

Abstract
Background and purpose:
Sphingosine kinases (SKs) convert sphingosine to sphingosine 1-phosphate (S1P), which is a bioactive lipid that regulates a variety of cellular processes including proliferation, differentiation and migration.
Experimental approach:
We used the human endothelial cell line EA.hy926 to investigate the effect of nitric oxide (NO) donors on SK-1 expression, and on cell migration and tube formation.
Key results:
We showed that exposure of EA.hy926 cells to Deta-NO (125–1000 µM) resulted in a time- and concentration-dependent up-regulation of SK-1 mRNA and protein expression, and activity with a first significant effect at 250 µM of Deta-NO. The increased SK-1 mRNA expression resulted from an enhanced SK-1 promoter activity. A similar effect was also seen with various other NO donors. In mechanistic terms, the NO-triggered effect occurred independently of cGMP, but involved the classical mitogen-activated protein kinase cascade because the MEK inhibitor U0126 abolished the NO-induced SK-1 expression. The effect of NO was also markedly reduced by the thiol-reducing agent N-acetylcysteine, suggesting a redox-dependent mechanism. Functionally, Deta-NO triggered an increase in the migration of endothelial cells in an adapted Boyden chamber assay, and also increased endothelial tube formation in a Matrigel assay. These responses were both abolished in cells depleted of SK-1.
Conclusions and implications:
These data show that NO donors up-regulate specifically SK-1 expression and activity in human endothelial cells, and SK-1 in turn critically contributes to the migratory capability and tube formation of endothelial cells. Thus, SK-1 may be considered an attractive novel target to interfere with pathological processes involving angiogenesis.
Keywords: nitric oxide, sphingosine 1-phosphate, sphingosine kinase 1, endothelial cells, angiogenesis
Introduction
Nitric oxide (NO) is a gaseous radical with multiple physiological, but also pathophysiological, functions. The ability of NO to diffuse freely across biological membranes enables it to exert cellular actions in an autocrine and paracrine manner. NO is synthesized from the amino acid l-arginine by specific enzymes, the NO synthases (NOS). When produced in physiological concentrations, mainly by the constitutively expressed neuronal (nNOS) and endothelial (eNOS) enzymes, it is involved in blood pressure regulation, nerve cell transmission and protection against injuries. On the other hand, excessive NO production derived mainly from the inducible NOS (iNOS) is linked to a variety of pathophysiological conditions, such as septic shock, neurodegeneration, arthritis and stroke and killing of pathogens (Moncada and Higgs, 1993; Kröncke et al., 1998; Beck et al., 1999). In addition, NO has been shown to be a potent angiogenic factor (Chin et al., 1997). Enhanced angiogenesis can lead to accelerated tumour growth and promote the process of metastasis (Weidner et al., 1991; Folkman, 1997). On the other side, the pro-angiogenic potential of NO may also have beneficial effects such as in ischaemia-induced angiogenesis (Amano et al., 2003) and in wound-healing processes (Luo and Chen, 2005).
During the last decade, it has become apparent that sphingolipids are not only essential components of cell membranes, but also serve as important signalling molecules involved in the regulation of cellular functions. The most important bioactive sphingolipid metabolites, sphingosine 1-phosphate (S1P), sphingosine and ceramide are forming a so-called sphingolipid rheostat (Spiegel and Milstien, 2002). It is proposed that the dynamic balance between cellular S1P, sphingosine and ceramide determines a cell's fate by triggering opposing signalling pathways. Whereas ceramide accumulates under stress conditions and leads to cell death, S1P promotes cell survival and proliferation. Sphingosine kinases (SKs) catalyse the conversion of sphingosine to S1P and therefore play a central role in balancing the sphingolipid rheostat. So far, two different SKs, denoted as SK-1 and SK-2, have been cloned and characterized. Although both enzymes generate the same product, that is, S1P, different biological functions have been appointed to the different SKs (Alemany et al., 2007; Hofmann et al., 2008). SK-1 expression is stimulated by various growth factors and cytokines (Alemany et al., 2007), whereas not much is known about the activation mechanism of SK-2, although EGF has been reported to enhance SK-2 activity in the breast cancer cell line MCF-7 (Hait et al., 2005). The common product of the activation of SK, S1P, is known to act extracellularly via binding to five G protein-coupled receptors of the lysophospholipid receptor family (S1P1–5) (Alexander et al., 2008), but it has also been suggested that it has intracellular effects and acts as a ‘second messenger’. However, the intracellular targets of S1P have not been identified.
SK-1 and S1P have both been appointed a critical role in endothelial cell migration and the process of angiogenesis (Panetti, 2002). In a previous study, we showed that reduced oxygen levels (hypoxia) can induce endothelial cell migration, which strictly depended on SK-1 expression and activity (Schwalm et al., 2008). Furthermore, S1P was shown to activate vascular cells to produce vasoactive molecules including NO and prostacyclin (Nofer et al., 2004; Rodríguez et al., 2009).
In this study, we show for the first time that exposure of human endothelial cells to NO-releasing compounds results in an up-regulation of SK-1 mRNA, and protein expression and sustained activity. This up-regulation involves redox regulation and the classical MAPK/ERK pathway, but is independent of cGMP formation. Most importantly, we present evidence that the process of NO-triggered migration and tube formation of endothelial cells also critically involves functional SK-1.
Methods
Cell culture
The human endothelial cell line EA.hy 926 was provided by Dr Edgell (University of North Carolina, Chapel Hill, NC, USA) (Edgell et al., 1983) and cultured exactly as previously described (Huwiler et al., 2006). Primary cultures of human umbilical vein endothelial cells (HUVECs) were obtained from PromoCell (Heidelberg, Germany). Prior to stimulation, cells were rendered quiescent for 24–48 h in Dulbecco's modified Eagle's medium (DMEM) that included 0.1 mg·mL−1 of fatty acid-free BSA. HUVECs were deprived of nutrients for 24 h in 0.5 % fetal bovine serum prior to stimulation.
Western blot analysis
Stimulated cells were homogenized in lysis buffer (Huwiler et al., 2006) and centrifuged for 10 min at 14 000× g. The supernatant was taken for protein determination, and 30 µg of protein was separated by SDS–PAGE, transferred to nitrocellulose membrane and subjected to Western blot analysis, as previously described (Huwiler et al., 2006), using the antibodies indicated in the figure legends.
SK activity assay
In vitro SK-1 activity assays were performed exactly as previously described (Huwiler et al., 2006; Schwalm et al., 2008).
Quantitative Real-time PCR (TAQMAN)
One microgram of total RNA isolated with TRIZOL reagent was used for reverse transcriptase–PCR (First Strand Synthesis Kit, MBI Fermentas, St-Leon-Roth, Germany); a random hexamer primer was utilized for amplification. The real-time PCR reaction was carried out exactly as described previously (Huwiler et al., 2006; Schwalm et al., 2008). All primers were from Applied Biosystems (Darmstadt, Germany). The fold induction values were obtained according to the ΔΔCT method, after normalization to the housekeeping gene 18S RNA.
Promoter studies
A 2217 bp fragment of the human SK-1 promoter was cloned by PCR as previously described (Schwalm et al., 2008). For promoter analyses, EA.hy 926 cells were seeded into 12-well plates (1.5 × 105 cells per well) 1 day before transfection. Cells were transfected with 400 ng of plasmid DNA plus 100 ng Renilla luciferase DNA per well by use of Effectene transfection reagent, according to the manufacturer's recommendations. Twenty-four hours after transfection, cells were rendered serum free for an additional 24 h and then stimulated for 16 h. Promoter reporter assays were performed using the Dual Luciferase assay kit (Promega Gmbh, Mannheim, Germany). Luciferase activities were measured with a Lumat LB9507 luminometer (Berthold Detection Systems, Pforzheim, Germany), and values for the relative SK-1 promoter activities were calculated from the ratio of firefly/Renilla luciferase activities.
siRNA transfections
For gene silencing, specific siRNA sequences of human SK-1 and a scrambled sequence were used as previously described (Huwiler et al., 2006). The cells, 5200 per cm2, were seeded 24 h before transfection with 200 nM of the 21-nucleotide siRNAs using oligofectamine as recommended by the manufacturer. The silencing effect was verified by Western blot analysis as previously described (Huwiler et al., 2006).
