

Abstract
BACKGROUND AND PURPOSE
The c-Jun N-terminal kinase (JNK) and tubulin are, frequently, targets for developing anti-cancer drugs. A major obstacle to successful development is P-glycoprotein (P-gp)-mediated resistance. Here, we have assessed a compound that inhibited growth of cancer cells, for effects on JNK and tubulin and as a substrate for P-gp.
EXPERIMENTAL APPROACH
Several pharmacological and biochemical assays were used to characterize signalling pathways of 2-phenyl-5-(pyrrolidin-1-yl)-1-(3,4,5-trimethoxybenzyl)-1H-benzimidazole (PPTMB), a benzimidazole analogue, in prostate cancer cells.
KEY RESULTS
PPTMB inhibited proliferation of several human prostate cancer cell lines. It displayed similar activity against a P-gp-rich cell line, indicating that PPTMB was not a substrate for P-gp. PPTMB induced G2/M arrest of the cell cycle and subsequent apoptosis, using flow cytometry. Tubulin polymerization assays and Western blot analysis showed that PPTMB directly acted on tubulin and caused disruption of microtubule dynamics, inducing mitotic arrest and sustained high levels of cyclin B1 expression and Cdk1 activation. Subsequently, mitochondria-related apoptotic cascades were induced, including Bcl-2 and Bcl-xL phosphorylation, Mcl-1 down-regulation, truncated Bad formation and activation of caspase-9 and -3. PPTMB stimulated JNK phosphorylation at Thr183/Tyr185. SP600125, a specific JNK inhibitor, significantly inhibited apoptotic signalling, indicating that JNK plays a key role in PPTMB action. PPTMB showed a 10-fold higher potency against prostate cancer cells than normal prostate cells.
CONCLUSIONS AND IMPLICATIONS
PPTMB is an effective anti-cancer agent. It disrupted microtubule dynamics, leading to mitotic arrest of the cell cycle and JNK activation, which in turn stimulated the mitochondria-related apoptotic cascades in prostate cancer cells.
Keywords: JNK, microtubule, cell cycle progression, prostate cancer, intrinsic apoptotic pathway
Introduction
Chemotherapeutic agents that inhibit tubulin polymerization or stabilize microtubule assembly are one of the most effective categories of anti-cancer agents. Therefore, targeting tubulin continues to provide many drug discoveries in anti-cancer research. The mechanism of action caused by disruption of normal tubulin function lies in the induction of cell cycle arrest at the mitotic phase, leading to the activation of sophisticated molecular events that ultimately cause apoptotic cell death (Bhalla, 2003; Marinho et al., 2008). Many molecular signals have been identified as being triggered by tubulin-targeting agents, including the phosphorylation and degradation of anti-apoptotic Bcl-2 family members, depolarization of mitochondrial membrane potential, release of cytochrome c and formation of apoptosomes and activation of caspases (Pellegrini and Budman, 2005; Vitale et al., 2007; Marinho et al., 2008).
The c-Jun N-terminal kinase (JNK), a member of the mitogen-activated protein kinase (MAPK) family, is activated by numerous types of stress, including UV and γ-irradiation, toxins, pharmacological agents and inflammatory cytokines (Carboni et al., 2008). JNK has been extensively implicated in various cellular functions, such as cytokine release reaction (Ciallella et al., 2005), cell differentiation (Chang et al., 2008), proliferation and apoptosis (Li et al., 2007; Moon et al., 2008). Several lines of evidence reveal that JNK is capable of phosphorylating many downstream effectors in addition to c-Jun, such as Bcl-2, Bcl-xL, 14-3-3, p53 and c-Myc (Weston and Davis, 2002; Nishina et al., 2004; Sunayama et al., 2005). Recently, JNK has been proposed to mediate the apoptotic cell death induced by tubulin-targeting agents (Stone and Chambers, 2000; Kolomeichuk et al., 2008). In contrast, some studies have demonstrated JNK-independent apoptosis in several types of cancer cells (Wang et al., 1999; Muscarella and Bloom, 2008). Accordingly, the interaction between JNK activity and tubulin polymerization remains unclear and needs further investigation.
Emerging evidence reveals that targeting tubulin is a promising approach for cancer chemotherapy. However, most of the tubulin-binding agents are derived from natural products with complex chemical structures that restrict chemical modification. Therefore, active compounds with relatively simple chemical structures could be valuable candidates for further development. Several studies have revealed that a variety of therapeutic drugs are derived from benzimidazole analogues, including a class of anti-helminthic drugs (Critchley et al., 2005). The benzimidazole structure also occurs in naturally, being part of the vitamin B12 molecule. Based on these considerations, we synthesized a series of small molecule benzimidazole derivatives to meet the goal of developing anti-cancer agents. After an extensive functional screening test, one compound, 2-phenyl-5-(pyrrolidin-1-yl)-1-(3,4,5-trimethoxybenzyl)-1H-benzimidazole (PPTMB), was clearly differentiated from more than 100 derivatives.
In the present work, we have made several biochemical assessments of the action of PPTMB in cell lines derived from human prostate cancer. Particularly, we analysed the complex interactions between the regulation of cell cycle progression and apoptotic signalling cascades. We also elucidated the effects of PPTMB on tubulin interactions and on signalling pathways involving JNK. We have also assessed PPTMB in primary cultures of normal human prostate gland cells.
Methods
Tissue explants and cell culture
All human tissue samples were obtained following informed consent of the donors and after full review by the Ethics Review Committee at National Taiwan University Hospital. Human hyperplastic prostates were from men by transurethral resection of the prostate in National Taiwan University Hospital. All patients had histories of prostatism and were diagnosed to have benign prostate hyperplasia by rectal digital examination, transrectal sonography of prostate and urodynamic studies. Isolation of human prostatic cells from prostatic tissue explants was described in the previous study (Guh et al., 1998). Human prostate cancer cell lines (PC-3, DU-145 and LNCaP) were from American Type Culture Collection (Rockville, MD, USA). The cells were cultured in RPMI1640 medium with 10% fetal bovine serum (FBS) (v/v) and penicillin (100 units·mL–1)/streptomycin (100 µg·mL−1). Cultures were maintained in a humidified incubator at 37°C in 5% CO2/95% air.
Assays for cell proliferation with sulphorhodamine B (SRB assay)
Cells were seeded in 96-well plates in medium with 5% FBS. After 24 h, the cells were fixed with 10% trichloroacetic acid (TCA) to represent cell population at the time of drug addition (T0). After additional incubation with dimethyl sulphoxide (DMSO) or PPTMB for 48 h, the cells were fixed with 10% TCA, and SRB at 0.4% (w/v) in 1% acetic acid was added to stain cells. Unbound SRB was washed out by 1% acetic acid, and SRB-bound cells were solubilized with 10 mM Trizma base. The absorbance was read at a wavelength of 515 nm. Using these absorbance measurements, taken at zero time (T0), after incubation under control conditions (C), and after incubation in the presence of PPTMB (Tx), the percentage growth was calculated at each concentration of the compound, as 100 −[(Tx−T0)/(C−T0)]× 100. The concentration giving growth inhibition of 50% (IC50) was also calculated from the concentration–inhibition data.
