.05 in the absence of diltiazem, and P < 0.01 with the presence of diltiazem, one-way anova and Newman–Keuls test). Importantly, diltiazem shifts the V0.5 of Cav1.2SM and Cav1.2b more than Cav1.2CM channels to negative potentials. The difference of ΔV0.5 for Cav1.2CM was 7.9 mV, while for Cav1.2SM and Cav1.2b channels, ΔV0.5s were 16.2 mV and 12.7 mV (Figure 3D, inset).
Figure 3.
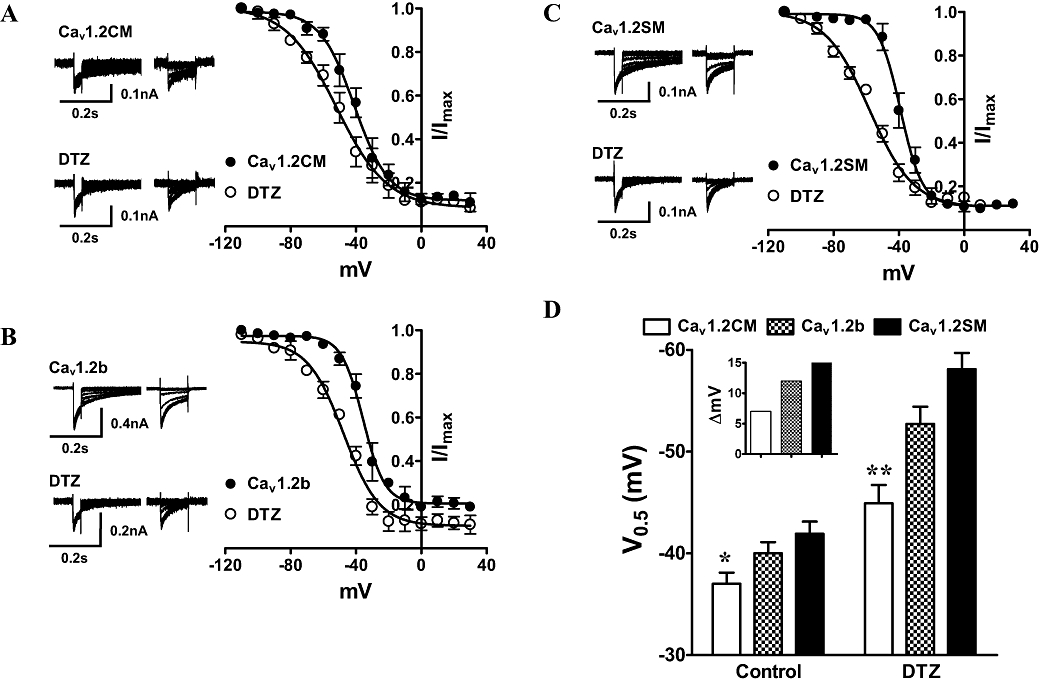
Inactivation kinetics of smooth and cardiac muscle Cav1.2 channels in the absence or presence of 100 µM diltiazem (DTZ). Ca2+ (1.8 mM) was used as the charge carrier. (A–C) The steady-state inactivation (SSI) curves were measured from a holding potential of −90 mV to a 30 ms normalization pulse to 10 mV, followed by a family of 5 s long prepulses from −110 to 30 mV and a final 104 ms test pulse to 10 mV. The normalization pulse can eliminate the rundown effect during recording. Each test pulse was normalized to the peak current of individual normalizing pulse. The SSI curve was obtained from a single Boltzmann equation: Inormalized =Imin+ (Imax−Imin)/(1 + exp((V0.5−V)/k), where Inormalized is the normalized current; V0.5 is the half potential of inactivation and k is the slope factor. (D) Summary of V0.5. *P < 0.05 and **P < 0.01, one-way anova. Inset shows the V0.5 (ΔmV) difference between the presence and absence of DTZ.
