

Abstract
BACKGROUND AND PURPOSE
Sorafenib is an inhibitor of several intracellular signalling kinases with anti-proliferative, anti-angiogenic and pro-apoptotic effects in tumour cells. Sorafenib is used in the therapy of advanced renal cell carcinoma, and several phase II clinical trials are being carried out in patients with urothelial carcinomas.
EXPERIMENTAL APPROACH
Using a panel of human bladder cancer cell lines (RT4, T24, J82), we characterized systematically the effects of sorafenib on intracellular signalling, migration, proliferation and apoptosis.
KEY RESULTS
We demonstrated that at low concentrations (<1 µM), sorafenib is capable of significantly stimulating migration and proliferation of the bladder cancer cells. We hypothesize that these stimulatory effects on tumour cell functions might be explained by an activation of the Ras/ERK-1/2 signal transduction pathway. In addition, the comparison of different bladder cancer cell lines not only revealed a different biology (e.g. cell migration), but also a differential susceptibility to the anti-apoptotic effects of sorafenib. Finally, we confirmed in different bladder cancer cell lines the known inhibitory actions of sorafenib in pharmacological concentrations (≥3 µM) on ERK-1/2 phosphorylation, migration and proliferation, as well as the pro-apoptotic effects of the compound.
CONCLUSIONS AND IMPLICATIONS
Taken together, these findings suggest that although sorafenib has the potential to be used in the treatment of urothelial carcinoma, this compound might also activate bladder cancer cells at low concentrations. This should be relevant for dosing regiments to optimize the treatment with this promising anti-tumour drug.
Keywords: bladder cancer, urothelial cancer, sorafenib, ERK
Introduction
Current chemotherapy for metastatic bladder cancer is based on gemcitabine and cisplatin (von der Maase et al., 2005). Although many therapeutical concepts have been developed, no real progression has been achieved during the last 20 years, and the median survival is 14 months (vom Dorp et al., 2008).
Over-expression of the receptor for the epidermal growth factor (EGFR) is known to correlate with poor prognosis in advanced urothelial cancer (Nicholson et al., 2001; Dominguez-Escrig et al., 2004). However, as the combination of standard anti-tumour therapy with the EGFR inhibitor gefitinib did not provide any significant benefit (Philips et al., 2008), new therapeutical concepts are clearly needed to improve the prognosis of patients suffering from urothelial cancers.
Sorafenib is a bis-aryl urea which inhibits a variety of receptor tyrosine kinases (RTKs), such as those associated with receptors for vascular endothelial growth factor receptor (VEGFR)-2/-3, for platelet-derived growth factor receptor-beta (PDGFR-β), Flt-3 and c-Kit, but also – based on a potent inhibition of Raf-1 – targets the Raf/MEK/ERK signalling pathways (Wilhelm and Chien, 2002). In vitro and in vivo studies have demonstrated that this multi-kinase inhibitor reduces tumour growth and disrupts tumour microvasculature through anti-proliferative, anti-angiogenic and/or pro-apoptotic effects (Liu et al., 2006; Plastaras et al., 2007; Rosato et al., 2007; Ammoun et al., 2008; Ding et al., 2008; Wilhelm et al., 2008). Sorafenib is used in the therapy of advanced renal cell carcinoma (Gradinetti and Goldspiel, 2007). Currently, several phase II clinical trials are being carried out in patients with urothelial carcinomas (http://clinicaltrials.gov). However, only limited data on the effects of sorafenib on urothelial carcinoma cells have been published thus far.
Using a panel of human bladder cancer cell lines, we aimed to characterize systematically the in vitro effects of sorafenib on intracellular signalling, migration, proliferation and apoptosis.
Methods
Cell lines
The human bladder cancer cell lines RT4, T24 (containing the constitutively active H-ras oncogene) and J82 were obtained from the American Type Culture Collection (Manassas, VA, USA) and maintained in McCoy's 5a medium (RT4, T24) or MEM (J82), each containing 10% fetal calf serum (FCS, Invitrogen, Carlsbad, CA, USA) and 1% penicillin–streptomycin (Invitrogen) at 37°C in 5% CO2. The human renal carcinoma cell lines A-498 and Caki-1 were obtained from the American Type Culture Collection and maintained in MEM (A-498) and McCoy's 5a medium (Caki-1), each containing 10% FCS (Invitrogen) and 1% penicillin–streptomycin (Invitrogen) at 37°C in 5% CO2.
