

Abstract
BACKGROUND AND PURPOSE
Inflammation of the extraplacental membranes plays a key role in the pathogenesis of preterm labour. The aim of this study was to screen a number of commercially available small molecule nuclear factor-kappa B inhibitors to identify candidates suitable for clinical evaluation as anti-inflammatory agents for the prevention of preterm birth.
EXPERIMENTAL APPROACH
Nine inhibitors were evaluated across a range of concentrations for their ability to inhibit lipopolysaccharide (LPS)-stimulated cytokine production in primary term choriodecidual cells in culture without affecting cell viability. Expression of 112 inflammation- and apoptosis-related genes was evaluated using boutique oligonucleotide arrays.
KEY RESULTS
Two IKKβ inhibitors were found to be highly effective and non-toxic inhibitors of choriodecidual cytokine production: parthenolide and [5-(p-fluorophenyl)-2-ureido] thiophene-3-carboxamide (TPCA-1). Both compounds also inhibited LPS-stimulated nuclear translocation of p65/RelA. Expression of 38 genes on the arrays (34%) was significantly (P < 0.05) inhibited by TPCA-1 or parthenolide. Of the 14 genes significantly stimulated by LPS, all were inhibited by TPCA-1 and 12 were inhibited by parthenolide. Overall, gene expression was more robustly inhibited by TPCA-1 than parthenolide; however, expression of two genes was only inhibited by parthenolide. Neither compound significantly altered the expression profile of anti-apoptosis genes on the arrays.
CONCLUSIONS AND IMPLICATIONS
These studies provide evidence that pharmacological inhibition of IKKβ activity holds promise as a potential strategy for the prevention and/or treatment of inflammation-driven preterm birth. TPCA-1 appeared the most promising compound among those tested in this study. Different inhibitors may have subtly different effect profiles despite having similar modes of action.
Keywords: inflammation, lipopolysaccharide, NF-κB, I-κB kinase, choriodecidua, gene expression, preterm labour
Introduction
Intrauterine inflammation plays an important role in the mechanism of parturition, both at term and preterm (Romero et al., 2007a). Term labour is associated with increased infiltration of leukocytes into the cervix, uterus and fetal membranes and increased expression of inflammation-associated genes in these tissues is necessary for cervical ripening and uterine activation (Romero et al., 2006). With spontaneous preterm labour, however, the inflammatory process is often exaggerated, and is manifested as chorioamnionitis and funisitis, accompanied by elevated concentrations of cytokines, chemokines, prostanoids and metalloproteases in amniotic and cervicovaginal fluids with preterm rupture of membranes (Keelan et al., 2003; Romero et al., 2007a). There is abundant evidence that pathological intrauterine inflammation resulting in preterm birth can be a consequence of intrauterine infection, particularly in early preterm births less than 32 weeks gestation (Romero et al., 2007a; DiGiulio et al., 2008). Fetal inflammatory response syndrome and a range of fetal morbidities are thought to be the results of fetal exposure to hyperinflammation in utero (Gotsch et al., 2007; Murthy and Kennea, 2007). However, it is also apparent that in a sizeable proportion of preterm births complicated by intrauterine inflammation there is no clinical evidence of pathological intrauterine infection (Moutsopoulos and Madianos, 2006; Romero et al., 2007a; Lee et al., 2008). It has been hypothesized that activation of the innate immune system in the decidua and fetal membranes by bacterial products or other non-bacterial agonists might be the triggering agents in such cases (Elovitz, 2006; Lee et al., 2008). In support of this, intra-amniotic administration of lipopolysaccharide (LPS), an agonist of the Toll-like receptor (TLR)4 receptor, or pro-inflammatory cytokines alone can trigger preterm labour and birth in primate models (Sadowsky et al., 2006; Adams Waldorf et al., 2008).
The nuclear factor-kappa B (NF-κB) transcription factor is at the nexus of cellular inflammatory response (Lu et al., 2008). In the absence of inflammatory stimuli, NF-κB p50/p65 subunits are retained in an inactive complex in the cytoplasm by the chaperone-like protein I-κBα. With exposure to pro-inflammatory stimuli, such as a TLR4 agonist or pro-inflammatory cytokines, phosphorylation of I-κB occurs, leading to its degradation and subsequent release of p50/p65 homo-and hetero-dimers to translocate to the nucleus, driving inflammatory gene expression (Kawai and Akira, 2007). Phosphorylation of I-κB is performed by the IκB kinase (IKK) complex, which consist of two catalytic subunits, IKKα, IKKβ, and a regulatory subunit IKKγ (NEMO) (Schmid and Birbach, 2008). IKKβ is 20-fold more active than IKKα in terms of I-κB phosphorylation and is the primary kinase responsible for inflammatory NF-κB activation in vivo (Kawai and Akira, 2007; Schmid and Birbach, 2008). For this reason small-molecular IKKβ inhibitors are the focus of active investigation as potential anti-inflammatory therapeutics in a number of clinical settings (Calzado et al., 2007; Strnad and Burke, 2007). However, NF-κB activation also regulates expression of several pro-survival genes; hence, IKK inhibition can also provoke apoptosis, particularly in neoplastic tissue (Calzado et al., 2007).
Several anti-inflammatory compounds have been evaluated in a variety of models for their capacity to block intrauterine inflammation. Using human choriodecidual membranes, in vitro studies have demonstrated efficacy of the non-selective anti-inflammatory/anti-oxidant N-acetylcysteine (NAC) (Lappas et al., 2003; 2004;). This drug has also been shown to be effective in rodent models (Beloosesky et al., 2006; Paintlia et al., 2008), and very recently it has been trialled in women at high risk of preterm birth with encouraging results (Shahin et al., 2009). However, its unpalatability and lack of potency and selectivity are potential drawbacks to its administration in pregnancy. The salicylate drug sulphasalazine is another potential anti-inflammatory candidate that inhibits NF-κB activation in intra-uterine tissues (Lappas et al., 2004, 2005); it has been administered to pregnant women for decades as a treatment for inflammatory bowel disease, and although there is evidence that it is safe in pregnancy (Norgard et al., 2007; Rahimi et al., 2008), a recent cohort study suggests a need for caution (Norgard et al., 2007). We have recently evaluated sulphasalazine in an ex vivo membrane model and found that while it is an effective inhibitor of inflammation at high concentrations, it also induces apoptosis in the chorion (Keelan et al., 2009), a potential concern for drugs acting via inhibition of NF-κB signalling (Karin, 2008).