FACScan flow cytometric assay
After the treatment, the cells were harvested by trypsinization, fixed with 70% (v/v) alcohol at 4°C for 30 min and washed with phosphate-buffered saline (PBS). After centrifugation, the cells were incubated in 0.1 M of phosphate–citric acid buffer (0.2 M NaHPO4, 0.1 M citric acid, pH 7.8) for 30 min at room temperature. Then, the cells were centrifuged and resuspended with 0.5 mL propidium iodide (PI) solution containing Triton X-100 (0.1% v/v), RNase (100 µg·mL−1) and PI (80 µg·mL−1). DNA content was analysed with the FACScan and CellQuest software (Becton Dickinson, Mountain View, CA, USA).
Tubulin turbidity assay
Tubulin polymerization was detected by the use of CytoDYNAMIX Screen 03 kit (Cytoskeleton, Inc., Denver, CO, USA). Tubulin proteins (>99% purity) were suspended in G-PEM buffer containing 80 mM PIPES, 2 mM MgCl2, 0.5 mM EDTA, 1.0 mM GTP (pH 6.9) and 5% glycerol with or without the compound. Then, the mixture was transferred to a 96-well plate, and the absorbance was measured at 340 nm (37°C) for 1 h (SpectraMAX Plus, Molecular Devices Inc., Sunnyvale, CA, USA).
Tubulin polymerization assay
After the treatment, the cells were harvested by trypsinization and collected by centrifugation. The cells were lysed with 0.1 mL of hypotonic buffer (1 mM MgCl2, 2 mM EGTA, 0.5% NP-40, 2 mM phenylmethylsulphonylfluoride (PMSF), 200 U·mL−1 aprotinin, 100 µg·mL−1 soy bean trypsin inhibitor, 5.0 mM ε-amino caproic acid, 1 mM benzamidine and 20 mM Tris–HCl, pH 6.8). The cytosolic and cytoskeletal fraction of cell lysate were separated by centrifugation at 16 000×g for 15 min. The supernatant contained cytosolic tubulin. The pellet representing the particulate fraction of polymerized tubulin was resuspended in 0.1 mL hypotonic buffer. Tubulin contents in both fractions were detected by Western blotting.
Western blotting
After the treatment, the cells were washed twice with ice-cold PBS, and reaction was terminated by the addition of 100 µL ice-cold lysis buffer (10 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 mM EGTA, 1 mM PMSF, 10 µg·mL−1 aprotinin, 10 µg·mL−1 leupeptin and 1% Triton X-100). For Western blot analysis, the amount of proteins (40 µg) was separated by electrophoresis in a 10 or 15% polyacrylamide gel, and transferred to a nitrocellulose membrane. After an overnight incubation at 4°C in PBS/5% non-fat milk, the membrane was washed with PBS/0.1% Tween 20 for 1 h and immuno-reacted with the indicated antibody for 2 h at room temperature. After four washings with PBS/0.1% Tween 20, the anti-mouse or anti-rabbit IgG (diluted 1:2000) was applied to the membranes for 1 h at room temperature. The membranes were washed with PBS/0.1% Tween 20 for 1 h, and the detection of signal was performed with an enhanced chemiluminescence detection kit (Amersham Biosciences, Piscataway, NJ, USA).
Enzyme assays
The enzyme activities of mitogen-activated protein kinase kinase 1 (MEK1) and Abl tyrosine kinase were detected according to established assay methods (Farley et al., 1992; Buchdunger et al., 1996; Favata et al., 1998). The enzyme sources were from human (MEK1) and mouse recombinant Escherichia coli (Abl). For MEK1 assay, after a 15 min incubation of PPTMB (or 1% DMSO) with the substrate, myelin basic protein (MBP, 50 µg·mL−1) containing [γ-32P]ATP in the buffer (20 mM MOPS, pH 7.2, 5 mM EGTA, 20 mM MgCl2, 1 mM dithiothreitol (DTT), 25 mM β-glycerolphosphate, 1 mM Na3VO4) at 37°C, the enzyme was added for another 30 min incubation and the level of [32P]MBP was determined. For Abl assay, after a 15 min incubation of PPTMB (or 1% DMSO) with 10 µg·mL−1 poly(Glu : Tyr) in the buffer (50 mM HEPES, pH 7.4, 5 mM EGTA, 20 mM MgCl2, 1 mM DTT, 0.2 mM Na3VO4) at 37°C, the enzyme was added for another 60 min incubation, and the level of poly(Glu : Tyr-P) was determined by elisa quantification.
Confocal microscopic examination of mitotic spindle organization
Cells were seeded in eight-well chamber slides. After the treatment, the cells were fixed with 100% methanol at −20°C for 5 min, and incubated in 1% BSA containing 0.1% Triton X-100 at 37°C for 30 min. The cells were washed twice with PBS for 5 min and incubated with anti-tubulin antibody at 37°C for 1 h. The cells were washed twice with PBS and incubated with fluorescein isothiocyanate (FITC, 1:100) conjugated secondary antibody at 37°C for 40 min. The nuclei were recognized by staining with 4,6-diamidino-2-phenylindole dihydrochloride (DAPI) (1 µg·mL−1). The labelled targets in cells were detected by a confocal laser microscopic system (Leica TCS SP2, Mannheim, Germany).
Data analysis
Data are presented as the mean ± SEM for the indicated number of separate experiments. Statistical analysis of data was performed with one-way anova followed by Bonferroni t-test, and P values < 0.05 were considered significant.
Materials
RPMI 1640 medium, FBS, penicillin, streptomycin and all other tissue culture reagents were obtained from Gibco/BRL Life Technologies (Grand Island, NY, USA). Antibodies to Bcl-2, Bcl-xL, Mcl-1, Bak, Bax, Bad, GAPDH, PARP, cyclin A, E and B1, cyclin-dependent kinase 1 (Cdk1), Cdk2 and anti-mouse and anti-rabbit IgGs were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Antibodies to p27Kip1, caspase-3 and -9, mitogen-activated protein kinase kinase 1/2 (MEK1/2), phospho-MEK1/2 (Ser217/221), JNK and phospho-JNK (Thr183/Tyr185) were from Cell Signaling Technology (Boston, MA, USA). Antibodies to β-tubulin and mitotic protein monoclonal 2 (MPM-2) were from BD Biosciences PharMingen (San Diego, CA, USA) and Upstate Biotechnology (Lake Placid, NY, USA) respectively. Taxol, vincristine, DAPI, EDTA, leupeptin, DTT, SP600125, PMSF, SRB, PI and all of the other chemical reagents were obtained from Sigma-Aldrich (St Louis, MO, USA). PPTMB was synthesized and provided by our colleagues (Dr Chih-Shiang Chang, Figure 1A). The purity is more than 96% by examination of HPLC and NMR. The compound was dissolved in DMSO. The final concentration of DMSO was 0.1% in cells.
Figure 1.
