

Abstract
Background and purpose:
Increased glutamatergic innervation of the substantia nigra pars reticulata (SNpr) and pars compacta (SNpc) may contribute to the motor deficits and neurodegeneration, respectively, in Parkinson's disease (PD). This study aimed to establish whether activation of pre-synaptic group III metabotropic glutamate (mGlu) receptors reduced glutamate release in the SN, and provided symptomatic or neuroprotective relief in animal models of PD.
Experimental approach:
Broad-spectrum group III mGlu receptor agonists, O-phospho-l-serine (l-SOP) and l-2-amino-4-phosphonobutyrate (l-AP4), were assessed for their ability to inhibit KCl-evoked [3H]-d-aspartate release in rat nigral prisms or inhibit KCl-evoked endogenous glutamate release in the SNpr in vivo using microdialysis. Reversal of akinesia in reserpine-treated rats was assessed following intranigral injection of l-SOP and l-AP4. Finally, the neuroprotective effect of 7 days' supra-nigral treatment with l-AP4 was examined in 6-hydroxydopamine (6-OHDA)-lesioned rats.
Key results:
l-SOP and l-AP4 inhibited [3H]-d-aspartate release by 33 and 44% respectively. These effects were blocked by the selective group III mGlu antagonist (RS)-α-cyclopropyl-4-phosphonophenylglycine (CPPG). l-SOP also reduced glutamate release in the SNpr in vivo by 48%. Injection of l-SOP and l-AP4 into the SNpr reversed reserpine-induced akinesia. Following administration above the SNpc, l-AP4 provided neurochemical, histological and functional protection against 6-OHDA lesion of the nigrostriatal tract. Pretreatment with CPPG inhibited these effects.
Conclusions and implications:
These findings highlight group III mGlu receptors in the SN as potential targets for providing both symptomatic and neuroprotective relief in PD, and indicate that inhibition of glutamate release in the SN may underlie these effects.
Keywords: akinesia, group III mGlu receptor, 6-hydroxydopamine lesion, metabotropic glutamate receptors, neuroprotection, nigrostriatal tract, Parkinson's disease, reserpine-treated rat, substantia nigra, symptomatic relief
Introduction
Parkinson's disease (PD) is a debilitating disorder in which degeneration of dopaminergic neurones in the nigrostriatal tract evokes a myriad of motor symptoms, such as tremor, postural instability and akinesia. Current treatments for PD rely heavily on reinstating dopaminergic transmission either with l-dopa or dopamine agonists. While extremely effective at treating the symptoms of the disease, these approaches do little to combat the progressive degeneration. This failure affects patients' long-term health because the increasing doses of drug required to stabilize worsening symptoms often result in disabling adverse effects, such as l-dopa induced dyskinesia and psychosis (Stocchi et al., 1997).
In PD, loss of striatal dopamine evokes downstream changes within the basal ganglia, leading, among other things, to increased firing of the subthalamic nucleus (STN) (Vila et al., 1999). The subsequent increased release of glutamate in STN target areas, such as the substantia nigra pars reticulata (SNpr) and internal globus pallidus, leads to downstream inhibition of thalamocortical feedback, which contributes to the generation of motor deficits associated with PD. The parallel increase in glutamate release from STN terminals in the substantia nigra pars compacta (SNpc) (Smith et al., 1990; Iribe et al., 1999) may, on the other hand, contribute to the progressive SNpc degeneration, characteristic of PD pathology (Rodriguez et al., 1998), through a potential excitotoxic route. Accordingly, reducing or modulating STN firing via subthalamotomy or deep brain stimulation effectively reduces symptoms in animal models and PD patients (Peppe et al., 2004; Alvarez et al., 2005; Windels et al., 2005). Moreover, surgical modulation of STN firing has been shown to reduce the degree of SNpc degeneration apparent in the 6-hydroxydopamine (6-OHDA) lesion rat model of PD (Piallat et al., 1996). However, given these surgical procedures are available in only a few centres worldwide, alternative strategies for combating the effects of increased STN activity are desirable. Pharmacological inhibition of glutamate release from STN terminals in the SN is one strategy worth pursuing, and group III metabotropic glutamate (mGlu) receptors hold great promise in this area.
Group III mGlu receptors, including mGlu4, mGlu7 and mGlu8 (receptor nomenclature follows Alexander et al., 2009), play important neuromodulatory roles in the brain (Conn and Pin, 1997). These Gi/Go-coupled receptors are found predominantly on pre-synaptic elements of both GABAergic and glutamatergic synapses, where their activation in regions such as the thalamus, superior colliculus and globus pallidus has already been shown to reduce transmitter release (Turner and Salt, 1999; Pothecary et al., 2002; MacInnes and Duty, 2008). Electrophysiological studies further indicate that activation of group III mGlu receptors in the SN with the broad-spectrum agonist, l-2-amino-4-phosphonobutyrate (l-AP4) can lead to inhibition of glutamate transmission across the subthalamonigral synapse, measured as reduced STN-evoked excitatory post-synaptic currents (EPSCs) in the SNpr (Wittmann et al., 2001b) and SNpc (Wigmore and Lacey, 1998; Valenti et al., 2005). Preliminary evidence supports a potential symptomatic and neuroprotective role of group III mGlu receptors in the SN. For example, it has been shown that the broad-spectrum agonist O-phospho-l-serine (l-SOP) reverses reserpine-induced akinesia in the rat following direct injection into the SNpr (MacInnes et al., 2004), while intranigral injection of l-AP4 provides histological and neurochemical protection against 6-OHDA-induced nigrostriatal tract lesion in rats (Vernon et al., 2007). Despite these promising findings, it remains to be seen whether group III mGlu receptor-mediated inhibition of glutamate release in vitro will be confirmed using alternative, non-electrophysiological methods, and more importantly whether such inhibition is observed in vivo. Furthermore, it is not known whether agonists other than l-SOP can reverse akinesia in the reserpine-treated rat in a receptor-dependent manner, or whether the level of neuroprotection offered by group III mGlu receptor activation is sufficient to preserve motor function in these animals.
The aims of this study were therefore to establish whether activation of group III mGlu receptors could inhibit glutamate release in the SN, both in vitro and in vivo; whether the broad-spectrum agonist l-AP4 could offer symptomatic relief in the way of reversal of reserpine-induced akinesia; and whether the neuroprotective relief afforded by l-AP4 in the 6-OHDA-lesioned rat model of PD was accompanied by preservation of motor behaviours.
The findings of this study support the conclusion that targeting group III mGlu receptors in the SN is an effective way to bring about inhibition of glutamate release in the STN target areas, and that activation of these receptors can provide both symptomatic relief and functional neuroprotection in animal models of PD.
Methods
Animals
All animal care and procedures conformed to the UK Animals (Scientific Procedures) Act, 1986, and every effort was made to minimize animal numbers and suffering. Male Sprague Dawley rats (B & K or Harlan, UK) weighing 270–300 g were used in these studies. Food and water were provided ad libitum. The animals were housed in a temperature- and humidity-controlled environment with a 12 h light/dark cycle.
Experimental protocol for [3H]-d -aspartate release in the SN
To avoid complications of separating out radiolabelled glutamate used in metabolism (e.g. GABA synthesis) compared with that used as a transmitter, the non-metabolized analog d-aspartate, which shares the same neuronal transporter as endogenous glutamate and is released in a similar manner (Ferkany and Coyle, 1983; Savage et al., 2001), was used in these studies. The animals were killed by stunning and cervical dislocation, the brains rapidly removed and the SN (containing both SNpr and SNpc) dissected out and chopped into 350 µm prisms. Following equilibration in Krebs solution (composition in mM: NaCl 134, CaCl2 1.3 mM, MgSO4 1, NaHCO3 25, KCl 5, KH2PO4 1.24, glucose 10; pH 7.4; 95% O2/5% CO2, 37°C) with tissues from three animals pooled for any given experimental run, prisms of SN were loaded for 45 min in Krebs solution containing 33 µM [3H]-d-aspartate (0.222 MBq). After three washes, the tissues were suspended in 1.2 mL Krebs solution and 100 µL aliquots transferred into a 12-chamber Brandel perfusion system (Semat, St Albans, UK), perfused at 1 mL·min–1 with aerated Krebs solution at 37°C. Following a 1 h equilibration, 2 min perfusate fractions were collected into vials containing 3 mL of scintillation fluid. Basal [3H]-d-aspartate release, determined over the first three fractions was followed by a 2 min stimulation (S1) with Krebs containing 25 mM KCl (with equimolar reduction of NaCl to 114 mM) to evoke [3H]-d-aspartate release. An 18 min washout period preceded a second 2 min stimulation with 25 mM KCl (S2). Group III mGlu receptor agonists, l-SOP (1–1000 µM) or l-AP4 (3–300 µM), or vehicle were added to the perfusate 8 min prior to and during S2. When examined, the group III mGlu receptor antagonist (RS)-α-cyclopropyl-4-phosphonophenylglycine (CPPG; 100 µM), was added 4 min prior to and during agonist exposure. Release fractions and remaining tissues were analysed for [3H]-d-aspartate content by liquid scintillation spectroscopy.