Western blot
Cells were grown to subconfluency in six-well chambers, serum-deprived for 24 h and then treated with sorafenib for 2 h. After treatment, the cells were lysed in RIPA buffer containing 150 mM NaCl, 10 mM Tris (pH 8), 1% (m/v) deoxycholic acid, 1% (v/v) NP-40, 0.1% (m/v) SDS, 4 mM EDTA, 20 µg·mL−1 benzamidine, 2 mM phenylmethylsulphonyl fluoride, 20 µg·mL−1 soya bean inhibitor, 2 µg·mL−1 aprotinin and 1 µg·mL−1 leupeptin. Protein concentrations were measured using the BCA Protein Assay Kit (Thermo Scientific, Franklin, MA, USA) according to the manufacturer's protocol. Equal amounts of protein were fractionated in either 8 or 10% SDS–polyacrylamide gels and transferred to nitrocellulose membranes (Whatman, Maidstone, Kent, UK). The membranes were probed with primary antibodies (1:1000, 4°C, overnight) followed by incubation with horseradish peroxidase-conjugated secondary antibodies (1:5000, 20°C, 1 h) (Sigma-Aldrich, St Louis, MO, USA). Bands were visualized by chemiluminescence (PerkinElmer Life Sciences, Waltham, MA, USA). Quantification was performed by scanning the films with a grey scale-calibrated scanning system (HP Scanjet G4050, Phoretix Power Scan V 2003.01, Nonlinear Dynamics, Newcastle upon Tyne, UK). Band intensity was calculated with LabImage 1D V 4.0 (Kapelan, Leipzig, Germany).
Migration
Cells were grown to confluency in 24-well chambers and then serum deprived for 24 h. A mechanotransductor (pipette tip) was used to scratch the cell monolayer in a standardized fashion (Hermann et al., 2002). After washing of the cells twice with phosphate-buffered saline (PBS), the wells were refilled with medium containing 10% FCS and 1 mM hydroxyurea (Sigma-Aldrich) to inhibit cell division. The wells were marked on the underside across the wound to ensure that the same region was assessed microscopically. All experiments have been performed in quadruplicate. Photographs were taken at 0 h and after 24 h using an Axiovert S100 phase contrast microscope (Carl-Zeiss, Jena, Germany). The area, invaded by tumour cells, was calculated using the AxioVision Software V461.0 (Carl-Zeiss Imaging Solutions).
Nuclear staining
Cells were seeded in 24-well chambers and allowed to attach for 2 h in a medium containing 10% FCS. The cells were serum deprived for 24 h and then treated with sorafenib. After 24 h incubation with sorafenib, the supernatants were removed and the cells were washed with PBS and fixed with ice-cold methanol. Mounting and DNA staining were performed using the Vectashield Mounting Medium and DAPI (Vector Labaratories, Burlingame, CA, USA). Photographs were taken with an Axiovert S100 fluorescence microscope (Carl-Zeiss).
Cell proliferation
Cells were seeded at a concentration of 2 × 105 per well in six-well chambers and allowed to attach overnight in a medium containing 10% FCS. The cells were serum deprived for 24 h and then treated with sorafenib for another 24 and 48 h respectively. After removing the supernatant, the cells were washed with PBS, detached with 0.25% trypsin/1 mM EDTA. After staining with 0.25% Trypan blue solution (Biochrom, Berlin, Germany), living cells were counted in a Neubauer chamber.
Annexin V binding
Cells were washed twice with PBS and resuspended in annexin V binding buffer [100 µM HEPES (pH 7.4), 1.4 M NaCl and 25 mM CaCl2]. After incubation with annexin V–Alexa Fluor 488 conjugate (Invitrogen) for 15 min, the amount of Alexa Fluor-labelled cells (% positive) was determined by flow cytometry (Cytomics FC 500, Beckman Coulter, Brea, CA, USA) as previously described.
Ras activation assay
Ras activation was studied using a pull-down assay with Raf1 fused to GST and bound to glutathione sepharose beads (#BK008, Cytoskeleton, Denver, CO, USA) according to the manufacturer's instructions. The amount of activated Ras was determined by Western blotting using a Ras antibody.
Statistical analysis
Data are expressed as means ± SEM. Statistical significance was determined using the one-way anova test followed by the Bonferroni test (GraphPad Prism V302 GraphPad Software, La Jolla, CA, USA). P < 0.05 was considered significant. In most experiments, concentration–response curves using a range of sorafenib concentrations have been performed. In order to minimize multiple comparisons, statistical comparisons with the control were performed with one pre-selected low (0.1 µM) and one pre-selected high (10 µM) concentration of sorafenib.
Materials
Sorafenib (Nexavar, Bayer, Leverkusen, Germany) was dissolved in dimethyl sulphoxide (DMSO) (10 mM) and was stored at −80°C. In all experiments, equal amounts of DMSO were used as a vehicle control. Phorbol 12-myristate 13-acetate (PMA) was from Sigma-Aldrich. P44/42 MAP kinase antibody (#4695), phospho-p44/42 MAP kinase antibody (#9101), phospho-p38 MAP kinase antibody (#9215), phospho-Akt antibody (#4060), A-Raf antibody (#4432), B-Raf antibody (#9434), c-Raf antibody (#9422) and poly ADP ribose polymerase (PARP) antibody (#9542) were all purchased from Cell Signaling Technology (Danvers, MA, USA). U0126 was from Calbiochem (#662005) (San Diego, CA, USA).