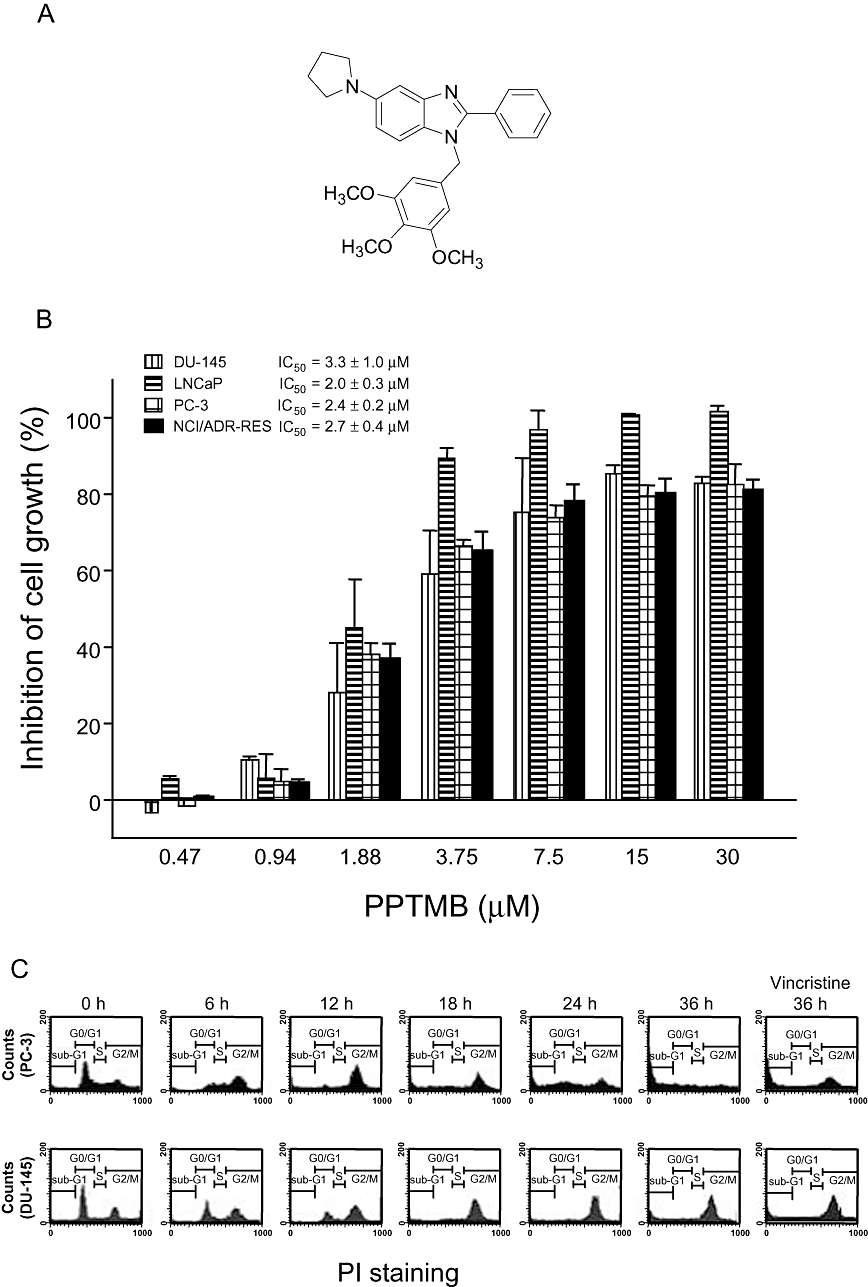
Effects of PPTMB on cell proliferation and cell cycle progression. (A) The chemical structure of PPTMB. (B) Various human prostate cancer cell lines were treated with PPTMB for 48 h. Cell proliferation was examined by SRB assay. Data are expressed as mean ± SEM of four determinations. (C) The cells were incubated in the absence or presence of PPTMB (10 µM) for the indicated times. Then, the cells were fixed and stained with PI to analyse DNA content by FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA). Data are representatives of three independent experiments. Vincristine (positive control), 0.1 µM.
Results
Effect of PPTMB on cell proliferation and cell cycle progression
The exposure of several human cancer cell lines, including prostate cancer cells (DU-145, LNCaP and PC-3) and P-glycoprotein (P-gp)-rich NCI/ADR-RES cells, to PPTMB resulted in a concentration-dependent inhibition of cell proliferation with similar IC50 values of 3.3, 2.0, 2.4 and 2.7 µM, respectively (Figure 1B). In contrast, the anti-proliferative effects of taxol and vincristine were reduced in NCI/ADR-RES cells. The resistance factor (RF) was determined based on the ratio of IC50 in NCI/ADR-RES cells to that in other cancer cell lines. The average RF of PPTMB was 1.1 (a range from 0.8 to 1.4), which was much lower than that of taxol (RF 925) and vincristine (RF 382) (Table 1).
Table 1.
Comparison of drug resistance of PPTMB, taxol and vincristine in several cancer cell lines
| NCI/ADR-RES | PC-3 | DU-145 | LNCaP | |||||
|---|---|---|---|---|---|---|---|---|
| Agent | IC50 | RF | IC50 | RF | IC50 | RF | IC50 | RF |
| PPTMB | 2.7 ± 0.4 | – | 2.4 ± 0.2 | 1.1 | 3.3 ± 1.1 | 0.8 | 2.0 ± 0.3 | 1.4 |
| Taxol | 3.7 ± 0.5 | – | 0.004 ± 0.001 | 925 | 0.006 ± 0.001 | 617 | 0.003 ± 0.001 | 1233 |
| Vincristine | 4.5 ± 0.5 | – | 0.011 ± 0.002 | 409 | 0.019 ± 0.004 | 237 | 0.009 ± 0.002 | 500 |
Different cell lines were treated with various concentrations of the indicated agent for 48 h, and the anti-proliferative effects of these agents were examined by SRB assays. The IC50 (µM) values were obtained and compared between NCI/ADR-RES and the other cell lines. Data are expressed as mean ± SEM of three to five determinations (each in triplicate). The RF represents the ratio between the IC50 in NCI/ADR-RES cells and that in the other cell lines.
The effect of PPTMB on cell cycle progression was determined by FACScan flow cytometric analysis of PI staining in PC-3 and DU-145, two androgen-independent prostate cancer cell lines. The data showed that PPTMB induced an increase of cell population at G2/M phase, followed by a subsequent increase of hypodiploid (apoptotic sub-G1 phase) population of the cell cycle. The data revealed that PC-3 cells were much more vulnerable to apoptosis when G2/M arrest occurred. Vincristine induced very similar effects in these cells (Figure 1C).
Effect of PPTMB on tubulin polymerization and mitotic spindle organization
The turbidity assay for tubulin polymerization was used to examine if PPTMB displayed a direct interaction with tubulins. As demonstrated in Figure 2A, in the presence of GTP at 37°C, the tubulins were triggered to polymerize in a time-dependent fashion. Taxol (a microtubule stabilization agent) was used as a reference agent. As a result, taxol significantly increased, while PPTMB inhibited, tubulin polymerization (Figure 2A). We also performed an in vitro microtubule assembly assay in PC-3 cells, which separated assembly and disassembly microtubules in particulate and soluble fractions respectively. The data showed that a 24 h exposure of PPTMB significantly reduced the particulate fraction of polymerized tubulins, confirming the inhibitory effect on microtubule assembly (Figure 2B).
Figure 2.