Adapted Boyden chamber assay
To measure undirected endothelial cell migration, an adapted Boyden chamber assay was performed exactly as previously described (Schwalm et al., 2008).
Tube formation assay
Cells were cultured for 24 h under serum-free conditions. Then, 10 µL of growth factor-reduced Matrigel (BD Discovery Labware, Bedford, MA, USA) was placed into the lower chambers of µ-slide angiogenesis wells (ibidi GmbH, Munich, Germany) and allowed to polymerize for 30 min at 37°C. Next, 104 cells per well were seeded on the Matrigel and stimulated for 20 h in the absence or presence of Deta-NO. Images were taken with an LSM 510 confocal microscope (Carl Zeiss AG, Göttingen, Germany) using the appended software (ZEN). The images were analysed by a Web-based image analysis system with the tube formation module of S.CORE (S.CO LifeScience GmbH, Garching, Germany; http://www.sco-lifescience.eu/technology.php5).
Statistical analysis
Statistical analysis was performed by one-way anova. For multiple comparisons with the same control group, the limit of significance was divided by the number of comparisons according to Bonferroni.
Chemicals and materials
[32P-γ]-ATP (specific activity, >5000 Ci mmol−1), secondary horseradish peroxidase-coupled IgGs, Hyperfilm MP and enhanced chemiluminescence reagents were from GE Healthcare Systems GmbH (Freiburg, Germany); U0126 and histamine were from Calbiochem (Schwalbach, Germany); (Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl) amino] diazen-1-ium-1,2-diolate (Deta-NO) (Z)-1-{N-[6-(N-methylammoniohexyl)amino)} diazen-1-ium 1,2-diolate (MAHMA-NO), 3-morpholino-sydnonimine (SIN-1) (Z)-1-{N-[3-aminopropyl]-N-[4-(3-aminopropylammonio)butyl]amino} diazen-1-ium 1,2-diolate (spermine-NO), sodium nitroprusside (SNP), and S-nitroso-N-acetyl-penicillamine (SNAP), 1H-[1,2,4]oxadiazolo[4,3,a]quinoxaline-1-one (ODQ), 4H-8-bromo-1,2,4-oxadiazolo(3,4-d)benz(b)(1,4)oxazin-1-one (NS2028), 8-bromo-cGMP and 3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole (YC-1) were all purchased from Alexis Biochemicals (Läufelfingen, Switzerland); N-acetylcysteine (NAC) was from Sigma Aldrich Fine Chemicals (Taufkirchen, Germany); Effectene was from Qiagen AG (Hilden, Germany); oligofectamine and all cell culture nutrients were from Life Technologies (Karlsruhe, Germany). An affinity-purified antibody against human SK-1 was generated and characterized as previously described (Döll et al., 2005; Huwiler et al., 2006).
Results
Treatment of the human endothelial cell line EA.hy 926 for 24 h with Deta-NO, a compound that induces the slow release of NO, resulted in a concentration-dependent increase of SK-1 activity with a maximal, 3.5-fold, increase at 500 µM Deta-NO, the highest concentration tested (Figure 1A). The up-regulation of SK-1 activity by Deta-NO also occurred in a time-dependent manner, and, with 500 µM Deta-NO, after an initial delay of approximately 8 h, the response peaked at 24 h (Figure 1B). To see whether this delayed increase of SK-1 activity is due to increased protein expression, Western blot analyses were performed. Deta-NO up-regulated the expression of SK-1 protein in a dose- and time-dependent manner (Figure 2A,B respectively). According to our previous studies (Huwiler et al., 2006; Schwalm et al., 2008), EA.hy 926 cells express two variants of SK-1, that is, SK-1a and SK-1b, with sizes of 42 and 51 kDa respectively. Both of these variants were up-regulated by Deta-NO (Figure 2A,B).
Figure 1.
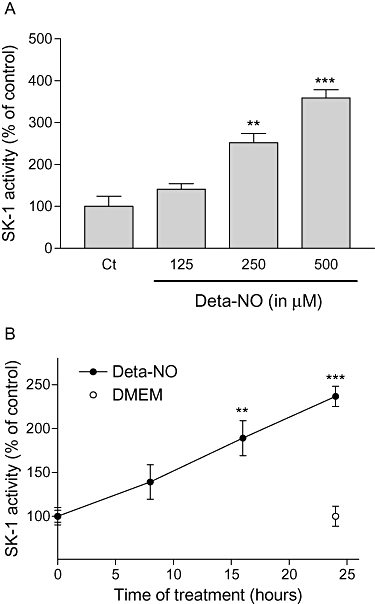
Concentration- and time-dependent effects of Deta-NO on SK-1 activity in EA.hy 926 cells. (A) EA.hy 926 cells were stimulated for 24 h with either vehicle (Ct) or the indicated concentrations of Deta-NO. (B) Cells were stimulated for the indicated time periods with either vehicle (DMEM) or Deta-NO (500 µM). Thereafter, cell lysates containing 30 µg of protein were taken for an in vitro SK-1 activity assay as described in the Methods section. The [32P]-S1P generated was analysed on an Imaging System. Data are expressed as % of control values and are means ± SD (n = 3–4); **P < 0.01, ***P < 0.001, significantly different when compared to the respective control values.
Figure 2.
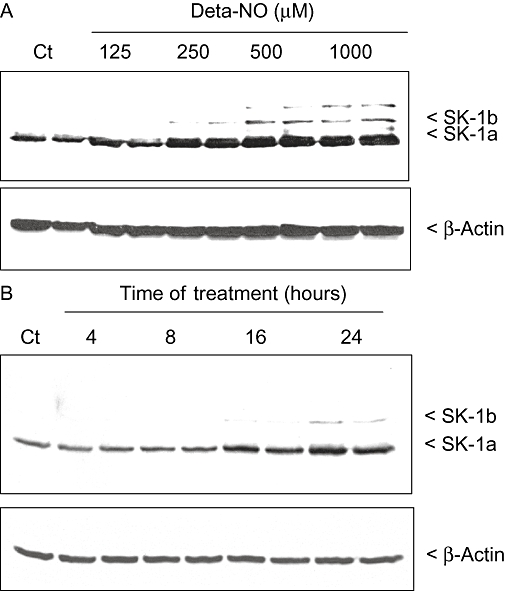
Concentration- and time-dependent effects of Deta-NO on SK-1 protein expression in EA.hy 926 cells. (A) EA.hy 926 cells were stimulated for 24 h with either vehicle (Ct) or the indicated concentrations of Deta-NO. (B) Cells were stimulated for the indicated time periods with either vehicle (Ct) or Deta-NO (500 µM). Thereafter, cell lysates containing 30 µg of protein were separated by SDS–PAGE, transferred to nitrocellulose and subjected to Western blot analysis using antibodies against SK-1 (dilution of 1:3000, upper panels) and β-actin (1:2000, lower panels). Data are representative of three independent experiments giving similar results.
In order to evaluate whether the NO-mediated up-regulation of SK-1 protein expression is preceded by enhanced synthesis of mRNA, we performed quantitative PCR analysis. Primers against SK-1 that recognized both splice variants were generated. Exposure of EA.hy 926 cells for 24 h to increasing concentrations of Deta-NO led to a dose-dependent enhancement of SK-1 mRNA expression (Figure 3A), whereas SK-2 mRNA expression was not affected by Deta-NO (Figure 3A). The effect on SK-1 mRNA also occurred in a time-dependent manner; after 4 h of exposure to Deta-NO, SK-1 mRNA steady-state levels were already significantly enhanced, and maximal values were reached after 16–24 h (Figure 3B). Again, SK-2 mRNA expression was not affected over a time period of 24 h (Figure 3B). Importantly, the up-regulating effect of NO on the expression of SK-1 mRNA seen in EA.hy 926 cells was also found in primary cultures of HUVECs (Figure 3C).