Drug and molecular target nomenclature in this paper follows Alexander et al. (2009).
Results
Sorafenib is known to inhibit the signalling kinase Raf-1 (c-Raf), as well as wild-type and V599E mutant B-Raf (Wilhelm et al., 2004). Thus, we first characterized the expression of the different Raf isoforms in three different human bladder cell lines (RT4, T24, J82). B-Raf and Raf-1 were strongly expressed in all cell lines examined, whereas A-Raf was not detectable in the RT4 cell line (Figure 1).
Figure 1.
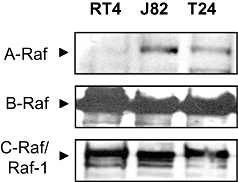
Western blots demonstrating the expression of Raf isoforms in three different bladder carcinoma cell lines (RT4, T24, J82).
We next studied the effects of sorafenib on ERK-1/2 phosphorylation in the human bladder cancer cell lines. As expected, at pharmacological concentrations (≥3 µM), the compound inhibited both basal (Figure 2) and stimulated (data not shown) ERK-1/2 phosphorylation, resulting in a significant inhibition of cell migration (Figure 3) and proliferation (Figure 4). In addition, sorafenib stimulated apoptosis (as measured by annexin V binding and PARP cleavage) in T24 and J82 cells, but not in RT4 cells (Figure 5). Interestingly, staurosporine (2 µM)-induced apoptosis did not differ between the three cell lines (not shown).
Figure 2.
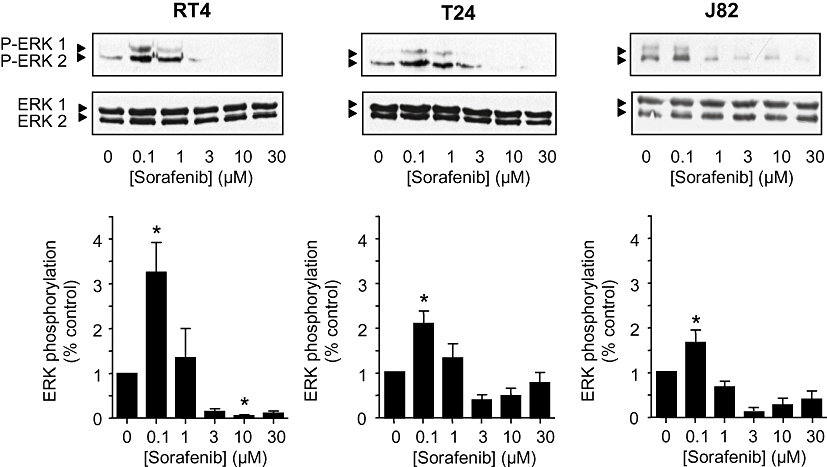
Effects of sorafenib (0.1–30 µM) on ERK-1/2 phosphorylation (2 h) in different human bladder carcinoma cell lines (RT4, T24, J82). Total and phosphorylated ERK-1/2 was detected by Western blotting. Original tracings and densitometrical analysis (means ± SEM) of five experiments are shown. *P < 0.05 versus control (anova /Bonferroni).
Figure 3.
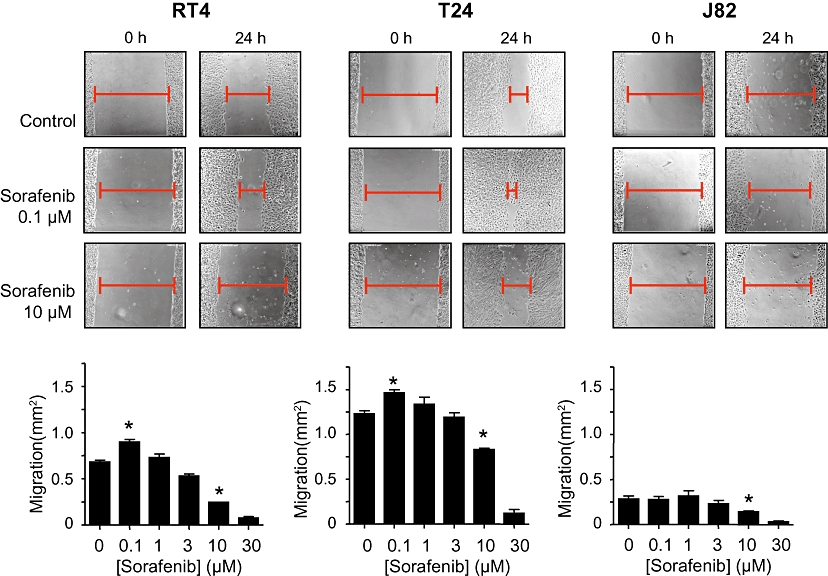
Effects of sorafenib (0.1–30 µM) on migration of different human bladder carcinoma cell lines (RT4, T24, J82). After a scratch injury, microscopic analysis of the cell-free area was performed at 0 h and after 24 h. The area, invaded by tumour cells, was calculated. Original photographs (cell-free area indicated) and quantitative analysis (means ± SEM) of five experiments are shown. *P < 0.05 versus control (anova /Bonferroni).