Experimental protocol for microdialysis and glutamate HPLC
A total of 16 rats were used for these studies. Given that l-SOP and l-AP4 produced near identical profiles of inhibition of release in the above in vitro studies, in vivo dialysis studies were performed with l-SOP alone. Under terminal general anaesthesia (chloral hydrate, 130 mg·kg–1 induction; 16 mg·kg–1 maintenance doses given when indicated through regular pedal withdrawal testing), animals were placed on a Kopf stereotaxic frame, with the incisor bar at 3.3 mm below the interaural line. Microdialysis probes (concentric design, 1 mm tip, Hospal AN 69 membrane, Huntingdon, UK; Wright et al., 1992) were implanted into the right SNpr [anteroposterior (AP), –5.5 mm; mediolateral (ML), –2.2 mm; dorsoventral (DV), –8.3 mm from bregma; (Paxinos and Watson, 1998)] and secured using dental cement and microscrews anchored into the skull. Dialysis probes were perfused (1 µL·min–1) with artificial CSF (aCSF; composition in mM: NaCl 140, KCl 3, CaCl2 2.4, MgCl2 1, NaH2PO4 0.25, Na2HPO4 1.2; pH 7.4) and after a 1 h equilibration, collection of 20 min dialysate fractions commenced. After five baseline fractions, 100 mM KCl in aCSF was infused for 10 min via the probe to evoke depolarization-dependant glutamate release (S1). Five fractions later, l-SOP (100 or 300 µM in aCSF; n = 4 per group) or vehicle (aCSF; n = 4) was infused via the probe for 20 min with 100 mM KCl added during the last 10 min to evoke glutamate release (S2). Sample collection continued for a further five fractions. Probe location was confirmed in cryostat cut sections by light microscopic visualization of Trypan blue dye that was injected through the probe at the end of the experiment. Microdialysis samples were assayed for glutamate using HPLC with electrochemical detection with a Prodigy 5 µ ODS3 column (Phenomenex, Macclesfield, Cheshire, UK). Then, 20 µL aliquots of samples and glutamate standards (10, 20 or 40 pmol in aCSF) were mixed with 25 µL derivatization agent (5% absolute ethanol, 50 mM sodium sulphite, 0.1 mM sodium tetraborate; 16 mM O-phthaldialdehyde) for 5 min at 9°C before injection into the mobile phase (0.1 M NaH2PO4,0.5 mM EDTA, 25% methanol; pH 4.2), which was pumped at 0.65 mL·min–1 without recycling. Samples were detected by electrochemical detector with the Ag/AgCl reference electrode maintained at +0.80 V, and the chromatograms were recorded and analysed by Alexys data system (Antec Leyden, NV Zoeterwoude, the Netherlands).
Experimental protocol for supra-nigral cannulation to facilitate drug/toxin delivery
Under general anaesthesia (isofluorane, 4–5% induction and 2–3% maintenance in 95% O2/5% CO2) 23-gauge stainless steel guide cannulae were stereotaxically implanted, 2 mm above either the SNpr (AP, –5.5 mm; ML, ±2.2 mm; DV, –6.3 mm) or the SNpc [AP, –4.8 mm; ML, ±2.0 mm; DV, –6.3 mm; relative to skull surface at Bregma (Paxinos and Watson, 1998)]. Bilateral cannulation doubled the chances of obtaining a patent cannula in each animal, thereby reducing the number of animals required. Cannulae were secured into position as per dialysis probes, and 30-gauge stainless steel stylets were inserted to prevent blockage.
Experimental protocol for monitoring reversal of akinesia in reserpine-treated rats following intranigral drug administration
A total of 78 rats were used for these studies. A minimum of 4 days following cannulation above the SNpr, the animals were injected with reserpine [4 mg·kg–1, s.c.; in 0.85% (v/v) glacial acetic acid] under light anaesthesia (3% isofluorane in 95% O2/5% CO2). Eighteen hours later, when akinesia had developed, pre-acclimatized animals were placed into 40 cm diameter, flat-bottomed hemispherical bowls, and baseline activity was video-recorded for 30 min. The animals (n = 6 per group) then received a single, unilateral injection (1 µL·min–1) of l-SOP (250, 500 or 750 nmol in 2.5 µL), l-AP4 (3–300 nmol in 2 µL) or vehicle (2.5 µL sterile PBS) via a 30-gauge needle inserted into the SNpr, and locomotor activity was video-recorded for 60 min (MacInnes et al., 2004). Given that any reversal of akinesia will occur on the injected side only, net 360° contraversive rotations were quantified as an index of relief of akinesia. When examined, CPPG (75 nmol in 2.5 µL PBS) or vehicle (n = 6 per group) was injected into the SNpr 30 min prior to l-SOP or l-AP4.
Experimental protocol for monitoring neuroprotection in the 6-OHDA-lesioned rat following supra-nigral L-AP4 injections
A total of 77 rats were used for these studies which commenced a minimum of 7 days after cannulation. Given that the responses to l-SOP and l-AP4 were similar in the reserpine model of PD, these more intensive neuroprotective studies were performed with l-AP4 alone. The animals were pretreated with desipramine (25 mg·kg–1 i.p.) and pargyline (5 mg·kg–1 i.p.) 30 min before receiving, under brief isofluorane-induced anaesthesia (as above), a unilateral injection of 6-OHDA (12 µg in 2.5 µL of 0.2% ascorbic acid in 0.9% saline; 1.25 µL·min–1) via a 30-gauge needle into the SNpc. While conscious, the animals received unilateral supra-nigral infusions of vehicle or l-AP4 (0.1–30 nmol in 4 µL of PBS; 2 µL·min–1) 1 mm above the SNpc 1 h prior to and daily for 7 days after 6-OHDA injection. When examined, CPPG (75 nmol; dissolved in 4 µL 0.1 M NaOH) or vehicle was infused above the SNpc 30 min prior to l-AP4. Drug injections were made 1 mm above the SNpc to avoid causing mechanical damage to this structure from the repeated injections. In a pilot study (n = 2 rats), Davidson's dye was stereotaxically injected at this site and the animals were killed 1 h later. Histological verification of staining in 6 µm sections from fixed, paraffin-embedded midbrain blocks containing the SN confirmed that the dye had reached its intended target site, the SNpc.
The extent of motor impairment in animals following a 6-OHDA lesion was assessed using three behavioural tests. The cylinder test of forelimb akinesia was performed pre- and 4 days post-lesion. Animals were placed in a perspex cylinder (21 cm d× 34 cm h) and the number of upward reaches, in 5 min, video-recorded. Use of the contralateral paw was assessed as a percentage of total reaches made by observers unaware of the treatment. The adjusted steps test measured contralateral forelimb use 1 day prior to and 6 days post-lesion. The rat was held with the torso slightly raised, and the hind limbs and ipsilateral forelimb gently restrained. The number of steps made by the animal's contralateral paw was recorded as it was moved sideways in backhand and forehand directions across a 90 cm distance with the contralateral forelimb bearing weight. The average use of the contralateral paw on three repeat runs each day was taken and post-lesion score was expressed as a percentage of pre-lesion score. Amphetamine-induced rotational behaviour was assessed 7 days post-lesion. Full 360° ipsiversive rotations were video-recorded in 5 min intervals for up to 60 min following injection of d-amphetamine (5 mg·kg–1 i.p.) for subsequent assessment by observers unaware of the treatment. On the final day of l-AP4 dosing (day 8), some animals were terminally anaesthetized using pentobarbital (100 mg·kg–1) then trans-cardially perfused with 0.1 M PBS, followed by 4% paraformaldehyde (PFA) in 0.1 M PBS. The brains were removed and stored in PFA at 4°C until further processing for tyrosine hydroxylase (TH) and dopa decarboxylase (DDC) immunohistochemistry. Other animals were killed by CO2 asphyxiation, the brains removed and freshly dissected striata snap frozen on card-ice before storing at –80°C until required for HPLC measurement of dopamine levels.
Immunohistochemistry protocols
Coronal sections (6 µm) were taken from paraffin-embedded brains at three levels of the striatum (+1.2, +0.2 and –0.26 mm AP from Bregma) and SN [–4.8, –5.3 and –5.8 mm AP from Bregma; (Paxinos and Watson, 1998)]. Sections were processed in triplicate at each level for each animal. Following removal of paraffin (in xylene and 100% industrial methylated spirit), block of endogenous peroxide activity (in 3% H2O2) and standard antigen retrieval with 1 mM citric acid, sections were incubated for 10–20 min with blocking buffer (TH: 1% BSA in 0.1 M PBS and 10% sodium azide; DDC: 1% normal goat serum in 0.1 M PBS), then for either 2 h with rabbit-α-TH antibody (1:1250; Chemicon) or overnight at r.t. with rabbit polyclonal DDC antibody (1:1500; Chemicon, Millipore, Watford, UK). After 1 h incubation with secondary antibody (goat anti-rabbit IgG-biotinylated 1:200; Sigma-Aldrich, Somerset, UK), sections were incubated for 30 min with an ABC kit (Vector Laboratories, Peterborough, UK) and the signal developed in 10% diaminobenzidine tetrahydrochloride in Tris-buffered saline for 10 min. Digital images of the striata were captured on a mounted CCD Nikon camera (Kingston, Surrey, UK), and densitometric analysis of the level of TH or DDC staining was performed using Scion Image Software (Scion Corporation, Frederick, MD, USA). For the SNpc, sections were viewed on a Zeiss apotome microscope and recorded using Axiovision LE software (Carl Zeiss Ltd, Hertfordshire, UK). Only viable TH-positive cells (i.e. intact round cells displaying a clear nucleus and cytoplasm) were counted in each hemisphere at ×50 magnification using an image analysis software (Image J). For each animal, the number of cells in three adjacent sections of SNpc was counted in both ipsilateral and contralateral hemispheres at each rostrocaudal level.
