

Abstract
Background and purpose:
The aim of this study was to determine whether the apelin/APJ system is altered in human cardiovascular disease by investigating whether the expression of apelin or its receptor is altered at the protein level.
Experimental approach:
Radioligand binding studies were used to determine apelin receptor density in human cardiac tissues. Apelin peptide levels in cardiovascular tissues were determined by radioimmunoassay. In vitro pharmacology was used to assess vasoactive properties of apelin in human coronary artery. Localization of apelin and its receptor in coronary artery was determined using immunohistochemistry.
Key results:
Apelin receptor density was significantly decreased in left ventricle from patients with dilated cardiomyopathy or ischaemic heart disease compared with controls, but apelin peptide levels remained unchanged. Apelin was up-regulated in human atherosclerotic coronary artery and this additional peptide localized to the plaque, colocalizing with markers for macrophages and smooth muscle cells. Apelin potently constricted human coronary artery.
Conclusions and implications:
We have detected changes in the apelin/APJ system in human diseased cardiac and vascular tissue. The decrease in receptor density in heart failure may limit the positive inotropic actions of apelin, contributing to contractile dysfunction. The contribution of the increased apelin levels in atherosclerotic coronary artery to disease progression remains to be determined. These data suggest a potential role for the apelin/APJ system in human cardiovascular disease.
Keywords: apelin, APJ, dilated cardiomyopathy, ischaemic heart disease, atherosclerosis, coronary artery, human, G-protein coupled receptor
Introduction
The apelin receptor (APJ) is a class A G-protein coupled receptor (GPCR), discovered in 1993 by homology cloning (O'Dowd et al., 1993) and designated an ‘orphan’ until 1998 when its endogenous ligand was identified as apelin (Tatemoto et al., 1998). Since this pairing, a number of roles for the apelin/APJ system have emerged including regulation of fluid homeostasis, the adipoinsular axis and the cardiovascular system (Kleinz and Davenport, 2005; Masri et al., 2005; Lee et al., 2006; Carpénéet al., 2007; Japp and Newby, 2008; Ladeiras-Lopes et al., 2008; Pitkin et al., 2010).
In the human cardiovascular system, apelins have been shown to modulate cardiac contractility (Maguire et al., 2009) and vascular tone in vitro, both as an endothelium-dependent vasodilator and endothelium-independent vasoconstrictor (Katugampola et al., 2001; Salcedo et al., 2007; Maguire et al., 2009). Apelin has also been shown to cause vasodilatation when infused into the human forearm (Japp et al., 2008). Apelin is a potent angiogenic factor (Kasai et al., 2004; Cox et al., 2006) and mitogen of endothelial (Kasai et al., 2004; Masri et al., 2004) and vascular smooth muscle cells (Li et al., 2008) in vitro.
There is evidence for a role of the apelin/APJ system in cardiovascular disease. There have been a number of reports of changes in plasma apelin levels in patients with heart failure, with somewhat disparate results. Overall plasma apelin appears to rise in early heart failure (Chen et al., 2003), but normalize or decrease in later stages (Chen et al., 2003; Chong et al., 2006; Miettinen et al., 2007). The source of plasma apelin is unclear and it is not known how this relates to tissue levels.
Cardiac apelin is up-regulated by hypoxia (Ronkainen et al., 2007) and the expression of both apelin and APJ is increased in ischaemic heart failure in rats (Atluri et al., 2007; Sheikh et al., 2008). Apelin appears to be beneficial in heart failure in animal models, protecting against ischaemia reperfusion injury (Simpkin et al., 2007; Zeng et al., 2009), having positive inotropic effects when administered acutely and improving cardiac function without evidence of hypertrophy when chronically administered (Ashley et al., 2005). Conversely, mice lacking the gene encoding apelin develop impaired cardiac contractility in response to ageing or pressure overload (Kuba et al., 2007). In man, APLNR was found to be the most significantly up-regulated gene after mechanical offloading of failing myocardium (Chen et al., 2003) and the G212A variant of this gene is associated with slower heart failure progression (Sarzani et al., 2007). Foldes et al. (2003) found a decrease in apelin receptor mRNA in human cardiac tissue from patients with ischaemic heart disease (IHD) or idiopathic dilated cardiomyopathy (DCM). However, it is unknown whether this is reflected in changes in the apelin/APJ system at the protein level in man.
There is recent evidence of a role for the apelin/APJ system in atherosclerosis in mice. Apelin receptor deficient mice, on an ApoE−/− background, have reduced atherosclerotic burden compared with ApoE−/− mice (Hashimoto et al., 2007). This contrasts with the findings of Chun et al. (2008), that apelin signalling opposed angiotensin II-induced atherosclerosis in ApoE−/− mice. However, the involvement of the apelin/APJ system in atherosclerosis in man has not been investigated.
This study has revealed a significant down-regulation of apelin receptor protein in the myocardium of failing hearts and an up-regulation of apelin peptide in atherosclerotic coronary artery. This implicates the apelin/APJ system in the pathogenesis of both atherosclerosis and heart failure in man. Preliminary data were presented to the British Pharmacological Society (Pitkin et al., 2007; 2008a,b; 2009;)
Methods
Tissue collection
Human tissue was collected with local ethical approval and informed consent. Human heart, coronary artery and epicardial adipose tissues were from patients undergoing cardiac transplantation for DCM or IHD, or from control hearts from donors where there was no suitable recipient. Cardiac transplant patients were on a range of drugs including diuretics, positive inotropes, anti-arrhythmics, anticoagulants and vasodilators (see Tables S1–S7). Mammary artery, saphenous vein and atrial appendage were from patients undergoing coronary artery bypass graft surgery, who were on a variety of drugs including β-blockers, diuretics, statins, anti-coagulents and vasodilators (see Tables S1–S7). General patient characteristics are shown in Table 1. Tissues for radioligand binding, Western blotting, immunohistochemistry and radioimmunoassay were stored at −70°C until use. Tissues for in vitro pharmacology were collected in Krebs solution (mM: NaCl, 90; NaHCO3, 45; KCl, 5; MgSO4.7H2O, 0.5; Na2HPO4.2H2O, 1; CaCl2, 2.25; fumaric acid, 5; glutamic acid, 5; glucose, 10; sodium pyruvate, 5; pH 7.4), kept at 4°C and used within 24 h of surgery.
Table 1.
General patient characteristics
| Patient group | n | Age (years) |
Sex |
||
|---|---|---|---|---|---|
| Male | Female | Unknown | |||
| Control | 22 | 49.0 (17–62) | 12 | 6 | 4 |
| DCM | 30 | 52.0 (24–62) | 21 | 9 | 0 |
| IHD | 20 | 55.5 (35–62) | 19 | 1 | 0 |
| CABG | 40 | 71.0 (44–85) | 36 | 3 | 1 |
Values are median (range).
CABG, coronary artery bypass graft surgery; DCM, dilated cardiomyopathy; IHD, ischaemic heart disease.