Figure 3.
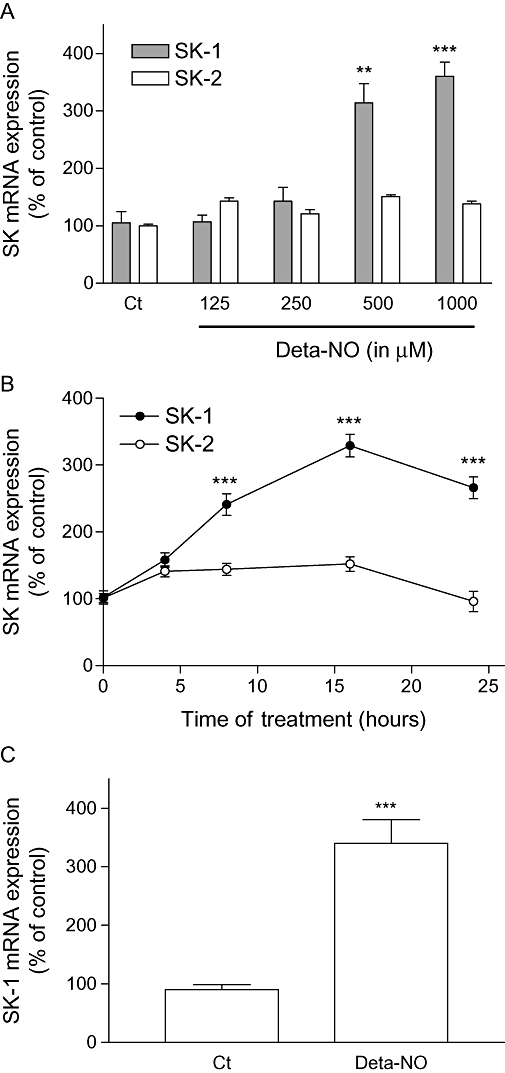
Effect of Deta-NO on SK-1 and SK-2 mRNA expression in EA.hy 926 cells and HUVECs. (A) EA.hy 926 cells were stimulated for 24 h with either vehicle (Ct) or the indicated concentrations of Deta-NO. (B) EA.hy 926 cells were stimulated for 24 h with vehicle (0) or for the indicated time periods with Deta-NO (500 µM). (C) HUVEC cells were stimulated for 24 h with either vehicle (Ct) or Deta-NO (500 µM). Thereafter, RNA was extracted and subjected to quantitative PCR analysis using primers of human SK-1, SK-2 and 18S RNA. ΔΔCt values were calculated as described in the Methods section, and results are expressed as % of control values and are means ± SD (n = 3). **P < 0.01, ***P < 0.001, significantly different when compared to the control values.
To see whether the enhanced expression of SK-1 mRNA is due to stimulation of SK-1 promoter activity, luciferase reporter gene assays were performed using a 2217 bp fragment of the human SK-1 promoter which was cloned as previously described (Schwalm et al., 2008). As shown in Figure 4, Deta-NO dose-dependently enhanced SK-1 promoter activity in EA.hy 926 cells transfected with the promoter construct.
Figure 4.
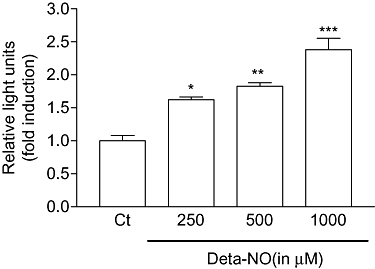
Effect of Deta-NO on human SK-1 promoter activity in EA.hy 926 cells. Subconfluent EA.hy 926 cells were co-transfected with a 2217 bp SK-1 promoter DNA plus the plasmid coding for Renilla luciferase; 24 h after transfection, cells were exposed for an additional 16 h to either vehicle (Ct) or the indicated concentrations of Deta-NO. The ratio between beetle luciferase activity and Renilla luciferase activity was calculated and depicted as relative luciferase activity. Data show the fold induction and are means ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 significantly different when compared to the control values.
All these data strongly suggest that cellular S1P levels should increase upon NO treatment. However, mass spectrometric quantification of S1P in cellular lipid extracts did not reveal a significant change upon Deta-NO treatment (data not shown). Possible explanations could be that either S1P is rapidly interconverted to other sphingolipid species or that S1P is only generated at very specific subcellular sites, which do not alter the total cellular levels of S1P.
We further tested whether the effect of Deta-NO on SK-1 expression is also seen with other NO donors, which have different kinetics of NO release. To this end, the very rapidly NO-releasing compounds MAHMA-NO (half-life 1.3 min) and spermine-NO (half-life 73 min), and the more slowly releasing compounds SNAP (half-life 3 h) and SNP (half-life 12 h) were compared with Deta-NO (half-life 20 h) (Mooradian et al., 1995). After 24 h of stimulation using the same concentration of 500 µM for all NO donors, all the compounds triggered a comparable up-regulation of SK-1 protein expression (Figure 5A). Additionally, the NO donor 3-morpholino-sydnonimine (SIN-1), which simultaneously releases NO and superoxide radicals leading to the generation of peroxynitrite, was tested. However, this compound, at concentrations up to 3 mM, had no significant effect on SK-1 expression (Figure 5B).
Figure 5.
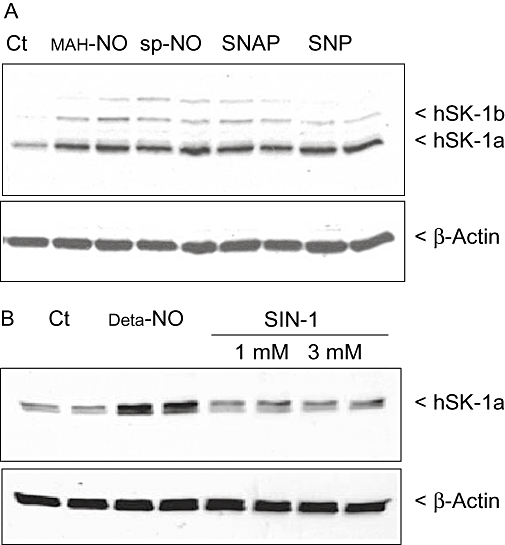
Effect of different NO donors on SK-1 protein expression in EA.hy 926 cells. Cells were incubated for 24 h with either vehicle (Ct) or 500 µM of MAHMA-NO (MAH-NO, A), spermine-NO (sp-NO, A), SNAP (A), SNP (A), Deta-NO (B) and 1 and 3 mM of 3-morpholino-sydnonimine (SIN-1, B) as indicated. Thereafter, cell lysates containing 30 µg of protein were separated by SDS–PAGE, transferred to nitrocellulose and subjected to Western blot analysis using antibodies against SK-1 (dilution 1:3000, upper panels) and β-actin (dilution 1:2000; lower panels). Bands were stained with the ECL methods as recommended by the manufacturer. Data are representative of two independent experiments giving similar results.
Furthermore, we investigated whether the NO-triggered up-regulation of SK-1 expression is mechanistically mediated by activation of soluble guanylyl cyclase (sGC) and subsequent cGMP production. To this end, the cell-permeable cGMP analog 8-Br-cGMP and the sGC activator YC-1 (Ko et al., 1994) were tested. However, we found that these two agents did not stimulate the expression of SK-1 (Figure 6). Rather, 8-Br-cGMP reduced the basal expression of SK-1 protein. Additionally, the two sGC inhibitors ODQ (Schrammel et al., 1996) and NS2028 (Olesen et al., 1998) were unable to reduce the increased expression of SK-1 triggered by Deta-NO (data not shown). Collectively, these data indicate that NO acts by a cGMP-independent mechanism probably involving redox regulation (Pfeilschifter et al., 2001) to up-regulate SK-1 expression. Indeed, we found that the thiol-reducing agent and antioxidant NAC reduced the effect of Deta-NO (Figure 7). In addition, the MEK inhibitor U0126 (Favata et al., 1998) completely blocked the effect of Deta-NO, suggesting the critical involvement of the classical MAPK/ERK cascade (Figure 7). We further tested whether the SK-1 up-regulating effect seen with exogenously applied NO donors also occurs through endogenously generated NO. To this end, histamine was used, as we have previously identified it as a potent inducer of SK-1 in EA.hy 926 cells (Huwiler et al., 2006). Histamine is also known to be an activator of eNOS in endothelial cells (Venema et al., 1997), and it is very likely that histamine-triggered NO formation participates in the up-regulation of SK-1. As shown in Figure 8, the up-regulating effect of histamine on SK-1 protein expression was not reduced in the presence of the NOS inhibitor NG-monomethyl l-arginine (l-NMMA); this indicates either that NO is not involved in this effect of histamine or that the concentration of eNOS-derived endogenous NO was not sufficient to exert an effect.