Figure 4.
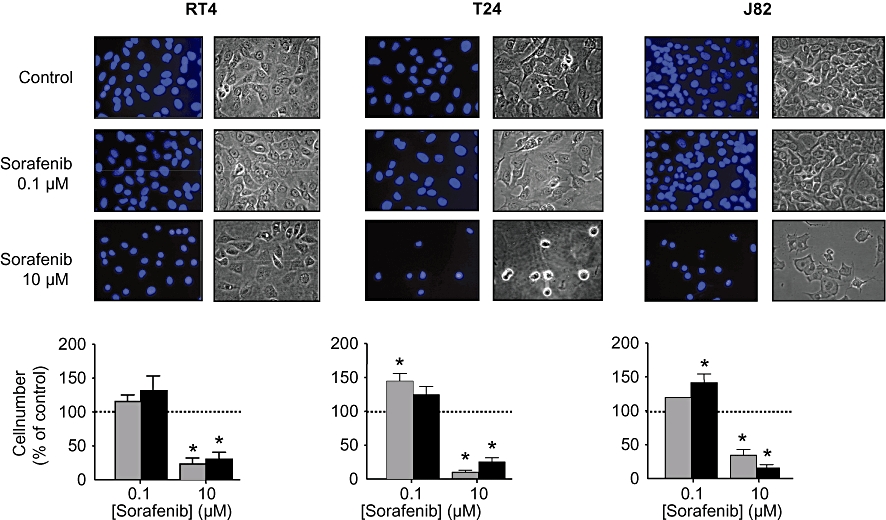
Effects of sorafenib (0.1 and 10 µM) on proliferation of different human bladder carcinoma cell lines (RT4, T24, J82). For the assessment of nuclear morphology, cells were incubated for 24 or 48 h, and stained with DAPI. For cell counts, cells were detached with trypsin/EDTA, stained with Trypan blue and counted in a Neubauer chamber. Original photographs and the quantitative analysis (means ± SEM) of five experiments are shown. *P < 0.05 versus control (anova /Bonferroni).
Figure 5.
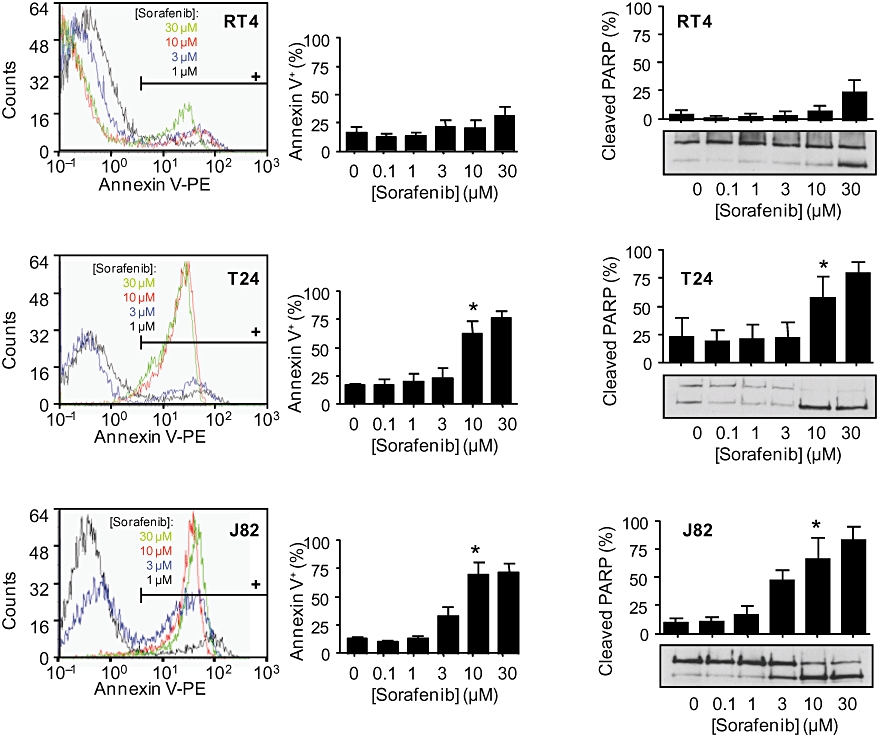
Effects of sorafenib (0.1–30 µM) on apoptosis of different human bladder carcinoma cell lines (RT4, T24, J82). Annexin V binding was measured by flow cytometry. PARP cleavage was assessed by Western blotting. Original FACS histograms and quantitative analysis of annexin V binding (left panel) and original Western blots with densitometrical analyses (right panel) are shown. Data are means ± SEM of five experiments. *P < 0.05 versus control (anova /Bonferroni).