A large number of potent and selective small molecule NF-κB inhibitors have been developed in recent years and tested in a variety of models (Pande and Ramos, 2005; Strnad and Burke, 2007). Although many are available commercially, few if any have been evaluated in placental tissues. We hypothesized that more selective inhibitors of NF-κB activation may provide advantages for the prevention of inflammation-driven preterm birth, over historic so-called anti-inflammatory drugs, in terms of more favourable pharmacodynamics and fewer side effects. Such compounds, assuming they crossed the placenta, might also offer some protection against the deleterious effects of inflammation in the fetus and neonate (Gotsch et al., 2007; Murthy and Kennea, 2007; Romero et al., 2007b), and therefore could improve fetal outcomes independent of any effects on rates of preterm birth. Here we report the results of a screening study of a number of agents, shown to be effective inhibitors of NF-κB activation in various animal and in vitro models, for their ability to inhibit choriodecidual inflammatory activation. Their effects on both inflammation and apoptosis in primary cultures of human choriodecidual cells were investigated with the intention of identifying potential agents suitable for subsequent evaluation in pre-clinical and clinical trials. We identified two IKK inhibitors that exhibited effective and non-toxic inhibition of inflammatory activation in choriodecidual cells, and concluded that IKKβ is a promising therapeutic target for prevention of inflammation-driven preterm birth.
Methods
Cell culture and treatments
Primary choriodecidual (CD) cell cultures were prepared from placentas obtained by Caesarean section at term prior to the onset of labour according to previously published methods (Keelan and Mitchell, 1998). Tissues were collected with informed maternal consent in accordance with the approval of the local Human Ethics committee. In brief, the choriodecidua was manually separated from the reflected amnion, washed in phosphate buffered saline (PBS), dissected and digested for 1 h at 37°C in 0.012% collagenase/0.25% dispase. DNase I (4 mg·L−1) was added for the final 15 min of incubation. The liberated cells were isolated and fractionated on a 5–60% discontinuous Percoll gradient. Cells lying between the 60–20% layers were recovered by aspiration, washed and plated at 2.5 × 105 cells per cm2 in M199 medium supplemented with 10% FCS, glutamax and antibiotic/antimycotics. Cells were cultured at 37°C in humidified 5% CO2/95% air for 2–3 days prior to addition of treatments (Keelan and Mitchell, 1998). Cell composition was determined by staining with anti-vimentin and anti-cytokeratin 7 antibodies to determine the relative proportions of decidual and chorionic cells respectively (Figure S1); 20 ± 10% of the cells stained positive for the trophoblast marker cytokeratin 7.
Cytokine assays
Concentrations of IL-6 and TNF-α in conditioned media were determined by ELISA using commercially available capture and detection antibodies according to the manufacturer's instructions (PeproTech, NJ, USA). Plate reading and curve fitting was performed on a SpectraMax plate reader using SoftMax ProV software (Molecular Dynamics/GE Healthcare, Sunnyvale, CA, USA), or a Synergy 2 Multi Mode Microplate reader using Gene 5 software (BioTek Instruments Inc., VT, USA). Cytokine production rates were normalized to total cellular protein, as determined by the BCA method calibrated against BSA (Redinbaugh and Turley, 1986).
Assessment of cell viability
The viability of cells plated at 5 × 104 cells per well of a 96-well plate was assessed by MTT assay (Denizot and Lang, 1986). After 24 h treatment with LPS or inhibitors, MTT reagent (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was added to a final concentration of 0.5 mg·ml-1 and the cells incubated for 2-4 h. The formazan dye generated was dissolved in acidified isopropanol and absorbance at 570 nm was measured using a Spectramax plate spectrophotometer (Molecular Dynamics/GE Healthcare).
Cell lysis and nuclear extraction
Cells were homogenized in lysis buffer (10 mM HEPES pH 7.4, 10 mM KCL, 0.1 mM EDTA, 0.1 mM EGTA, 2 mM dithiothreitol, 1% NP-40 and protease inhibitors). Lysates were centrifuged for 1 min at 3800× g at 4°C and the supernatant containing cytoplasmic proteins removed. The pellet was resuspended in 200 µL high-salt resuspension buffer (10 mM HEPES pH 7.4, 400 mM NaCl, 10 mM KCL, 0.1 mM EDTA, 0.1 mM EGTA, 2 mM dithiothreitol, 1% NP-40, protease inhibitors), incubated on ice for 15 min, then centrifuged for 5 min at 13 200 rpm at 4°C. Protein concentrations of the resulting supernatants containing nuclear proteins were then determined by a RC-DC protein assay, calibrated against BSA.
Immunoblot analysis
Nuclear and cytoplasmic protein extracts were subjected to SDS polyacrylamide gel electrophoresis on a 4–15% gradient gel under reducing conditions (NuPage System, Invitrogen/Life Technologies, Carlsbad, CA, USA) and transferred onto PVDF membranes. Membranes were blocked for 1 h in 1–5% non-fat dried milk in TBS-T (10 mM Tris, 150 mM NaCl and 0.1% Tween-20) and incubated overnight at 4°C with either anti-p65 (1:1000), anti-p50 (1:1000), anti-pyruvate kinase (1:1000), anti-histone H1 (1:5000) or anti-β-actin (1:20 000) antibodies. After washing, the membranes were incubated for 1-2 h with HRP-conjugated secondary antibody (1:2500), washed, and immunoreactive bands visualized by chemiluminescence using Supersignal West Dura ECL reagent followed by exposure to X-ray film (Agfa-Gevaert NV, Mortsel, Belgium).
Immunofluorescence staining
Choriodecidual cells were cultured in four-well chamber slides and stimulated with LPS with or without inhibitors for 2 h. Cells were then fixed for 20 min with 4% paraformaldehyde in 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA and 10 mM PIPES pH 6.8, then 1 min exposure to acetone at −20°C followed by PBS washes. Cells were next pre-incubated with 4% BSA in PBS for 1 h, then incubated with anti-p65 antibody (1:100) for 1 h, washed and incubated with FITC-labelled anti-rabbit IgG (1:500) for 1 h. Cells were then washed again and mounted using DAPI-containing aqueous mounting medium. Visualization was carried out using a TH4-200 microscope equipped with a DP70 digital camera and DPManager software (Olympus Corp., Tokyo, Japan).
RNA extraction and quantitative RT-PCR
Pilot experiments showed that TNF-α mRNA expression peaked at 4 h post-LPS. Therefore, total RNA was extracted from cells (n = 6 placentas) after 4 h of treatment with either vehicle control, LPS (0.1 µg·mL−1), LPS plus [5-(p-fluorophenyl)-2-ureido] thiophene-3-carboxamide (TPCA-1) (7 µM) or LPS plus parthenolide (40 µM) using PureLink™ Micro-to-Midi Total RNA Purification kits (Invitrogen/Life Technologies). RNA yield and quality was determined by measuring 260:280 nm ratios using a Nanodrop 1000 (Nanodrop Technologies, DE, USA). First–strand cDNA synthesis was performed using a Superscript III reverse transcription kit with random hexamer priming, followed by treatment with DNase-I to remove genomic DNA contamination. All RNA samples were initially screened for expression of two cytokine markers, IL-6 and TNF-α, by qRT-PCR using an ABI 7700 Sequence Detector (Applied Biosystems/Life Technologies, CA, USA) using SybrGreen detection. Relative expression was computed based on the 2[–ΔΔCt] method, relative to the housekeeping gene GAPDH. Following array analysis, another six genes were selected for qRT-PCR confirmation of changes in expression (PTGS1/COX-2, NOD2, CCL4/MIP-1β, MMP-2, BIRC5/survivin). PCR primers (Table 1) were designed using OligoPerfect™ Designer (Invitrogen/Life Technologies).