Effect of diltiazem on current decay
Diltiazem can accelerate current decay with Ba2+ as the charge carrier (Kraus et al., 1998; Dilmac et al., 2003). Here, we investigated whether 100 µM diltiazem could accelerate current decay in the three channels with 5 mM Ba2+. Figure 4A shows that in the absence of 100 µM diltiazem, Cav1.2SM and Cav1.2b channels exhibited faster current decay than Cav1.2CM channels. The current inactivation was well fitted with a single-exponential function. The time constant (τ) was calculated, and as shown in Figure 4C, the τ-value was significantly longer in Cav1.2CM than in Cav1.2b and Cav1.2SM (P < 0.05, one-way anova and Newman–Keuls test). There was no difference between Cav1.2b and Cav1.2SM channels (P > 0.05, one-way anova and Newman–Keuls test). After 100 µM diltiazem was added to the bath solution, a robust current decay could be clearly identified in all three channels (Figure 4B). From the sample traces, we found that the Cav1.2CM channel exhibited slower current decay than the other two channels, and this was verified by fitting to a double exponential function. The fast time constants (τfast) and slow time constants (τslow) were calculated. τfast of Cav1.2CM channel was longer than the other two channels (P = 0.0022, one-way anova and Newman–Keuls test). Again, there is no difference between the two smooth muscle channels. Similar results were found in the measurement of τslow. Cav1.2CM channel exhibited longer τslow than the other two channels (P = 0.0052, one-way anova and Newman–Keuls test), and there is no difference between the two smooth muscle channels (Figure 4C). Thus, we showed here that diltiazem could accelerate current decay, and Cav1.2CM channel had a slower current decay in the presence or absence of diltiazem, compared with the vascular smooth muscle channels.
Figure 4.
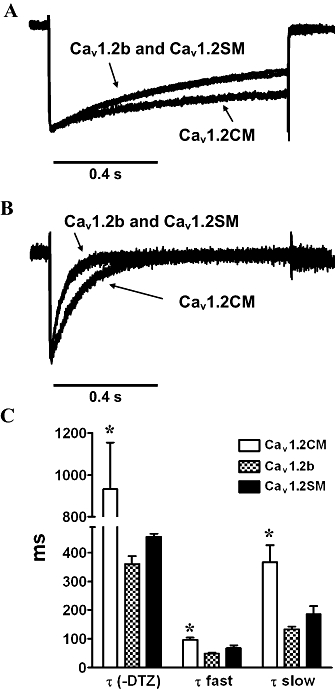
Effect of diltiazem (DTZ) on current decay. (A) Representative traces of normalized Ba2+ currents in the absence of DTZ. A test pulse of 10 mV for 0.9 s was applied from a holding potential of −90 mV. Cav1.2b and Cav1.2SM channels displayed similar current decay. (B) Representative traces of normalized Ba2+ currents after 100 µM DTZ treatment. The current decays in Cav1.2b and Cav1.2SM were almost identical. (C) Summary of the time constants in three channels before and after DTZ treatment. Single-exponential function was applied to fit the current without DTZ, while two-exponential fitting was used on channels after DTZ treatment. *P < 0.05, one-way anova.
Effect of diltiazem on recovery from inactivation
Diltiazem was shown to delay the recovery of Cav1.2 channels from inactivation (Dilmac et al., 2003). To investigate the effect of diltiazem on the three splice variants from cardiac and smooth muscles, we performed experiments using a standard two-pulse protocol. A 2 s inactivation pulse to 10 mV was applied first from a holding potential of −90 mV. With bath solution containing 1.8 mM Ca2+, we observed an almost full inactivation of the channels. Following a recovery time interval between 50 ms to 7 s at holding potential, a test pulse to 10 mV was given to record the fractions of recovery. For experiments with the presence of 100 µM diltiazem, the same protocol was applied following equilibration of drug block. The fraction of recovery was fitted to a double exponential equation. We observed a great delay of current recovery from inactivation when diltiazem was present (Figure 5A–C). The fast (τfast) time constants were significantly shorter in all three channels in the absence of 100 µM diltiazem. When diltiazem was present, τfast was increased over 10 times (Figure 5D). The slow (τslow) time constants were also shorter in Cav1.2b and Cav1.2CM channels when compared with 100 µM diltiazem treated channels (data not shown). Furthermore, the diltiazem affects the three channels differently. At the seventh second, less than 50% currents were recovered in Cav1.2CM channels, while over 60% currents were recovered in the two smooth muscle channels (Figure 5A–C). The τfast of Cav1.2CM channels was over twice as long as those of the other two channels (Figure 5D, P = 0.0041, one-way anova and Newman–Keuls test).
Figure 5.