Radioligand binding studies
Unless otherwise stated, radioligand binding studies were carried out with plastics coated using sigmacote. Sections, 10 µm, of human left ventricle were thaw mounted on to gelatine-coated slides and stored at −70°C until use. Sections were thawed and preincubated for 15 min with assay buffer (50 mM Tris, 5 mM MgCl2, pH 7.4) and then incubated for 90 min with 0.05 nM [Glp65,Nle75,Tyr77][125I]-apelin-13 (=[Nle75,Tyr77][125I]-(Pyr1)apelin-13) in assay buffer. Non-specific binding was defined using 1 µM (Pyr1)apelin-13. Sections were subject to three 1 min washes in an excess of Tris-HCl buffer (50 mM, pH 7.4, 4°C), wiped from the slides using filter paper and counted using a Cobra II E5003 gamma counter (Canberra UK Ltd, Didcot, UK). The specificity of [Glp65,Nle75,Tyr77][125I]-apelin-13 binding was verified by incubating sections in the presence of 1 µM apelin-related or unrelated peptides. For association binding studies, sections were incubated with the radioligand for varying amounts of time (1–360 min). For dissociation binding studies, sections were washed at 4°C for increasing periods of time (1–1440 min). For saturation binding studies, sections were incubated with increasing concentrations of [Glp65,Nle75,Tyr77][125I]-apelin-13 (0.001–0.5 nM). Protein concentrations were determined using the Bio-Rad DC 96 well microtiter plate system.
Western blotting
Human atherosclerotic coronary artery samples were homogenized in lysis buffer (50 mM Tris, 5 mM MgCl2, 5 mM EDTA, 1 mM EGTA, pH 7.5, 1:1000 protease inhibitor cocktail) and the membrane fraction was collected. These protein lysates (5–20 µg) were separated on 10% SDS-polyacrylamide gels by electrophoresis, transferred onto polyvinylidene membranes and incubated with rabbit anti-APJ(357–377) diluted 1:100 000 in TBS/T (Tris buffered saline 0.1% Tween-20) overnight at 4°C. The membranes were then incubated with ECL (enhanced chemiluminescence) peroxidase conjugated goat anti-rabbit diluted 1:5000 for 1 h at room temperature, followed by ECL reagent for 5 min. Bands were visualized by apposition to ELC hyperfilm.
Peroxidase anti-peroxidase immunohistochemistry
Sections of human coronary artery (30 µm) were thaw-mounted on to gelatine-coated slides and stored at −70°C. When required, sections were dried overnight at room temperature and fixed in ice-cold acetone for 10 min. Sections were blocked with 5% non-immunized swine serum (SS) in phosphate buffered saline (PBS) for 2 h at room temperature. They were then incubated with either rabbit anti-APJ(357–377) (1:100 in PBS 0.1% Tween-20 0.1% SS (1% SS PBS/T)) for 48 h or rabbit anti-apelin-12 (diluted 1:500 in 1% SS PBS/T) for 72 h at 4°C. In adjacent sections, primary antisera were omitted as a negative control. Slides were washed in PBS/T (three 5 min washes, 4°C) and incubated with swine anti-rabbit serum (diluted 1:200 in 1% SS PBS/T) for 1 h at room temperature. Slides were washed as before and incubated with rabbit peroxidase/anti-peroxidase complex (diluted 1:400 in 1% SS PBS/T) for 1 h at room temperature. Following a final wash step, sections were incubated with a 0.6 mg·mL−1 DAB (3,3′-diaminobenzidine tetrahydrochloride) solution in 0.05 M Tris-HCl buffer, pH 7.6, containing 0.03% hydrogen peroxide. The chromogenic reaction was stopped by immersion in deionized water. Sections were dehydrated using a graded alcohol series and cleared in xylene for 1 h at room temperature. Sections were mounted using DePeX-Gurr mounting medium and examined using a standard bright field microscope (Olympus UK Ltd, Southend-on-Sea, UK). Images were captured using a U-TV1-X digital camera and CellD software (Olympus UK Ltd, Southend-on-Sea, UK).
Dual-labelling fluorescence immunohistochemistry
Sections of human coronary artery (30 µm) were thaw-mounted onto gelatine-coated slides and stored at −70°C until use. Sections were dried overnight at room temperature and fixed in ice-cold acetone for 10 min. Sections were blocked with 5% normal goat serum in PBS and incubated for 48 h at 4°C with either rabbit anti-APJ(357–377) (1:100 in 1% GS PBS/T) or rabbit anti-apelin-12 (1:200 in 1% GS PBS/T) together with either mouse anti-human von-Willebrand factor (1:100), mouse anti-smooth muscle α-actin (1:100), mouse anti-CD3 (1:100) or mouse anti-CD68 (1:2000). In adjacent sections, primary antisera were omitted as a negative control. Slides were washed in PBS/T (three 5 min washes, 4°C) and incubated with Alexafluor568 conjugated goat anti-mouse and Alexafluor488 conjugated goat anti-rabbit antisera (1:100 in 1% GS PBS/T) for 1 h at room temperature. Sections were washed as above and mounted using ProLong Gold and visualized using laser scanning confocal microscopy (DM RXA, Leica Microsystems (UK) Ltd, Milton Keynes, UK).
Radioimmunoassay
Cardiac tissue and epicardial adipose tissue were homogenized using a Polytron PT.K homogeniser in 10 mL·g−1 tissue 0.5 M acetic acid. Blood vessels were homogenized using the FastPrep instrument in 3 mL·g−1 tissue 0.5 M acetic acid. 10 mL·g−1 tissue 0.5 M acetic acid was then added to these homogenates. All tissue homogenates were heated to 100°C for 15 min and centrifuged at 48 000×g for 1 h at 4°C. The resulting supernatants and plasma samples were acidified with an equal volume of 0.1% trifluoroacetic acid (TFA) and solid phase extracted using Sep – Pac Vac C18 cartridges. Cartridges were conditioned with 2 mL 80% methanol/0.1% TFA and washed with 9 mL 0.1% TFA prior to addition of sample. After being washed with 12 mL 0.1% TFA, the peptides were eluted with 4 mL 80% methanol/0.1% TFA. Eluent was evaporated to dryness using an evacuated centrifuge and stored at −70°C. Lyophilized samples were reconstituted in 250 µL radioimmunoassay (RIA) buffer (40 mM Na2HPO4, 10 mM NaH2PO4, 8 mM sodium azide, 0.05% Tween 20, pH 7.4).
One hundred microlitres of sample or standard peptide was incubated with 100 µL of rabbit anti-apelin-36 serum for 16–24 h at 4°C; 100 µL [125I]-(Pyr1)apelin-13 (10 000 counts·min−1) was added and the mixture incubated for 16–24 h at 4°C. Bound tracer was separated from free using Amerlex-M according to the manufacturer's instructions. Bound [125I](Pyr1)-apelin-13 in the pellet was detected using a Cobra II E5003 gamma counter (Canberra UK Ltd, Didcot, UK).
In vitro pharmacology
Rings of human histologically normal coronary artery, 4 mm [from patients with DCM (Dec and Fuster, 1994) or control hearts], were mounted in 5 mL organ baths containing oxygenated modified Krebs solution at 37°C (mM: NaCl, 90; NaHCO3, 45; KCl, 5; MgSO4.7H2O, 0.5; Na2HPO4.2H2O, 1; CaCl2, 2.25; fumaric acid, 5; glutamic acid, 5; glucose, 10; sodium pyruvate, 5; pH 7.4; gassed with 95% O2/5% CO2) and isometric tension recorded using F30 force transducers (Hugo Sachs, March – Hugstetten, Germany). After a 1 h equilibration period, optimum basal tension was determined by constricting vessels with KCl (0.1 M) at increasing levels of basal tension until no further increase in response was obtained. The absence of functional endothelium was confirmed by constricting vessels to 10 µM phenlyephrine (PE) and testing for lack of endothelium-dependent relaxation (<15%) to 1 µM acetylcholine (ACh). Cumulative concentration-response curves were constructed to (Pyr1)apelin-13 (0.001–300 nM) or angiotensin II (0.01–100 nM) for comparison and when no further response was obtained, experiments were terminated with the addition of 0.1 M KCl (post KCl response) to determine the maximal constrictor response of each vessel.