Protocol for HPLC measurement of tissue monoamines
Frozen striata were homogenized in ice-cold buffer (0.1 M PCA, 0.1 mM EDTA, 2.5 mg·L–1 ascorbate), then centrifuged at 20 000× g for 15 min at 4°C. The supernatant was filtered, then assayed for dopamine by HPLC with electrochemical detection using a Hypersil BDS C18 analytical column (Fisher Scientific, Loughborough, UK). The mobile phase (0.1 M NaH2PO4, 0.1 M H3PO4, 2 mM 1-octane sulphonic acid, 1 mM EDTA, 13% methanol; pH 2.8) was pumped at 200 µL·min–1 without recycling. The limit of electrochemical detection by the Antec reference electrode (+0.75 V) was approximately 0.1 nM, and chromatographic analysis was carried out using Millennium 32 software.
Data handling and statistical analysis
All data are expressed as mean ± SEM, where n represents the number of animals in each experimental group. Statistical analyses were performed using GraphPad Prism (version 5.0), for each set of results as described below.
In vitro release
The amount of [3H]-d-aspartate present in each release fraction was calculated as % of total present in the sample at the start of that fraction. The total [3H]-d-aspartate content of S1 and S2 was calculated, from graphs of % release over time, by integrating the area under the respective peaks using Origin 7 Peak-fitting software (Aston Scientific, Buckinghamshire, UK). The mean S2/S1 ratio (an index of drug effectiveness) was calculated by pooling data from at least two independent experimental runs for that condition (minimum of six tissues from six rats). Differences in the S2/S1 ratio between treatment groups were analysed using a one-way anova with a Student–Newman–Keuls (SNK) post hoc test.
Microdialysis
Absolute glutamate levels were based on peak height from the chromatogram and compared to a 3-point calibration plot completed prior to every experiment. Baseline glutamate levels were averaged from the first three fractions and subsequent fractions expressed as % of baseline. Release evoked at S1 and S2 was calculated, as above, by integrating the area under the respective peaks and the S2/S1 ratio calculated to allow comparison between different groups using a one-way anova with an SNK post hoc test.
Reversal of reserpine-induced akinesia
The number of 360° contraversive rotations in 10 min intervals was counted from videotaped recordings and plotted as a time-course. Differences at each time point between drug and vehicle were analysed using a two-way anova and SNK post hoc test. The total number of rotations over 60 min was compared between different treatments using one-way anova and SNK post hoc test. The ability of CPPG to block responses to l-SOP or l-AP4 was analysed using an unpaired two-tailed t-test.
Measures of motor function in 6-OHDA-lesioned rats
Use of the contralateral paw in the cylinder test, expressed as a % of total reaches, was compared pre- and post-lesion between treatments using a two-way anova and Bonferroni post hoc test. In the adjusted stepping test, group differences in use of the contralateral paw post-lesion, expressed as a % pre-lesion, were compared using one-way anova with a Bonferroni post hoc test. Amphetamine-induced rotations in 60 min were also compared between groups using a one-way anova with a Bonferroni post hoc test.
Immunohistochemistry
For both TH and DDC staining in the striatum, the average optical density (OD) of three sections, corrected for background cortical staining, was obtained for each animal in each quadrant and at each rostrocaudal level. Preliminary analysis revealed no significant differences in staining either between striatal quadrants or between rostrocaudal levels for any given treatment group. Therefore, for final analysis, the OD of TH or DDC staining in the whole striatum was averaged across the three rostrocaudal levels for each animal. The mean OD of the lesion side, expressed as % of the contralateral intact side, was compared between treatment groups using a one-way anova with a Bonferroni post hoc test. For each animal, the number of TH-positive cells in three adjacent sections of SNpc was averaged to give a mean cell count per section for the ipsilateral and contralateral hemispheres at each level. The mean number of cells over the three rostrocaudal regions was then pooled so final numbers represent cell counts in the equivalent of three sections. TH-positive cell counts in the 6-OHDA-lesioned side, expressed as % of the contralateral intact hemisphere, were compared between treatment groups using a one-way anova with a Bonferroni post hoc test.
Dopamine HPLC
Dopamine content of striatal samples (ng·g–1 protein) was converted from peak areas of the chromatogram using a calibration curve of pure reference standards. Dopamine content of the 6-OHDA-lesioned side, expressed as a % of the contralateral intact side, was compared between treatment groups using a one-way anova with a Bonferroni post hoc test.
Materials
l-SOP, L-AP4 and CPPG were obtained from Tocris Cookson, Bristol, UK. [3H]-d-aspartate was obtained from Amersham, Little Chalfont, Bucks, UK. All other reagents were obtained from Sigma-Aldrich. For in vitro studies, l-SOP, l-AP4 and CPPG were dissolved in Krebs buffer.
Results
[3H]-d-aspartate release
Basal release of [3H]-d-aspartate from the SN prisms (∼1%) was in keeping with levels reported in studies using [3H]-d-aspartate as an index of glutamate release in other brain regions, such as the cortex (Meldrum et al., 1992). The Ca2+-dependent proportion of KCl-evoked [3H]-d-aspartate release (∼70%; data not shown) was also in agreement with others (Palmer and Reiter, 1994), and indicates that the majority of [3H]-d-aspartate released was likely to be of neuronal origin.
Both broad-spectrum group III mGlu receptor agonists inhibited 25 mM KCl-evoked [3H]-d-aspartate release in tissue prisms of the rodent SN. The profile of this inhibition can be seen for the optimum concentration of each agonist in Figure 1A,B. l-SOP produced a concentration-dependent inhibition of release (quantified as S2/S1 ratio), reaching significance at 30 (P < 0.05), 100 and 300 µM (P < 0.01; one-way anova with an SNK post hoc test) (Figure 1C). Maximal inhibition of 33 ± 10.8% (mean ± SEM, n = 6) was achieved with 100 µM l-SOP. At the highest concentration tested, 1000 µM, the inhibitory effects of l-SOP were lost or masked. l-AP4 produced a similar bell-shaped concentration-dependent inhibition of [3H]-d-aspartate release, reaching significance at 10 (P < 0.05) and 30 µM (P < 0.01; one-way anova with an SNK post hoc test) (Figure 1D), and a maximal inhibition of 44 ± 4.5% (mean ± SEM, n = 6) with 30 µM l-AP4. A loss of efficacy was again seen at the higher concentrations tested (100 and 300 µM). Pretreatment of tissues with CPPG (100 µM) had no effect on basal levels of [3H]-d-aspartate release, but almost completely abolished the ability of both l-SOP (100 µM) and l-AP4 (30 µM) to inhibit KCl-evoked [3H]-d-aspartate release (P < 0.001; one-way anova with an SNK post hoc test; Figure 1E,F), confirming that l-SOP and l-AP4 exert their effects via activation of group III mGlu receptors.
Figure 1.
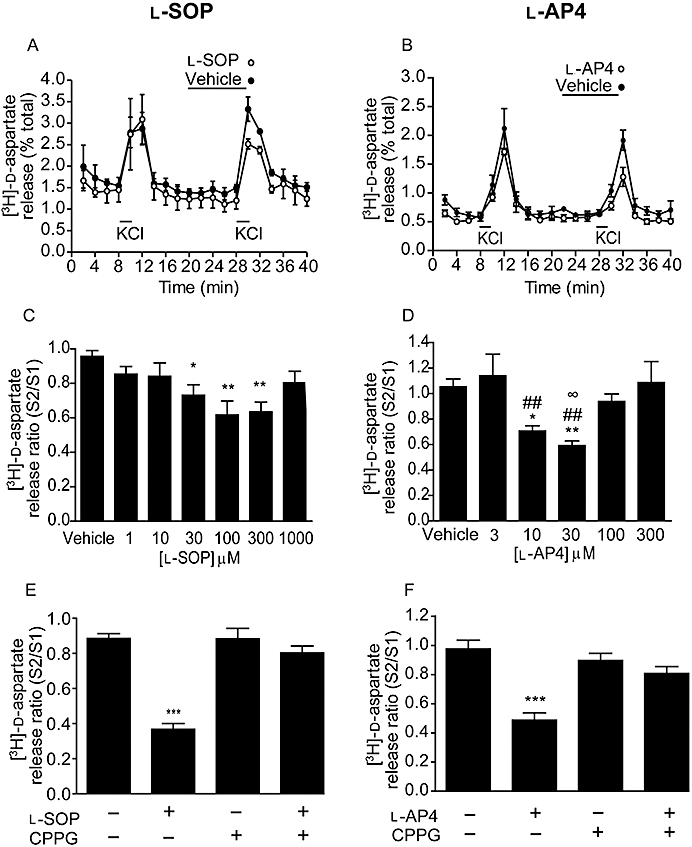
Effect of l-SOP (A,C,E) or l-AP4 (B,D,F) on 25 mM KCl-evoked [3H]-d-aspartate release in rat nigral tissue prisms. (A,B) Release profile showing effect of optimum concentration of l-SOP or l-AP4 on release evoked by the second KCl stimulus (S2). Horizontal bars indicate the periods of contact with KCl or drug/vehicle. Data are mean ± SEM (n = 3, from a single experimental run). (C,D) Graphs of S2/S1 ratio showing concentration-dependent effects of l-SOP or l-AP4 on [3H]-d-aspartate release. Data are mean ± SEM (n = 6); *P < 0.05 and **P < 0.01, significantly different from vehicle; ##P < 0.01 and ∞P < 0.05 significantly different from 3 and 100 µM l-AP4 respectively. (E,F) Effect of pre-incubation with the group III mGlu receptor antagonist, CPPG (100 µM) on responses to l-SOP (100 µM) or l-AP4 (30 µM). The presence (+) or absence (–) of drugs is indicated. Data are mean ± SEM (n = 6); ***P < 0.001 significantly different from other groups.