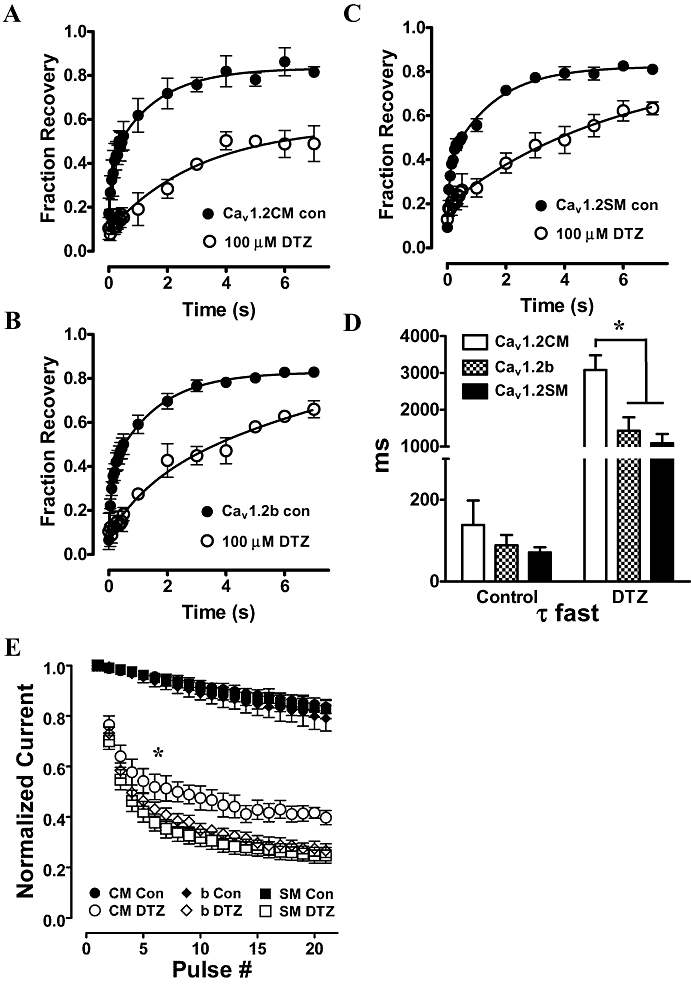
Diltiazem (DTZ) slows recovery from inactivation. (A–C) Recovery from inactivation in the absence or presence of 100 µM DTZ. Inactivation was achieved by a 2 s depolarization of 10 mV from a holding potential of −90 mV. The fraction of recovery was plotted against the time interval and fitted with a two-exponential equation. (D) The fast time constant of recovery (τfast). (E) Frequency dependence of DTZ block at 5 Hz. Whole-cell Ba2+ currents were recorded in the absence and presence of 100 µM DTZ. Twenty-one consecutive pulses were given as control. The same train of pulses were given again after incubation of the cells with 100 µM DTZ to reach equilibrium. Each pulse is normalized to the first pulse. *P < 0.01, one-way anova.
Use-dependent block by diltiazem
Diltiazem is used to inhibit channels during high-frequency stimulation, as observed in cardiac arrhythmias. We next examined the frequency-dependent block by diltiazem in the three channels with Ba2+ as the charge carrier. We first applied a 21-pulse, 5 Hz train of 100 ms depolarization to 0 mV from a holding potential of −90 mV in the absence of diltiazem. All three channels exhibited similar responses (Figure 5E). One hundred micromolar diltiazem was then added to the bath solution. After reaching equilibrium, the same protocol was applied. Both the two smooth muscle channels responded similarly to frequency-dependent block by diltiazem. However, frequency-dependent block was significantly reduced in Cav1.2CM channels (Figure 5E). This was obvious at the seventh pulse (P < 0.05, one-way anova). The results show that Cav1.2CM channels were less sensitive to diltiazem, under conditions of use-dependent block.
Effect of diltiazem on activation gating
To study whether the activation gating of the three channels can be affected by diltiazem, we carried out current–voltage (I–V) experimentations on the three channels in the absence and presence of 100 µM diltiazem (Figure 6). Without diltiazem treatment, Cav1.2CM showed the most positive half-activation potential (Figure 6D, P = 0.0003, one-way anova). The V0.5 of Cav1.2SM was 2.1 mV more negative than that of Cav1.2b. The P-value was slightly above 0.05. After the application of 100 µM diltiazem, all three I–V curves were shifted to negative potentials (Figure 6). Interestingly, the effect of diltiazem on activation gating was different among the three channels. Both Cav1.2SM and Cav1.2b smooth muscle channels showed a mild shift of about −3 mV. However, the negative shift of the V0.5 of Cav1.2CM channels by diltiazem was as large as −7.9 mV (Figure 6D, P < 0.001, Student's t-test).
Figure 6.