Data analysis
Data are expressed as the median value (range) unless otherwise stated and n-values are the number of patients from whom tissue was obtained. Radioligand binding data were analysed using the iterative non-linear curve fitting programmes KINETIC, EBDA and LIGAND (KELL package, Biosoft, Cambridge, UK). Kinetic analysis gave values for observed association (Kobs) and dissociation (K−1) constants, which were then used to derive values for half-time (t1/2) for association and dissociation. Saturation binding data were analysed in order to obtain values for dissociation constant (KD), maximum receptor density (Bmax) and Hill coefficient (nH). Whether a one site or two site fit was preferred was determined using the F-test in LIGAND. For radioimmunoassay, standards curves were fitted to a four parameter logistic equation (FigP, Biosoft, Cambridge, UK) and unknowns were interpolated from this curve. For in vitro pharmacology, data were expressed as percentage of the post KCl response and fitted to a four parameter logistic equation (FigP, Biosoft, Cambridge, UK) to obtain values for potency (pD2: negative logarithm of the EC50, the concentration required to produce half-maximal response) and maximum response (Emax).
Bmax data and apelin peptide levels in cardiac tissue were compared between disease states using the Kruskal-Wallis test followed by Dunn's Multiple Comparison test. Apelin peptide levels in atherosclerotic coronary artery were compared with those in histologically normal coronary artery using the Mann-Whitney U-test. Statistical tests were performed using GraphPad Prism (GraphPad Software, CA, USA). A P-value less than 0.05 was considered significant.
Materials
All reagents were obtained from Sigma (Poole, UK) unless otherwise stated. [Glp65,Nle75,Tyr77][125I]-apelin-13 was from PerkinElmer (MA, USA). [125I]-(Pyr1)apelin-13, Amerlex-M and ECL plus reagents and hyperfilm were from Amersham Biosciences (Little Chalfont, UK), rabbit anti-APJ(357–377), rabbit anti-apelin-12 and rabbit anti-apelin-36 serum were from Phoenix Pharmaceuticals (CA, USA). Swine anti-rabbit serum, rabbit peroxidase/anti-peroxidase complex, mouse anti-human von-Willebrand factor, mouse anti-smooth muscle α-actin, mouse anti-CD3 and mouse anti-CD68 were from Dako (Glostrup, Denmark). The Bio-Rad DC 96 well microtiter plate system was from Bio-Rad Laboratories (Hemel Hempstead, UK). Sep – Pac Vac C18 cartridges were from Waters Ltd. (Elstree, UK), Alexafluor568 conjugated goat anti-mouse, Alexafluor488 conjugated goat anti-rabbit antisera and ProLong Gold were from Molecular Probes, Invitogen (Paisley, UK).
Results
Characterization of [Glp65,Nle75,Tyr77][125 I]-apelin-13 binding in human left ventricle
(Pyr1)apelin-13, apelin-13 and apelin-36 competed for [Glp65,Nle75,Tyr77][125I]-apelin-13 binding in human left ventricle, whereas angiotensin I-IV and a range of structurally unrelated peptides (all 1 µM) failed to compete (Figure 1A). [Glp65,Nle75,Tyr77][125I]-apelin-13 binding to human left ventricle was time-dependent, with a median association rate constant (Kobs) of 0.038 (0.037–0.048) min−1 and a half-time for association (t1/2) of 18.27 (14.41–18.74) min (n = 3) (Figure 1B). Binding of the radioligand was reversible, with a relatively slow rate of dissociation. The median dissociation rate constant (K−1) was 0.021 (0.010–0.023) min−1, giving a half-time for dissociation (t1/2) of 33.67 (30.77–68.93) min (n = 3) (Figure 1C).
Figure 1.
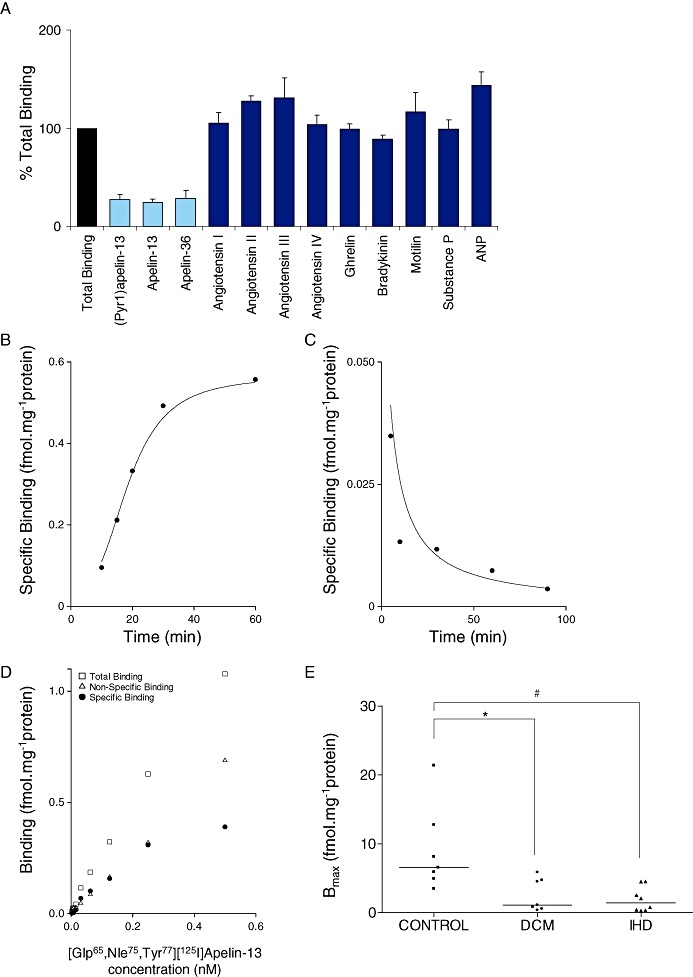
[Glp65,Nle75,Tyr77][125I]-apelin-13 binding in human left ventricle. (A) Specificity of [Glp65,Nle75,Tyr77][125I]-apelin-13 binding (mean ± SEM, n = 3). ANP, atrial natriuretic peptide. (B) Time-dependent association of [Glp65,Nle75,Tyr77][125I]-apelin-13 binding; a representative curve is shown, with an association rate constant (Kobs) of 0.038 min−1. (C) Time-dependent dissociation of [Glp65,Nle75,Tyr77][125I]-apelin-13 binding; a representative curve is shown, with a dissociation rate constant (K−1) of 0.023 min−1. (D) Example saturation binding curve with a dissociation constant (KD) of 0.29 nM, maximal receptor density (Bmax) of 0.55 fmol·mg−1 protein and a Hill coefficient (nH) of 1.03. (E) Maximal receptor density (Bmax) in left ventricle from control hearts (n = 7) and patients with dilated cardiomyopathy (DCM, n = 6) or ischaemic heart disease (IHD, n = 8); horizontal lines represent median values; *P < 0.05; #P < 0.01. DCM, dilated cardiomyopathy; IHD, ischaemic heart disease.