Table 1.
PCR primer sequences
| Locus ID gene bank | Gene name | Primer name | Sequence (5′ to 3′) | Amplicon size (bp) |
|---|---|---|---|---|
| NM_003998 | NF-κB1/p50 | Forward | CTG GAA GCA CGA ATG ACA GA | 172 |
| Reverse | TGA GGT CCA TCT CCT TGG TC | |||
| NM_002984 | CCL4/MIP-1β | Forward | GTA GCT GCC TTC TGC TCT CC | 196 |
| Reverse | TCA CTG GGA TCA GCA CAG AC | |||
| NM_022162 | NOD2 | Forward | TCT GTC CAG ACC CTG CTC IT | 148 |
| Reverse | CCT GTT CAG AGA AGC CCT TG | |||
| NM_000962 | PTGS1/COX-2 | Forward | TGA GCA TCT ACG GTT TGC TG | 158 |
| Reverse | TGC TTG TCT GGA ACAACT GC | |||
| NM_004530 | MMP2 | Forward | AGG GCA CAT CCT ATG ACA GC | 186 |
| Reverse | ATT TGT TGC CCA GGA AAG TG | |||
| NM_001168 | BIRC5/Survivin | Forward | AAA TGA CTT GGC TCG ATG CT | 219 |
| Reverse | CAC CCT GCA GCT CTA TGA CA | |||
| NM_000600 | IL-6 | Forward | ATG ACT CCT TCT CCA CAA GCG | 287 |
| Reverse | AGC CAT CTT TGG AAG GTT CAG G | |||
| NM_000594 | TNF-α | Forward | TGC TTG TTC CTC AGC CTC TTC T | 276 |
| Reverse | TAT CTC TCA GCT CCA CGC CAT T | |||
| NM_002046 | GAPDH | Forward | GAG TCA ACG GAT TTG GTC GT | 185 |
| Reverse | GAC AAG CTT CCC GTT CTC AG |
Oligonucleotide array processing and analysis
Custom boutique oligonucleotide arrays (‘superarrays’) were fabricated by SABiosciences Corp. (Frederick, MD, USA). Each membrane contained 112 genes (4 spots per gene) related either to apoptosis, NF-κB signalling or inflammation. Five housekeeping genes, including GAPDH, β-actin and β2-macroglobulin, were included on the arrays. RPS27A (ribosomal protein 27a) and BAS2C (biotinylated artificial sequence 2 complementary) served as positive detection controls; negative controls and blanks were also included. The full list of genes is detailed in Table S1.
From n = 6 independent experiments, three representative sets of RNA samples were selected for array analysis based on (i) RNA quality; (ii) RNA yield; and (iii) representative response to LPS, as determined by increased expression of IL-6 and TNF-α. Synthesis of cDNA, cRNA and labelling of probes from 0.5 µg sample RNA was carried out using a TrueLabeling-AMP™ 2.0 kit. Amplification, hybridization and signal development were performed according to the manufacturer's instructions; 4 µg of cRNA was used for each hybridization. Signal from the arrays was determined by chemiluminescence, visualized by exposure to X-ray film using a range of exposure times (1–15 min) to obtain optimal images.
X-ray films were scanned and analysed by densitometry using ImageQuant software (GE Healthcare, Uppsala, Sweden), by employing the array analysis option and ‘spot surface minimum’ background subtraction. Residual blanks were then subtracted and spot intensities were normalized to GAPDH. Prior to statistical analysis of pooled data, to correct for experiment-to-experiment variations in overall intensity, the overall mean signals of each set of arrays was adjusted so that the overall signal of the three control arrays were equivalent. Mean and standard deviation for each gene from the n = 3 experiments was then calculated and these data used for subsequent analysis.
Materials and reagents
Nuclear factor-kappa B inhibitors were purchased from Calbiochem/Merck KGaA, Darmstadt, Germany. Lipopolysaccharide (Escherichia coli LPS, serotype 055:B5), bicinchoninic acid (BCA) reagent and anti-β-actin antibody were from Sigma-Aldrich, St Louis, MO, USA. IL-6 and TNF-α ELISA development reagents were obtained from PeproTech, NJ, USA. M199 medium, fetal calf serum (FCS), glutamax, DNase 1, antibiotic/antimycotic solution, MTT reagent, PCR primers, PureLink™ Micro-to-Midi RNA purification kits, Superscript III kits, SybrGreen Mastermix and NuPage™ precast polyacrylamide gels were supplied by Invitrogen/Life Technologies (Carlsbad, CA, USA). Custom OligoGE Array™ membranes and TrueLabelling-AMP™ 2.0 kits were purchased from SABioscience Corporation, Frederick, MD, USA. Anti-p65/RelA antibody was from Santa Cruz Biotechnology Inc. (CA, USA); anti-histone H1, anti-pyruvate kinase and were purchased from Abcam, Cambridge, MA, USA. SuperSignal West Dura extended Duration Substrate and goat anti-rabbit IgG-peroxidase conjugate were purchased from Pierce/Thermo Scientific, Rockford IL, USA. Complete protease inhibitor tablets, biotin-16-dUTP and dispase II were purchased from Roche Diagnostics, Mannheim, Germany. Percoll and PVDF transfer membranes were supplied by GE Healthcare Bio-Sciences AB, Uppsala, Sweden. RC-DC protein assay reagents were obtained from Bio-Rad Laboratories Pty Ltd, NSW, Australia. Vectashield™ mounting medium was supplied by Vector Laboratories, Burlinghame, CA, USA.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5.0 or InStat 3.0 (GraphPad Software, La Jolla, CA, USA). Significant differences between group means were determined by one-way anova with post-hoc testing. P≤ 0.05 was considered significant. Data are represented as mean ± SD.
Results
Screening of NF-κB inhibitors
Primary choriodecidual cells were initially stimulated over an 18 h incubation period with 100 ng·mL−1 LPS together with one of nine different NF-κB inhibitors or vehicle control. The inhibitors tested were selected based on their published anti-inflammatory efficacy from in vitro and in vivo models, relative specificity, and mode of action. Concentrations for testing were estimated from published IC50 data and were selected to achieve approximately 75% maximal effect: SN50, an inhibitor of NF-κB translocation (36 µM); SC514 (5-(thien-3-yl)-3-aminothiophene-2-carboxamide), a selective and competitive IKKβ inhibitor (5 µM); BMS 345541 (4(2′-aminoethyl)amino-1,8-dimethylimidazo(1,2-a)quinoxaline), a selective allosteric IKKβ inhibitor (5 µM); evodiamine (21-methyl-3,13,21-triazapentacyclohenicosa-2(10),4,6,8,15,17,19-heptaen-14-one), an IKK inhibitor (1 µM); wedelolactone (7-methoxy-5,11,12-trihydroxycoumestan), a non-selective IKK inhibitor (50 µM); butein (3,4,2′,4′-tetrahydroxychalcone), a partially selective IKKβ inhibitor (50 µM); CAPE (caffeic acid phenylmethyl ester), an inhibitor of NF-κB translocation (35 µM); parthenolide, an inhibitor of the IKK complex (40 µM), and TPCA-1, a selective IKKβ inhibitor (5–7 µM).