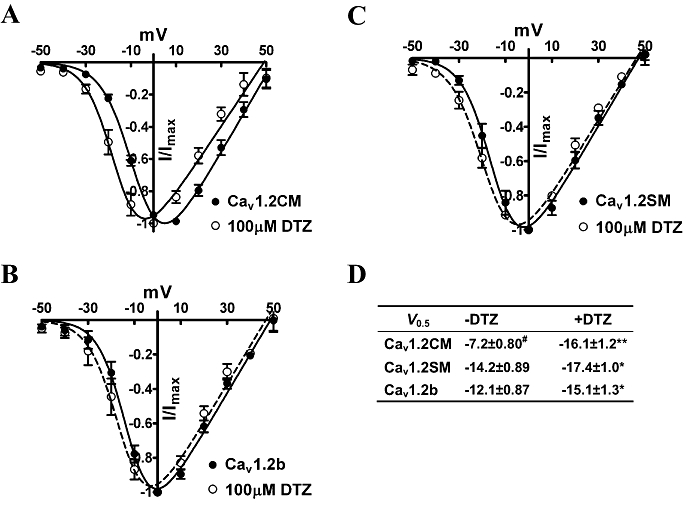
(A–C) An ensemble of current–voltage curves from three Cav1.2 channels treated with or without 100 µM diltiazem (DTZ) in 5 mM Ba2+. The cells were first held at −90 mV and then stepped to various potentials from −50 to 50 mV. The normalized I–V curves were obtained by fitting to the Boltzmann function. (D) Summary of V0.5 values. *P < 0.05, **P < 0.001, Student's t-test, as compared with –DTZ. #P = 0.0003, one-way anova, as compared with Cav1.2SM and Cav1.2b channels.
Alternative splicing affects diltiazem sensitivity between cardiac and vascular smooth muscle channels
The vascular two smooth muscle isoforms of Cav1.2 channels are more sensitive to diltiazem block than the cardiac isoform under closed-channel state and use-dependent block. We sought to understand the molecular structures that underlie the diltiazem sensitivity. The smooth muscle Cav1.2 channel isoforms are different from cardiac Cav1.2CM at three major loci: N-terminus, IS6 transmembrane segment and I–II loop (Figure 1). We generated chimeric channels from Cav1.2CM and Cav1.2b to determine the critical exons that are important in modulating diltiazem sensitivity. It has been reported that the mutually exclusive exons 8/8a at IS6 transmembrane segment determine the sensitivity to nisoldipine, between cardiac and smooth muscle isoforms (Welling et al., 1997). As such, we tested whether exon 8 or 8a may influence diltiazem sensitivity by exchanging exon 8a for 8 in the Cav1.2CM channels to generate the chimeric construct Cav1.2CM-8 (Figure 7). The IC50 of Cav1.2CM-8 was 77 ± 12 µM, similar to Cav1.2b, which is 87 ± 13 µM and significantly different from that of Cav1.2CM (see Figure 2C). The result shows that exon 8 plays a significant role in modulating diltiazem sensitivity. We then generated Cav1.2CM-1. This construct contains exon 1, while Cav1.2CM contains exon 1a (Figure 7). Unexpectedly, Cav1.2CM-1 had an IC50 of 77 ± 13 µM, similar to that of Cav1.2b (87 ± 13 µM) and Cav1.2SM (75 ± 13 µM). Thus, exon 1 alone can also increase diltiazem sensitivity without the presence of exon 8. To determine the effect of exon 9* at I–II loop on diltiazem block, we generated Cav1.2CM-1-8. This construct is different from Cav1.2b as it did not contain exon 9*. The IC50 of Cav1.2CM-1-8 was 112 ± 11 µM, a value that was between the IC50s of Cav1.2b and Cav1.2CM channels. However, at concentrations higher than 100 µM, diltiazem blocked Cav1.2CM-1-8 channels as effectively as Cav1.2b channels. Thus, exon 9* also influenced diltiazem sensitivity but to a lesser degree than exon 1 and exon 8. Our conclusion is that all three loci for alternative splicing contribute to modulating diltiazem sensitivity of cardiac and vascular smooth muscle Cav1.2 channels, but that exon 1/1a and 8/8a have a larger effect than exon 9*.
Figure 7.
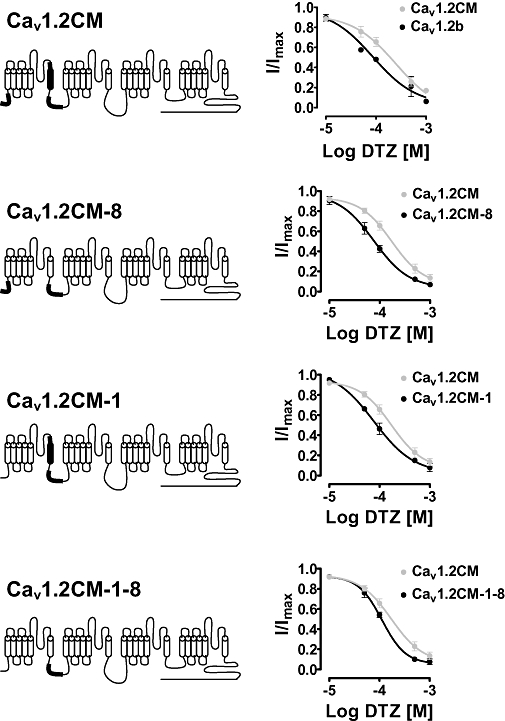
Structure of chimeras and dose–response curves to diltiazem (DTZ) block under the same conditions as in Figure 2. The three loci of Cav1.2CM that are different from Cav1.2b are labelled as dark transmembrane segment and/or bold lines.