Figure 6.
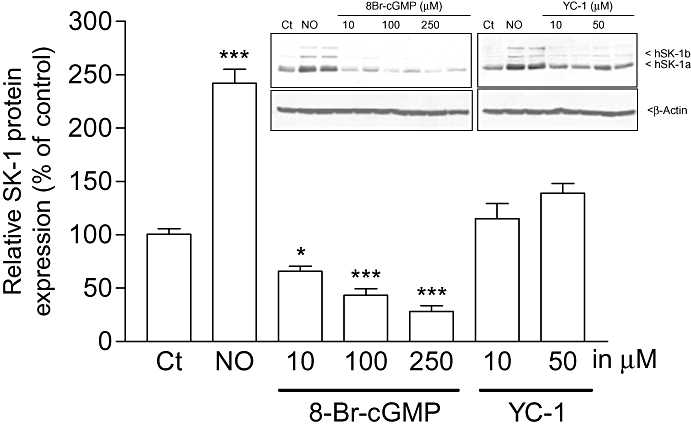
Effects of a cGMP mimetic and an sGC activator on SK-1 expression in EA.hy 926 cells. Cells were treated for 24 h with either vehicle (Ct), Deta-NO (NO, 500 µM) or the indicated concentrations of 8-Br-cGMP or the sGC activator YC-1. Thereafter, cell lysates containing 30 µg of protein were separated by SDS–PAGE, transferred to nitrocellulose and subjected to Western blot analysis using antibodies against SK-1. Bands corresponding to SK-1a were densitometrically evaluated. Data are expressed as % of control and are means ± SD (n = 2–4). *P < 0.05, ***P < 0.001, significantly different when compared to the control values. The inset shows one representative Western blot of SK-1 (upper panel) and β-actin as a loading control (lower panel).
Figure 7.
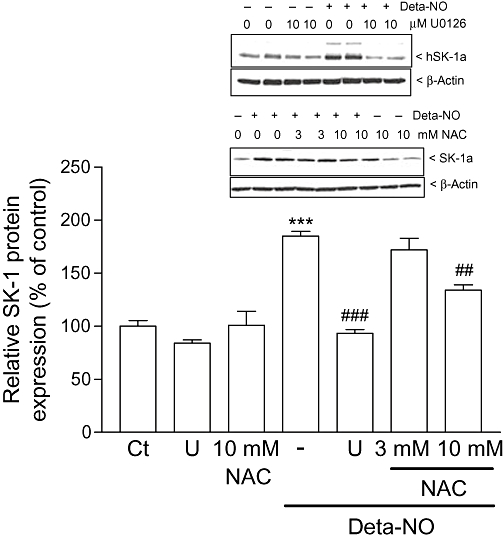
Effect of the antioxidant NAC and the MEK inhibitor U0126 on Deta-NO-induced SK-1 expression in EA.hy 926 cells. Cells were pretreated for 1 h with either U0126 (10 µM), or NAC (3 and 10 mM) prior to stimulation for 24 h with either DMEM (Ct) or Deta-NO (500 µM). Thereafter, cell lysates containing 30 µg of protein were separated by SDS–PAGE, transferred to nitrocellulose and subjected to Western blot analysis using antibodies against SK-1. Bands corresponding to SK-1a were densitometrically evaluated. Data are expressed as % of control and are means ± SD (n = 3–4). ***P < 0.001, significantly different when compared to the control values. ##P < 0.01, ###P < 0.001, significantly different when compared to the Deta-NO-stimulated values. The inset shows one representative Western blot of SK-1 (upper panels) and β-actin (lower panels).
Figure 8.
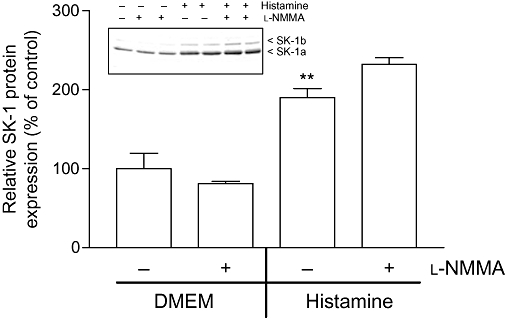
Effect of endogenous NO production on SK-1 protein expression in EA.hy 926 cells. Cells were incubated for 24 h with either vehicle (DMEM, −) or histamine (1 µM) in the absence (−) or presence of l-NMMA (1 mM, +). Thereafter, cell lysates containing 30 µg of protein was separated by SDS-PAGE, transferred to nitrocellulose and subjected to Western blot analysis using antibodies against SK-1 (dilution 1:3000). Bands corresponding to SK-1a were densitometrically evaluated. Results are expressed as % of control and are means ± SD (n = 3). The inset shows one representative Western blot.
As a next step, the functional consequence of SK-1 induction by NO was addressed. NO is known to act as an angiogenic factor on endothelial cells (Ziche et al., 1994; 1997; Murohara et al., 1999). Therefore, we tested the effect of Deta-NO on EA.hy 926 cell migration by using an adapted Boyden chamber assay. Consistent with previous reports (Ziche et al., 1997; Murohara et al., 1999), Deta-NO enhanced the migration of the endothelial cells (Figure 9). This effect was completely abolished after SK-1 had been down-regulated by siRNA transfection. The silencing efficiency of the siRNA transfection was verified by Western blot analysis of SK-1 (Figure 9, inset).
Figure 9.
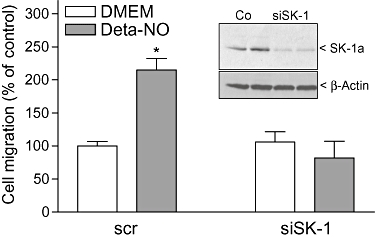
Effect of SK-1 depletion on NO-induced migration of EA.hy 926 cells. EA.hy 926 cells were transfected with either a scrambled siRNA (scr) or a siRNA specific for SK-1 (siSK-1) as described in the Methods section. (A) Cells were then seeded onto 8 µm polycarbonate insets and treated for 20 h with either DMEM (Ct) or Deta-NO (500 µM). Migrated cells were analysed as described in the Methods section. Data are expressed as % of control values and are means ± SD (n = 4). *P < 0.05 considered statistically significant when compared to the control values. Inset: scrambled transfected cells (Ct) and siSK-1-transfected cells (siSK-1) were taken for determination of SK-1 protein expression levels by Western blot analysis.
Finally, endothelial tube formation was measured in a Matrigel assay. To this end, EA.hy 926 cells were stably depleted of SK-1 by transducing the cells with a lentiviral shRNA construct of SK-1 (Figure 10B, inset). Again, this procedure significantly reduced Deta-NO-stimulated tube formation (Figure 10A,B).
Figure 10.