Lipopolysaccharide induced a significant increase in IL-6 and TNF-α production with no decrease in cell viability (Figure 1). Out of nine inhibitors tested, only evodiamine and SN-50 failed to exert any inhibitory effects on LPS-stimulated cytokine production at the concentrations tested. SC514, wedelolactone, BMS 345541, CAPE and butein exerted modest inhibitory effects on the two cytokines measured (Figure 1A and B). TPCA-1 and parthenolide were the most effective, significantly inhibiting (P < 0.01) both IL-6 and TNF-α production by greater than 50%. None of the compounds tested showed evidence of toxicity as judged by MTT assay (Figure 1C).
Figure 1.

Initial evaluation of anti-inflammatory properties of nine commercially available NF-κB inhibitors in primary human choriodecidual cells stimulated with LPS (0.1 µg·mL−1) for 18 h. Candidate inhibitors were tested for their ability to inhibit production of IL-6 (A) and TNF-α (B) as assessed by ELISA. Cytokine data were normalized to cellular protein and expressed relative to LPS control. Cell viability was also determined by MTT assay (C), normalized to vehicle control. Inhibitor concentrations were as follows: SN50 (36 µM); SC514 (5 µM); BMS 345541 (5 µM); evodiamine (1 µM); wedelolactone (50 µM); butein (50 µM); caffeic acid phenylmethyl ester [CAPE] (35 µM); TPCA-1 (7 µM); parthenolide (40 µM). Data represent mean ± SD of n = 3–4 experiments performed in triplicate. *P < 0.05, **<0.01 versus LPS alone by anova with Dunnett's test post hoc.
TPCA-1 and parthenolide both inhibited LPS-induced IL-6 and TNF-α production in CD cells in a concentration-dependent manner (Figure 2). IC50 values for inhibition of IL-6 production by TPCA-1 and parthenolide were 7.05 µM and 3.82 µM respectively. However, TPCA-1 was a more potent inhibitor of TNF-α production, with an IC50 value 2.35 µM compared with 4.16 µM for parthenolide. No effects on cell viability were detected at any concentration.
Figure 2.

Concentration-dependent effects of TPCA-1 and parthenolide on LPS-stimulated cytokine production by choriodecidual cells. Cells were treated with LPS (0.1 mg·mL−1) for 18 h together with inhibitor or vehicle for 18 h; production of IL-6 and TNF−α was normalized to cellular protein and expressed as percentage of vehicle control. MTT activity was measured to monitor effects on cell viability. Data represent mean ± SD of n = 3–4 experiments performed in triplicate. *P < 0.05, **P < 0.01 versus vehicle control by anova with Dunnett's test post hoc.
Time course studies (4–24 h) of the effect of TPCA-1 (7 µM) and parthenolide (40 µM) on LPS-induced IL-6 production revealed that the inhibitory effects of both compounds increased with duration of exposure (Figure 3); no significant effects were observed at 4 h, but by 16 h inhibition had reached maximal levels. We also investigated the effects of time of addition of inhibitors (−1 h, 0 h, +1 h relative to LPS addition) but this did not significantly influence the efficacy of either inhibitor (data not shown).
Figure 3.
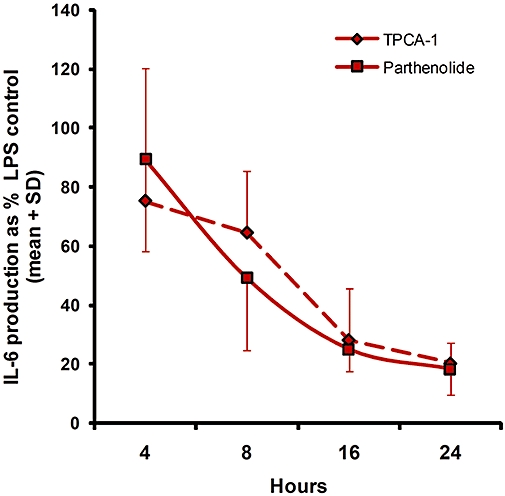
Time-course study of the effects of TPCA-1 (7 µM) and parthenolide (40 µM) on LPS-sitimulated IL-6 production. The data are the mean ± SD of n = 3 experiments performed in triplicate.
Effects of TPCA-1 and parthenolide on NF-κB activation by LPS
To confirm the mode of action of the two NF-κB inhibitors, we first investigated by immunofluorescence changes in the cellular localization of p65/RelA subunit in response to LPS (Figure 4). Absence of cross-contamination of the cytosolic and nuclear fractions was confirmed using immunoblotting for pyruvate kinase (cytosolic marker) and histone H1 (nuclear marker). In control cells, p65 was mainly localized in the cytoplasm. At 2 h, addition of LPS triggered translocation of p65 into the nucleus. In the presence of inhibitors, nuclear staining was partially blocked by parthenolide and completely abrogated by TPCA-1. The effects of the inhibitors on nuclear translocation of p65 were also investigated by immunoblotting of nuclear protein extracts prepared six after treatment (Figure 5). A single band that migrated at 65 kDa was detected in the nuclear protein fraction of untreated cells and its intensity was increased markedly with LPS treatment. In cells co-treated with TPCA-1 and parthenolide the intensity of the p65 band was diminished to below control levels.
Figure 4.

Immunoflourescence localization of p65/RelA cellular localization using FITC-labelled detection antibody in choriodecidual cells stimulated with LPS (0.1 µg·mL−1) for 2 h in the presence or absence of TPCA-1 (7 µM) and parthenolide (40 µM). Nuclei were visualized by DAPI staining (right-hand panel). LPS induced marked nuclear p65 staining, which was inhibited by both IKK inhibitors. The results are representative of n = 3 experiments.
Figure 5.

Immunoblotting analysis of nuclear translocation of p65/RelA in LPS-stimulated choriodecidual cells treated with IKK inhibitors. Cells were treated with LPS and either TPCA-1 (7 µM) or parthenolide (40 µM) for 6 h, then lysed and nuclear proteins extracted and immunoblotted; p65 was detected by chemiluminescence, visualized after exposure to X-ray film. Equal amounts of protein were loaded on each lane. A representative experiment of n = 4 is shown.