Discussion
Our results provide insight into the mechanism by which alternative splicing acts as a molecular switch to modulate inhibition by diltiazem of the cardiac and vascular smooth muscle isoforms of the L-type Cav1.2 calcium channel. Diltiazem has been used to treat disorders both in the heart and blood vessels. It can decrease coronary resistance and therefore has been used to manage angina pectoris. Diltiazem can also lower diastolic blood pressure during long-term oral administration to treat patients with hypertension (Abernethy and Schwartz, 1999). However, the negative inotropic effect of diltiazem is mild in the cardiac muscle. The pharmacological action of diltiazem is not on cardiac contractility, but rather on negative chronotropy as an oxygen-consumption decreasing effect, which is due to the inhibition of action potentials in the sino-atrial node. This vascular selectivity of diltiazem is supported by the finding that the IC50 for diltiazem to inhibit papillary muscle is almost twice to that of the tail artery in rats (Sun and Triggle, 1995).
Cardiac muscle and vascular smooth muscle share the same Cav1.2 channels that serve as the major target of diltiazem. However, there exist minor but critical differences between the two types of channels generated by alternative splicing (Soldatov, 1994; Welling et al., 1997; Liao et al., 2005; 2007;). Such differences of Cav1.2 calcium channels do not change the domains at IIIS6 and IVS6 regions that bind to diltiazem (Hering et al., 1996; Hockerman et al., 2000). Here we showed that under closed-channel block (0.03 Hz), cardiac Cav1.2CM channel is less sensitive to diltiazem than the two vascular smooth muscle channels with both Ca2+ and Ba2+. The time interval between two pulses (0.03 Hz) is longer than that previously used (0.05 Hz) by Dilmac et al. (2003) to make sure the channels are not in an inactivated state. The results from both Ca2+ and Ba2+ studies show that alternatively spliced exons determine the lower sensitivity of cardiac Cav1.2CM channel to diltiazem block. However, the IC50 of diltiazem in Ba2+ is smaller than that in Ca2+ in our study, which is inconsistent with a previous report showing that Ca2+ can potentiate diltiazem block (Dilmac et al., 2003). This could be due to the different concentrations of charge carriers that were used. Our experiment was carried out in 1.8 mM Ca2+ or 5 mM Ba2+, whereas 10 mM of both Ca2+ and Ba2+ was used in the previous study. We further carried out experimentations on chimeras. Unexpectedly, all three alternative spliced sites participate in diltiazem block. Exon 1/1a and 8/8a have a larger modulatory effect than exon 9*. The vascular smooth muscle exons 1 and 8 were more sensitive to diltiazem block than exons 1a and 8a.
There are a number of studies using HEK cells to study the effects of diltiazem on cloned channels. The IC50 of diltiazem was around 100 µM (Hering et al., 1996; Kraus et al., 1998; Hockerman et al., 2000; Dilmac et al., 2003). We looked at those clones carefully and found that the exon combination of the Cav1.2 channel variant was neither cardiac nor smooth muscle. For example, the rat Cav1.2 cDNA clone used by Dilmac et al. (2003) and Hockerman et al. (2000) contains exons 1 and 8a and does not contain exon 9* (Snutch et al., 1991). The reported IC50 of diltiazem for this clone is 65 µM (Dilmac et al., 2003). The IC50 of diltiazem in these reports is close to our values for the smooth muscle clones but different from the cardiac clones. This is not surprising as the molecular structures of those rat clones are closer to smooth muscle channels.