Apelin receptor density in normal and diseased human left ventricle
[Glp65,Nle75,Tyr77][125I]-apelin-13 binding in human left ventricle was saturable (Figure 1D) with sub-nanomolar affinity (Table 2) over the concentration range tested (0.001–0.5 nM). In all the tissues examined, a one-site fit was preferred to a two-site model, as determined by the F-test, and Hill coefficients were close to unity (Table 2). Apelin receptor density (Bmax) was significantly lower in left ventricle from patients with DCM [1.10 (0.36–5.85) fmol·mg−1 protein, n = 6] or IHD [1.41 (0.28–4.50) fmol·mg−1 protein, n = 8] compared with left ventricle from control hearts [6.56 (3.46–21.36) fmol·mg−1 protein, n = 7] (P < 0.05 control vs. DCM, P < 0.01 control vs. IHD, Kruskal-Wallis test followed by Dunn's Multiple Comparison test). There was no significant difference in Bmax between left ventricle from patients with DCM and those with IHD (P > 0.05 Kruskal-Wallis test follwed by Dunn's Multiple Comparison Test) (Figure 1E, Table 2).
Table 2.
Saturation binding assay data for [Glp65,Nle75,Tyr77][125I]-apelin-13 binding to human left ventricle
| Tissue | KD (nM) | Bmax (fmol·mg−1protein) | nH | n |
|---|---|---|---|---|
| Control left ventricle | 0.40 ± 0.07 | 6.56 (3.46–21.36) | 1.01 (1.00–1.03) | 7 |
| DCM left ventricle | 0.30 ± 0.10 | 1.10 (0.36–5.85)* | 1.02 (0.92–1.13) | 7 |
| IHD left ventricle | 0.28 ± 0.07 | 1.41 (0.28–4.50)# | 0.99 (0.88–1.10) | 8 |
Values are median (range), except KD values which are mean ± SEM (SEM calculated by LIGAND).
P < 0.05 compared with control left ventricle.
P < 0.01 compared with control left ventricle.
Bmax, maximal density of receptors; DCM, dilated cardiomyopathy; IHD, ischaemic heart disease; KD, dissociation constant; nH, Hill coefficient.
Apelin peptide levels in human normal and diseased cardiovascular tissues
A novel RIA was developed for detection of apelin peptide. Standard curves were constructed to apelin-36 (10–1280 pg·mL−1) in each assay and a representative curve is shown in Figure 2A. An atrial tissue sample diluted in parallel with the standard curve (Figure 2A), indicating that the immunoreactivity detected in tissue samples was apelin peptide. The linear portion of the curve ranged from 15–150 pg·mL−1 and IC50 values ranged from 54–122 pg·mL−1. The sensitivity of detection (defined as the amount of standard peptide required to reduce B0 by two standard deviations, where B0 is equivalent to 100% [125I]-(Pyr1)apelin-13 bound) was 11.9 pg·mL−1. The intra-assay variation (including the variation caused by solid phase extraction) was 14.1%, the intra-assay variation (only) was 8.3% and the inter-assay variation was 12.3%. The cross-reactivity profile of the RIA is shown in Table 3. The percentage recovery from solid phase extraction, determined using an apelin-36 (25 pg) spiked atrial appendage sample, was 86.8%.
Figure 2.
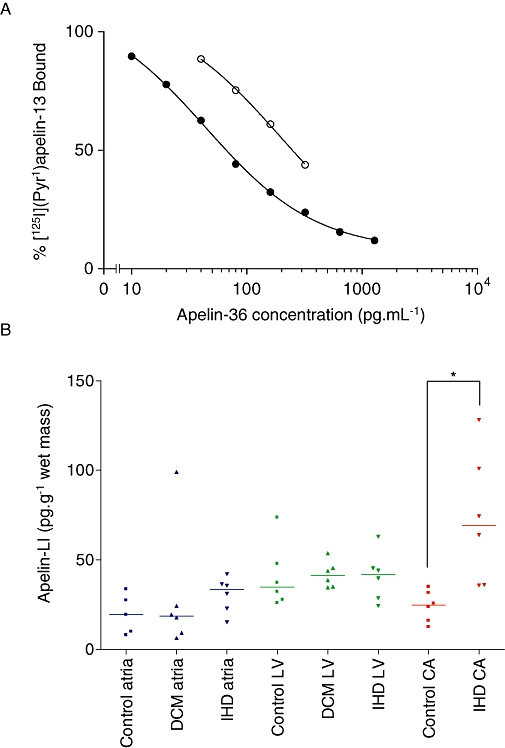
(A) Typical standards curve (solid symbols) and dilutions of an atrial tissue sample (open symbols); expressed as mean of duplicate determinations. (B) Apelin-like immunoreactivity (LI) levels in human cardiac tissue and coronary artery (n = 5–6). CA, coronary artery; DCM, dilated cardiomyopathy; IHD, ischaemic heart disease; LV, left ventricle. *P < 0.01.
Table 3.
Cross-reactivity profile of apelin radioimmunoassay
| Antigen | Cross-reactivity (%) |
|---|---|
| Synthetic apelin peptides | |
| Apelin-36 | 100.00 |
| (Pyr1)apelin-13 | 43.76 |
| Apelin-13 | 39.90 |
| Unrelated vasoactive peptides | |
| Angiotensin II | BLL |
| Angiotensin III | BLL |
| Angiotensin IV | BLL |
| Ghrelin | BLL |
| Endothelin-1 | BLL |
| Bradykinin | BLL |
| Motilin | BLL |
| Substance P | BLL |
| Atrial natriuretic peptide | BLL |
Cross-reactivities of synthetic apelin peptides are expressed as percentage relative immunoreactivity compared with apelin-36, based on the EC50 values of their standard curves. The cross-reactivities of unrelated vasoactive peptides were determined by testing a 1 µM solution of each peptide.
BLL, below lower limit of detection.
Apelin-like immunoreactivity (LI) was detected in all human cardiac and vascular tissues tested (Table 4). Levels were also detected in epicardial adipose tissue (Table 4). Apelin-LI was significantly higher in atherosclerotic coronary artery [69.1 (35.8–128.1) pg·g−1 wet mass, n = 6] compared with control coronary artery [24.7 (12.5–35.0) pg·g−1 wet mass, n = 6] (P < 0.01 Mann-Whitney U-test) (Figure 2B, Table 4). There was no significant difference in apelin-LI between atria or ventricle from control, DCM and IHD patients (P > 0.05 Kruskal-Wallis test) (Figure 2B, Table 4).
Table 4.
Apelin-LI levels in human cardiovascular tissues
| Tissue | Apelin-LI (pg·g−1wet mass) | n |
|---|---|---|
| Saphenous vein | 52.7 (27.0–88.8) | 6 |
| Mammary artery | 14.8 (9.8–44.6) | 5 |
| Epicardial adipose tissue | 2.1 (1.7–5.0) | 6 |
| Atrial appendage | 62.8 (56.8–94.0) | 29# |
| Control atria | 19.4 (8.0–33.6) | 5 |
| DCM atria | 18.6 (6.3–99.0) | 6 |
| IHD atria | 33.3 (15.1–42.0) | 6 |
| Control left ventricle | 34.7 (25.9–73.7) | 6 |
| DCM left ventricle | 41.3 (34.5–53.7) | 6 |
| IHD left ventricle | 41.9 (24.3–63.0) | 6 |
| Control coronary artery | 24.7 (12.5–35.0) | 6 |
| IHD coronary artery | 69.1 (35.8–128.1)* | 6 |
Values are median (range).
P < 0.01 compared with normal coronary artery.
n = 29 in three pooled samples.