The most intriguing finding, however, was the demonstration of significant stimulatory effects of sorafenib at low concentrations (<1 µM) on migration of RT4 and T24 cells (Figure 3). No stimulatory effects were observed in J82 cells, possibly reflecting the very low general migratory potency of this cell line. In addition, a significant stimulation of cell proliferation by sorafenib (0.1 µM) was observed in T24 and J82 cells (in RT4 cells, there was a trend for an increased mitogenesis). Because no anti-apoptotic effects of sorafenib (0.1 µM) were detected (Figure 5), the observed stimulation of cell proliferation by sorafenib most likely depends on the activation of mitogenic events.
As sorafenib is currently approved for the treatment of advanced renal carcinoma, we were interested if similar stimulatory effects of sorafenib could also be detected in renal carcinoma cell lines. Therefore, we studied the effects of sorafenib on ERK-1/2 phosphorylation and migration in the human renal carcinoma cell lines A-498 and Caki-1. In contrast to our results in bladder carcinoma cells, no stimulation of ERK-1/2 could be detected at low concentrations (<1 µM) of sorafenib, indicating a possible organ specificity. As expected, at higher concentrations, the phosphorylation of ERK-1/2 was clearly inhibited (data not shown).
Next, we aimed to elucidate if the observed stimulatory effects of sorafenib at low concentrations in human bladder cancer cells might be explained mechanistically by a significant activation of ERK-1/2 phosphorylation (Figure 2). Basal, PMA-stimulated and sorafenib (0.1 µM)-stimulated ERK-1/2 phosphorylation were inhibited by the MEK inhibitor U0126 (Figure 6), indicating a stimulatory mechanism of sorafenib located upstream of MEK. Both basal and sorafenib-stimulated cell migration were also significantly inhibited by MEK inhibitor U0126 (Figure 7). However, MEK inhibition only partially (about 50%) inhibited cell migration. Interestingly, in the presence of U0126, the stimulatory effects on migration of sorafenib were completely blunted.
Figure 6.
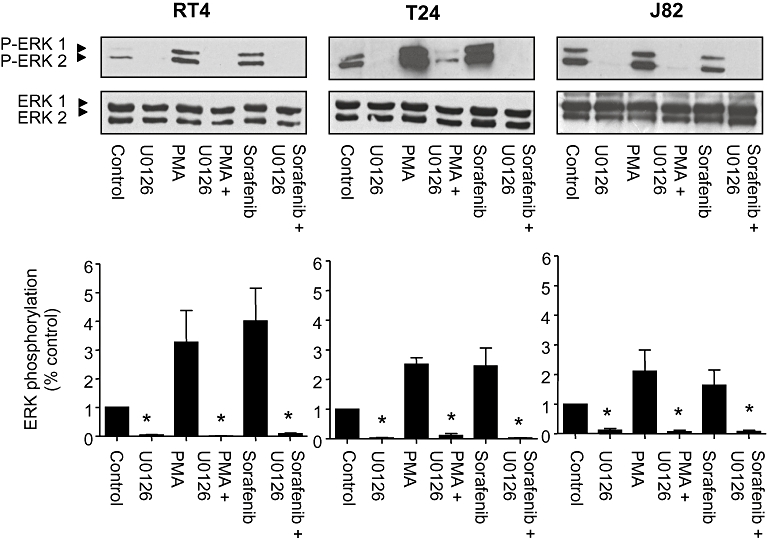
Effects of the MEK inhibitor U0126 (10 µM) on PMA (100 nM) versus sorafenib (0.1 µM)-induced ERK-1/2 phosphorylation in different human bladder carcinoma cell lines (RT4, T24, J82). Original photographs and the quantitative analysis (means ± SEM) of five experiments are shown. *P < 0.05 versus control (anova /Bonferroni).
Figure 7.
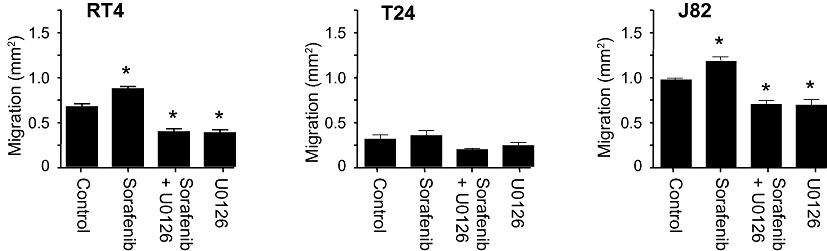
Effects of the MEK inhibitor U0126 (10 µM) on sorafenib (0.1 µM)-induced migration of different human bladder carcinoma cell lines (RT4, T24, J82). The quantitative analysis (means ± SEM) of five experiments are shown. *P < 0.05 versus control (anova /Bonferroni).