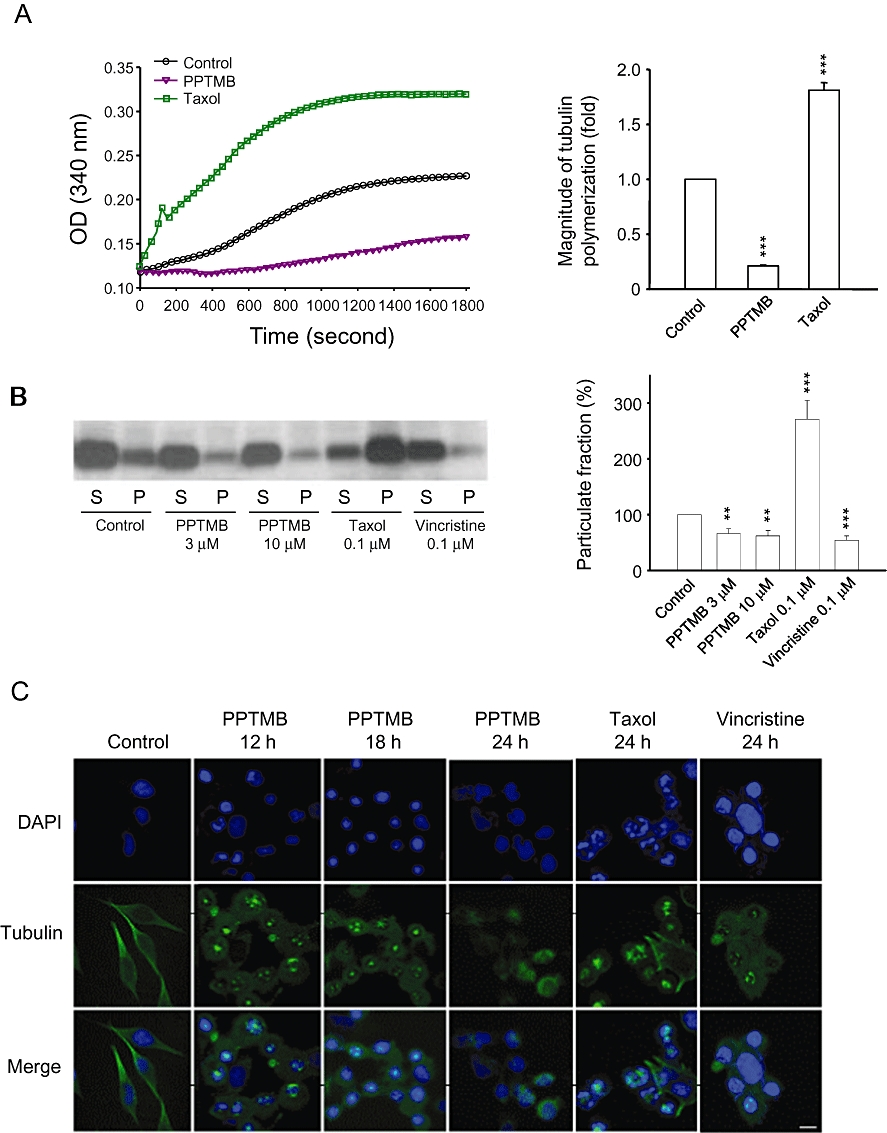
Effect of PPTMB on tubulin polymerization assays. (A) Purified tubulins were incubated at 37°C with GTP in the absence (control) or presence of PPTMB (30 µM) or taxol (0.1 µM). Tubulin polymerization was examined turbidimetrically. Data are expressed as mean ± SEM of three determinations. (B) PC-3 cells were incubated with vehicle, PPTMB, taxol or vincristine for 24 h. Then, the cells were harvested and separated into soluble (S, tubulin monomer) and particulate form (P, tubulin polymers), and the proteins were separated by Western blot analysis, and α-tubulin was detected. The expression of particulate fraction was quantified using the computerized image analysis system ImageQuant (Amersham Biosciences). Data are expressed as mean ± SEM of three determinations. **P < 0.01 and ***P < 0.001 compared with the control. (C) PC-3 cells were incubated with vehicle, PPTMB (10 µM), taxol (0.1 µM) or vincristine (0.1 µM) for the indicated times. Then, the cells were fixed and stained with primary antibody to α-tubulin. Then, FITC-labelled secondary antibodies were used (green fluorescence), and the protein was detected by a confocal laser microscopic system. The nuclei were detected by DAPI staining (blue fluorescence). Data are representatives of two independent experiments. Scale bar: 20 µM.
The organization and formation of intracellular microtubules were also detected by immunofluorescence microscopy of PC-3 cells. The in situ labelling of tubulins and chromosomes demonstrated that taxol induced the formation of multipolar spindles and multiple nuclei (Figure 2C). Notably, PPTMB induced a dynamic change of abnormal microtubule organization from mono- and multipolar mitotic spindle formation to disassembly of microtubule. Similar effects also occurred in vincristine-treated cells (Figure 2C). The data suggested that there might be more than one mechanism that directly conjugated to tubulins attributing to microtubule dynamics.
Effect of PPTMB on MEK1/2 phosphorylation and kinase activities
There is increasing evidence that MEK1/2 plays a crucial role in microtubule assembly and spindle organization (Sun et al., 2008). As PPTMB caused abnormal spindle organization and chromosome alignment, its effects on MEK1/2 were examined. As shown in Figure 3, PPTMB induced a time-related inhibition of MEK1/2 phosphorylation at Ser217/221 (Figure 3). To determine if PPTMB directly affected MEK1/2 activity, an enzyme activity assay was carried out. In these assays, PPTMB (10 µM) did not induce inhibition of MEK1/2 activity (data not shown). We subsequently tested Abl kinase activity because this tyrosine kinase has been widely suggested to be responsible for the regulation of MEK1/2 activity (Mizuchi et al., 2005; Coppo et al., 2006). The data showed that PPTMB (10 µM) induced a 65% inhibition of Abl kinase activity by kinase assays.
Figure 3.

Effect of PPTMB on MEK1/2 and phospho-MEK1/2 expression. PC-3 cells were incubated in the absence or presence of PPTMB (10 µM) for the indicated times. The cells were harvested and lysed for the detection of protein expression by Western blot analysis. The expression was quantified using the computerized image analysis system ImageQuant (Amersham Biosciences). Data are expressed as mean ± SEM of three determinations. **P < 0.01 and ***P < 0.001 compared with the control.
Effect of PPTMB on cell cycle- and apoptosis-relevant regulators
The progression of cell cycle is tightly regulated by cyclins and their binding partners, the Cdks. Cyclin D and E and the associated Cdk4 and Cdk2 regulate G1-phase progression of the cell cycle. Cyclin A/Cdk2 complex activity dominates S and G2 phases, and the progression from G2- to M-phase is driven by the activation of cyclin B1/Cdk1 complex (Vermeulen et al., 2003). PPTMB caused an up-regulation of cyclin B protein expression with a down-regulation of cyclin E and A. The expression of p27Kip1, a potent inhibitor of cyclin D1/Cdk4, was also decreased by PPTMB (Figure 4A). These data confirmed the G2/M arrest of the cell cycle. Moreover, the dramatic appearance of mitotic epitope MPM-2 further identified the mitotic arrest caused by PPTMB (Figure 4A).
Figure 4.