Microdialysis and glutamate HPLC
Pilot studies (n = 4 rats; data not shown) confirmed that 100 mM KCl increased extracellular glutamate levels in the SNpr approximately 2.5-fold over basal, in line with that of previous studies (Paulsen and Fonnum, 1989; Silverstein and Naik, 1991). Twenty minute incubations with l-SOP (100 and 300 µM) reduced KCl-evoked glutamate release at S2, while having no effect on basal glutamate levels (Figure 2A). Mean S2/S1 ratios revealed the inhibition of release with 300 µM l-SOP, which amounted to 48 ± 4.4% of control release (mean ± SEM; n = 4) was significant when compared to vehicle (P < 0.05; one-way anova with an SNK post hoc test; Figure 2B).
Figure 2.
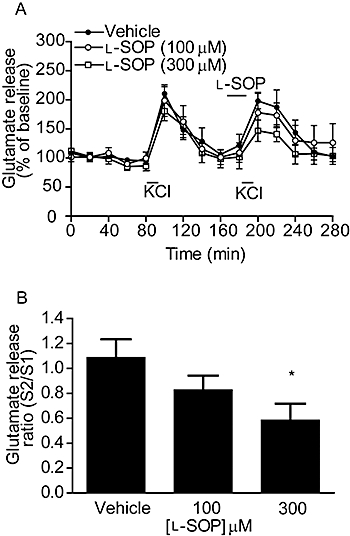
Effect of l-SOP on 100 mM KCl-evoked glutamate release in the rodent SNpr in vivo. (A) Release profile showing the effect of vehicle or l-SOP on release evoked by the second KCl stimulus, S2. Horizontal bars indicate the periods of contact with KCl or l-SOP/vehicle. (B) Graph of S2/S1 ratio showing concentration-dependent effect of l-SOP on glutamate release. In both cases, data are mean ± SEM (n = 4). *P < 0.05 significantly different from vehicle.
Reversal of akinesia in reserpine-treated rats following intranigral drug administration
l-SOP was examined over a range of doses selected on the basis of previously published data from this laboratory (MacInnes et al., 2004), while the lower doses of l-AP4 chosen reflect the lower concentrations of this agent required above to produce significant inhibition of [3H]-d-aspartate release in vitro. Intranigral injection of the maximally effective dose of l-SOP (750 nmol) produced net contraversive rotations which were significantly increased compared to vehicle (PBS) at 10 and 20 min post-injection (P < 0.001; two-way anova and SNK post hoc test), returning to baseline values by 40 min (Figure 3A). A similar profile of reversal of akinesia was seen with the maximally effective dose of l-AP4 (300 nmol; Figure 3B). Quantification of the total rotations produced in 60 min (Figure 3C,D) revealed that responses to both l-SOP (250–750 nmol) and l-AP4 (3–300 nmol) were dose dependent, reaching a maximum of 66 ± 18 rotations with 750 nmol l-SOP and 37 ± 5 rotations with 300 nmol l-AP4 (mean ± SEM, n = 6; P < 0.01 vs. respective vehicle controls; one-way anova with an SNK post hoc test). While administration of CPPG alone produced no change in net contraversive rotations compared to vehicle (data not shown), 30 min pretreatment with CPPG (75 nmol) significantly reduced the subsequent number of net contraversive rotations produced by 300 nmol l-AP4 and 750 nmol l-SOP by 70 and 80%, respectively (P < 0.001, unpaired two-tailed Student's t-test; Figure 3E,F).
Figure 3.
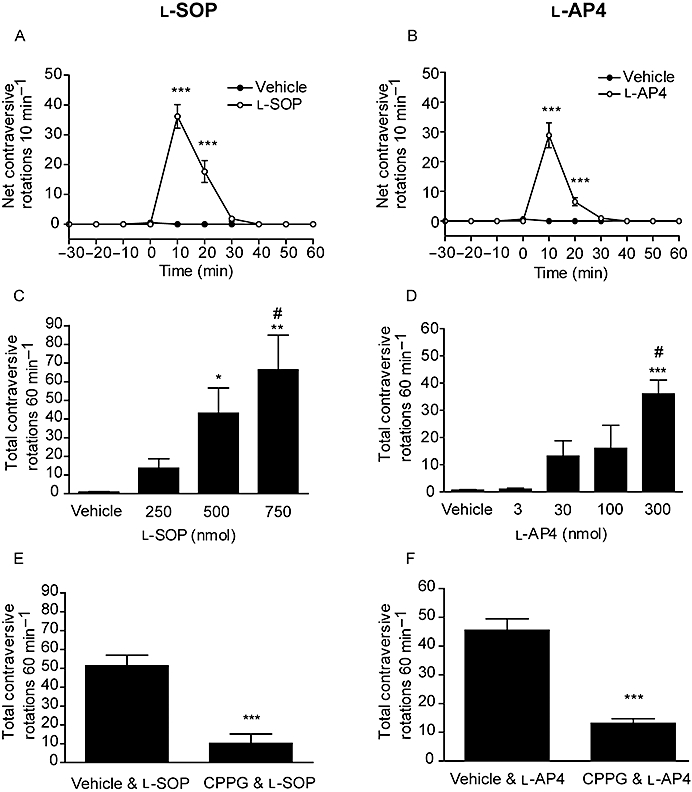
Effect of unilateral injection of l-SOP (A,C,E) or l-AP4 (B,D,F) in the SNpr on reversal of akinesia in the reserpine-treated rat. (A,B) Time-course of locomotor activity induced by the maximally effective dose of l-SOP or l-AP4. Data are mean ± SEM (n = 6); ***P < 0.001 significantly different from vehicle. (C,D) Effect of l-SOP or l-AP4 on rotational activity over 60 min. Data are mean ± SEM (n = 6). In (C), *P < 0.05 and **P < 0.01 significantly different from vehicle, #P < 0.05 significantly different from 250 nmol dose. In (D), ***P < 0.001 significantly different from vehicle and 3 nmol: #P < 0.05 significantly different from 30 and 100 nmol. (E,F) Effect of pretreatment with CPPG (75 nmol) on the rotational responses to l-SOP (750 nmol) or l-AP4 (300 nmol). Data are mean ± SEM (n = 6). ***P < 0.001 significantly different from vehicle pretreatment.
Behavioural indices of neuroprotection in the 6-OHDA-lesioned rat following supra-nigral injection of l-AP4
Supra-nigral injection of l-AP4 provided functional protection against 6-OHDA-induced loss of motor function. In the cylinder reaching test, pre-lesion use of the contralateral paw alone amounted, as expected, to ∼30% of total reaches made with contralateral paw alone plus ipsilateral paw alone plus both paws together. At 4 days post 6-OHDA lesion, use of the contralateral paw fell significantly in vehicle-treated animals (n = 9; P < 0.05; two-way anova and Bonferroni post hoc test; Figure 4A). In contrast, animals treated with l-AP4 (0.1–30 nmol) showed no reduction in contralateral paw use post-lesion (Figure 4A). In the adjusted steps test performed 6 days post 6-OHDA lesion, use of the contralateral paw in vehicle-treated animals fell to about 40% of pre-lesion use, for either forehand or backhand responses (n = 9; Figure 4B). l-AP4 treatment protected against loss of contralateral paw use in a dose-dependent manner, reaching significance at 3 nmol l-AP4 compared to vehicle treatment with contralateral paw, in forehand and backhand directions (n = 7; P < 0.05 (forehand) and P < 0.01 (backhand); one-way anova with a Bonferroni post hoc test). The number of amphetamine-induced ipsiversive rotations 7 days post-lesion was also significantly reduced in 6-OHDA-lesioned animals treated with l-AP4 (P < 0.05; one-way anova with a Bonferroni post hoc test; Figure 4C).
Figure 4.
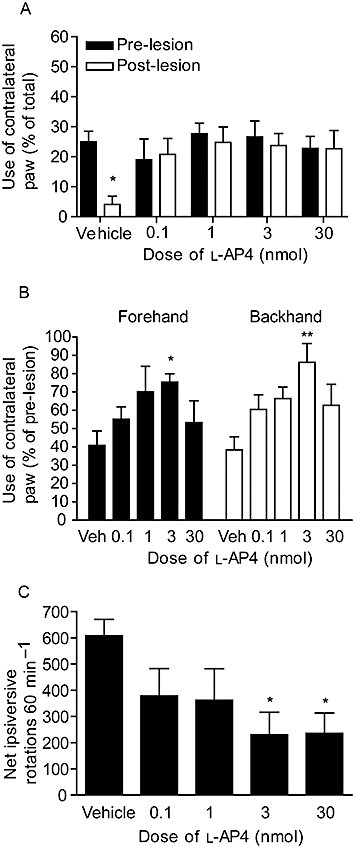
Effect of repeated l-AP4 treatment against 6-OHDA-induced (A) reduced contralateral paw use in the cylinder test at 4 days post-lesion, (B) reduced contralateral paw use in forehand or backhand adjusted stepping test at 6 days post-lesion and (C) increased amphetamine-induced ipsiversive rotations at 7 days post-lesion. Data are mean ± SEM (n = 9, vehicle; n = 6–7, l-AP4). *P < 0.05 and **P < 0.01, significantly different from (A) pre-lesion score or (B,C) vehicle.