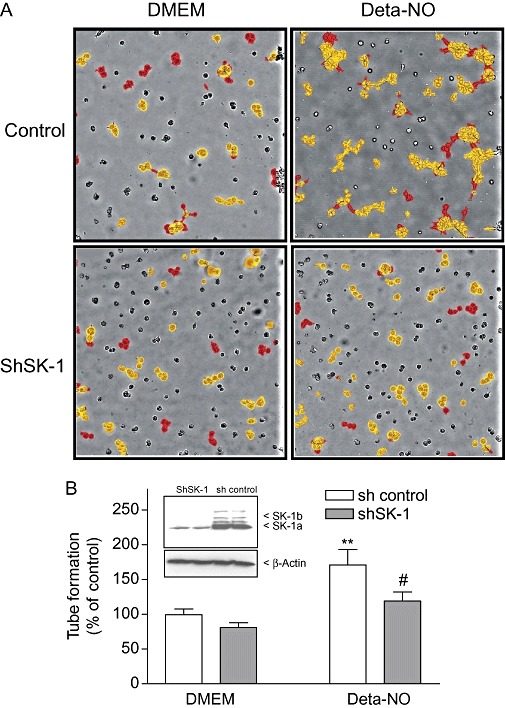
Effect of SK-1 depletion on NO-induced tube formation. EA.hy 926 cells, which were either transduced with a control shRNA construct (sh control) or transduced with a lentiviral shRNA construct to stably deplete SK-1 (shSK-1), were plated onto Matrigel-coated dishes and treated for 20 h with either DMEM or Deta-NO (500 µM). Thereafter, cells were photographed and tube formation was quantified as described in the Methods section. Red coloured structures represent well-developed tubes, whereas yellow structures represent poorly developed tubes. Data in (A) show one representative sample of each group. Results in (B) are expressed as % of control and are means ± SD (n = 8–10). **P < 0.01, significantly different when compared to the DMEM sh-control values. #P < 0.05 when compared to the Deta-NO-stimulated sh-control values. The inset shows the expression of SK-1 in control EA.hy 926 cells (sh control) and in stably SK-1 depleted cells (shSK-1).
Finally, we investigated whether Deta-NO had an impact on the expression of S1P receptors. The expression of the mRNA of S1P receptor subtypes was quantified by real-time PCR analyses. Interestingly, Deta-NO triggered a dose-dependent fivefold increase of S1P3 receptor mRNA (Figure 11). In contrast, S1P1 receptor mRNA was reduced, whereas S1P2 receptor expression was unaffected by Deta-NO (Figure 11).
Figure 11.
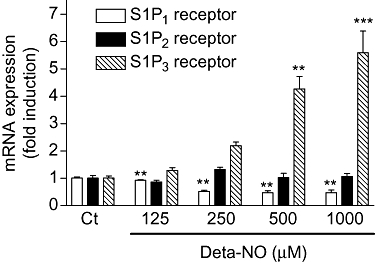
Effect of NO on S1P receptor subtype mRNA expression in EA.hy 926 cells. EA.hy 926 cells were stimulated for 24 h with either vehicle (Ct) or the indicated concentrations of Deta-NO. Thereafter, RNA was extracted and subjected to quantitative PCR analysis using primers of human S1P1, S1P2, and S1P3 receptors. ΔΔCt values were calculated as described in the Methods section. Results are expressed as fold induction and are means ± SD (n = 3). **P < 0.01, ***P < 0.001, significantly different when compared to the control values.
Discussion
In this study, we showed, for the first time, that NO donors can trigger a sustained activation of SK-1 in human endothelial cells. This is mechanistically due to an activation of the SK-1 promoter, resulting in increased gene transcription and de novo protein synthesis. The effect is specifically exerted by the NO radical, because various NO donors, which only differ in their kinetics to release NO, showed a similar effect on SK-1 expression. However, SIN-1, which simultaneously releases NO and superoxide leading to the scavenging of both radicals and supposedly to peroxynitrite formation, as well as superoxide-generating agents, had no significant effect on SK-1 expression (Figure 5B). Hence, the mechanism by which NO activates the SK-1 promoter and especially which transcription factors contribute to this effect is still unclear. However, the classical MAPK/ERK cascade seems to be a key enzyme in the transcriptional regulation of SK-1 (Döll et al., 2005; 2007; Huwiler et al., 2006; Schwalm et al., 2008; Ren et al., 2009). The human SK-1 promoter was first cloned and characterized in the MEG-O1 leukaemia cell line by Nakade et al. (2003). These authors identified the phorbol ester as a potent inducer of the SK-1 promoter. More detailed promoter sequence analyses revealed potential transcription factor-binding sites for SP-1, AP-2 and AP-4 (Nakade et al., 2003). However, whether these sites are functional and involved in the phorbol ester-induced gene transcription were not addressed. Our own previous studies in EA.hy 926 endothelial cells, the breast cancer cell line MCF-7 and glomerular podocytes revealed additional important and functional binding sites such as a hypoxia responsive element (HRE), which mediates the effect of hypoxia on SK-1 transcription in EA.hy 926 cells (Schwalm et al., 2008), a STAT-5 binding element, which mediates the effect of prolactin on SK-1 in MCF-7 cells (Döll et al., 2007), and two Smad-binding elements, which mediate the actions of TGF-β on SK-1 in glomerular podocytes (Ren et al., 2009).
In the past, extensive work has been performed to characterize NO-triggered signalling events. The physiologically most relevant action of NO is the activation of sGC, which occurs through direct nitrosylation of its haem moiety and results in an increased generation of cGMP (Ignarro, 1990). However, in our study, the possibility that NO induces SK-1 expression through a sGC/cGMP-dependent mechanism can be excluded. This is based on the following findings: (i) the cell-permeable cGMP analog 8-Br-cGMP and (ii) the direct sGC activator YC-1 did not increase the expression of SK-1 (Figure 6); and (iii) the two sGC inhibitors ODQ and NS2028 failed to reduce the NO-triggered increase in SK-1 expression (data not shown). Strikingly, in our study, 8-Br-cGMP even significantly suppressed the expression of SK-1. However, because the sGC activator YC-1 showed no such suppression, the possibility that this effect of 8-Br-cGMP was non-specific cannot be excluded.
Alternatively, NO may act as a redox mediator and affect gene expression in a wide variety of cells and influencing many target genes (Pfeilschifter et al., 2001; 2002;). In fact, this mechanism of action is supported by our finding that the thiol-reducing agent and antioxidant NAC down-regulated the effect of NO on SK-1 expression (Figure 7). In this context, NO was shown to affect a large panel of protein kinase cascades which ultimately leads to altered gene transcription. It has been reported that NO can activate all three MAPK cascades, that is the classical MAPK/ERK, the stress-activated protein kinase SAPK/JNK and the p38-MAPK in various cell types including T-cells (Lander et al., 1996), glomerular endothelial and mesangial cells (Pfeilschifter and Huwiler, 1996; Callsen et al., 1998). Indeed, in our study, we found that the classical MAPK/ERK cascade had a key role in the effect of NO, because the MEK inhibitor U0126 completely blocked the up-regulation of SK-1 mediated by NO (Figure 7). Mechanistically, the activation of the MAPK cascade by NO may occur by direct activation of the small GTP-binding protein p21Ras located upstream of MAPK. The activation of p21Ras by NO has been reported to be due to the direct S-nitrosation of a critical cysteine residue of p21Ras (Lander et al., 1995; Yun et al., 1998). Other potential direct targets of NO also located upstream of the classical MAPK/ERKs are heterotrimeric G proteins. Previously, it was shown that NO accelerated the GTPase activity of Gsα and Giα in intact T-cells, and also in vitro when using recombinant proteins (Lander et al., 1993).