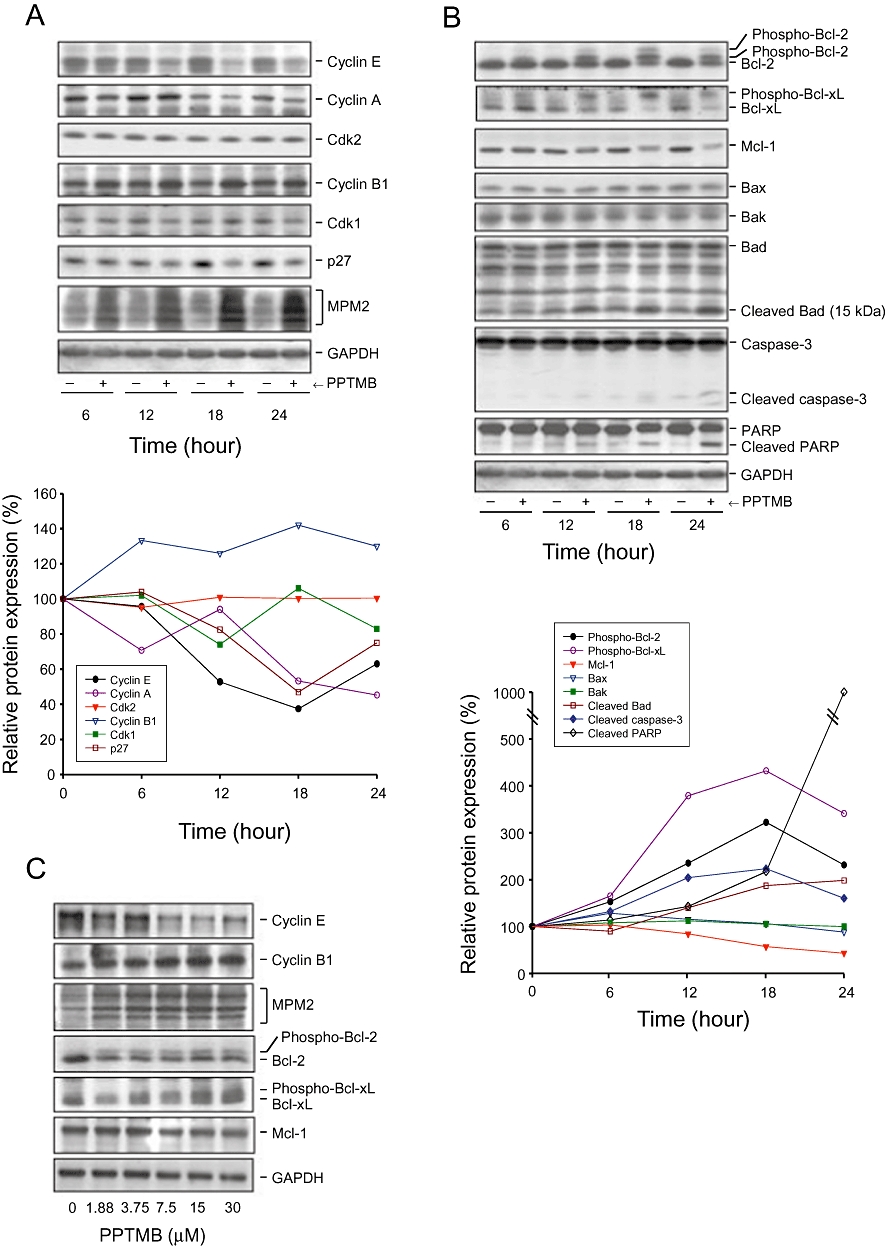
Effect of PPTMB on cell cycle regulators and apoptosis-related proteins. PC-3 cells were incubated in the absence or presence of PPTMB for the time-dependent effect (10 µM, A and B) or concentration-dependent effect (12 h, C). The cells were harvested and lysed for the detection of protein expression by Western blot analysis. Data are representatives of three independent experiments. The expression was quantified using the computerized image analysis system ImageQuant (Amersham Biosciences).
The members of Bcl-2 family are important regulators of apoptotic cell death. In this family, several pro-apoptotic proteins, including Bax, Bak and Bad, stimulate the release of cytochrome c, whereas anti-apoptotic proteins, such as Bcl-2, Bcl-xL and Mcl-1, are capable of antagonizing pro-apoptotic members, preventing the loss of mitochondrial membrane potential and upholding mitochondrial integrity (Reed et al., 1996). In this study, PPTMB augmented the phosphorylation of Bcl-2 and Bcl-xL, and decreased the protein levels of Mcl-1 (Figure 4B). The data revealed the loss of function of the anti-apoptotic members. On the contrary, truncated Bad (a promoter of apoptosis) was increased by PPTMB. The outcome was consistent with the activation of caspase-3 and PARP cleavage, by PPTMB action (Figure 4B). Moreover, the effects of PPTMB on several proteins were found to be concentration dependent (Figure 4C).
Determination of the involvement of JNK pathway
The intracellular kinase, JNK, participates in many signalling pathways mediating a range of cellular events, including cell transformation, differentiation, proliferation and apoptosis. In this study, PPTMB stimulated a profound increase of JNK phosphorylation at Thr183/Tyr185, indicating the activation of JNK (Figure 5A). SP600125, a specific JNK inhibitor, was used to determine the functional role of JNK. As demonstrated in Figure 5B, SP600125 (20 µM) significantly reduced PPTMB-mediated apoptosis of PC-3 cells. As well, several intracellular signalling pathways affected by PPTMB were markedly inhibited by SP600125 (Figure 5C). Of note, the abnormal mitotic spindle organization induced by PPTMB was dramatically inhibited by 79% (Figure 5D). The data suggest that JNK plays a key role in the apoptotic signalling cascades.
Figure 5.
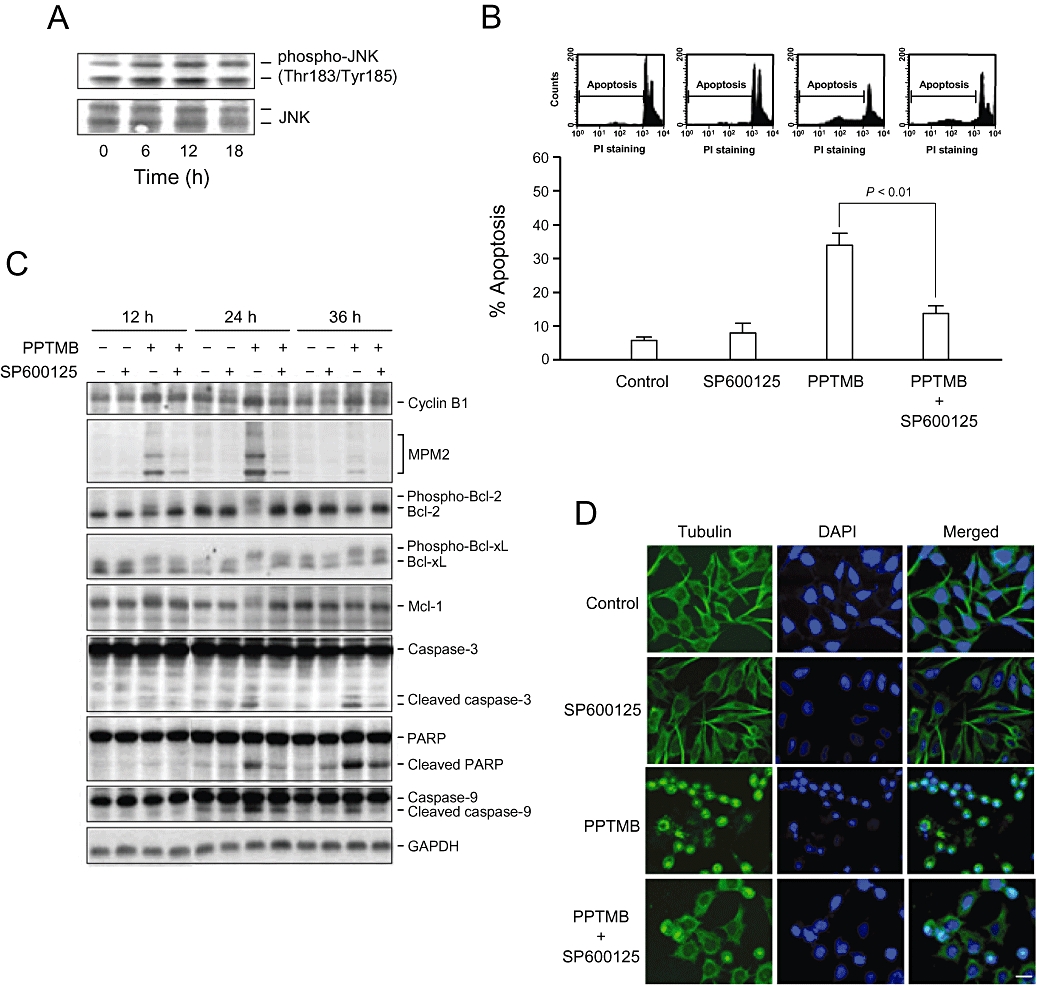
Determination of the functional role of JNK on PPTMB-mediated effects. (A) PC-3 cells were incubated in the absence or presence of PPTMB (10 µM) for the indicated times. Then, the cells were harvested and lysed for the detection of protein expression by Western blot analysis. Data are representatives of three independent experiments. (B) PC-3 cells were incubated in the indicated agent (PPTMB, 10 µM; SP600125, 20 µM) for 24 h. The cells were analysed for DNA content by FACScan flow cytometer. Data are expressed as mean ± SEM of four determinations. (C) The cells were harvested and lysed for the detection of protein expression by Western blot analysis. Data are representatives of three independent experiments. (D) PC-3 cells were incubated with the indicated agent (PPTMB, 10 µM; SP600125, 20 µM) for 12 h. The cells were fixed and stained with primary antibody to α-tubulin. FITC-labelled secondary antibodies were used (green fluorescence), and the protein was detected by a confocal laser microscopic system. The nuclei were detected by DAPI staining (blue fluorescence). Data are representatives of two independent experiments. Scale bar: 20 µM.