DCM, dilated cardiomyopathy; IHD, ischaemic heart disease.
Localization of apelin receptor-LI to human coronary artery
Western blot using rabbit anti-APJ(357–377) revealed a single band of approximately 60 kDa in human atherosclerotic coronary artery (n = 3) (Figure 3A), consistent with an N-glycosylated form of the receptor, as reported previously (Puffer et al., 2000). This band was attenuated on pre-absorption of the antibody with the immunizing peptide (rat APJ 357–377) (Figure 3A). Immunohistochemistry localized apelin receptor-LI to the endothelium and smooth muscle layer of histologically normal coronary artery (Figure 3B), as previously reported (Kleinz et al., 2005), whereas additional staining of the plaque was observed in atherosclerotic coronary artery (Figure 3D). Dual-labelling fluorescence immunohistochemistry showed that apelin receptor-LI within the atherosclerotic plaque colocalized with markers for smooth muscle cells (SMαA) (Fig. 3F) and macrophages (CD68) (Figure 3G), but there was little evidence of colocalization with a T-cell marker (CD3) (Figure 3H). No staining was observed in adjacent sections where the primary antibody was omitted as a negative control (Figure 3C,E, data not shown for fluorescence immunohistochemistry).
Figure 3.
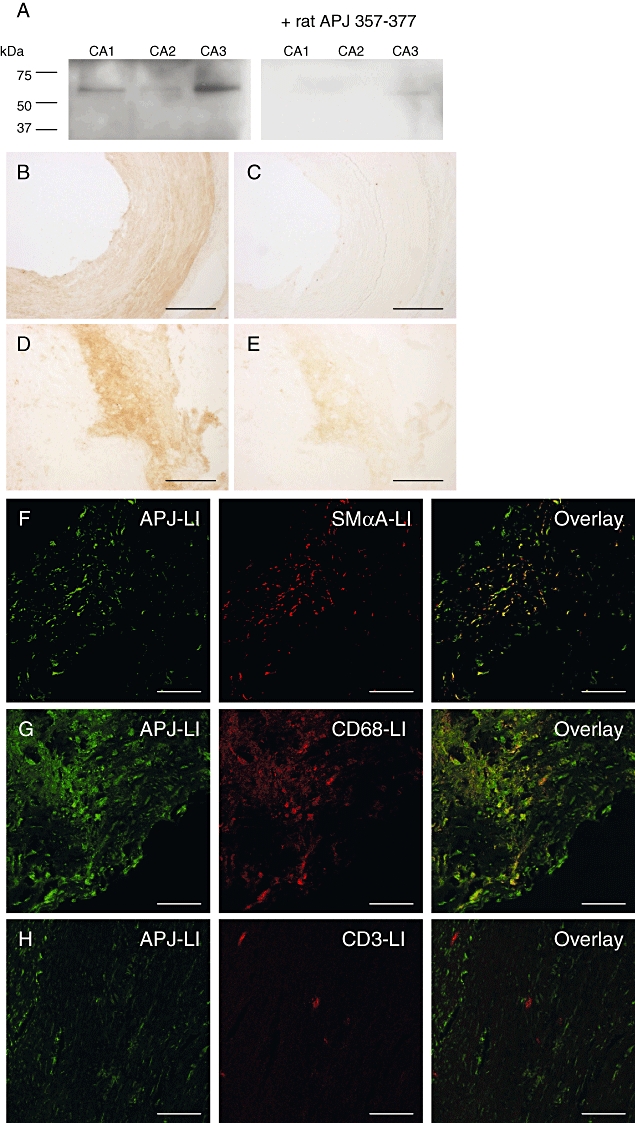
(A) Western blot using rabbit anti-APJ(357–377) showing a single band of approximately 60 kDa in human atherosclerotic coronary artery (CA, n = 3), attenuated following pre-absorption with the immunizing peptide. (B–H) Representative photomicrographs showing APJ-like immunoreactivity (LI) in human histologically normal (B, C) and atherosclerotic (D–H) coronary artery. (C, E) Primary antibody omitted. APJ-LI within the atherosclerotic plaque colocalizes with smooth muscle α-actin (SMαA)-LI (F) and CD68-LI (G), but not with CD3-LI (H). All n = 3. Scale bar 200 µm (B–E) or 50 µm (F–H).
Localization of apelin-LI to human coronary artery
In sections of human, histologically normal coronary artery, apelin-LI was restricted to the endothelium (Figure 4A), consistent with previous reports (Kleinz and Davenport, 2004). Intense staining of the plaque was observed in atherosclerotic coronary artery (Figure 4C). Apelin-LI within the plaque colocalized with markers for smooth muscle cells (Figure 4E) and macrophages (Figure 4F), but not with a marker for T-cells (Figure 4G). Staining was absent in sections where the primary antibody was omitted (Figure 4B,D, data not shown for fluorescence immunohistochemistry).
Figure 4.
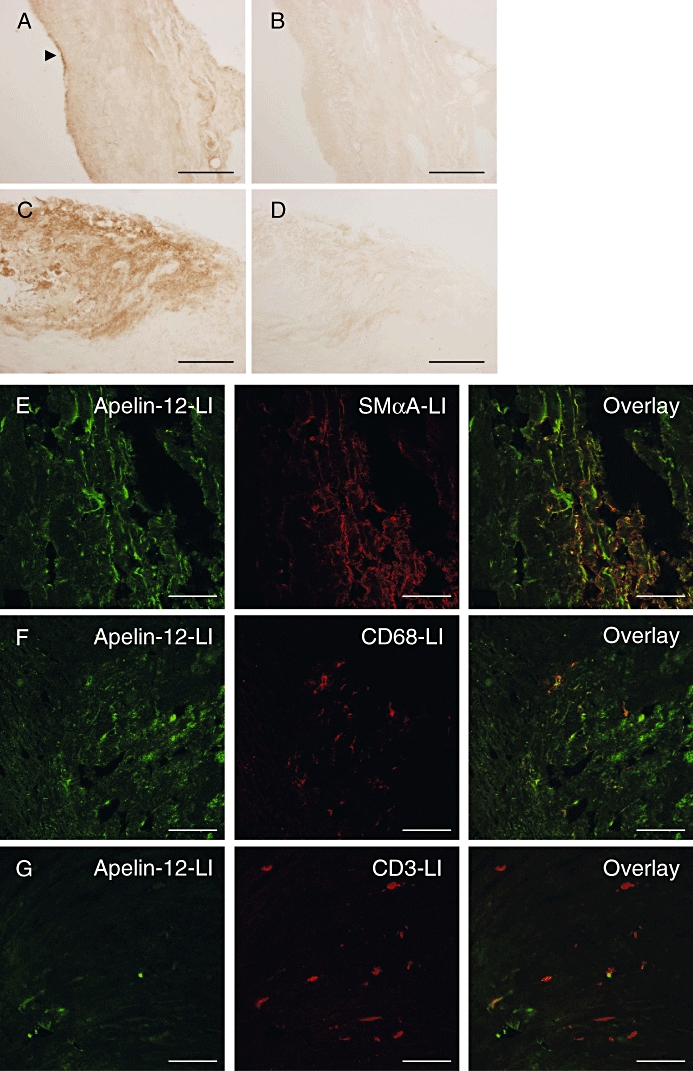
Representative photomicrographs showing apelin-like immunoreactivity (LI) in human histologically normal (A, B) and atherosclerotic (C–G) coronary artery. (B, D) Primary antibody omitted. Within the atherosclerotic plaque apelin-LI colocalizes with smooth muscle α-actin (SMαA)-LI (E) and CD68-LI (F), but not with CD3-LI (G). All n = 3. Scale bar 200 µm (A–D) or 50 µm (E–G). Arrow indicates endothelial staining.