Array analysis of the effects of TPCA-1 and parthenolide on NF-κB-associated gene expression
In order to more fully assess the effects of TPCA-1 and parthenolide on LPS-stimulated choriodecidual gene expression profiles, boutique pathway-specific arrays were designed and probed with biotin-dUTP-labelled RNA from three representative sets of experiments. A 4 h time-point was chosen, based on preliminary studies showing that the TNF-α mRNA response to LPS was maximal at this time. An example of a set of images is shown in Figure S2. All positive controls and housekeeping genes gave strong signals, while blanks and negative controls were devoid of spots. Analysis of the background-corrected and normalized mean signal from the arrays showed that 38 genes (34%) were consistently stimulated by LPS in all three experiments (mean stimulation approximately threefold), of which 14 were statistically significant (P < 0.05, anova) (Figure 6). The hybridization signals for housekeeping genes and positive controls were not significantly altered by LPS treatment. Genes induced by LPS included inflammatory signalling proteins (A20, p65/RelA, I-κBα, IRAK2/4, NOD1/2, TAB1-3, TAK1, TLR4, TRAF2), cytokines/chemokines (CSF2, IL-1β, IL-6, TNF-α, MIP-1α, MIP-1β), apoptosis-related genes (bcl2, BIRC5/survivin, PYCARD, caspase-8) and parturition-related inflammatory genes (SP-A1/SFTPA1, PTGES, PLA2 IV4B, PLA2VI, MMP-2, MMP-13). On the other hand, a number of genes that were expected to be stimulated by LPS were unresponsive. These included PTGS-2/COX-2, CCL2/MCP-1, CCL22/MDC and IL-8, although it is worth noting that robust stimulation of these genes was observed on two of the three arrays. No genes on the arrays were significantly inhibited by LPS, although RIP-2 and TRADD were modestly inhibited in all three array experiments.
Figure 6.

Identification of LPS-responsive genes in choriodecidual cells stimulated for 4 h with LPS (0.1 µg·mL−1) using custom oligonucleotide arrays. Expression is expressed as percentage of vehicle control after normalization to GAPDH following background subtraction. Data shown (mean ± SD) represent those genes consistently stimulated in all three experiments; the solid black columns are significantly different from LPS control. Significance was determined by anova with Tukey's test post ho c. *P < 0.05; **P < 0.01; ***P < 0.001 versus control. Note log y-axis.
The two IKK inhibitors exhibited effective and widespread inhibition of LPS-responsive and non-responsive genes. 38 genes were significantly down-regulated by one or more of the inhibitors (Table 2). Of these, 36 were inhibited by TPCA-1 (mean inhibition: 60 ± 17.5%), while parthenolide inhibited 20 (mean inhibition: 45.1 ± 20.1%). Of the 14 genes that were significantly stimulated by LPS (displayed in Figure 6), all were significantly down-regulated by either or both IKK inhibitors. Both TPCA-1 and parthenolide significantly reduced the expression of A20, BIRC5/survivin, I-κBα, MAPK3, MIP-1α, MIP-1β, MMP-2, MMP-13, p50, PLA2G6, TAB2, TAB3, TIRAP/Mal, TNF-α and TRAF2 (Table 2). In addition, a number of genes that were not significantly up-regulated by LPS were also inhibited by TPCA-1 or parthenolide: AKT1, AP1, CARD11, COX-2, I-κBβ, MAPK3, MCP-1, MIP-1δ, MYD88, p50, RelB, SARM1, TICAM1, TIRAP/Mal, TNFR1 and TRAM1. While TPCA-1 inhibited the greatest number of genes, two genes were significantly inhibited by parthenolide alone: TAK1 and TRAM1. Notable genes that were not inhibited by either inhibitor included bcl2, bcl10, BIRC4/XIAP, caspase-8, FADD, IL-10, IKKα, IKKβ, IKKγ, MALT1, PYCARD, RIP1, TAB1 and TLR4.
Table 2.
Inhibition of choriodecidual gene expression by TPCA-1 or parthenolide
| Gene name | TPCA-1 (7 µM) % change | Parthenolide (40 µM) % change |
|---|---|---|
| A20 | −85.30 ± 9.0*** | −26.3 ± 6.9** |
| AKT1 | −43.40 ± 32.8* | −37.0 ± 35.9 (NS) |
| AP-1 | −55.7 ± 37.6* | −19.5 ± 35.9 (NS) |
| BCL3 | −64.6 ± 2.2* | 18.2 ± 35.9 (NS) |
| BIRC5/survivin | −66.1 ± 8.0*** | −46.7 ± 6.9*** |
| CARD11 | −62.3 ± 32.4* | −24.9 ± 39.0 (NS) |
| COX-2/PTGS2 | −45.9 ± 28.9* | −40.9 ± 36.7** |
| CSF2 | −52.6 ± 36.9* | −33.6 ± 7.3 (NS) |
| I-kBα | −89.6 ± 2.25*** | −54.7 ± 10.6*** |
| I-kBβ | −53.5 ± 7.1** | −43.5 ± 9.2 (NS) |
| MAPK3 | −42.5 ± 17.0* | −42.3 ± 10.8* |
| MCP-1 | −72.8 ± 15.4** | −4.9 ± 28.2 (NS) |
| MDC | −41.5 ± 13.0* | −12.4 ± 35.1 (NS) |
| MIP-1α | −50.3 ± 30.1** | −61.1 ± 15.5** |
| MIP-1β | −69.6 ± 32.5** | −80.2 ± 10.7** |
| MIP-1δ | −67.6 ± 33.6* | −62.3 ± 24.0 (NS) |
| MMP13 | −76.8 ± 5.9*** | −59.2 ± 10.5** |
| MMP2 | −50.4 ± 25.1* | −46.6 ± 26.0* |
| MYD88 | −22.8 ± 3.7* | −12.5 ± 14.4 (NS) |
| NOD1 | −61.8 ± 17.1* | −43.7 ± 46.5 (NS) |
| NOD2 | −72.6 ± 20.2* | −26.3 ± 63.8 (NS) |
| p50 | −52.3 ± 13.4** | −33.0 ± 24.1** |
| p65 | −75.0 ± 23.5* | −49.9 ± 48.1 (NS) |
| PLA2G6 | −57.7 ± 28.2* | −69.8 ± 10.9** |
| PTGES | −36.6 ± 30.1* | −19.4 ± 32.7 (NS) |
| RELB | −56.2 ± 37.5* | −40.9 ± 26.4 (NS) |
| RIP2 | −26.9 ± 16.0* | 15.2 ± 13.0 (NS) |
| SARM1 | −83.4 ± 12.8* | −16.5 ± 45.2 (NS) |
| SP-A1 | −75.8 ± 10.0* | −37.0 ± 46.1 (NS) |
| TAB2 | −75.0 ± 4.5*** | −44.0 ± 19.5* |
| TAB3 | −60.6 ± 13.7* | −51.3 ± 33.2* |
| TAK1 | −47.1 ± 22.9 (NS) | −61.4 ± 23.7* |
| TICAM1 | −87.2 ± 12.2** | −35.6 ± 48.8 (NS) |
| TIRAP/Mal | −48.5 ± 13.8* | −48.5 ± 18.2* |
| TNF-α | −89.5 ± 10.6*** | −63.7 ± 28.0** |
| TNFR1 | −31.8 ± 17.7** | −17.8 ± 13.2 (NS) |
| TRAF2 | −58.8 ± 25.0* | −56.7 ± 39.7* |
| TRAM1 | 0.3 ± 51.1 (NS) | −68.7 ± 20.4** |
The data are presented as the mean ± SD percentage change in expression compared with LPS-stimulated controls (n = 3). Significance was determined by anova with Tukey's test post hoc.