Functionally, we showed that NO is involved in the process of endothelial migration, which is an important event in angiogenesis. However, controversial data exist about the involvement of NO in angiogenesis. On the one hand, NO can trigger increased endothelial cell migration and thereby positively contribute to angiogenesis. Thus, Ziche et al. (1997) showed that the NOS inhibitor l-NAME blocked VEGF-induced angiogenesis in postcapillary endothelial cells in vitro and in vivo in rabbits, suggesting that NO is a downstream mediator of the action of VEGF. Also in human glioblastoma cells and in HepG2 liver carcinoma cells, NO donors were found to increase VEGF synthesis (Chin et al., 1997), and, thereby, indirectly promote endothelial cell migration and new vessel formation. Furthermore, in an in vivo mouse model of angiogenesis in which vascular sprouting in the cornea was measured, l-NAME reduced vascular sprouting in response to an angiogenic stimulus (Kon et al., 2003). Similar to the classical angiogenic factor VEGF, endothelin has also been shown to induce angiogenesis, which was shown to involve eNOS activation and NO synthesis, because depletion of eNOS abolished endothelin-induced endothelial cell migration (Noiri et al., 1997; 1998; Goligorsky et al., 1999). On the other hand, various groups have reported data that indicate a negative effect of NO on angiogenesis. Thus, in an embryo chorioallantoic membrane (CAM) chicken model, the NO donor SNP inhibited angiogenesis, whereas the NOS inhibitor l-NMMA promoted angiogenesis (Pipili-Synetos et al., 1993). In addition, the NO-producing nitrovasodilators isosorbide mononitrate and isosorbide dinitrate were shown to reduce angiogenesis in the CAM model, and reduce the growth and metastatic properties of Lewis lung carcinoma cells implanted into mice (Pipili-Synetos et al., 1995). More recently, it was shown that in a transcellular system, NO produced by the endothelial eNOS inhibited smooth muscle cell migration induced by PDGF and angiotensin II; migration of smooth muscle cells is an important event in vascular remodelling. Mechanistically, it was proposed that NO inhibited the migration of smooth muscle cells by blocking the RhoA pathway (Largiadèr et al. 2008; Suzuki et al., 2009). Obviously, the effects of NO on migratory events occur in a tissue- and cell type-specific manner.
Similar to NO, the sphingolipid S1P has also been accredited to have angiogenic potential. It is now clear that the contribution of S1P to cell migration may either be positive or negative, depending on the cell type and the S1P receptor subtypes expressed. In endothelial cells, S1P stimulates cell migration by activating the S1P1 and S1P3 receptors (Kimura et al., 2000). In contrast, the S1P2 receptor mediates a negative effect on endothelial cell migration, because blocking the S1P2 receptor with an antagonist up-regulated S1P-induced migration (Osada et al., 2002; Skoura and Hla, 2009). Most interestingly, it became evident that the S1P1 receptor is essential for in vivo embryogenesis, as it mediates smooth muscle cell migration, and thereby vessel maturation. Thus, S1P1 receptor-deficient mouse embryos die at day 14 of embryogenesis due to a failure in vessel maturation resulting in vascular bleeding (Allende et al., 2003). Our data revealed that expression of the S1P3 receptor mRNA especially is highly up-regulated by Deta-NO treatment, whereas S1P1 receptor mRNA was reduced. These data make it tempting to suggest that the S1P3 receptor is mediating the migratory response towards NO. However, because mRNA expression of receptors is not necessarily indicative of receptor functioning, further studies are clearly needed to unravel a clear contribution of the S1P receptor subtypes to endothelial cell migration.
The intracellular mechanisms leading to cell migration are still not fully understood. However, the small G-proteins Rho, Rac and cdc42 seem to play a key role (Muñoz-Chápuli et al. 2004). Notably, S1P has been shown to activate small G-proteins in cell culture experiments (Okamoto et al., 2000). Small G-proteins lead to a rearrangement of cytoskeletal proteins with the formation of lamellipodia and membrane ruffles, and inhibition of the formation of stress fibres (Burridge and Wennerberg, 2004). However, the exact sequence of events taking place from SK-1 activation by NO to cell movement needs to be addressed in further studies. Clearly, it would be important to determine whether cellular S1P levels indeed increase upon NO treatment. So far, we were unable to see clear increases in cellular S1P levels by quantitative mass spectrometry, which suggests that either the S1P generated is rapidly eliminated again, for example, by a constitutively active S1P lyase or phosphatase, or that there is only a spacially restricted generation of S1P which does not significantly alter the total cellular levels.
Another interesting question that arises from our findings is whether SK-1 also mediates other cell responses such as the crucially important vasodilatator action of NO at blood vessels. In this regard, Nofer et al. (2004) showed that in mouse isolated aortas, S1P via activation of the S1P3 receptor triggered vessel relaxation, which was abolished in eNOS-deficient mice. These data support the hypothesis that S1P has an anti-atherosclerotic effect, an effect that has also been attributed to NO (Wohlfart et al., 2008). However, whether S1P is indeed anti-atherogenic or rather is pro-atherogenic is still controversial (Okajima, 2002). Very recently, McDonald et al. (2010) showed that the SK inhibitor N,N-dimethylsphingosine drastically reduced neointimal growth in an in vivo porcine model of coronary artery balloon injury, suggesting a role for S1P in neointimal growth.
Furthermore, Suzuki et al. (2007) recently showed that iNOS-derived NO is involved in the expression of SK-1 in the liver, and contributes to a hepatoprotective effect; fumonisin B1-induced hepatotoxicity was significantly enhanced in iNOS-deficient mice, and this correlated with a loss of SK-1 expression.
The effects of NO are strictly dependent on the microenvironmental conditions, such as the availability of oxygen and reactive oxygen species that modulate the ability of NO to affect hypoxia sensing and the redox state of a cell respectively. In this context, NO has been reported to affect the expression of many important genes and mediators derived therefrom (Pfeilschifter et al., 2001; 2002;). The involvement of SK-1 and possibly S1P downstream of NO thus adds another intriguing facet to the multitude of actions of this versatile gaseous mediator.
In summary, we have shown that NO donors can stimulate the chronic activation of SK-1 in endothelial cells. This, in turn, critically contributes to NO-induced endothelial cell migration and tube formation. Finally, these observations provide the framework to understand how mediators like NO, targeting lipid signalling cascades like SK-1, can be harnessed for drug development and clinical applications.
Acknowledgments
This work was supported by the Swiss National Foundation (3100A0-111806), the German Research Foundation (FOG784, SPP1267, GRK757, GRK1172, SFB 815, PF361/6-1, HU842/4-1, EXC147), the EC (FP6: LSHM-CT-2004-005033), the Wilhelm Sander-Foundation and the Novartis-Foundation.
Glossary
Abbreviations:
- Deta-NO
(Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl) amino] diazen-1-ium-1,2-diolate
- DMEM
Dulbecco's modified Eagle's medium
- HUVEC
human umbilical vein endothelial cell
- MAHMA-NO
(Z)-1-{N-[6-(N-methylammoniohexyl)amino)} diazen-1-ium 1,2-diolate
- NAC
N-acetylcysteine
- NO
nitric oxide
- NS2028
4H-8-bromo-1,2,4-oxadiazolo(3,4-d)benz(b)(1,4)oxazin-1-one
- ODQ
1H-[1,2,4]oxadiazolo[4,3,a]quinoxaline-1-one
- S1P
sphingosine 1-phosphate
- SIN-1
3-morpholino-sydnonimine
- SK
sphingosine kinase
- SNAP
S-nitroso-N-acetyl-penicillamine
- SNP
sodium nitroprusside
- spermine-NO
(Z)-1-{N-[3-aminopropyl]-N-[4-(3-aminopropylammonio)butyl]amino} diazen-1-ium 1,2-diolate
- YC-1
3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole
Conflicts of interest
None.