Other signalling pathways, such as p38 MAPK, Akt or ERK-5 phosphorylation, were not activated by sorafenib (data not shown).
In order to characterize the upstream effectors that activate ERK-1/2 upon treatment with low concentrations of sorafenib, we performed pull-down experiments to detect activated Ras. These experiments demonstrated that at concentrations corresponding to concentrations leading to ERK-1/2 activation, sorafenib also activated Ras (Figure 8).
Figure 8.
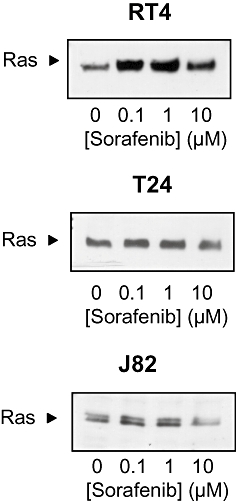
Effects of sorafenib (0.1–10 µM) on Ras activation in different human bladder carcinoma cell lines (RT4, T24, J82). Representative blots from three independent experiments are shown.
Discussion
The standard therapeutic concept of urothelial cancer is based on a cisplatin chemotherapy. However, during the last decades, no real progress could be achieved, although the cancer, over-expressing VEGFRs (Xia et al., 2006), might represent an interesting potential target for new drugs. In addition, studies have shown that the large majority of bladder tumours express activated H-Ras (Fontana et al., 1996; Vageli et al., 1996; Przybojewska et al., 2000), and that this oncogenic activation is an important tumourigenic factor (Adjei, 2001).
Sorafenib, an oral multi-kinase inhibitor, inhibiting serine/threonine Raf kinases and RTKs such as VEGFR and PDGFR-β, is currently used for the therapy of advanced renal carcinoma. Furthermore, anti-tumour activity and an improved outcome have also been shown for patients with other carcinomas such as hepatocellular carcinoma (Llovet et al., 2008), non-small cell lung cancer (Okamoto et al., 2009) and metastatic breast cancer (Bianchi et al., 2009).
Phosphorylated ERK is the key downstream target of the Ras/Raf/MEK/ERK signalling pathway, and dysregulation of this pathway occurs in around one-third of all human cancers (for review, see Dhillon et al., 2007). In a phase II study in patients with advanced, inoperable hepatocellular carcinoma, the pretreatment tumour levels of phosphorylated ERK-1/2 were correlated with the time to tumour progression (Abou-Alfa et al., 2006). Furthermore, recently it was suggested that phosphorylated ERK-1/2 might be a potential predictive marker of sensitivity to sorafenib in hepatocellular carcinoma. The compound inhibited ERK-1/2 phosphorylation, dependent on the degree of basal expression level of phosphorylated ERK-1/2 (Zhang et al., 2009).
Currently, several phase II clinical trials of sorafenib are being carried out in patients with urothelial carcinomas. Therefore, we focused in our study on the effects of sorafenib on bladder cancer cells. We studied the phorsphorylation status of ERK-1/2 as the key downstream component of the Ras/Raf/MEK/ERK signalling pathway, as well as functional effects such as migration and proliferation.
As described for a variety of different tumour types, pharmacological concentrations (≥3 µM) of sorafenib decreased the phosphorylation level of ERK-1/2. Unexpectedly, we found a significant stimulatory effect of sorafenib at low concentrations (<1 µM) on ERK-1/2 phosphorylation, as well as on migration and proliferation in human bladder cancer cells.
As sorafenib is currently approved for the treatment of advanced renal carcinoma in several countries, we were interested if similar activatory effects could also be detected in renal cancer cells. However, in contrast to our results in bladder cancer cells, no stimulatory action of low concentrations of sorafenib could be detected in the human renal carcinoma cell lines A-498 and Caki-1 (data not shown).
To further elucidate the underlying signalling pathways, we used the MEK inhibitor U0126. We could show that cell migration was also dependent on ERK-independent mechanisms as the compound inhibited cell migration only about 50%. The sorafenib-induced migration was completely blunted by the MEK inhibitor, thereby indicating that this pathway is responsible for the observed stimulation of cell migration.
However, the systematic comparison of different bladder cancer cell lines, as presented in this study, revealed marked differences in cell biology (e.g. cell migration), but also a differential susceptibility to the inhibitory effects of sorafenib (e.g. apoptosis). These differences might also partially explain the different biology of bladder cancers in vivo, as well as possible inter-individual differences in the responsiveness to chemotherapy, including sorafenib (Dreicer et al., 2009). However, these data are in accordance with previous reports, demonstrating inhibitory effects of sorafenib on different tumour cell types (Wilhelm et al., 2008) and might indicate that tumour cell stimulation by sorafenib might be restricted to specific tumour types. Different basal levels of ERK-1/2 phosphorylation of different tumour cell types might be of importance for the different susceptibility to the compound (Zangh et al., 2009), as well as other cell type-specific characteristics. These should be explored in detail in future studies.