Selectivity of PPTMB effects for cancer cells
We were able to make primary cultures of normal human prostate cells. The doubling time of the primary cultured cells was 33.9 ± 2.1 h (n = 4). Three prostate cancer cell lines showed a range of doubling time (PC-3, 24.7 ± 1.9 h; DU-145, 29.1 ± 2.1 h; LNCaP, 31.7 ± 2.3 h, n = 4). PPTMB also displayed inhibitory effects on cell proliferation in primary prostate cells with an IC50 value of 26.7 µM, which was about 8- to 13-fold higher for normal cells than those for prostate cancer cells (Figure 6A and Table 1). With flow cytometry, PPTMB (10 µM) caused an extensive apoptosis of PC-3 cells after a 36 h treatment, whereas a higher concentration (50 µM) of the compound induced negligible increase of apoptosis in primary prostate cells (Figure 6B).
Figure 6.
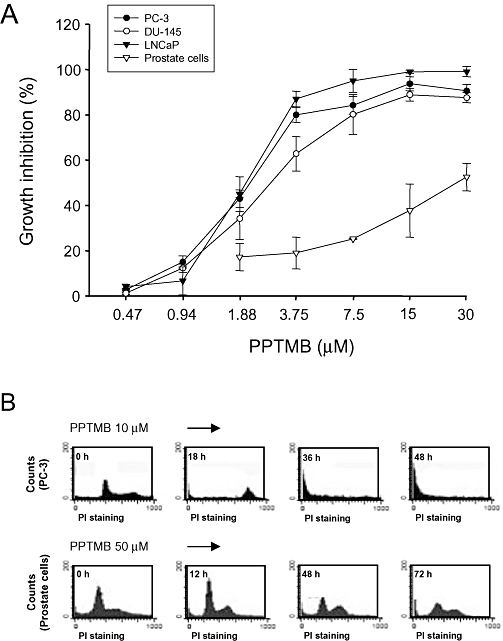
Selectivity of PPTMB for cancer cells. (A) Prostate cancer cells or primary prostate cells were treated with PPTMB at the indicated concentrations for 48 h. Cell proliferation was examined by SRB assay. Data are expressed as mean ± SEM of four determinations. (B) The cells were treated with PPTMB, and the DNA content was analysed by FACScan flow cytometer. Data are representatives of two independent experiments.
Discussion and conclusions
Microtubules are important cellular components participating in the maintenance of cell shape and many cellular processes including cytokinesis and mitosis. Microtubules are highly dynamic, and they are regulated by polymerization and depolymerization of the constituent tubulin. Microtubule dynamics are therefore vulnerable to tubulin-binding agents, and, because of its involvement in mitosis, the microtubule become a substantial target for the development of cancer chemotherapeutic agents (Bhalla, 2003; Marinho et al., 2008). From the tubulin turbidity and polymerization assays, our data revealed that PPTMB displayed a direct interaction with tubulins. In contrast to the naturally occurring tubulin-binding agents that have complex chemical structures, PPTMB is a benzimidazole analogue with a relatively simple chemical structure, which meets one criterion as a therapeutic candidate. Additionally, PPTMB was not a P-gp substrate, as its efficacy in suppressing cell growth was similar between P-gp-deficient and P-gp-rich cancer cell lines. These results supported the developmental potential as P-gp confers multiple drug resistance on a variety of cells by pumping out numerous other tubulin-binding drugs, such as vincristine, vinblastine, docetaxel and taxol (Wils et al., 1994; Metzinger et al., 2006).
The intracellular efficacy of the compound was higher than that on the turbidity test which uses pure tubulin, in the absence of microtubule-associated proteins. One reason could be that PPTMB may interfere with certain microtubule-associated proteins, and exhibited better efficacy in cells. Another possibility is that the intracellular tubulin-related signalling cascades may potentiate the effects induced by PPTMB. It is worth noting that PPTMB induced a dynamic change of abnormal microtubule organization from mono- and multipolar mitotic spindle formation to disassembly of microtubule. There are several lines of evidence suggesting that phosphorylated MEK1/2 co-localizes with microtubules and regulates spindle organization. UO126, a specific MEK1/2 inhibitor, induces abnormal spindle arrangement and chromosome misalignment in oocytes (Sun et al., 2008). In this study, PPTMB significantly blocked MEK1/2 phosphorylation (i.e. its activation), but did not directly inhibit the enzyme activity. Notably, in the kinase activity assay, PPTMB was able to directly inhibit Abl tyrosine kinase. The inhibitory ability was comparable to that on MEK1/2 phosphorylation. There is a large body of evidence showing that Abl tyrosine kinase can stimulate MEK1/2 activation (Mizuchi et al., 2005; Coppo et al., 2006). The inhibition of Abl kinase activity by PPTMB may thus lead to the blockade of MEK1/2 phosphorylation. In addition, there are several studies indicating the anti-cancer potential of combination therapy between Abl kinase inhibitors and anti-tubulin agents (Greene et al., 2007). PPTMB, by itself, displayed anti-tubulin activity and inhibition of Abl indicating a potential for anti-cancer development.
Activation of cyclin B1/Cdk1 complex at the end of G2-phase is critical for initiating mitotic entry. For successful mitosis, cyclin B1 should be degraded efficiently by the 26S proteasome in metaphase (van Leuken et al., 2008). Once the checkpoint is interrupted, the mitosis will be arrested, leading to the initiation of apoptotic signalling cascades. PPTMB caused a down-regulation of cyclin E and cyclin A expressions, while sustaining a high level of cyclin B1 protein. Together with the abnormal microtubule organization and mitotic arrest by flow cytometry, the data indicated that the PC-3 cells were compelled to stay in an unsuccessful mitosis by PPTMB. Subsequently, an apoptotic signalling cascade was initiated, and the cell apoptosis was identified by the detection of caspase-3 activation and PARP cleavage.