References
- Alemany R, van Koppen CJ, Danneberg K, Ter Braak M, Meyer zu Heringdorf D. Regulation and functional roles of sphingosine kinases. Naunyn Schmiedebergs Arch Pharmacol. 2007;374:413–428. doi: 10.1007/s00210-007-0132-3. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. 2008;153(Suppl. 1):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allende ML, Yamashita T, Proia RL. G-protein-coupled receptor S1P1 acts within endothelial cells to regulate vascular maturation. Blood. 2003;102:3665–3667. doi: 10.1182/blood-2003-02-0460. [DOI] [PubMed] [Google Scholar]
- Amano K, Matsubara H, Iba O, Okigaki M, Fujiyama S, Imada T, et al. Enhancement of ischemia-induced angiogenesis by eNOS overexpression. Hypertension. 2003;41:156–162. doi: 10.1161/01.hyp.0000053552.86367.12. [DOI] [PubMed] [Google Scholar]
- Beck KF, Eberhardt W, Frank S, Huwiler A, Messmer UK, Mühl H, et al. Inducible NO synthase: role in cellular signalling. J Exp Biol. 1999;202:645–653. doi: 10.1242/jeb.202.6.645. [DOI] [PubMed] [Google Scholar]
- Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- Callsen D, Pfeilschifter J, Brüne B. Rapid and delayed p42/p44 mitogen-activated protein kinase activation by nitric oxide: the role of cyclic GMP and tyrosine phosphatase inhibition. J Immunol. 1998;161:4852–4858. [PubMed] [Google Scholar]
- Chin K, Kurashima Y, Ogura T, Tajiri H, Yoshida S, Esumi H. Induction of vascular endothelial growth factor by nitric oxide in human glioblastoma and hepatocellular carcinoma cells. Oncogene. 1997;15:437–442. doi: 10.1038/sj.onc.1201201. [DOI] [PubMed] [Google Scholar]
- Döll F, Pfeilschifter J, Huwiler A. The epidermal growth factor stimulates sphingosine kinase-1 expression and activity in the human mammary carcinoma cell line MCF7. Biochim Biophys Acta. 2005;1738:72–81. doi: 10.1016/j.bbalip.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Döll F, Pfeilschifter J, Huwiler A. Prolactin upregulates sphingosine kinase-1 expression and activity in the human breast cancer cell line MCF7 and triggers enhanced proliferation and migration. Endocr Relat Cancer. 2007;14:325–335. doi: 10.1677/ERC-06-0050. [DOI] [PubMed] [Google Scholar]
- Edgell CJ, McDonald CC, Graham JB. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci U S A. 1983;80:3734–3737. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Folkman J. Angiogenesis and angiogenesis inhibition: an overview. EXS. 1997;79:1–8. doi: 10.1007/978-3-0348-9006-9_1. [DOI] [PubMed] [Google Scholar]
- Goligorsky MS, Budzikowski AS, Tsukahara H, Noiri E. Co-operation between endothelin and nitric oxide in promoting endothelial cell migration and angiogenesis. Clin Exp Pharmacol Physiol. 1999;26:269–271. doi: 10.1046/j.1440-1681.1999.03029.x. [DOI] [PubMed] [Google Scholar]
- Hait NC, Sarkar S, Le Stunff H, Mikami A, Maceyka M, Milstien S, et al. Role of sphingosine kinase 2 in cell migration toward epidermal growth factor. J Biol Chem. 2005;280:29462–29469. doi: 10.1074/jbc.M502922200. [DOI] [PubMed] [Google Scholar]
- Hofmann LP, Ren S, Schwalm S, Pfeilschifter J, Huwiler A. Sphingosine kinase 1 and 2 regulate the capacity of mesangial cells to resist apoptotic stimuli in an opposing manner. Biol Chem. 2008;389:1399–1407. doi: 10.1515/BC.2008.160. [DOI] [PubMed] [Google Scholar]
- Huwiler A, Döll F, Ren S, Klawitter S, Greening A, Römer I, et al. Histamine increases sphingosine kinase-1 expression and activity in the human arterial endothelial cell line EA.hy 926 by a PKC-alpha-dependent mechanism. Biochim Biophys Acta. 2006;1761:367–376. doi: 10.1016/j.bbalip.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ. Haem-dependent activation of guanylate cyclase and cyclic GMP formation by endogenous nitric oxide: a unique transduction mechanism for transcellular signaling. Pharmacol Toxicol. 1990;67:1–7. doi: 10.1111/j.1600-0773.1990.tb00772.x. [DOI] [PubMed] [Google Scholar]
- Kimura T, Watanabe T, Sato K, Kon J, Tomura H, Tamama K, et al. Sphingosine 1-phosphate stimulates proliferation and migration of human endothelial cells possibly through the lipid receptors, Edg-1 and Edg-3. Biochem J. 2000;348:71–76. [PMC free article] [PubMed] [Google Scholar]
- Ko FN, Wu CC, Kuo SC, Lee FY, Teng CM. YC-1, a novel activator of platelet guanylate cyclase. Blood. 1994;84:4226–4233. [PubMed] [Google Scholar]
- Kon K, Fujii S, Kosaka H, Fujiwara T. Nitric oxide synthase inhibition by N(G)-nitro-l-arginine methyl ester retards vascular sprouting in angiogenesis. Microvasc Res. 2003;65:2–8. doi: 10.1016/s0026-2862(02)00011-0. [DOI] [PubMed] [Google Scholar]
- Kröncke KD, Fehsel K, Kolb-Bachofen V. Inducible nitric oxide synthase in human diseases. Clin Exp Immunol. 1998;113:147–156. doi: 10.1046/j.1365-2249.1998.00648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander HM, Sehajpal PK, Novogrodsky A. Nitric oxide signaling: a possible role for G proteins. J Immunol. 1993;151:7182–7187. [PubMed] [Google Scholar]
- Lander HM, Ogiste JS, Pearce SF, Levi R, Novogrodsky A. Nitric oxide-stimulated guanine nucleotide exchange on p21ras. J Biol Chem. 1995;270:7017–7020. doi: 10.1074/jbc.270.13.7017. [DOI] [PubMed] [Google Scholar]
- Lander HM, Jacovina AT, Davis RJ, Tauras JM. Differential activation of mitogen-activated protein kinases by nitric oxide-related species. J Biol Chem. 1996;271:19705–19709. doi: 10.1074/jbc.271.33.19705. [DOI] [PubMed] [Google Scholar]
- Largiadèr T, Eto M, Payeli SK, Greutert H, Viswambharan H, Lachat M, et al. Endothelial nitric oxide synthase gene transfer inhibits human smooth muscle cell migration via inhibition of Rho A. J Cardiovasc Pharmacol. 2008;52:369–374. doi: 10.1097/FJC.0b013e31818953d0. [DOI] [PubMed] [Google Scholar]
- Luo JD, Chen AF. Nitric oxide: a newly discovered function on wound healing. Acta Pharmacol Sin. 2005;26:259–264. doi: 10.1111/j.1745-7254.2005.00058.x. [DOI] [PubMed] [Google Scholar]
- McDonald RA, Pyne S, Pyne NJ, Grant A, Wainwright CL, Wadsworth RM. The sphingosine kinase inhibitor N,N-dimethylsphingosine inhibits neointimal hyperplasia. Br J Pharmacol. 2010;159:543–553. doi: 10.1111/j.1476-5381.2009.00533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada S, Higgs A. The l-arginine–nitric oxide pathway. N Engl J Med. 1993;329:2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- Mooradian DL, Hutsell TC, Keefer LK. Nitric oxide (NO) donor molecules: effect of NO release rate on vascular smooth muscle cell proliferation in vitro. J Cardiovasc Pharmacol. 1995;25:674–678. [PubMed] [Google Scholar]
- Muñoz-Chápuli R, Quesada AR, Angel Medina M. Angiogenesis and signal transduction in endothelial cells. Cell Mol Life Sci. 2004;61:2224–2243. doi: 10.1007/s00018-004-4070-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murohara T, Witzenbichler B, Spyridopoulos I, Asahara T, Ding B, Sullivan A, et al. Role of endothelial nitric oxide synthase in endothelial cell migration. Arterioscler Thromb Vasc Biol. 1999;19:1156–1161. doi: 10.1161/01.atv.19.5.1156. [DOI] [PubMed] [Google Scholar]