Early in 1983, Lee and Tsien reported that blockade of inactivated channels is of particular importance for diltiazem (Lee and Tsien, 1983). Thus, we focused on the gating properties of the cardiac and smooth muscle channels, particularly inactivation properties. SSI fitting showed that cardiac Cav1.2CM has less negative half-inactivation potentials than the two smooth muscle channels (Figure 3). Diltiazem shifts the SSI curves to more negative values in all three isoforms, but this shift was greater in smooth muscle channels. In Cav1.2SM and Cav1.2b, diltiazem shifts V0.5 to negative potentials of 16.2 and 12.7 mV, while in Cav1.2CM channels, this shift is only 7.9 mV. This indicates that under conditions of depolarized membrane potentials, there will be more smooth muscle channels than cardiac channels in the inactivated state, and after binding to diltiazem, more smooth muscle channels are inactivated. Secondly, the current of Cav1.2CM channels decayed more slowly than that of both vascular smooth muscle channels (Figure 4). Therefore, upon sustained depolarization, less Cav1.2CM channels become inactivated than smooth muscle channels. Both lines of evidence support the notion that more smooth muscle calcium channels are inactivated upon membrane depolarization. A well known physiological finding (not shown here) is that the blood vessels exhibit less hyperpolarized membrane potentials than cardiac muscle (Nelson et al., 1988; Hadley and Lederer, 1991). This helps to inactivate the calcium channels in smooth muscle even at resting membrane potential. Considering that diltiazem exerts its blocking effect through binding to inactivated channels, we suggest that the different inactivation properties could also make the smooth muscle Cav1.2 channels more sensitive to diltiazem at arterial resting membrane potentials than the cardiac Cav1.2 channels at cardiac membrane potentials.
We showed here the different inactivation properties between cardiac and smooth muscle channels. One major difference between cardiac and smooth muscle channels is exon 9* located at the I–II loop (Figure 1) And this I–II loop is known to play a critical role in channel inactivation (Zamponi et al., 1996; Herlitze et al., 1997). The well known mutations in patients with Timothy syndrome have also been located at the beginning of the I–II loop (Splawski et al., 2004; 2006;). Channels with Timothy syndrome mutation exhibit loss of voltage-dependent inactivation. It is likely that exon 9* could alter the difference in inactivation properties between the different cardiac and smooth muscle channels.
Dilmac et al. (2003) found that diltiazem could slow the recovery of wild-type Cav1.2 channels from inactivation. We also found similar effects of diltiazem on all three channels tested. Without diltiazem, the three channels showed similar recovery after 2 s inactivation. However, after diltiazem treatment, cardiac Cav1.2CM channels recovered more slowly than the other two channels. Thus, although cardiac channels are less sensitive to diltiazem, binding of the drug will keep the channels inactivated for longer. We carried out studies on use-dependent block to reveal the effects of the different recovery from inactivation properties. A frequency of 5 Hz was used to mimic the fast heartbeat under arrhythmia. Unexpectedly, cardiac Cav1.2CM channels showed lower sensitivity to diltiazem block from the seventh test pulse with Ba2+ as the charge carriers (Figure 5E). Use-dependent block indicates how inactivation of the channels affects diltiazem block. The lower sensitivity observed from use-dependent block in Cav1.2CM channels contrasted with their slower recovery from inactivation upon diltiazem block. A possible explanation is that the current decay in Cav1.2CM channels is also slower upon diltiazem treatment. Furthermore, the hyperpolarized shift of SSI by the binding of diltiazem is less in Cav1.2CM channels than in the two smooth muscle channels. Thus, under the train of test pulses to 10 mV, overall inactivated Cav1.2CM channels are still less than the inactivated smooth muscle channels.
The activation properties of the three channels also showed differences. The two smooth muscle channels were activated at relatively more negative membrane potentials, while the cardiac Cav1.2CM channel was activated at the most positive potential without diltiazem treatment. Unexpectedly, the I–V curves of all three channels were shifted to negative potentials in the presence of diltiazem. This is in contrast to the positive shift triggered by binding of DHPs (Striessnig et al., 1998; Liao et al., 2004). Diltiazem and DHPs share a common binding site located on the IIIS6 and IVS6 segments of Cav1.2 channels (Striessnig, 1999). The opposite activation properties suggest that the mechanism for kinetic changes of Cav1.2 channels is modulated differently by DHPs and diltiazem. Interestingly, the negative shift of the I–V curves in Cav1.2CM channels by diltiazem was more prominent and significant than for the two vascular smooth muscle channels. The molecular difference in IS6 encoded by the mutually exclusive exons 8 or 8a could influence the effect diltiazem has on activation, as IS6 is critical for gating of the Cav1.2 channels and lines the pore (Zhang et al., 1994). However, we could not rule out the role of the cytoplasmic N-terminus encoded by the mutually exclusive exons 1a or 1, which is close to the IS1 segments. These segments affect channel activation but do not form part of the diltiazem-binding site (Ren et al., 1998).