P < 0.05
P < 0.01
P < 0.001 versus LPS alone.
LPS, lipopolysaccharide; NS, not significant; TPCA-1, [5-(p-fluorophenyl)-2-ureido] thiophene-3-carboxamide.
To validate the array findings, we used qRT-PCR to examine expression of a selection of high- and low-expression genes with a range of responsiveness to LPS: IL-6, TNF-α, MMP2, p50, BIRC5/survivin, PTGS2/COX-2 and NOD2 (Figure 7). In general, the qRT-PCR results corroborated the array results in terms of response to LPS and IKK inhibition. The effects of parthenolide, however, were much less evident in the qRT-PCR data. Unexpectedly, MMP2 expression showed no response to LPS by RT-PCR, in contrast to the twofold increase demonstrated in the array data.
Figure 7.

Comparison of gene expression data using oligonucleotide arrays (A) and quantitative real-time PCR analysis (B). A selection of low and high abundance genes were chosen for analysis. Array data (A) are taken from the complete set of genes shown in Figure 6 (n = 3 experiments). For real-time PCR, relative expression was computed based on the 2[−ΔΔCt] method relative to the housekeeping gene GAPDH. PCR data are presented as mean ± SD (n = 4 experiments). *P < 0.05; **P < 0.01 versus LPS control by anova.
Discussion
The primary objective of this study was to screen a range of NF-κB inhibitors for their anti-inflammatory effects in human primary choriodecidual cells, in order to identify candidate agents suitable for further evaluation for the prevention and treatment of inflammation-driven preterm birth. Additional objectives were to identify favourable molecular pathways or targets amenable to pharmacological manipulation, and assess the differences in the effects of different inhibitors on expression of inflammatory/apoptosis-related genes. From a total of nine potential candidate compounds tested, we identified two IKK inhibitors that effectively inhibited the production of inflammatory cytokines and the expression of genes associated with inflammatory signalling and activation without reducing cell viability: TPCA-1 and parthenolide. We conclude that IKKβ inhibition holds promise as an effective pharmacological approach for the treatment of intra-uterine inflammation-driven preterm birth.
It is now well established that IKK activation is a convergence point in inflammatory signalling via the NF-κB pathway (Schmid and Birbach, 2008) and, as such, has been of interest for over two decades because of its attraction as a therapeutic target for the treatment of chronic inflammatory disorders, such as inflammatory bowel disease, ulcerative colitis and arthritis (Strnad and Burke, 2007; Schmid and Birbach, 2008). The IKK complex contains two related kinases, IKKα and IKKβ, which, despite their homology, are independently regulated and functionally distinct (Schmid and Birbach, 2008). IKKα plays an important role in the non-canonical (p52/RelB) NF-κB pathway which is central to keratinocyte differentiation (Descargues et al., 2008), receptor-induced apoptosis and the negative regulation of NF-κB activation (Strnad and Burke, 2007). It can translocate to the cell nucleus and phosphorylate nuclear proteins including histone H3, SRC-3 and CBP (Chariot, 2009). On the other hand, IKKβ activation is key to the canonical (inflammatory) pathway and regulates the expression of many inflammation-associated genes. IKKβ is approximately 20-fold more efficient than IKKα at phosphorylating Iκ-Bα, and knockout studies in mice have shown that IKKβ, not IKKα, is essential for the activation of NF-κB by TNF-α, IL-1β or LPS (Li et al., 1999). IKKβ also phosphorylates other targets in addition to Iκ−Bα; these include NF-κB signalling proteins such as p65, p105, Bcl10, CARMA1, CYLD and NEMO, as well as other important cell regulators such as β-catenin, IRS-1, FOXO3a and TSC1 (Chariot, 2009). Therefore, selective inhibition of IKK activity could theoretically influence a wide variety of cellular processes in addition to those mediated through NF-κB activation.
We identified two IKK inhibitors, TPCA-1 and parthenolide, as effective and non-toxic anti-inflammatory agents. This was not unexpected, although it is interesting that several other IKK inhibitors were tested [SC514 (Kishore et al., 2003), BMS 345541 (Burke et al., 2003), evodiamine (Takada et al., 2005), wedelolactone (Kobori et al., 2004), butein (Pandey et al., 2007)] but none of these exerted more than a modest inhibition of IL-6 and TNF-α production. The widely utilized cell permeable peptide SN-50, which acts via blockade of NF-κB translocation (Lin et al., 1995), was also without effect. While we evaluated these compounds at concentrations reported to be effective in other tissues, it is possible that higher concentrations might have had more effect. However, in previous pilot studies we tested some other NF-κB inhibitors using choriodecidual explants and, surprisingly, these were also without effect, although strong inhibition was seen in other cell types (J.A. Keelan et al., unpublished observations). On the other hand, we and others have reported inhibition of NF-κB activity in choriodecidual tissues by salicylates (which are low potency IKKβ inhibitors) and the free radical scavenger/NF-κB inhibitor NAC (Lappas et al., 2002, 2003; Beloosesky et al., 2006; Keelan et al., 2009). These findings suggest that the NF-κB pathway has some unusual characteristics in choriodecidual tissue that make it refractory to pharmacological inhibition. Studies are currently underway to explore this possibility further. Regardless of the underlying mechanisms, the present results indicate that the efficacy of NF-κB inhibitors in these tissue requires careful experimental determination and cannot be accurately predicted from studies in other models.
TPCA-1 is a thiophene carboxamide shown to inhibit LPS-stimulated TNF-α, IL-6 and IL-8 production by macrophages (IC50– 0.3 µM). In murine models it has been shown to block LPS-induced airway inflammation (Birrell et al., 2005), and reduce the severity of collagen-induced arthritis with efficacy similar to that of etanercept (Kondo et al., 2008). It is a potent inhibitor of IKKβ with 22-fold selectivity over IKKα (Yamamoto and Gaynor, 2001). Parthenolide is a sesquiterpene lactone extract of the medicinal plant feverfew, and an inhibitor of the IKK complex (Hehner et al., 1999); it does not, however, exert direct effects on either IKKα or β kinase activity (Hehner et al., 1999). It has also been shown to inhibit tyrosine phosphorylation and the activity of other kinases including MAPK (Hwang et al., 1996). Parthenolide inhibits inflammatory gene expression in vitro (Hwang et al., 1996; Smolinski and Pestka, 2003), although in vivo it elicits a much weaker response, with only IL-6 expression being reported to be mildly suppressed by parthenolide in murine spleen and liver (Smolinski and Pestka, 2005). Hence, our findings of more widespread and robust inhibition of inflammatory gene expression by TPCA-1 are consistent with published data. The differences in specificity, potency and mode of action probably explain the observed differences in genes inhibited by the two compounds. We speculate that the two genes responsive only to parthenolide inhibition are likely to be driven by other kinases (e.g. MAPK) that are responsive to the effects of parthenolide (Hwang et al., 1996).