Post-mortem indices of nigrostriatal tract integrity
Within the SNpc, the mean total number of TH-positive cells in three sections obtained across three rostrocaudal levels declined from 313 ± 16.7 in the intact hemisphere (i.e. ∼104 cells per section) to 62.6 ± 21.4 in the lesioned hemisphere (i.e. ∼21 per section) of vehicle-treated 6-OHDA-lesioned animals (n = 10), representing a reduction of around 80%. l-AP4 treatment produced a dose-dependent protection against this cell loss. Maximum protection was observed with 3 nmol l-AP4 when the number of TH-positive cells remaining in the lesioned hemisphere was 53.6 ± 5.2% of the intact hemisphere (n = 7), compared to an equivalent 20 ± 6.6% remaining in vehicle-treated animals (n = 10; P < 0.05; one-way anova with a Bonferroni post hoc test; Figure 5A,C). TH levels were also preserved within the striatum of l-AP4-treated animals (Figure 5B,C). In vehicle-treated animals (n = 10), TH levels in the lesioned striatum declined to 8.6 ± 1.7% of the intact side. In contrast, TH levels remained significantly higher (>67% of intact side) in all l-AP4 treatment groups (n = 6–8; P < 0.001 vs. vehicle treatment; one-way anova with a Bonferroni post hoc test). l-AP4 also prevented the decline in a second marker of dopaminergic neurones, DDC, confirming that the preserved levels of TH did not merely reflect increased expression of that marker (Figure 5D). When assayed using HPLC, striatal dopamine content was also found to be preserved following l-AP4 treatment. In vehicle-treated 6-OHDA-lesioned animals (n = 6), striatal dopamine content fell from 13 048 ± 1770 ng·g–1 in the intact hemisphere to 1433 ± 759 ng·g–1 in the lesioned hemisphere, representing a decline to 11.1 ± 5.6% of the intact hemisphere levels. l-AP4 treatment produced a dose-dependent preservation of striatal dopamine content, which reached significance compared to vehicle at the 3 nmol dose when dopamine content in the lesioned hemisphere remained at 78.9 ± 4.6% of intact hemisphere (P < 0.01; one-way anova with a Bonferroni post hoc test).
Figure 5.
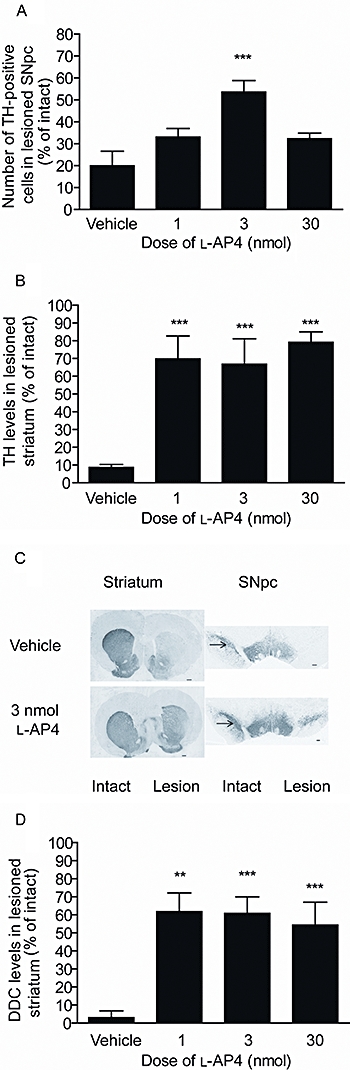
Effect of repeated l-AP4 treatment against 6-OHDA-induced (A) loss of TH-positive cells in the SNpc in the lesion hemisphere, (B) reduction in TH levels in the lesion striatum and (D) reduction in DDC levels. Data are mean ± SEM (A,B) n = 10 for vehicle; n = 6–7 for l-AP4; (D) n = 10 for vehicle; n = 4,7 and 8 for increasing doses of l-AP4. **P < 0.01 and ***P < 0.001 significantly different from vehicle (one-way anova with a Bonferroni post hoc test). (C) Photomicrographs showing TH staining in the striatum and SNpc of animals treated with the optimum dose of l-AP4 (3 nmol) or vehicle; lesion side on right. Scale bar: 500 µm for striatum; 200 µm for SNpc.
In a separate study (Figure 6), the protective effects of 3 nmol l-AP4 treatment against 6-OHDA-induced: (i) loss of contralateral paw function in the cylinder test at 4 days post-lesion and the adjusted steps test at 6 days post-lesion; (ii) increase in amphetamine-induced rotations at day 7 post-lesion; and (iii) loss of striatal dopamine content in the lesion hemisphere were found to be significantly inhibited by pretreatment with 75 nmol CPPG 30 min before each dose of l-AP4 (n = 6–7; P < 0.05; one-way anova with a Bonferroni post hoc test).
Figure 6.
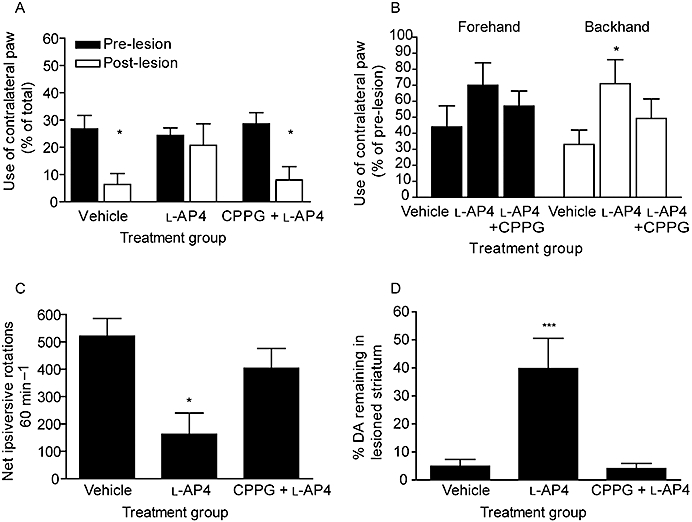
Effect of pretreatment with 75 nmol CPPG or vehicle on the protection afforded by 3 nmol l-AP4 against 6-OHDA-induced (A) reduced contralateral paw use in the cylinder test at 4 days post-lesion, (B) reduced contralateral paw use in forehand or backhand adjusted stepping test at 6 days post-lesion, (C) increased amphetamine-induced ipsiversive rotations at 7 days post-lesion and (D) reduction in striatal dopamine content. X-axis labels: vehicle, vehicle treatment; l-AP4, l-AP4 treatment following vehicle pretreatment; CPPG +l-AP4, l-AP4 treatment following CPPG pretreatment. Data are mean ± SEM (n = 6, vehicle; n = 7 l-AP4 and l-AP4 + CPPG). (A) *P < 0.05, significantly different from pre-lesion score (two-way anova and Bonferroni post hoc test). (B,C,D) *P < 0.05 and ***P < 0.001 significantly different from vehicle treatment and from CPPG pretreatment followed by l-AP4 (one-way anova with Bonferroni post hoc test).
Discussion
The present study shows that targeting group III mGlu receptors using the broad-spectrum agonists l-SOP or l-AP4 is an effective way to inhibit glutamate release in the SN, both in vitro and, more importantly, in vivo. Given that increased glutamate release in SNpr and SNpc is linked to Parkinsonian symptom generation and progressive nigral cell degeneration, respectively, it is encouraging to find that injection of l-SOP or l-AP4 into the SNpr reversed akinesia in the reserpine-treated rat model of PD, while infusion of l-AP4 above the SNpc prevented both the motor deficits and nigrostriatal tract degeneration in the 6-OHDA-lesioned rat model of PD. These responses were each inhibited by pretreatment with the group III mGlu receptor antagonist CPPG, confirming they were mediated by activation of group III mGlu receptors.
Effects of group III mGlu receptor activation on glutamate release in the SN
Under the present experimental conditions, ∼70% of the KCl-evoked [3H]-d-aspartate release in rat SN tissue prisms (comprising SNpc and SNpr) was Ca2+ dependent, indicating a neuronal origin. The source of the remaining fraction is unknown, but a glial origin is unlikely because external K+ concentrations of 50–55 mM are required for glial amino acid transmitter release (Bernath, 1992), and only 25 mM KCl was used here. The ability of l-SOP and l-AP4 to inhibit [3H]-d-aspartate release therefore suggests the presence of functional group III mGlu receptors on pre-synaptic terminals of excitatory neurones in the SN. Glutamatergic STN efferents comprise the major excitatory input to this region (Parent and Hazrati, 1995), leading to the conclusion that l-SOP and l-AP4 inhibit release from these so-called subthalamonigral neurones. This idea is consistent with the ability of l-AP4 to reduce STN-evoked EPSCs generated in both the SNpr (Wittmann et al., 2001b; 2002;), albeit at concentrations higher than those used here (100–1000 µM) and in the SNpc (Wigmore and Lacey, 1998; Valenti et al., 2005) at concentrations similar to those used here (between 1 and 100 µM). In further support of the subthalamonigral pathway being a key site of action of l-SOP and l-AP4, in situ hybridization studies have revealed mRNA encoding multiple group III mGlu receptors (mGlu4, mGlu7 and mGlu8) within the STN (Testa et al., 1994; Kosinski et al., 1999; Messenger et al., 2002), with corresponding immunoreactivity for at least two of these receptors (mGlu4 and mGlu7) reported on presumed glutamatergic terminals in the SNpr (Kosinski et al., 1999; Corti et al., 2002).