Mitochondria, found in the cytoplasm of every eukaryotic cell, are the most susceptible organelles in response to a variety of stresses that halt the cell cycle progression. The collapse and dysfunction of mitochondria trigger a series of apoptotic pathways. A large body of evidence reveals that the Bcl-2 family of proteins plays a central role in the determination of mitochondrial membrane permeability and cell survival (Reed et al., 1996). Our data demonstrated that PPTMB induced phosphorylation of Bcl-2, an intracellular process that results in the loss of its anti-apoptotic function (Haldar et al., 1995; Kutuk and Letai, 2008). Additionally, Bcl-xL phosphorylation was induced by PPTMB. It is suggested that Bcl-xL phosphorylation plays a role in regulating its function and participates in the determination of cancer chemotherapeutic efficacy of tubulin-binding agents (Poruchynsky et al., 1998). The Bad protein was cleaved into a 15 kDa fragment by PPTMB. Several studies show that the apoptotic stimuli may induce Bad cleavage into a 15 kDa truncated fragment, which is a more potent apoptosis inducer than the wild-type protein (Condorelli et al., 2001). The data indicate that the mitochondria-related mechanism of action explains PPTMB-induced apoptosis.
JNK is activated by many cellular stresses and extracellular signals. However, JNKs have been documented to play opposing roles of inducing apoptosis or enhancing cell survival in different cell types, depending on the upstream stimuli and downstream effectors (Bode and Dong, 2007; Brozovic and Osmak, 2007). In recent studies, the association of JNK with microtubules has been documented. Several microtubule-associated proteins are reported to be involved in the interaction, including kinesin-related motor molecules and several stathmin family proteins (Nagata et al., 1998; Tararuk et al., 2006). By using the specific JNK inhibitor SP600125, the data demonstrated that PPTMB-induced apoptotic signals were almost completely abolished, revealing the central role of JNK.
The primary culture of human prostate cells was used to determine the effect of PPTMB on normal prostate cells. The data showed that PPTMB displayed higher efficacy against prostate cancer cells than normal prostate cells with a 10-fold selectivity. Notably, PPTMB could scarcely induce apoptosis in normal prostate cells even at a fivefold high dose. It has been suggested that different doubling time may contribute to the susceptibility of cells to anti-cancer drugs that target on cell cycle. Although primary prostate cells showed longer doubling time than both PC-3 and DU-145 cells, the doubling time was similar between primary and LNCaP cells. Therefore, the different doubling time could not explain the differential between primary and prostate cancer cells. It is critical to identify the cellular targets that determine the selectivity of PPTMB, and several factors may be involved, including the ability of tubulin conjugation, susceptibility of mitochondria-mediated events and JNK activity. The mechanisms related to the selectivity await further investigation.
In conclusion, our data suggest that PPTMB is an effective anti-cancer agent. PPTMB causes the disturbance of microtubule dynamics, leading to mitotic arrest of the cell cycle and JNK activation, which in turn stimulates the subsequent mitochondria-related apoptotic signalling pathways. PPTMB also shows a preferential efficacy against human prostate cancer cells compared with normal prostate cells.
Acknowledgments
We acknowledge the support provided by the National Science Council of the Republic of China (NSC 96-2323-B-002-004 and NSC 97-2323-B-002-006). We thank Dr Shih-Chieh Chueh (Department of Urology, National Taiwan University Hospital) for helping us with the primary culture of prostate cells.
Glossary
Abbreviations
- Cdk
cyclin-dependent kinase
- CMC
carboxymethyl cellulose
- DAPI
4,6-diamidino-2-phenylindole dihydrochloride
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- MEK
mitogen-activated protein kinase kinase
- MPM-2
mitotic protein monoclonal 2
- PBS
phosphate-buffered saline
- P-gp
P-glycoprotein
- PI
propidium iodide
- PMSF
phenylmethylsulphonylfluoride
- PPTMB
2-phenyl-5-(pyrrolidin-1-yl)-1-(3,4,5-trimethoxybenzyl)-1H-benzimidazole
- SRB
sulphorhodamine B
- TCA
trichloroacetic acid
Conflicts of interest
The authors state no conflict of interest.
References
- Bhalla KN. Microtubule-targeted anticancer agents and apoptosis. Oncogene. 2003;22:9075–9086. doi: 10.1038/sj.onc.1207233. [DOI] [PubMed] [Google Scholar]
- Bode AM, Dong Z. The functional contrariety of JNK. Mol Carcinog. 2007;46:591–598. doi: 10.1002/mc.20348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brozovic A, Osmak M. Activation of mitogen-activated protein kinases by cisplatin and their role in cisplatin-resistance. Cancer Lett. 2007;251:1–16. doi: 10.1016/j.canlet.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Buchdunger E, Zimmermann J, Mett H, Meyer T, Müller M, Druker BJ, et al. Inhibition of the Abl protein–tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996;56:100–104. [PubMed] [Google Scholar]
- Carboni S, Boschert U, Gaillard P, Gotteland JP, Gillon JY, Vitte PA. AS601245, a c-Jun NH2-terminal kinase (JNK) inhibitor, reduces axon/dendrite damage and cognitive deficits after global cerebral ischaemia in gerbils. Br J Pharmacol. 2008;153:157–163. doi: 10.1038/sj.bjp.0707574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EJ, Ha J, Huang H, Kim HJ, Woo JH, Lee Y, et al. The JNK-dependent CaMK pathway restrains the reversion of committed cells during osteoclast differentiation. J Cell Sci. 2008;121:2555–2564. doi: 10.1242/jcs.028217. [DOI] [PubMed] [Google Scholar]
- Ciallella JR, Saporito M, Lund S, Leist M, Hasseldam H, McGann N, et al. CEP-11004, an inhibitor of the SAPK/JNK pathway, reduces TNF-alpha release from lipopolysaccharide-treated cells and mice. Eur J Pharmacol. 2005;515:179–187. doi: 10.1016/j.ejphar.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Condorelli F, Salomoni P, Cotteret S, Cesi V, Srinivasula SM, Alnemri ES, et al. Caspase cleavage enhances the apoptosis-inducing effects of BAD. Mol Cell Biol. 2001;21:3025–3036. doi: 10.1128/MCB.21.9.3025-3036.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppo P, Flamant S, De Mas V, Jarrier P, Guillier M, Bonnet ML, et al. BCR-ABL activates STAT3 via JAK and MEK pathways in human cells. Br J Haematol. 2006;134:171–179. doi: 10.1111/j.1365-2141.2006.06161.x. [DOI] [PubMed] [Google Scholar]
- Critchley J, Addiss D, Gamble C, Garner P, Gelband H, Ejere H, et al. Albendazole for lymphatic filariasis. Cochrane Database Syst Rev. 2005;19:CD003753. doi: 10.1002/14651858.CD003753.pub3. [DOI] [PubMed] [Google Scholar]