Expression of a variety of pro-inflammatory cytokines and chemokines was markedly inhibited by TPCA-1, many of which were also responsive to LPS stimulation. TPCA-1 also blocked the expression of a number of proteins involved in the canonical signalling pathway, such as p65, MyD88, TAB2, TAB3, TICAM1/TRIF and TIRAP/Mal, plus other inflammation-associated proteins AKT1, CARD11/CARMA1 and the intracellular peptidoglycan sensors NOD1 and NOD2 (Verstrepen et al., 2008). We would expect that the overall effects of these changes would be effective and sustained inhibition of the inflammatory response. TPCA-1 also inhibited the expression of a number of key signalling proteins involved in the activation of NF-κB by TNF-α: RIP2, TRAF2 and TAK1, as well as the type I TNF receptor and TNF-α itself. TNF-α is a key pro-inflammatory cytokine in intra-uterine tissues and mediates much of the effects of LPS in choriodecidual tissues (Sato et al., 2003). Amniotic administration of a TNF antagonist has recently been shown to prevent LPS-induced preterm labour in a rodent model (Holmgren et al., 2008). Blockade of TNF production and actions by TPCA-1 is likely to further enhance its anti-inflammatory efficacy. On the other hand, TPCA-1 also inhibited expression of several genes involved in the negative regulation of NF-κB activation: A20, Bcl3, IκBα/β/ζ (Sun and Ley, 2008). This may have implications for its efficacy long term, as inflammatory gene expression changes with time and typically undergoes a resolution phase via induction of negative feedback pathways. Inhibition of these negative regulators by TPCA-1 might result in excessive or dysregulated inflammation rather than the opposite. Extended duration time-course studies would be required to address this possibility.
Nuclear factor-kappa B is also an important driver of anti-apoptotic protein expression, particularly in transformed cells, and as such its inhibition has been investigated for the treatment of various cancers (Karin, 2008). Previous studies with sulphasalazine in full thickness fetal membranes (Keelan et al., 2009) highlighted the possibility that chorionic trophoblasts might be susceptible to apoptosis following NF-κB inhibition. However, in the present studies none of the inhibitors tested affected cell viability as determined by MTT assay over 24 h exposure. Finally, a number of genes known to be important mediators of parturition were inhibited by TPCA-1 and parthenolide, specifically genes involved in prostanoid biosynthesis (prostaglandin H and E synthases, phospholipases A2) and collagenolysis/membrane rupture (MMP2, 9, 13). Inhibition of prostaglandin biosynthesis and metalloprotease activity would further enhance the anti-parturogenic effects of the IKK inhibitors.
From a clinical perspective, our findings suggest that IKKβ inhibition has potential as a therapeutic target for prevention and treatment of inflammation-driven preterm labour and birth. Several studies in animal models have supported the use of anti-inflammatory modalities to prevent LPS-driven preterm birth, although caution should be exercised when comparing these data to human preterm labour due to the nature of the stimuli (Buhimschi et al., 2003; Xu et al., 2005; Holmgren et al., 2008). The LPS model commonly employed revolves around activation of inflammatory responses via TLR4, which elicits a specific signal transduction pathway resulting in characteristic gene expression profile. Our data would be applicable to these models. Intra-uterine inflammation, however, could be triggered by different agents (a range of different microbes, pathogen-activated molecular patterns or non-microbial TLR agonists), resulting in activation of different signalling pathways; hence, selective pharmacological inhibitors may or may not be effective in such cases. Studies using inhibitors to treat tissues delivered following spontaneous preterm delivery are currently planned to address this issue. The ultimate goal of any treatment modality is improved fetal outcomes, not just prevention of prematurity. In this regard, the use of anti-inflammatory IKKβ inhibitors may hold a significant advantage over other pharmacological approaches, such as prostaglandin synthase inhibitors or progesterone analogues. Fetal inflammation is believed to play a central role in the pathophysiology of a variety of neonatal morbidities associated with preterm birth, including cerebral palsy, necrotizing enterocolitis and periventricular leukomalacia (Romero et al., 2007b). In theory, treatment of the fetus via maternal administration of an IKK inhibitor should prevent damage caused by fetal inflammation and improve neonatal and perinatal outcomes, independent of any effect on timing of delivery. In a recently published trial by Shahin et al. (2009) the effects of administration of NAC and 17-hydroxyprogesterone caproate (17-OHPC) in mid pregnancy were compared with 17-OHPC alone in a group of 280 high risk women with bacterial vaginosis and prior PTB. Remarkably, NAC administration resulted in significantly fewer preterm deliveries (35%), an increase in birth weight (∼400 g) and a reduction in neonatal morbidity/mortality compared with 17-OHPC alone (Shahin et al., 2009). NAC also appeared to interfere with normal labour, increasing the requirement for induced labour more than fourfold. (Shahin et al., 2009). These findings are encouraging in terms of the efficacy and benefits of an anti-inflammatory agent on both prematurity rates and neonatal outcomes. However, NAC is unpleasant to take and there was a high drop-out rate in the treatment arm of the study. A more palatable and targeted drug would be expected to have better compliance and more positive outcomes.
While our findings suggest that IKKβ is an attractive target for inhibition, this approach could also suffer from potential drawbacks and unwanted side effects. The foremost concern would be that blocking NF-κB activation might prevent proper activation of the host innate and adaptive immune system and increase maternal susceptibility to infections. This would be of particular concern in pregnant women with an already suppressed immune response (Luppi, 2003). However, this same possibility applies to glucocorticoids, salicylates and TNF biologics, and appears to be more of a theoretical concern rather than a clinical reality. Nevertheless, the period of administration would need to be kept to a minimum; a 3 month treatment window for asymptomatic high-risk women between 20 and 32 weeks gestation might be a reasonable starting point. On the other hand, somewhat counter-intuitively, chronic IKKβ inhibition has been reported to induce hyper-stimulation, augmenting IL-1β secretion by macrophages in response to LPS, raising concerns about the risk of septic shock with long-term IKKβ inhibitor administration (Greten et al., 2007). This effect does not appear to be mediated through inhibition of expression of negative regulators of NF-κB activation such as A20 and IκB proteins, but through inhibition of NF-κB-mediated negative regulation of pro-IL-1β processing (Greten et al., 2007). The effects of maternal administration of IKKβ inhibitors on the fetus would also be of concern, depending on whether the agent crosses the placenta and significant concentrations were achieved in fetal circulation. There are no published studies we are aware of describing the teratogenic effects of IKKβ inhibitors, although in mice, IKKβ gene deletion is embryonically lethal as a consequence of massive hepatic apoptosis (Li et al., 1999), while conditional disruption of the IKKβ locus in endothelial cells prevents embryonic hepatic vascular development (Hou et al., 2008). However, IKKβ−/+ heterozygotes are born fully developed, healthy and with normal livers, despite ∼50% reduced IKKβ activity (Li et al., 1999; Strnad and Burke, 2007). It is also reassuring to note that currently employed anti-inflammatory drugs prescribed in pregnancy such as amino-salicylates and infliximab, all of which effectively cross the placenta, do not appear to have any significant foetotoxicity or teratogenicity (Brar and Einarson, 2008; Rahimi et al., 2008). The trial of NAC administration in pregnancy did not report any increase in fetal abnormalities (Shahin et al., 2009), although numbers were too small to detect an increase in a rare occurrence.