The inhibitory effects of l-SOP and l-AP4 on [3H]-d-aspartate release were lost at the highest concentrations examined (1000 and 300 µM respectively). Such a bell-shaped concentration–response relationship may reflect a loss of pharmacological specificity at the higher doses because l-AP4, which shares near-identical steric structure to l-SOP, is known to activate excitatory group I mGlu receptors with an EC50 of >1000 µM, compared to between 0.2 and 100 µM at the different group III mGlu receptors (Cartmell and Schoepp, 2000). However, given that activation of group I mGlu receptors has itself been shown to produce a similar functional inhibition of excitatory synaptic transmission in the SNpr at least (Wittmann et al., 2001a), this explanation seems unlikely. Increasing concentrations of l-AP4 will undoubtedly recruit activation of mGlu7 receptors (l-AP4 exhibits potencies of between 160 and 1300 µM at recombinant rat mGlu7; Schoepp et al., 1999) in addition to mGlu4 receptors (potencies of 0.2–1 µM). However, our preliminary data (reviewed in Duty, 2010) indicate that activation of both these receptor subtypes leads to a similar functional outcome so this more widespread receptor stimulation is again unlikely to be the cause of the loss of efficacy observed at higher concentrations. The most plausible explanation at this stage is that the loss of effect at higher concentrations may be a result of receptor desensitization, as suggested to explain the similar bell-shaped nature of electrophysiological responses of l-AP4 at the striatopallidal synapse (Valenti et al., 2003). Certainly, l-AP4 has been shown to induce rapid internalization of at least two of the group III mGlu receptors in cell lines, mGlu4 (at 100 µM; Iacovelli et al., 2004) and mGlu7 (at 400 µM; Pelkey et al., 2005), indicating that desensitization is a likely contributing factor. However, why such an effect is observed here, but not in the electrophysiological studies noted above (Wigmore and Lacey, 1998; Wittmann et al., 2001b; 2002; Valenti et al., 2005), remains to be established.
The microdialysis data showing that l-SOP (300 µM) inhibits depolarization-evoked endogenous glutamate release in the SNpr of anaesthetized rats are consistent with the above in vitro effects, and, most importantly, confirm that this inhibitory effect of group III mGlu receptor activation is preserved in vivo. Although not directly examined here, the source of glutamate release affected in these studies is again most likely neuronal, because the high KCl stimulus used (100 mM) evokes increases in transmitter release that are both Ca2+ and tetrodotoxin sensitive (Paulsen and Fonnum, 1989; Campbell et al., 1993).
Having demonstrated the ability of group III mGlu receptor agonists to reduce glutamate release in the SN, subsequent studies examined whether group III mGlu receptors in this region were useful targets for combating some of the physiological and pathological consequences of overactive glutamate transmission in PD.
Anti-Parkinsonian effects of group III mGlu receptor activation in the SNpr
In line with previous findings from this laboratory (MacInnes et al., 2004), activation of group III mGlu receptors in the SNpr with l-SOP produced anti-Parkinsonian effects in the reserpine-treated rat which were reflected in a unilateral reversal of akinesia on the injected side and manifest as contraversive rotations. This study showed that a second agonist, l-AP4, produced an almost identical reversal of akinesia following intranigral injection. Although the group III mGlu receptor antagonist, CPPG (75 nmol), produced a marked inhibition of these responses, confirming receptor mediation, it remains to be determined whether higher doses of CPPG would have inhibited the residual component of these responses. Within 2 h of injection, reserpine produces ∼85% loss of dopamine in the SNpc, and >95% dopamine depletion in the striatum (Heeringa and Abercrombie, 1995). Although dopamine content in the SNpc returns to ∼30% by 24 h after reserpine injection (i.e. when behaviour was examined here) animals are still completely akinetic at this stage, suggesting that their behavioural responses closely mirror the striatal dopamine depletion which persists at >95% for at least 24 h (Heeringa and Abercrombie, 1995). Under these conditions, STN firing is increased approximately 50% (Robledo and Feger, 1991) and is predicted to elevate extracellular glutamate in the SNPr. Therefore, although direct measurements of glutamate release were not made here in the reserpine-treated rats, the reversal of akinesia following group III mGlu receptor activation most likely reflects inhibition of glutamate release from STN terminals in the SNpr.
Injections into the SNpr of another broad-spectrum agonist, 1-aminocyclo-pentane-1,3,4-tricarboxylic acid (ACPT-I), have also been shown to reduce Parkinsonian symptoms in the haloperidol-induced catalepsy model (Konieczny et al., 2007), adding further support to the target potential of group III mGlu receptors in the SNpr in PD. However, within the SNpr, group III mGlu receptors also exist on pre-synaptic terminals of the GABAergic striatonigral pathway, where their activation inhibits striatal-evoked inhibitory post-synaptic currents (IPSCs; Wittmann et al., 2001b). Such inhibition of GABAergic transmission is predicted to worsen Parkinsonian symptoms by further increasing firing of the SNpr, rather than improve them as seen here. This suggests that under conditions of striatal dopamine depletion with reserpine (present study), or dopamine receptor blockade with haloperidol (Konieczny et al., 2007), group III mGlu agonist actions on subthalamonigral terminals (resulting in reduced glutamatergic drive to the SNpr) override those on striatonigral terminals (that would inhibit GABAergic drive to the SNpr). This interpretation is wholly consistent with the in vitro findings of Wittmann et al. (2002). They demonstrated that in brain slices obtained from acute (2 h) reserpine-treated animals bathed continuously in reserpine in vitro, or in control slices incubated with haloperidol, while the ability of l-AP4 (500 µM) to inhibit STN-evoked EPSCs in the SNpr was retained, the ability of l-AP4 to inhibit striatal-evoked IPSCs was lost. Conversely, under conditions where dopamine levels remain sufficient, actions on the striatonigral pathway are believed to predominate. This distinction may help explain the findings of Lopez et al. (2007) who reported induction of catalepsy following injection of ACPT-I into the SNpr of naïve animals and a worsening of akinesia in partially lesioned rats, bearing only 60% loss of striatal dopamine innervation as assessed by [3H]mazindol binding. Clearly, it will be important in the future to identify the critical threshold level for striatal dopamine at which the beneficial effects seen here are lost, in order to fully understand the implications of these findings.
Neuroprotective effects of group III mGlu receptor activation in the SNpc
The final study was designed to examine the neuroprotective potential of targeting group III mGlu receptors in the SNpc. For these studies, we used a severe lesion model in which 6-OHDA was injected directly into the SNpc to induce rapid cell death. Under this paradigm, the majority of cell death occurs within the first 3 days of injection (Zuch et al., 2000), and for this reason neuroprotective drug interventions are often, as in this case, introduced just prior to toxin injection. Such a design does not of course reflect the timing of drug interventions in the clinic, and future studies should aim to reinforce these findings in a slower retrograde lesion model in which there is a sufficient time window to delay treatment.
Accepting these caveats, these studies revealed that l-AP4, administered 1 h before and for 7 days after 6-OHDA injection, protected against a nigrostriatal tract lesion. This was expressed by a preservation of TH-positive cell numbers in the SNpc, as well as a reduction in 6-OHDA-induced loss of striatal TH immunoreactivity and dopamine levels. These effects were maximal around 3 nmol l-AP4 and, in the case of TH-positive cell counts and striatal dopamine content, were lost at the highest doses tested. These data are consistent with those of Vernon et al. (2007) who found maximal histological and neurochemical protection against 6-OHDA lesion with 10 nmol l-AP4 and a similar loss of effect with higher doses. This bell-shaped dose–response relationship is in keeping with the above in vitro release data and may likewise reflect desensitization of group III mGlu receptors at the higher doses examined. However, given the difficulty of predicting the exact concentration of agonist reaching the receptors following intracerebral doses, such suggestions remain speculative at this stage. It is notable in this study that the protection afforded at the striatal level was greater and occurred at slightly lower doses than required to give significant protection at the nigral level. This outcome was not surprising as we introduced the toxin directly into the cell bodies here so it may be harder to protect that area compared to the terminals. Indeed, more robust protection is often seen in the 6-OHDA-lesioned rats at the striatal level [e.g. with AMPA potentiators (Murray et al., 2003) and nicotinic agonists (Visanji et al., 2006)]. Alternatively, this discrepancy may reflect the different parameters being measured in these regions: cell numbers in the SNpc versus density of terminal staining in the striatum. Given that there is huge compensation in the nigrostriatal system, it is possible that small changes at the level of the terminals can have effects on function, whereas the level of protection may need to be greater to see clear effects on TH-positive cell counts.
In addition to confirming the neuroprotective effects of l-AP4 at a histological and neurochemical level, this study demonstrated for the first time that the protection observed against a 6-OHDA lesion translated to preservation of motor function. Thus, a significant preservation of contralateral paw use was shown in the adjusted steps test and cylinder test of forelimb akinesia following l-AP4 treatment, as well as a reduction in the degree of amphetamine-induced rotations. These positive behavioural outcomes indicated that the improvement in striatal dopamine levels in the lesioned hemisphere reduced striatal dopamine asymmetry between this and the intact hemisphere sufficiently to improve motor function.