Because sorafenib is known to inhibit a variety of RTKs and, specifically, the Raf/Ras/MEK/ERK signalling pathway, the observed stimulatory effects on Ras and ERK-1/2 in human bladder carcinoma cell lines are surprising and indicate a dual (activatory and inhibitory) mode of action of this compound. Of course, our data confirmed the anti-migratory and anti-proliferatory effects of this compound as observed across a variety of tumour types, including renal cell, hepatocellular, breast and colorectal carcinomas (Wilhelm et al., 2008). Low (<1 µM) concentrations of this compound have either not been studied (Plastaras et al., 2007; Rosato et al., 2007; Ding et al., 2008) or no stimulatory effects have been observed (Ammoun et al., 2008).
Taken together, we demonstrated that the multi-kinase inhibitor sorafenib exhibits a dual (activatory and inhibitory) mode of action in a panel of human bladder cell lines. These activatory effects were not only restricted to intracellular signalling events (ERK-1/2 phosphorylation), but also resulted in a stimulation of tumour cell functions (migration, proliferation), relevant for tumour growth and metastasis. Although the link between ERK-1/2 activation by sorafenib and stimulation of cell migration has only been demonstrated using pharmacological tools, one might speculate that activation of the Raf/Ras signalling pathway by low concentrations of sorafenib might result in an ERK-1/2-dependent activation of cell migration and mitogenesis.
Clearly, our data demonstrate the anti-migratory and anti-proliferatory effects of sorafenib in different human bladder cancer cell lines. However, the stimulatory effects at low concentrations of sorafenib may have major consequences for the therapeutic use of this anti-tumour drug. This might be particularly relevant for selecting dosing regimens that avoid low plasma concentrations of sorafenib in order to optimize the treatment with this promising anti-tumour drug.
Acknowledgments
We thank Helgard Geldermann for excellent technical support.
Glossary
Abbreviations
- EGFR
epidermal growth factor receptor
- ERK
extracellular signal-regulated kinase
- MAPK
mitogen-activated protein kinase
- PARP
poly ADP ribose polymerase
- PDGFR-β
platelet-derived growth factor receptor-β
- PMA
phorbol myristate acetate
- RTKs
receptor tyrosine kinases
- VEGFR
vascular endothelial growth factor receptor
Conflict of interest
The authors state no conflict of interest.
References
- Abou-Alfa GK, Schwartz L, Ricci S, Amadori D, Santoro A, Figer A, et al. Phase II study of sorafenib in patients with advanced hepatocelluar carcinoma. J Clin Oncol. 2006;24:4293–4300. doi: 10.1200/JCO.2005.01.3441. [DOI] [PubMed] [Google Scholar]
- Adjei AA. Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst. 2001;93:1062–1074. doi: 10.1093/jnci/93.14.1062. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammoun S, Flaiz C, Ristic N, Schuldt J, Hanemann CO. Dissecting and targeting the growth factor-dependent and growth factor-independent extracellular signal-regulated kinase pathway in human schwannoma. Cancer Res. 2008;68:5236–5245. doi: 10.1158/0008-5472.CAN-07-5849. [DOI] [PubMed] [Google Scholar]
- Bianchi G, Loibl S, Zamagni C, Salvagni S, Raab G, Siena S, et al. Phase II multicenter, uncontrolled trial of sorafenib in patients with metastatic breast cancer. Anticancer Drugs. 2009;20:616–624. [PubMed] [Google Scholar]
- Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- Ding Q, Huo L, Yang JY, Xia W, Wei Y, Liao Y, et al. Down-regulation of myeloid cell leukemia-1 through inhibiting Erk/Pin 1 pathway by sorafenib facilitates chemosensitization in breast cancer. Cancer Res. 2008;68:6109–6117. doi: 10.1158/0008-5472.CAN-08-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Escrig JL, Kelly JD, Neal DE, King SM, Davies BR. Evaluation of the therapeutic potential of the epidermal growth factor receptor tyrosine kinase inhibitor gefitinib in preclinical models of bladder cancer. Clin Cancer Res. 2004;10:4874–4884. doi: 10.1158/1078-0432.CCR-04-0034. [DOI] [PubMed] [Google Scholar]
- vom Dorp F, Börgermann C, Rose A, Becker M, Rübben H. Targeted therapy for metastatic bladder cancer. Urologe. 2008;47:1311–1314. doi: 10.1007/s00120-008-1747-9. [DOI] [PubMed] [Google Scholar]