In conclusion, our results provide strong evidence that alternative splicing can act as a molecular switch to modulate diltiazem inhibition of Cav1.2 calcium channels. Binding of diltiazem could alter the properties of cardiac and vascular smooth muscle channel properties, differentially in many aspects. Such differences could play an important role in the pharmacological effects of diltiazem on the heart and blood vessels.
Acknowledgments
HY Zhang is supported by a grant from Sichuan Science and Technology Bureau (2009HH0026). This work is supported by grants from the National Medical Research Council and Biomedical Research Council of Singapore.
Glossary
Abbreviations:
- BTZ
benzothiazepine
- DHP
1,4-dihydropyridine
- PAA
phenylalkylamines
Conflict of interest
The authors state no conflict of interest.
References
- Abernethy DR, Schwartz JB. Calcium-antagonist drugs. N Engl J Med. 1999;341:1447–1457. doi: 10.1056/NEJM199911043411907. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budriesi R, Cosimelli B, Ioan P, Carosati E, Ugenti MP, Spisani R. Diltiazem analogues: the last ten years on structure activity relationships. Curr Med Chem. 2007;14:279–287. doi: 10.2174/092986707779941122. [DOI] [PubMed] [Google Scholar]
- Diebold RJ, Koch WJ, Ellinor PT, Wang JJ, Muthuchamy M, Wieczorek DF, et al. Mutually exclusive exon splicing of the cardiac calcium channel alpha 1 subunit gene generates developmentally regulated isoforms in the rat heart. Proc Natl Acad Sci USA. 1992;89:1497–1501. doi: 10.1073/pnas.89.4.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilmac N, Hilliard N, Hockerman GH. Molecular determinants of Ca2+ potentiation of diltiazem block and Ca2+-dependent inactivation in the pore region of cav1.2. Mol Pharmacol. 2003;64:491–501. doi: 10.1124/mol.64.2.491. [DOI] [PubMed] [Google Scholar]
- Gidh-Jain M, Huang B, Jain P, Battula V, el-Sherif N. Reemergence of the fetal pattern of l-type calcium channel gene expression in noninfarcted myocardium during left ventricular remodeling. Biochem Biophys Res Commun. 1995;216:892–897. doi: 10.1006/bbrc.1995.2705. [DOI] [PubMed] [Google Scholar]
- Hadley RW, Lederer WJ. Properties of l-type calcium channel gating current in isolated guinea pig ventricular myocytes. J Gen Physiol. 1991;98:265–285. doi: 10.1085/jgp.98.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering S, Aczel S, Grabner M, Doring F, Berjukow S, Mitterdorfer J, et al. Transfer of high sensitivity for benzothiazepines from l-type to class A (BI) calcium channels. J Biol Chem. 1996;271:24471–24475. doi: 10.1074/jbc.271.40.24471. [DOI] [PubMed] [Google Scholar]
- Herlitze S, Hockerman GH, Scheuer T, Catterall WA. Molecular determinants of inactivation and G protein modulation in the intracellular loop connecting domains I and II of the calcium channel alpha1A subunit. Proc Natl Acad Sci USA. 1997;94:1512–1516. doi: 10.1073/pnas.94.4.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockerman GH, Dilmac N, Scheuer T, Catterall WA. Molecular determinants of diltiazem block in domains IIIS6 and IVS6 of l-type Ca(2+) channels. Mol Pharmacol. 2000;58:1264–1270. doi: 10.1124/mol.58.6.1264. [DOI] [PubMed] [Google Scholar]
- Kraus RL, Hering S, Grabner M, Ostler D, Striessnig J. Molecular mechanism of diltiazem interaction with l-type Ca2+ channels. J Biol Chem. 1998;273:27205–27212. doi: 10.1074/jbc.273.42.27205. [DOI] [PubMed] [Google Scholar]
- Lee KS, Tsien RW. Mechanism of calcium channel blockade by verapamil, D600, diltiazem and nitrendipine in single dialysed heart cells. Nature. 1983;302:790–794. doi: 10.1038/302790a0. [DOI] [PubMed] [Google Scholar]
- Liao P, Yu D, Lu S, Tang Z, Liang MC, Zeng S, et al. Smooth muscle-selective alternatively spliced exon generates functional variation in Cav1.2 calcium channels. J Biol Chem. 2004;279:50329–50335. doi: 10.1074/jbc.M409436200. [DOI] [PubMed] [Google Scholar]