In light of the present release data and the electrophysiological studies showing that l-AP4 inhibits STN-evoked EPSCs in the SNpc (Wigmore and Lacey, 1998; Katayama et al., 2003; Valenti et al., 2005), the neuroprotective effects of l-AP4 may result from inhibition of glutamate release from STN terminals in the SNpc. However, additional mechanisms may also contribute to the neuroprotection following group III mGlu receptor activation (reviewed in Duty, 2010). For example, group III mGlu receptors are also expressed on glial cells (astrocytes and microglia) (Taylor et al., 2003; Yao et al., 2005), and activation of these receptors is believed to underpin the l-AP4-mediated protection of dopaminergic neurones in culture against complex I inhibitor MPP+ by driving glutamate re-uptake into astrocytes (Yao et al., 2005). Because considerable microglial activation occurs in the 6-OHDA lesion model of PD (Marinova-Mutafchieva et al., 2009), it is conceivable that glial-mediated actions may contribute to the neuroprotection observed. The likely contribution from multiple mechanisms may explain why protection with l-AP4 was maximal with a relatively low dose (3 nmol) compared to that required to produce maximal reversal of reserpine-induced akinesia (300 nmol l-AP4), which most probably relies on inhibition of glutamate release alone. Although there is little in the way of changed expression of group III mGlu receptors following a 6-OHDA lesion (Messenger et al., 2002), a post-lesion increase in sensitivity of the receptors may also explain why lower doses of l-AP4 were required in this model compared to the acute reserpine model.
In conclusion, this study reveals that targeting group III mGlu receptors may offer a unifying means of treating both the symptoms and progressive degeneration in PD, and highlights normalization of glutamate release in the SN as one key event in mediating these actions. Studies are currently underway to identify which specific group III mGlu receptors are involved in mediating these beneficial actions. Current evidence points towards the involvement of mGlu4 receptors in both symptom relief and neuroprotection (Marino et al., 2003; Battaglia et al., 2006), although whether mGlu7 or mGlu8 receptors play important roles awaits clarification.
Acknowledgments
P.J.A., M.J.B. and M.B. contributed equally to this work and were in receipt of studentship funding from the Medical Research Council, BBSRC and Guy's & St Thomas' Charitable Trustees respectively. M.J.B.'s award was a Case Studentship supported additionally by Eli Lilly & Co Ltd.
Glossary
Abbreviations:
- 6-OHDA
6-hydroxydopamine
- CPPG
(RS)-α-cyclopropyl-4-phosphonophenylglycine
- l-AP4
l-2-amino-4-phosphonobutyrate
- l-SOP
O-phospho-l-serine
- mGlu
metabotropic glutamate
- PD
Parkinson's disease
- SNpr/pc
substantia nigra pars reticulata/pars compacta
- STN
subthalamic nucleus
Conflicts of interest
None to declare.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez L, Macias R, Lopez G, Alvarez E, Pavon N, Rodriguez-Oroz MC, et al. Bilateral subthalamotomy in Parkinson's disease: initial and long-term response. Brain. 2005;128:570–583. doi: 10.1093/brain/awh397. [DOI] [PubMed] [Google Scholar]
- Battaglia G, Busceti CL, Molinaro G, Biagioni F, Traficante A, Nicoletti F, et al. Pharmacological activation of mGlu4 metabotropic glutamate receptors reduces nigrostriatal degeneration in mice treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J Neurosci. 2006;27:7222–7229. doi: 10.1523/JNEUROSCI.1595-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernath S. Calcium-independent release of amino acid neurotransmitters: fact or artefact? Prog Neurobiol. 1992;38:57–91. doi: 10.1016/0301-0082(92)90035-d. [DOI] [PubMed] [Google Scholar]
- Campbell K, Kalen P, Lundberg C, Wictorin K, Rosengren E, Bjorklund A. Extracellular γ-amino butyric acid levels in the rat caudate-putamen: monitoring the neuronal and glial contribution by intracerebral microdialysis. Brain Res. 1993;614:241–250. doi: 10.1016/0006-8993(93)91041-p. [DOI] [PubMed] [Google Scholar]
- Cartmell J, Schoepp DD. Regulation of neurotransmitter release by metabotropic glutamate receptors. J Neurochem. 2000;75:889–907. doi: 10.1046/j.1471-4159.2000.0750889.x. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Corti C, Aldegheri L, Somogyi P, Ferraguti F. Distribution and synaptic localisation of the metabotropic glutamate receptor 4 (mGlu receptor4) in the rodent CNS. Neurosci. 2002;110:403–420. doi: 10.1016/s0306-4522(01)00591-7. [DOI] [PubMed] [Google Scholar]
- Duty S. Therapeutic potential of targeting group III metabotropic glutamate receptors in the treatment of Parkinson's disease. Br J Pharmacol. 2010 doi: 10.1111/j.1476-5381.2010.00882.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferkany JW, Coyle JT. Evoked release of aspartate and glutamate: disparities between prelabeling and direct measurement. Brain Res. 1983;278:279–282. doi: 10.1016/0006-8993(83)90254-8. [DOI] [PubMed] [Google Scholar]
- Heeringa MJ, Abercrombie ED. Biochemistry of somatodendritic dopamine release in substantia nigra: an in vivo comparison with striatal dopamine release. J Neurochem. 1995;65:192–200. doi: 10.1046/j.1471-4159.1995.65010192.x. [DOI] [PubMed] [Google Scholar]
- Iacovelli L, Capobianco L, Iula M, Di Giorgi Gerevini V, Picascia A,, Blahos J, et al. Regulation of mGlu4 metabotropic glutamate receptor signaling by type-2 G-protein coupled receptor kinase (GRK2) Mol Pharmacol. 2004;65:1103–1110. doi: 10.1124/mol.65.5.1103. [DOI] [PubMed] [Google Scholar]
- Iribe Y, Moore K, Pang KC, Tepper JM. Subthalamic stimulation-induced synaptic responses in substantia nigra pars compacta dopaminergic neurons in vitro. J Neurophysiol. 1999;82:925–933. doi: 10.1152/jn.1999.82.2.925. [DOI] [PubMed] [Google Scholar]
- Katayama J, Akaike N, Nabekura J. Characterization of pre- and post-synaptic metabotropic glutamate receptor-mediated inhibitory responses in substantia nigra dopamine neurons. Neurosci Res. 2003;45:101–115. doi: 10.1016/s0168-0102(02)00202-x. [DOI] [PubMed] [Google Scholar]
- Konieczny J, Wardas J, Kuter K, Pilc A, Ossowska K. The influence of group III metabotropic glutamate receptor stimulation by (1S,3R,4S)-1-aminocyclo-pentane-1,3,4-tricarboxylic acid on the parkinsonian-like akinesia and striatal proenkephalin and prodynorphin mRNA expression in rats. Neurosci. 2007;145:611–620. doi: 10.1016/j.neuroscience.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Kosinski CM, Risso BS, Conn PJ, Levey AI, Landwehrmeyer GB, Penney JB, et al. Localization of metabotropic glutamate receptor 7 mRNA and mGlu receptor 7a protein in the rat basal ganglia. J Comp Neurol. 1999;415:266–284. [PubMed] [Google Scholar]
- Lopez S, Turle-Lorenzo N, Acher F, De LE, Mele A, Amalric M. Targeting group III metabotropic glutamate receptors produces complex behavioral effects in rodent models of Parkinson's disease. J Neurosci. 2007;27:6701–6711. doi: 10.1523/JNEUROSCI.0299-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacInnes N, Duty S. Group III metabotropic glutamate receptors act as hetero-receptors modulating evoked GABA release in the globus pallidus in vivo. Eur J Pharmacol. 2008;580:95–99. doi: 10.1016/j.ejphar.2007.10.030. [DOI] [PubMed] [Google Scholar]
- MacInnes N, Messenger MJ, Duty S. Activation of group III metabotropic glutamate receptors in selected regions of the basal ganglia alleviates akinesia in the reserpine-treated rat. Br J Pharmacol. 2004;141:15–22. doi: 10.1038/sj.bjp.0705566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino MJ, Williams DL, Jr, O'Brien JA, Valenti O, McDonald TP, Clements MK, et al. Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach to Parkinson's disease treatment. Proc Natl Acad Sci U S A. 2003;100:13668–13673. doi: 10.1073/pnas.1835724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinova-Mutafchieva L, Sadeghian M, Broom L, Davis JB, Medhurst AD, Dexter DT. Relationship between microglial activation and dopaminergic neuronal loss in the substantia nigra: a time course study in a 6-hydroxydopamine model of Parkinson's disease. J Neurochem. 2009;110:966–975. doi: 10.1111/j.1471-4159.2009.06189.x. [DOI] [PubMed] [Google Scholar]
- Meldrum MJ, Glenton P, Dawson R. [3H]d-aspartic acid release in brain slices of adult and aged Fischer 344 rats. Neurochem Res. 1992;17:151–156. doi: 10.1007/BF00966793. [DOI] [PubMed] [Google Scholar]
- Messenger MJ, Dawson LG, Duty S. Changes in metabotropic glutamate receptor 1–8 gene expression in the rodent basal ganglia motor loop following lesion of the nigrostriatal tract. Neuropharmacol. 2002;43:261–271. doi: 10.1016/s0028-3908(02)00090-4. [DOI] [PubMed] [Google Scholar]