Vasopressor effect of (Pyr1)apelin-13 on human coronary artery
Human isolated coronary artery lacked functional endothelium. (Pyr1)apelin-13 potently constricted coronary artery with a median pD2 value of 8.0 (6.7–10.0) and a maximum response of 22.7 (8.8–36.6)% of the terminal response to 100 mM KCl (n = 5) (Figure 5). Angiotensin II elicited a comparable response, with a median pD2 value of 8.9 (7.7–10.7) and a maximum response of 22.2 (7.3–59.7)% of the terminal response to 100 mM KCl (n = 9).
Figure 5.
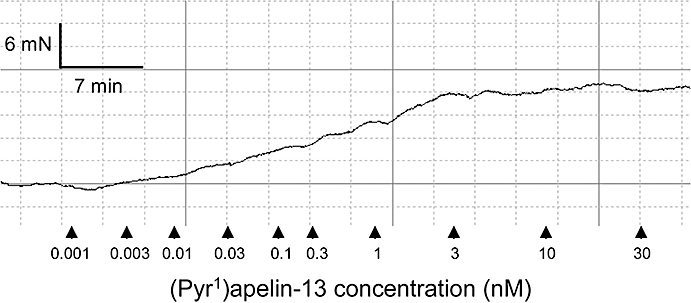
Representative cumulative concentration-response curve to (Pyr1)apelin-13 in human isolated coronary artery. Arrows indicate time points at which doses were added.
Discussion and conclusions
We report for the first time the binding characteristics of [Glp65,Nle75,Tyr77][125I]-apelin-13 in human cardiac tissue, fulfilling the criteria of high affinity, saturable, reversible and specific binding. We also validated a novel radioimmunoassay for the detection of apelin peptide levels in human tissues. These assays demonstrated a striking down-regulation of the apelin receptor in two types of heart failure, while apelin peptide levels were unchanged in these conditions in both ventricle and atria. We discovered a marked up-regulation of apelin in atherosclerotic coronary artery, with the additional peptide localizing to the atherosclerotic plaque. We also report that apelin is a potent constrictor of human coronary artery, with a comparable potency and maximum response to angiotensin II.
These data are consistent with a previous report showing no significant difference in apelin-36-LI between left ventricle from control, IHD and idiopathic DCM patients (Foldes et al., 2003). However, Foldes et al. (2003) reported a decrease in apelin-36-LI in atria from IHD patients compared with controls. The difference in our findings may be due to Foldes et al. (2003) using atrial appendage samples, rather than atrial tissue as in our study. We have shown that atrial appendage has markedly higher apelin content than atrial tissue, possibly due to its higher endocardial endothelial cell to cardiomyocyte ratio.
Foldes et al. (2003) demonstrated a significant decrease in apelin receptor mRNA in left ventricle from patients with DCM, but no difference in left ventricle from IHD patients compared with controls. We found a decrease in receptor protein in both conditions, suggesting that the down-regulation may be due to decreased transcription of the gene in DCM, but due to regulation at the post-transcriptional level in IHD. The apelin receptor has previously been shown to localize to cardiomyocytes and endocardial endothelial cells in healthy and diseased human left ventricle (Katugampola et al., 2001; Kleinz et al., 2005). Whether the apelin receptor is down-regulated in both cell types in DCM and IHD requires further investigation, but as cardiomyocytes greatly outnumber endocardial endothelial cells in the tissue sections used, the observed decrease in receptor density is likely to equate to a down-regulation of the apelin receptor in cardiomyocytes in these conditions.
A decrease in apelin receptor density in cardiomyocytes, without a compensatory up-regulation of apelin, in heart failure may contribute to contractile dysfunction, as the positive inotropic actions of apelin would presumably be reduced. Mechanical offloading of failing myocardium causes an up-regulation of the apelin receptor (Chen et al., 2003), suggesting that it may be stretch that triggers the down-regulation in disease. Studies in rats also reported a down-regulation of the apelin receptor in heart failure, attributed to AT1 receptor signalling (Iwanaga et al., 2006), suggesting that the increased angiotensin II levels observed in heart failure may cause the down-regulation of the apelin receptor.
The data presented here strengthen the case for the apelin receptor as a potential future drug target for heart failure. Chronic infusion of apelin into mice improved cardiac function without evidence of hypertrophy, while concomitantly decreasing preload and afterload (Ashley et al., 2005). Apelin treatment also protected against ischaemia reperfusion injury in rodents (Simpkin et al., 2007; Zeng et al., 2009), which may provide additional benefit to treating with an apelin receptor agonist. However, positive inotropic agents increase the oxygen demand of the myocardium and deleterious effects of positive inotropic agents such as milrinone and dobutamine have been demonstrated in clinical trials, giving an important caveat. We have shown that apelin is able to constrict human isolated coronary artery. Heart failure is associated with endothelial dysfunction; therefore, although apelin mediates endothelium-dependent vasodilator effects in the healthy vasculature (Japp et al., 2008), treatment of this patient group with an apelin agonist may contribute to detrimental coronary vasoconstriction. These possible deleterious effects will need careful investigation in pre-clinical and clinical trials.
We have also investigated changes in the apelin/APJ system in atherosclerosis, the primary cause of IHD. We have demonstrated that apelin peptide levels are significantly increased in human atherosclerotic coronary artery, and the additional peptide localizes to the atherosclerotic plaque, where it colocalizes with markers for smooth muscle cells and macrophages. The apelin receptor was also found to be present within the atherosclerotic plaque and to have a similar distribution to its ligand. We have previously shown that apelin receptor density is not altered in the media of atherosclerotic coronary artery compared with that of histologically normal coronary artery (Katugampola et al., 2002).
The role of the increased apelin within the plaque is unclear, with studies in mice reporting both beneficial (Chun et al., 2008) and detrimental (Hashimoto et al., 2007; Kojima et al., 2010) effects of the apelin/APJ system in atherosclerosis. Apelin has been shown to mediate oxidative stress in vascular tissue (Hashimoto et al., 2007), so the increased apelin within the plaque may contribute to atherogenesis. Alternatively, apelin may limit atherosclerosis progression by inhibiting angiotensin II effects on the vasculature (Chun et al., 2008). Within the plaque, both apelin and its receptor colocalized with markers for smooth muscle cells and macrophages. Apelin treatment decreases aortic aneurysm formation in mice by decreasing macrophage infiltration (Leeper et al., 2009), suggesting that apelin may be beneficial in human atherosclerosis by a similar mechanism. Apelin is a mitogen (Li et al., 2008) and increases migration (Kojima et al., 2010) of vascular smooth muscle cells, so may have detrimental effects by promoting smooth muscle cell proliferation and migration in to the neointima. Whether apelin receptor activation is beneficial or detrimental in atherosclerosis will require further investigation using agonists and particularly apelin receptor selective antagonists.
We observed a significant down-regulation of the apelin receptor in left ventricle from patients with DCM or IHD, with no change in apelin peptide levels. We have shown an increase in apelin expression in atherosclerotic coronary artery, with the additional peptide localizing to the atherosclerotic plaque. These data implicate the apelin/APJ system in heart failure and atherosclerosis in man, providing further evidence in support of the apelin receptor as a possible future drug target in these conditions.