- Dreicer R, Li H, Stein M, DiPaola R, Eleff M, Roth BJ, et al. Phase 2 trial of sorafenib in patients with advanced urothelial cancer. A trial of the Eastern Cooperative Oncology Group. Cancer. 2009;115:4090–4095. doi: 10.1002/cncr.24467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana D, Bellina M, Scoffone C, Cagnazzi E, Cappia S, Cavallo F, et al. Evaluation of c-ras oncogene product (p21) in superficial bladder cancer. Eur Urol. 1996;29:470–476. doi: 10.1159/000473799. [DOI] [PubMed] [Google Scholar]
- Gradinetti CA, Goldspiel BF. Sorafenib and sunitinib: a novel targeted therapies for renal cell cancer. Pharmacotherapy. 2007;27:1125–1144. doi: 10.1592/phco.27.8.1125. [DOI] [PubMed] [Google Scholar]
- Hermann A, Schrör K, Weber A-A. CD40 ligand (CD40L) does not stimulate proliferation of vascular smooth muscle cells. Eur J Cell Biol. 2002;81:213–221. doi: 10.1078/0171-9335-00240. [DOI] [PubMed] [Google Scholar]
- Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumour angiogenesis, and induces tumour cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66:11851–11858. doi: 10.1158/0008-5472.CAN-06-1377. [DOI] [PubMed] [Google Scholar]
- Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- von der Maase H, Sengelov L, Roberts JT, Ricci S, Dogliotti L, Oliver T, et al. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J Clin Oncol. 2005;23:4602–4608. doi: 10.1200/JCO.2005.07.757. [DOI] [PubMed] [Google Scholar]
- Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer. 2001;37(Suppl. 1):S9–15. doi: 10.1016/s0959-8049(01)00231-3. [DOI] [PubMed] [Google Scholar]
- Okamoto I, Miyazaki M, Morinaga R, Kaneda H, Ueda S, Hasegawa Y, et al. Phase I clinical and pharmacokinetic study of sorafenib in combination with carboplatin and paclitaxel in patients with advanced non-small cell lung cancer. Invest New Drugs. 2009 doi: 10.1007/s10637-009-9321-x. DOI: 10.1007/s10637-009-9321-x [Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips GK, Halabi S, Sanford BL, Bajorin D, Small EJ, Cancer and Leukaemia Group B. A phase II trial of cisplatin, fixed dose–rate gemcitabine and gefitinib for advanced urothelial tract carcinoma: results of the Cancer and Leukaemia Group B 90102. Br J Urol Int. 2008;101:20–25. doi: 10.1111/j.1464-410X.2007.07226.x. [DOI] [PubMed] [Google Scholar]
- Plastaras JP, Kim SH, Liu YY, Dicker DT, Dorsey JF, McDonough J, et al. Cell cycle dependent and schedule-dependent antitumour effects of sorafenib combined with radiation. Cancer Res. 2007;67:9443–9454. doi: 10.1158/0008-5472.CAN-07-1473. [DOI] [PubMed] [Google Scholar]
- Przybojewska B, Jagielo A, Jalmuzna P. H-RAS, K-RAS, and N-RAS gene activation in human bladder cancers. Cancer Genet Cytogenet. 2000;121:73–77. doi: 10.1016/s0165-4608(00)00223-5. [DOI] [PubMed] [Google Scholar]
- Rosato RR, Almenara JA, Coe S, Grant S. The multikinase inhibitor sorafenib potentiates TRAIL lethality in human leukemia cells in association with Mcl-1 and cFLIPL down-regulation. Cancer Res. 2007;67:9490–9500. doi: 10.1158/0008-5472.CAN-07-0598. [DOI] [PubMed] [Google Scholar]
- Vageli D, Kiaris H, Delakas D, Anezinis P, Cranidis A, Spandidos DA. Transcriptional activation of H-ras, K-ras, and N-ras proto-oncogenes in human bladder tumors. Cancer Lett. 1996;107:241–247. doi: 10.1016/0304-3835(96)04372-8. [DOI] [PubMed] [Google Scholar]
- Wilhelm S, Chien DS. BAY 43-9006: preclinical data. Curr Pharm Des. 2002;8:2255–2257. doi: 10.2174/1381612023393026. [DOI] [PubMed] [Google Scholar]
- Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. Bay 43-9006 exhibits broad spectrum oral antitumour activity and targets the Raf/MEK/ERK pathway and receptor tyrosine kinases involved in tumour progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signalling. Mol Cancer Ther. 2008;7:3129–3140. doi: 10.1158/1535-7163.MCT-08-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia G, Kumar SR, Hawes D, Cai J, Hassanieh L, Groshen S, et al. Expression and significance of vascular endothelial growth factor receptor 2 in bladder cancer. J Urol. 2006;175:1245–1252. doi: 10.1016/S0022-5347(05)00736-6. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Zhou X, Shen H, Wang D, Wang Y. Phosphorylated ERK is a potential predictor of sensitivity to sorafenib when treating hepatocellular carcinoma: evidence from an in vitro study. BMC Med. 2009;7:41. doi: 10.1186/1741-7015-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]