- Murray TK, Whalley K, Robinson CS, Ward MA, Hicks CA, Lodge D, et al. LY503430, a novel alpha-amino-3-hydroxy-5-methylisoxazole-4-propioninc acid receptor potentiator with functional, neuroprotective and neuroptrophic effects in rodent models of Parkinson's disease. J Pharmacol Exp Ther. 2003;306:752–762. doi: 10.1124/jpet.103.049445. [DOI] [PubMed] [Google Scholar]
- Palmer AM, Reiter CT. Comparison of the superfused efflux of preaccumulated d-[3H]aspartate and endogenous l-aspartate and l-glutamate from rat cerebrocortical minislices. Neurochem Int. 1994;25:441–450. doi: 10.1016/0197-0186(94)90020-5. [DOI] [PubMed] [Google Scholar]
- Parent A, Hazrati LN. Functional anatomy of the basal ganglia. II. The place of subthalamic nucleus and external pallidum in basal ganglia circuitry. Brain Res Rev. 1995;20:128–154. doi: 10.1016/0165-0173(94)00008-d. [DOI] [PubMed] [Google Scholar]
- Paulsen RE, Fonnum F. Role of glial cells for the basal and Ca2+-dependent K+-evoked release of transmitter amino acids investigated by microdialysis. J Neurochem. 1989;52:1823–1829. doi: 10.1111/j.1471-4159.1989.tb07263.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. New York: Academic Press; 1998. [Google Scholar]
- Pelkey KA, Lavezzari G, Racca C, Roche KW, McBain CJ. mGlu receptor7 is a metaplastic switch controlling bidirectional plasticity of feedforward inhibition. Neuron. 2005;46:89–102. doi: 10.1016/j.neuron.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Peppe A, Pierantozzi M, Bassi A, Altibrandi MG, Brusa L, Stefani A, et al. Stimulation of the subthalamic nucleus compared with the globus pallidus internus in patients with Parkinson disease. J Neurosurg. 2004;101:195–200. doi: 10.3171/jns.2004.101.2.0195. [DOI] [PubMed] [Google Scholar]
- Piallat B, Benazzouz A, Benabid AL. Subthalamic nucleus lesion in rats prevents dopaminergic nigral neuron degeneration after striatal 6-OHDA injection: behavioural and immunohistochemical studies. E J Neurosci. 1996;8:1408–1414. doi: 10.1111/j.1460-9568.1996.tb01603.x. [DOI] [PubMed] [Google Scholar]
- Pothecary CA, Jane DE, Salt TE. Reduction of excitatory transmission in the retino-collicular pathway via selective activation of mGlu8 receptors by DCPG. Neuropharmacol. 2002;43:231–234. doi: 10.1016/s0028-3908(02)00077-1. [DOI] [PubMed] [Google Scholar]
- Robledo P, Feger J. Acute monoaminergic depletion in the rat potentiates the excitatory effect of the subthalamic nucleus in the substantia nigra pars reticulata but not in the pallidal complex. J Neural Transm Gen Sect. 1991;86:115–126. doi: 10.1007/BF01250572. [DOI] [PubMed] [Google Scholar]
- Rodriguez MC, Obeso JA, Olanow CW. Subthalamic nucleus-mediated excitotoxicity in Parkinson's disease: a target for neuroprotection. Ann Neurol. 1998;44:S175–S188. doi: 10.1002/ana.410440726. [DOI] [PubMed] [Google Scholar]
- Savage DD, Galindo R, Queen SA, Paxton LL, Allan AM. Characterization of electrically evoked [3H]-d-aspartate release from hippocampal slices. Neurochem Int. 2001;38:255–267. doi: 10.1016/s0197-0186(00)00077-2. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Jane DE, Monn JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacol. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- Silverstein FS, Naik B. Effect of depolarization on striatal amino acid efflux in perinatal rats: an in vivo microdialysis study. Neurosci Let. 1991;128:133–136. doi: 10.1016/0304-3940(91)90777-q. [DOI] [PubMed] [Google Scholar]
- Smith Y, Hazrati LN, Parent A. Efferent projections of the subthalamic nucleus in the squirrel monkey as studied by the PHA-L anterograde tracing method. J Comp Neurol. 1990;294:306–323. doi: 10.1002/cne.902940213. [DOI] [PubMed] [Google Scholar]
- Stocchi F, Nordera G, Marsden CD. Strategies for treating patients with advanced Parkinson's diease and disastrous fluctuations and dyskinesias. Clinical Neuropharmacol. 1997;20:95–115. doi: 10.1097/00002826-199704000-00001. [DOI] [PubMed] [Google Scholar]
- Taylor DL, Diemel LT, Pocock JM. Activation of microglial group III metabotropic glutamate receptors protects neurons against microglial neurotoxicity. J Neurosci. 2003;23:2150–2160. doi: 10.1523/JNEUROSCI.23-06-02150.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa CM, Standaert DG, Young AB, Penney JB., Jr Metabotropic glutamate receptor mRNA expression in the basal ganglia of the rat. J Neurosci. 1994;14:3005–3018. doi: 10.1523/JNEUROSCI.14-05-03005.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JP, Salt TE. Group III metabotropic glutamate receptors control corticothalamic synaptic transmission in the rat thalamus in vitro. J Physiol. 1999;519(Pt 2):481–491. doi: 10.1111/j.1469-7793.1999.0481m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti O, Marino MJ, Wittmann M, Lis E, DiLella AG, Kinney GG, et al. Group III metabotropic glutamate receptor-mediated modulation of the striatopallidal synapse. J Neurosci. 2003;23:7218–7226. doi: 10.1523/JNEUROSCI.23-18-07218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti O, Mannaioni G, Seabrook GR, Conn PJ, Marino MJ. Group III metabotropic glutamate receptor-mediated modulation of excitatory transmission in the rodent substantia nigra pars compacta dopamine neurons. J Pharmacol Exp Ther. 2005;313:1296–1304. doi: 10.1124/jpet.104.080481. [DOI] [PubMed] [Google Scholar]
- Vernon AC, Zbarsky V, Datla KP, Dexter DT, Croucher MJ. Selective activation of group III metabotropic glutamate receptors by l-(+)-2-amino-4-phosphonobutyric acid protects the nigrostriatal system against 6-hydroxydopamine toxicity in vivo. J Pharmacol Exp Ther. 2007;320:397–409. doi: 10.1124/jpet.106.108159. [DOI] [PubMed] [Google Scholar]
- Vila M, Marin C, Ruberg M, Jimenez A, Raisman-Vozari R, Agid Y, et al. Systemic administration of NMDA and AMPA receptor antagonists reverses the neurochemical changes induced by nigrostriatal denervation in basal ganglia. J Neurochem. 1999;73:344–352. doi: 10.1046/j.1471-4159.1999.0730344.x. [DOI] [PubMed] [Google Scholar]
- Visanji NP, O'Neill MJ, Duty S. Nicotine, but neither the α4β2 ligand RJR2403 nor an α7 nAChR subtype selective agonist, protects against a partial 6-hydroxydopamine lesion of the rat median forebrain bundle. Neuropharmacol. 2006;51:506–516. doi: 10.1016/j.neuropharm.2006.04.015. [DOI] [PubMed] [Google Scholar]
- Wigmore MA, Lacey MG. Metabotropic glutamate receptors depress glutamate-mediated synaptic input to rat midbrain dopamine neurones in vitro. Br J Pharmacol. 1998;123:667–674. doi: 10.1038/sj.bjp.0701662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windels F, Carcenac C, Poupard A, Savasta M. Pallidal origin of GABA release within the substantia nigra pars reticulata during high-frequency stimulation of the subthalamic nucleus. J Neurosci. 2005;25:5079–5086. doi: 10.1523/JNEUROSCI.0360-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann M, Hubert GW, Smith Y, Conn PJ. Activation of metabotropic glutamate receptor 1 inhibits glutamatergic transmission in the substantia nigra pars reticulata. Neuroscience. 2001a;105:881–889. doi: 10.1016/s0306-4522(01)00254-8. [DOI] [PubMed] [Google Scholar]
- Wittmann M, Marino MJ, Bradley SR, Conn PJ. Activation of group III mGlu receptors inhibits GABAergic and glutamatergic transmission in the substantia nigra pars reticulata. J Neurophysiol. 2001b;85:1960–1968. doi: 10.1152/jn.2001.85.5.1960. [DOI] [PubMed] [Google Scholar]
- Wittmann M, Marino MJ, Conn PJ. Dopamine modulates the function of group II and group III metabotropic glutamate receptors in the substantia nigra pars reticulata. J Pharmacol Exp Ther. 2002;302:433–441. doi: 10.1124/jpet.102.033266. [DOI] [PubMed] [Google Scholar]
- Wright IK, Upton N, Marsden CA. Effect of established and putative anxiolytics on extracellular 5-HT and 5-HIAA in the ventral hippocampus of rats during behaviour on the elevated X-maze. Psychopharmacol (Berl) 1992;109:338–346. doi: 10.1007/BF02245882. [DOI] [PubMed] [Google Scholar]
- Yao HH, Ding JH, Zhou F, Wang F, Hu LF, Sun T, et al. Enhancement of glutamate uptake mediates the neuroprotection exerted by activating group II or III metabotropic glutamate receptors on astrocytes. J Neurochem. 2005;92:948–961. doi: 10.1111/j.1471-4159.2004.02937.x. [DOI] [PubMed] [Google Scholar]
- Zuch CJ, Nordstroem VK, Briedrick LA, Hoernig GR, Granholm AC, Bickford PC. Time course of degenerative alterations in nigral dopaminergic neurons following a 6-hydroxydopamine lesion. J Comp Neurol. 2000;427:440–454. doi: 10.1002/1096-9861(20001120)427:3<440::aid-cne10>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]