Acknowledgments
This work is supported by grants from the British Heart Foundation (PS/02/001, FS/06/017, PG/09/050/27734). We thank Jean Chadderton and the theatre and consultant staff of Papworth hospital for tissue collection.
Glossary
Abbreviations:
- Bmax
maximum receptor density
- DCM
dilated cardiomyopathy
- EC50
the concentration required to produce half-maximal response
- Emax
maximum response
- IHD
ischaemic heart disease
- K−1
observed dissociation constant
- KD
dissociation constant
- Kobs
observed association constant
- nH
Hill coefficient
- pD2
negative logarithm of the EC50
- t1/2
half-time
Conflicts of interest
None.
Note added in proof
A recent paper by Japp and colleagues has shown that acute administration of apelin in healthy volunteers and patients with heart failure causes peripheral and coronary vasodilatation and increases cardiac output, suggesting that an apelin agonist may be beneficial in this patient group (Japp et al., 2010).
Supporting information
Additional Supporting Information may be found in the online version of this article:
Table S1 Tissue Abbreviations.
Table S2 Experimental Abbreviations.
Table S3 Codes for pre-operative drugs.
Table S4 Control cardiac transplant tissue from donors for whom there was no suitable recipient.
Table S5 Cardiac transplant tissue from patients with dilated cardiomyopathy.
Table S6 Cardiac transplant tissue from patients with ischaemic heart disease.
Table S7 Tissue from coronary artery bypass graft patients.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Ashley EA, Powers J, Chen M, Kundu R, Finsterbach T, Caffarelli A, et al. The endogenous peptide apelin potently improves cardiac contractility and reduces cardiac loading in vivo. Cardiovasc Res. 2005;65:73–82. doi: 10.1016/j.cardiores.2004.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atluri P, Morine KJ, Liao GP, Panlilio CM, Berry MF, Hsu VM, et al. Ischemic heart failure enhances endogenous myocardial apelin and APJ receptor expression. Cell Mol Biol Lett. 2007;12:127–138. doi: 10.2478/s11658-006-0058-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpéné C, Dray C, Attane C, Valet P, Portillo MP, Churruca I, et al. Expanding role for the apelin/APJ system in physiopathology. J Physiol Biochem. 2007;63:359–373. [PubMed] [Google Scholar]
- Chen MM, Ashley EA, Deng DXF, Tsalenko A, Deng A, Tabibiazar R, et al. Novel role for the potent endogenous inotrope apelin in human cardiac dysfunction. Circulation. 2003;108:1432–1439. doi: 10.1161/01.CIR.0000091235.94914.75. [DOI] [PubMed] [Google Scholar]
- Chong KS, Gardner RS, Morton JJ, Ashley EA, McDonagh TA. Plasma concentrations of the novel peptide apelin are decreased in patients with chronic heart failure. Eu J Heart Fail. 2006;8:355–360. doi: 10.1016/j.ejheart.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Chun HJ, Ali ZA, Kojima Y, Kundu RK, Sheikh AY, Agrawal R, et al. Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis. J Clin Invest. 2008;118:3343–3354. doi: 10.1172/JCI34871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CM, D'Agostino SL, Miller MK, Heimark RL, Krieg PA. Apelin, the ligand for the endothelial G-protein-coupled receptor, APJ, is a potent angiogenic factor required for normal vascular development of the frog embryo. Dev Biol. 2006;296:177–189. doi: 10.1016/j.ydbio.2006.04.452. [DOI] [PubMed] [Google Scholar]
- Dec GW, Fuster V. Idiopathic dilated cardiomyopathy. N Engl J Med. 1994;331:1564–1575. doi: 10.1056/NEJM199412083312307. [DOI] [PubMed] [Google Scholar]
- Foldes G, Horkay F, Szokodi I, Vuolteenaho O, Ilves M, Lindstedt A, et al. Circulating and cardiac levels of apelin, the novel ligand of the orphan receptor APJ, in patients with heart failure. Bioch Biophys Res Commun. 2003;308:480–485. doi: 10.1016/s0006-291x(03)01424-4. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Kihara M, Imai N, Yoshida S, Shimoyamada H, Yasuzaki H, et al. Requirement of apelin-apelin receptor system for oxidative stress-linked atherosclerosis. Am J Pathol. 2007;171:1705–1712. doi: 10.2353/ajpath.2007.070471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwanaga Y, Kihara Y, Takenaka H, Kita T. Down-regulation of cardiac apelin system in hypertrophied and failing hearts: possible role of angiotensin II-angiotensin type 1 receptor system. J Mol Cell Cardiol. 2006;41:798–806. doi: 10.1016/j.yjmcc.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Japp AG, Newby DE. The apelin-APJ system in heart failure: pathophysiologic relevance and therapeutic potential. Biochem Pharmacol. 2008;75:1882–1892. doi: 10.1016/j.bcp.2007.12.015. [DOI] [PubMed] [Google Scholar]
- Japp AG, Cruden NL, Amer DAB, Li VKY, Goudie EB, Johnston NR, et al. Vascular effects of apelin in vivo in man. J Am Coll Cardiol. 2008;52:908–913. doi: 10.1016/j.jacc.2008.06.013. [DOI] [PubMed] [Google Scholar]
- Japp AG, Cruden NL, Barnes G, van Gemeren N, Mathews J, Adamson J, et al. Acute cardiovascular effects of apelin in humans: potential role in patients with chronic heart failure. Circulation. 2010;121:1818–1827. doi: 10.1161/CIRCULATIONAHA.109.911339. [DOI] [PubMed] [Google Scholar]
- Kasai A, Shintani N, Oda M, Kakuda M, Hashimoto H, Matsuda T, et al. Apelin is a novel angiogenic factor in retinal endothelial cells. Bioch Biophys Res Commun. 2004;325:395–400. doi: 10.1016/j.bbrc.2004.10.042. [DOI] [PubMed] [Google Scholar]
- Katugampola SD, Maguire JJ, Matthewson SR, Davenport AP. I-125 -(Pyr(1))Apelin-13 is a novel radioligand for localizing the APJ orphan receptor in human and rat tissues with evidence for a vasoconstrictor role in man. Br J Pharmacol. 2001;132:1255–1260. doi: 10.1038/sj.bjp.0703939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katugampola SD, Maguire JJ, Kuc RE, Wiley KE, Davenport AP. Discovery of recently adopted orphan receptors for apelin, urotensin II, and ghrelin identified using novel radioligands and functional role in the human cardiovascular system. Can J Physiol Pharmacol. 2002;80:369–374. doi: 10.1139/y02-029. [DOI] [PubMed] [Google Scholar]