- Farley K, Mett H, McGlynn E, Murray B, Lydon NB. Development of solid-phase enzyme-linked immunosorbent assays for the determination of epidermal growth factor receptor and pp60c-src tyrosine protein kinase activity. Anal Biochem. 1992;203:151–157. doi: 10.1016/0003-2697(92)90056-d. [DOI] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Greene LM, Kelly L, Onnis V, Campiani G, Lawler M, Williams DC, et al. STI-571 (imatinib mesylate) enhances the apoptotic efficacy of pyrrolo-1,5-benzoxazepine-6, a novel microtubule-targeting agent, in both STI-571-sensitive and -resistant Bcr-Abl-positive human chronic myeloid leukemia cells. J Pharmacol Exp Ther. 2007;321:288–297. doi: 10.1124/jpet.106.116640. [DOI] [PubMed] [Google Scholar]
- Guh JH, Hwang TL, Ko FN, Chueh SC, Lai MK, Teng CM. Antiproliferative effect in human prostatic smooth muscle cells by nitric oxide donor. Mol Pharmacol. 1998;53:467–474. doi: 10.1124/mol.53.3.467. [DOI] [PubMed] [Google Scholar]
- Haldar S, Jena N, Croce CM. Inactivation of Bcl-2 by phosphorylation. Proc Natl Acad Sci USA. 1995;92:4507–4511. doi: 10.1073/pnas.92.10.4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolomeichuk SN, Terrano DT, Lyle CS, Sabapathy K, Chambers TC. Distinct signaling pathways of microtubule inhibitors – vinblastine and Taxol induce JNK-dependent cell death but through AP-1-dependent and AP-1-independent mechanisms, respectively. FEBS J. 2008;275:1889–1899. doi: 10.1111/j.1742-4658.2008.06349.x. [DOI] [PubMed] [Google Scholar]
- Kutuk O, Letai A. Regulation of Bcl-2 family proteins by posttranslational modifications. Curr Mol Med. 2008;8:102–118. doi: 10.2174/156652408783769599. [DOI] [PubMed] [Google Scholar]
- van Leuken R, Clijsters L, Wolthuis R. To cell cycle, swing the APC/C. Biochim Biophys Acta. 2008;1786:49–59. doi: 10.1016/j.bbcan.2008.05.002. [DOI] [PubMed] [Google Scholar]
- Li M, Feurino LW, Li F, Wang H, Zhai Q, Fisher WE, et al. Thymosinalpha1 stimulates cell proliferation by activating ERK1/2, JNK, and increasing cytokine secretion in human pancreatic cancer cells. Cancer Lett. 2007;248:58–67. doi: 10.1016/j.canlet.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Marinho J, Pedro M, Pinto DC, Silva AM, Cavaleiro JA, Sunkel CE, et al. 4′-Methoxy-2-styrylchromone a novel microtubule-stabilizing antimitotic agent. Biochem Pharmacol. 2008;75:826–835. doi: 10.1016/j.bcp.2007.10.014. [DOI] [PubMed] [Google Scholar]
- Metzinger DS, Taylor DD, Gercel-Taylor C. Induction of p53 and drug resistance following treatment with cisplatin or paclitaxel in ovarian cancer cell lines. Cancer Lett. 2006;236:302–308. doi: 10.1016/j.canlet.2005.05.016. [DOI] [PubMed] [Google Scholar]
- Mizuchi D, Kurosu T, Kida A, Jin ZH, Jin A, Arai A, et al. BCR/ABL activates Rap1 and B-Raf to stimulate the MEK/Erk signaling pathway in hematopoietic cells. Biochem Biophys Res Commun. 2005;326:645–651. doi: 10.1016/j.bbrc.2004.11.086. [DOI] [PubMed] [Google Scholar]
- Moon DO, Kim MO, Kang SH, Lee KJ, Heo MS, Choi KS, et al. Induction of G2/M arrest, endoreduplication, and apoptosis by actin depolymerization agent pextenotoxin-2 in human leukemia cells, involving activation of ERK and JNK. Biochem Pharmacol. 2008;76:312–321. doi: 10.1016/j.bcp.2008.05.006. [DOI] [PubMed] [Google Scholar]
- Muscarella DE, Bloom SE. The contribution of c-Jun N-terminal kinase activation and subsequent Bcl-2 phosphorylation to apoptosis induction in human B-cells is dependent on the mode of action of specific stresses. Toxicol Appl Pharmacol. 2008;228:93–104. doi: 10.1016/j.taap.2007.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata K, Puls A, Futter C, Aspenstrom P, Schaefer E, Nakata T, et al. The MAP kinase kinase kinase MLK2 co-localizes with activated JNK along microtubules and associates with kinesin superfamily motor KIF3. EMBO J. 1998;17:149–158. doi: 10.1093/emboj/17.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishina H, Wada T, Katada T. Physiological roles of SAPK/JNK signaling pathway. J Biochem. 2004;136:123–126. doi: 10.1093/jb/mvh117. [DOI] [PubMed] [Google Scholar]
- Pellegrini F, Budman DR. Review: tubulin function, action of antitubulin drugs, and new drug development. Cancer Invest. 2005;23:264–273. doi: 10.1081/cnv-200055970. [DOI] [PubMed] [Google Scholar]
- Poruchynsky MS, Wang EE, Rudin CM, Blagosklonny MV, Fojo T. Bcl-xL is phosphorylated in malignant cells following microtubule disruption. Cancer Res. 1998;58:3331–3338. [PubMed] [Google Scholar]
- Reed JC, Zha H, Aime-Sempe C, Takayama S, Wang HG. Structure–function analysis of Bcl-2 family proteins. Regulators of programmed cell death. Adv Exp Med Biol. 1996;406:99–112. [PubMed] [Google Scholar]
- Stone AA, Chambers TC. Microtubule inhibitors elicit differential effects on MAP kinase (JNK, ERK, and p38) signaling pathways in human KB-3 carcinoma cells. Exp Cell Res. 2000;254:110–119. doi: 10.1006/excr.1999.4731. [DOI] [PubMed] [Google Scholar]
- Sun SC, Xiong B, Lu SS, Sun QY. MEK1/2 is a critical regulator of microtubule assembly and spindle organization during rat oocyte meiotic maturation. Mol Reprod Dev. 2008;75:1542–1548. doi: 10.1002/mrd.20891. [DOI] [PubMed] [Google Scholar]
- Sunayama J, Tsuruta F, Masuyama N, Gotoh Y. JNK antagonizes Akt-mediated survival signals by phosphorylating 14-3-3. J Cell Biol. 2005;170:295–304. doi: 10.1083/jcb.200409117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tararuk T, Ostman N, Li W, Björkblom B, Padzik A, Zdrojewska J, et al. JNK1 phosphorylation of SCG10 determines microtubule dynamics and axodendritic length. J Cell Biol. 2006;173:265–277. doi: 10.1083/jcb.200511055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003;36:131–149. doi: 10.1046/j.1365-2184.2003.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale I, Antoccia A, Cenciarelli C, Crateri P, Meschini S, Arancia G, et al. Combretastatin CA-4 and combretastatin derivative induce mitotic catastrophe dependent on spindle checkpoint and caspase-3 activation in non-small cell lung cancer cells. Apoptosis. 2007;12:155–166. doi: 10.1007/s10495-006-0491-0. [DOI] [PubMed] [Google Scholar]
- Wang TH, Popp DM, Wang HS, Saitoh M, Mural JG, Henley DC, et al. Microtubule dysfunction induced by paclitaxel initiates apoptosis through both c-Jun N-terminal kinase (JNK)-dependent and -independent pathways in ovarian cancer cells. J Biol Chem. 1999;274:8208–8216. doi: 10.1074/jbc.274.12.8208. [DOI] [PubMed] [Google Scholar]
- Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Genet Dev. 2002;12:14–21. doi: 10.1016/s0959-437x(01)00258-1. [DOI] [PubMed] [Google Scholar]
- Wils P, Phung-Ba V, Warnery A, Lechardeur D, Raeissi S, Hidalgo IJ, et al. Polarized transport of docetaxel and vinblastine mediated by P-glycoprotein in human intestinal epithelial cell monolayers. Biochem Pharmacol. 1994;48:1528–1530. doi: 10.1016/0006-2952(94)90580-0. [DOI] [PubMed] [Google Scholar]