- Liao P, Yong TF, Liang MC, Yue DT, Soong TW. Splicing for alternative structures of Cav1.2 Ca2+ channels in cardiac and smooth muscles. Cardiovasc Res. 2005;68:197–203. doi: 10.1016/j.cardiores.2005.06.024. [DOI] [PubMed] [Google Scholar]
- Liao P, Yu D, Li G, Yong TF, Soon JL, Chua YL, et al. A smooth muscle Cav1.2 calcium channel splice variant underlies hyperpolarized window current and enhanced state-dependent inhibition by nifedipine. J Biol Chem. 2007;282:35133–35142. doi: 10.1074/jbc.M705478200. [DOI] [PubMed] [Google Scholar]
- Liao P, Li G, Yu DJ, Yong TF, Wang JJ, Wang J, et al. Molecular alteration of Ca(v)1.2 calcium channel in chronic myocardial infarction. Pflugers Arch. 2009a;458:701–711. doi: 10.1007/s00424-009-0652-4. [DOI] [PubMed] [Google Scholar]
- Liao P, Zhang HY, Soong TW. Alternative splicing of voltage-gated calcium channels: from molecular biology to disease. Pflugers Arch. 2009b;458:481–487. doi: 10.1007/s00424-009-0635-5. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Standen NB, Brayden JE, Worley JF., 3rd Noradrenaline contracts arteries by activating voltage-dependent calcium channels. Nature. 1988;336:382–385. doi: 10.1038/336382a0. [DOI] [PubMed] [Google Scholar]
- Ren D, Xu H, Eberl DF, Chopra M, Hall LM. A mutation affecting dihydropyridine-sensitive current levels and activation kinetics in Drosophila muscle and mammalian heart calcium channels. J Neurosci. 1998;18:2335–2341. doi: 10.1523/JNEUROSCI.18-07-02335.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yu D, Hiel H, Liao P, Yue DT, Fuchs PA, et al. Alternative splicing of the Ca(v)1.3 channel IQ domain, a molecular switch for Ca2+-dependent inactivation within auditory hair cells. J Neurosci. 2006;26:10690–10699. doi: 10.1523/JNEUROSCI.2093-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snutch TP, Tomlinson WJ, Leonard JP, Gilbert MM. Distinct calcium channels are generated by alternative splicing and are differentially expressed in the mammalian CNS. Neuron. 1991;7:45–57. doi: 10.1016/0896-6273(91)90073-9. [DOI] [PubMed] [Google Scholar]
- Soldatov NM. Genomic structure of human l-type Ca2+ channel. Genomics. 1994;22:77–87. doi: 10.1006/geno.1994.1347. [DOI] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Splawski I, Yoo DS, Stotz SC, Cherry A, Clapham DE, Keating MT. CACNA1H mutations in autism spectrum disorders. J Biol Chem. 2006;281:22085–22091. doi: 10.1074/jbc.M603316200. [DOI] [PubMed] [Google Scholar]
- Striessnig J. Pharmacology, structure and function of cardiac l-type Ca(2+) channels. Cell Physiol Biochem. 1999;9:242–269. doi: 10.1159/000016320. [DOI] [PubMed] [Google Scholar]
- Striessnig J, Grabner M, Mitterdorfer J, Hering S, Sinnegger MJ, Glossmann H. Structural basis of drug binding to L Ca2+ channels. Trends Pharmacol Sci. 1998;19:108–115. doi: 10.1016/s0165-6147(98)01171-7. [DOI] [PubMed] [Google Scholar]
- Sun J, Triggle DJ. Calcium channel antagonists: cardiovascular selectivity of action. J Pharmacol Exp Ther. 1995;274:419–426. [PubMed] [Google Scholar]
- Triggle DJ. 1,4-Dihydropyridines as calcium channel ligands and privileged structures. Cell Mol Neurobiol. 2003;23:293–303. doi: 10.1023/A:1023632419813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welling A, Ludwig A, Zimmer S, Klugbauer N, Flockerzi V, Hofmann F. Alternatively spliced IS6 segments of the alpha 1C gene determine the tissue-specific dihydropyridine sensitivity of cardiac and vascular smooth muscle l-type Ca2+ channels. Circ Res. 1997;81:526–532. doi: 10.1161/01.res.81.4.526. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Soong TW, Bourinet E, Snutch TP. Beta subunit coexpression and the alpha1 subunit domain I-II linker affect piperidine block of neuronal calcium channels. J Neurosci. 1996;16:2430–2443. doi: 10.1523/JNEUROSCI.16-08-02430.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JF, Ellinor PT, Aldrich RW, Tsien RW. Molecular determinants of voltage-dependent inactivation in calcium channels. Nature. 1994;372:97–100. doi: 10.1038/372097a0. [DOI] [PubMed] [Google Scholar]