- Kleinz MJ, Davenport AP. Immunocytochemical localization of the endogenous vasoactive peptide apelin to human vascular and endocardial endothelial cells. Reg Pept. 2004;118:119–125. doi: 10.1016/j.regpep.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Kleinz MJ, Davenport AP. Emerging roles of apelin in biology and medicine. Pharmacol Ther. 2005;107:198–211. doi: 10.1016/j.pharmthera.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Kleinz MJ, Skepper JN, Davenport AP. Immunocytochemical localisation of the apelin receptor, APJ, to human cardiomyocytes, vascular smooth muscle and endothelial cells. Reg Pept. 2005;126:233–240. doi: 10.1016/j.regpep.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Kojima Y, Kundu R, Cox CM, Leeper NJ, Anderson JA, Chun HJ, et al. Upregulation of the apelin-APJ pathway promotes neointima formation in the carotid ligation model in mouse. Cardiovasc Res. 2010 doi: 10.1093/cvr/cvq052. DOI: 10.1093/cvr/cvq052 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuba K, Zhang L, Imai Y, Arab S, Chen M, Maekawa Y, et al. Impaired heart contractility in apelin gene deficient mice associated with aging and pressure overload. Circ Res. 2007;101:e32–e42. doi: 10.1161/CIRCRESAHA.107.158659. [DOI] [PubMed] [Google Scholar]
- Ladeiras-Lopes R, Ferreira-Martins J, Leite-Moreira AF. The apelinergic system: the role played in human physiology and pathology and potential therapeutic applications. Arq Bras Cardiol. 2008;90:343–349. doi: 10.1590/s0066-782x2008000500012. [DOI] [PubMed] [Google Scholar]
- Lee DK, George SR, O'Dowd BF. Unravelling the roles of the apelin system: prospective therapeutic applications in heart failure and obesity. Trends Pharmacol Sci. 2006;27:190–194. doi: 10.1016/j.tips.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Leeper NJ, Tedesco MM, Kojima Y, Schultz GM, Kundu RK, Ashley EA, et al. Apelin prevents aortic aneurysm formation by inhibiting macrophage inflammation. Am J Physiol Heart Circ Physiol. 2009;296:H1329–H1335. doi: 10.1152/ajpheart.01341.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Li L, Qin X, Pan W, Feng F, Chen F, et al. Apelin-induced vascular smooth muscle cell proliferation: the regulation of cyclin D1. Front Biosci. 2008;13:3786–3792. doi: 10.2741/2967. [DOI] [PubMed] [Google Scholar]
- Maguire JJ, Kleinz MJ, Pitkin SL, Davenport AP. [Pyr1]Apelin-13 identified as the predominant apelin isoform in the human heart: vasoactive mechanisms and inotropic action in disease. Hypertension. 2009;54:598–604. doi: 10.1161/HYPERTENSIONAHA.109.134619. [DOI] [PubMed] [Google Scholar]
- Masri B, Morin N, Cornu M, Knibiehler B, Audigier Y. Apelin (65–77) activates p70 S6 kinase and is mitogenic for umbilical endothelial cells. Faseb J. 2004;18:1909–1911. doi: 10.1096/fj.04-1930fje. [DOI] [PubMed] [Google Scholar]
- Masri B, Knibiehler B, Audigier Y. Apelin signalling: a promising pathway from cloning to pharmacology. Cell Signal. 2005;17:415–426. doi: 10.1016/j.cellsig.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Miettinen KH, Magga J, Vuolteenaho O, Vanninen EJ, Punnonen KR, Ylitalo K, et al. Utility of plasma apelin and other indices of cardiac dysfunction in the clinical assessment of patients with dilated cardiomyopathy. Reg Pept. 2007;140:178–184. doi: 10.1016/j.regpep.2006.12.004. [DOI] [PubMed] [Google Scholar]
- O'Dowd BF, Heiber M, Chan A, Heng HH, Tsui LC, Kennedy JL, et al. A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene. 1993;136:355–360. doi: 10.1016/0378-1119(93)90495-o. [DOI] [PubMed] [Google Scholar]
- Pitkin SL, Maguire JJ, Ashby MJ, Kuc RE, Davenport AP. Identification and measurement of endogenous apelin peptides in the human cardiovascular system. 2007. From the Brighton Winter 2007 Meeting: Proceedings of the British Pharmacological Society. [WWW document]. URL http://www.pa2online.org/abstract/abstract.jsp?abid=28999&author=pitkin&cat=-1&period=-1.
- Pitkin SL, Ashby MJ, Kuc RE, Maguire JJ, Davenport AP. Apelin peptides are upregulated in human atherosclerosis. Fundam Clin Pharmacol. 2008a;22:74 P081. [Google Scholar]
- Pitkin SL, Kuc RE, Maguire JJ, Davenport AP. Upregulation and localisation of apelin in human atherosclerosis. 2008b. From the Brighton Winter 2008 Meeting: Proceedings of the British Pharmacological Society. [WWW document]. URL http://www.pa2online.org/abstract/abstract.jsp?abid=29087&author=pitkin&cat=-1&period=-1.
- Pitkin SL, Macaluso NJM, Maguire JJ, Glen RC, Davenport AP. Role for apelin in human atherosclerosis and discovery of novel agonists for its receptor APJ. 2009. From the London Winter 2009 Meeting: Proceedings of the British Pharmacological Society. [WWW document]. URL http://www.pa2online.org/abstract/abstract.jsp?abid=29431&author=pitkin&cat=-1&period=-1.
- Pitkin SL, Maguire JJ, Bonner TI, Davenport AP. International Union of Basic and Clinical Pharmacology. LXXIV. Apelin Receptor Nomenclature, Distribution, Pharmacology and Function. Pharmacol Rev. 2010;62 doi: 10.1124/pr.110.002949. in press. [DOI] [PubMed] [Google Scholar]
- Puffer BA, Sharron M, Coughlan CM, Baribaud F, McManus CM, Lee B, et al. Expression and coreceptor function of APJ for primate immunodeficiency viruses. Virology. 2000;276:435–444. doi: 10.1006/viro.2000.0557. [DOI] [PubMed] [Google Scholar]
- Ronkainen VP, Ronkainen JJ, Hannninen SL, Leskinen H, Ruas JL, Pereira T, et al. Hypoxia inducible factor regulates the cardiac expression and secretion of apelin. Faseb J. 2007;21:1821–1830. doi: 10.1096/fj.06-7294com. [DOI] [PubMed] [Google Scholar]
- Salcedo A, Garijo J, Monge L, Fernandez N, Luis Garcia-Villalon A, Sanchez Turrion V, et al. Apelin effects in human splanchnic arteries. Role of nitric oxide and prostanoids. Reg Pept. 2007;144:50–55. doi: 10.1016/j.regpep.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Sarzani R, Forleo C, Pietrucci F, Capestro A, Soura E, Guida P, et al. The 212A variant of the apj receptor gene for the endogenous inotrope apelin is associated with slower heart failure progression in idiopathic dilated cardiomyopathy. J Card Fail. 2007;13:521–529. doi: 10.1016/j.cardfail.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Sheikh AY, Chun HJ, Glassford AJ, Kundu RK, Kutschka I, Ardigo D, et al. In vivo genetic profiling and cellular localization of apelin reveals a hypoxia-sensitive, endothelial-centered pathway activated in ischemic heart failure. Am J Physiol Heart Circ Physiol. 2008;294:H88–H98. doi: 10.1152/ajpheart.00935.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpkin JC, Yellon DM, Davidson SM, Lim SY, Wynne AM, Smith CC. Apelin-13 and apelin-36 exhibit direct cardioprotective activity against ischemia-reperfusion injury. Basic Res Cardiol. 2007;102:518–528. doi: 10.1007/s00395-007-0671-2. [DOI] [PubMed] [Google Scholar]
- Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX, et al. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Bioch Biophys Res Commun. 1998;251:471–476. doi: 10.1006/bbrc.1998.9489. [DOI] [PubMed] [Google Scholar]
- Zeng XJ, Zhang LK, Wang HX, Lu LQ, Ma LQ, Tang CS. Apelin protects heart against ischemia/reperfusion injury in rat. Peptides. 2009;30:1144–1152. doi: 10.1016/j.peptides.2009.02.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.