- Nakade Y, Banno Y, T-Koizumi K, Hagiwara K, Sobue S, Koda M, et al. Regulation of sphingosine kinase 1 gene expression by protein kinase C in a human leukemia cell line, MEG-O1. Biochim Biophys Acta. 2003;1635:104–116. doi: 10.1016/j.bbalip.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Nofer JR, van der Giet M, Tolle M, Wolinska I, von Wnuck Lipinski K, Baba HA, et al. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S. J Clin Invest. 2004;113:569–581. doi: 10.1172/JCI18004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noiri E, Hu Y, Bahou WF, Keese CR, Giaever I, Goligorsky MS. Permissive role of nitric oxide in endothelin-induced migration of endothelial cells. J Biol Chem. 1997;272:1747–1752. doi: 10.1074/jbc.272.3.1747. [DOI] [PubMed] [Google Scholar]
- Noiri E, Lee E, Testa J, Quigley J, Colflesh D, Keese CR, et al. Podokinesis in endothelial cell migration: role of nitric oxide. Am J Physiol. 1998;274:C236–C244. doi: 10.1152/ajpcell.1998.274.1.C236. [DOI] [PubMed] [Google Scholar]
- Okajima F. Plasma lipoproteins behave as carriers of extracellular sphingosine 1-phosphate: is this an atherogenic mediator or an anti-atherogenic mediator? Biochim Biophys Acta. 2002;1582:132–137. doi: 10.1016/s1388-1981(02)00147-6. [DOI] [PubMed] [Google Scholar]
- Okamoto H, Takuwa N, Yokomizo T, Sugimoto N, Sakurada S, Shigematsu H, et al. Inhibitory regulation of Rac activation, membrane ruffling, and cell migration by the G protein-coupled sphingosine-1-phosphate receptor EDG5 but not EDG1 or EDG3. Mol Cell Biol. 2000;20:9247–9261. doi: 10.1128/mcb.20.24.9247-9261.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen SP, Drejer J, Axelsson O, Moldt P, Bang L, Nielsen-Kudsk JE, et al. Characterization of NS2028 as a specific inhibitor of solubile guanylyl cyclase. Br J Pharmacol. 1998;123:299–309. doi: 10.1038/sj.bjp.0701603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada M, Yatomi Y, Ohmori T, Ikeda H, Ozaki Y. Enhancement of sphingosine 1-phosphate-induced migration of vascular endothelial cells and smooth muscle cells by an EDG-5 antagonist. Biochem Biophys Res Commun. 2002;299:483–487. doi: 10.1016/s0006-291x(02)02671-2. [DOI] [PubMed] [Google Scholar]
- Panetti TS. Differential effects of sphingosine 1-phosphate and lysophosphatidic acid on endothelial cells. Biochim Biophys Acta. 2002;1582:190–196. doi: 10.1016/s1388-1981(02)00155-5. [DOI] [PubMed] [Google Scholar]
- Pfeilschifter J, Huwiler A. Nitric oxide stimulates stress-activated protein kinases in glomerular endothelial and mesangial cells. FEBS Lett. 1996;396:67–70. doi: 10.1016/0014-5793(96)01070-8. [DOI] [PubMed] [Google Scholar]
- Pfeilschifter J, Eberhardt W, Beck KF. Regulation of gene expression by nitric oxide. Pflugers Arch. 2001;442:479–486. doi: 10.1007/s004240100586. [DOI] [PubMed] [Google Scholar]
- Pfeilschifter J, Beck KF, Eberhardt W, Huwiler A. Changing gears in the course of glomerulonephritis by shifting superoxide to nitric oxide-dominated chemistry. Kidney Int. 2002;61:809–815. doi: 10.1046/j.1523-1755.2002.00225.x. [DOI] [PubMed] [Google Scholar]
- Pipili-Synetos E, Sakkoula E, Maragoudakis ME. Nitric oxide is involved in the regulation of angiogenesis. Br J Pharmacol. 1993;108:855–857. doi: 10.1111/j.1476-5381.1993.tb13476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipili-Synetos E, Papageorgiou A, Sakkoula E, Sotiropoulou G, Fotsis T, Karakiulakis G, et al. Inhibition of angiogenesis, tumour growth and metastasis by the NO-releasing vasodilators, isosorbide mononitrate and dinitrate. Br J Pharmacol. 1995;116:1829–1834. doi: 10.1111/j.1476-5381.1995.tb16670.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren S, Babelova A, Moreth K, Xin C, Eberhardt W, Doller A, et al. Transforming growth factor-β2 upregulates sphingosine kinase-1 activity, which in turn attenuates the fibrotic response to TGF-β2 by impeding CTGF expression. Kidney Int. 2009;76:857–867. doi: 10.1038/ki.2009.297. [DOI] [PubMed] [Google Scholar]
- Rodríguez C, González-Díez M, Badimon L, Martínez-González J. Sphingosine-1-phosphate: a bioactive lipid that confers high-density lipoprotein with vasculoprotection mediated by nitric oxide and prostacyclin. Thromb Haemost. 2009;101:665–673. [PubMed] [Google Scholar]
- Schrammel A, Behrends S, Schmidt K, Koesling D, Mayer B. Characterization of 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one as a heme-site inhibitor of nitric oxide-sensitive guanylyl cyclase. Mol Pharmacol. 1996;50:1–5. [PubMed] [Google Scholar]
- Schwalm S, Döll F, Römer I, Bubnova S, Pfeilschifter J, Huwiler A. Sphingosine kinase-1 is a hypoxia-regulated gene that stimulates migration of human endothelial cells. Biochem Biophys Res Commun. 2008;368:1020–1025. doi: 10.1016/j.bbrc.2008.01.132. [DOI] [PubMed] [Google Scholar]
- Skoura A, Hla T. Regulation of vascular physiology and pathology by the S1P2 receptor subtype. Cardiovasc Res. 2009;82:221–228. doi: 10.1093/cvr/cvp088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel S, Milstien S. Sphingosine 1-phosphate, a key cell signaling molecule. J Biol Chem. 2002;277:25851–25854. doi: 10.1074/jbc.R200007200. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Riley RT, Sharma RP. Inducible nitric oxide has protective effect on fumonisin B1 hepatotoxicity in mice via modulation of sphingosine kinase. Toxicology. 2007;229:42–53. doi: 10.1016/j.tox.2006.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Kimura K, Shirai H, Eguchi K, Higuchi S, Hinoki A, et al. Endothelial nitric oxide synthase inhibits G12/13 and rho-kinase activated by the angiotensin II type-1 receptor: implication in vascular migration. Arterioscler Thromb Vasc Biol. 2009;29:217–224. doi: 10.1161/ATVBAHA.108.181024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venema VJ, Zou R, Ju H, Marrero MB, Venema RC. Caveolin-1 detergent solubility and association with endothelial nitric oxide synthase is modulated by tyrosine phosphorylation. Biochem Biophys Res Commun. 1997;236:155–161. doi: 10.1006/bbrc.1997.6921. [DOI] [PubMed] [Google Scholar]
- Weidner N, Semple JP, Welch WR, Folkman J. Tumor angiogenesis and metastasis – correlation in invasive breast carcinoma. N Engl J Med. 1991;324:1–8. doi: 10.1056/NEJM199101033240101. [DOI] [PubMed] [Google Scholar]
- Wohlfart P, Xu H, Endlich A, Habermeier A, Closs EI, Hübschle T, et al. Antiatherosclerotic effects of small-molecular-weight compounds enhancing endothelial nitric-oxide synthase (eNOS) expression and preventing eNOS uncoupling. J Pharmacol Exp Ther. 2008;325:370–379. doi: 10.1124/jpet.107.128009. [DOI] [PubMed] [Google Scholar]
- Yun HY, Gonzalez-Zulueta M, Dawson VL, Dawson TM. Nitric oxide mediates N-methyl-d-aspartate receptor-induced activation of p21ras. Proc Natl Acad Sci U S A. 1998;95:5773–5778. doi: 10.1073/pnas.95.10.5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziche M, Morbidelli L, Masini E, Amerini S, Granger HJ, Maggi CA, et al. Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance P. J Clin Invest. 1994;94:2036–2044. doi: 10.1172/JCI117557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziche M, Morbidelli L, Choudhuri R, Zhang HT, Donnini S, Granger HJ, et al. Nitric oxide synthase lies downstream from vascular endothelial growth factor-induced but not basic fibroblast growth factor-induced angiogenesis. J Clin Invest. 1997;99:2625–2634. doi: 10.1172/JCI119451. [DOI] [PMC free article] [PubMed] [Google Scholar]