In conclusion, we have tested a range of NF-κB inhibitors for their ability to prevent LPS-induced inflammation in human choriodecidual cells and identified two IKK inhibitors as effective and non-toxic agents with widespread effects on inflammation-associated gene expression. Our studies suggest that IKKβ inhibition is a potential therapeutic target for preventing inflammation in fetal membranes and as such merits investigation for the treatment prevention of preterm labour and birth. Studies are needed to determine the longer-term effects of IKKβ inhibitors and their ability to cross the placenta and act upon fetal organs. The potential risks and benefits of delivering anti-inflammatory agents to the fetus is a complex issue that will require careful investigation.
Acknowledgments
This study was funded in part by a Prematurity Initiative Program Grant No. 21-FY05-1247 from The March of Dimes Foundation, the Maurice and Phyllis Paykel Trust, and The University of Auckland. JAK is supported by the Women and Infants Research Foundation (WA); MDM is supported by a James Cook Research Fellowship from the Royal Society of New Zealand. The authors would like to thank Natalie Lorenz for technical assistance with generation of pilot data and the mothers at Auckland Central Hospital, Auckland, and King Edward Memorial Hospital, Perth, who kindly donated their placentas for this research.
Glossary
Abbreviations
- ABIN
A20-binding inhibitor of NF-κB
- AKT
ATP-dependent tyrosine kinase
- BCA
bicinchoninic acid
- BCL
B-cell CLL/lymphoma
- BIRC
baculoviral IAP repeat-containing
- CAPE
caffeic acid phenylmethyl ester
- CARD
caspase recruitment domain
- CARMA
caspase-recruitment domain (CARD) membrane-associated guanylate kinase protein 1
- CBP
CREB-binding protein
- CCL
chemokine (C-C motif) ligand
- CSF
colony stimulating factor
- CYLD
cylindromatosis
- DAPI
4′,6-Diamidino-2-phenylindole
- FADD
Fas-associated death domain
- FCS
fetal calf serum
- GAPDH
glyceraldehyde phosphate dehydrogenase
- I-κB
inhibitor of kappa B
- IKK
inhibitor of kappa B kinase
- IL
interleukin
- IRAK
insulin receptor-activated kinase
- IRS1
insulin receptor substrate 1
- LPS
lipopolysaccharide
- MALT1
mucosa associated lymphoid tissue lymphoma translocation gene 1
- MAPK
mitogen-activated kinase
- MDC
macrophage-derived chemokines
- MEKK3
mitogen-activated protein kinase kinase kinase 3
- MIP
macrophage inflammatory protein
- MMP
matrix metalloprotease
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrasodium bromide
- NAC
N-acetylcysteine
- NALP
NACHT, LRR and PYD containing protein
- NEMO
NF-κB essential modulator
- NF-κB
nuclear factor-kappa B
- PDGH
prostaglandin dehydrogenase
- PDLIM
PDZ and Lim domain
- PGLYRP
peptidoglycan recognition protein
- PLA2
phospholipase A2
- PTGS
prostaglandin endoperoxide synthase
- PVDF
polyvinylidine fluoride
- PYCARD
PYD and CARD domain containing
- RBCK1
RanBP-type and C3HC4-type zinc finger containing 1
- RIG
regulated in glioma
- RIP
receptor interacting protein
- SARM1
sterile alpha and TIR motif containing 1
- SP-A
surfactant protein-A
- SRC-3
steroid receptor co-activator-3
- TAB
TAK1-binding protein
- TAK
TGF-β-activated kinase
- TBS-T
Tris-buffered saline/0.1% Tween 20
- TICAM1
toll-like receptor adaptor molecule 1
- TIRAP
toll-interleukin 1 receptor (TIR) domain containing adaptor protein
- TLR
toll-like receptor
- TNF
tumour necrosis factor
- TNFR
TNF receptor
- TPCA-1
[5-(p-fluorophenyl)-2-ureido] thiophene-3-carboxamide
- TRAF
TNF receptor-associated factor
- TRAM
TRIF-related adapter molecule
- TSC1
tuberous sclerosis 1
- VISA
virus-induced signalling adaptor
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Immunocytochemical staining of choriodecidual cells. Cells were grown in 96-well tissue culture plates for 3 days then fixed for 10 min in ice-cold acetone : methanol (1:1). After washing in PBS/0.5% Tween (PBS-T), cells were incubated with anti-cytokeratin-7 monoclonal antibody (1:100; clone OV-TL 12/30, DakoCytomation, Denmark A/S) in immunobuffer (PBS-T / 1% BSA / 5% normal calf serum) for 1 h at room temperature with gentle shaking, then washed 3× in PBS-T. Immunostaining was developed with Cy3-labelled goat anti-mouse IgG (10 μg/mL, BioLegend, San Diego, CA, USA). Cells were then washed and incubated with anti-vimentin monoclonal antibody (1:200; clone V9, DakoCytomation), detected with DyLight488-labelled goat anti-mouse IgG (10 μg/mL BioLegend). Finally, nuclei were stained with Hoechst 33342 (4 μg/mL in PBS) for 10 min before washing and storing in PBS-T. Cells were imaged in Nikon Eclipse TiR inverted fluorescent microscope and photographed at 1:200 magnification using a Nikon CCD camera. Decidual cells (vimentin positive) are stained green; chorionic trophoblasts (cytokeratin-positive) are stained red-orange; nuclei are stained blue. Ratio of decidual : chorion cells was ∼4:1.
{kind=link}
Figure S2 Custom oligonucleotide array images. Representative images of 8 × 16 custom oligonucleotide arrays (SABiosciences Corp., Frederick, MD, USA) probed with biotin-labelled cRNA from choriodecidual cells stimulated (4 h) with LPS ± TPCA-1 or parthenolide. Arrays were visualized by chemiluminescence and exposed to X-ray film for 1 min. Films were then imaged by densitometry and saved as TIF files for image analysis using ImageQuant software (GE Healthcare). Spots on the left and bottom right corners are positive controls; housekeeping genes are the first six spots on the botton row.
{kind=link}
Table S1 Genes present on custom oligonucleotide arrays.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Adams Waldorf KM, Persing D, Novy MJ, Sadowsky DW, Gravett MG. Pretreatment with toll-like receptor 4 antagonist inhibits lipopolysaccharide-induced preterm uterine contractility, cytokines, and prostaglandins in rhesus monkeys. Reprod Sci. 2008;15:121–127. doi: 10.1177/1933719107310992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloosesky R, Gayle DA, Amidi F, Nunez SE, Babu J, Desai M, et al. N-acetyl-cysteine suppresses amniotic fluid and placenta inflammatory cytokine responses to lipopolysaccharide in rats. Am J Obstet Gynecol. 2006;194:268–273. doi: 10.1016/j.ajog.2005.06.082. [DOI] [PubMed] [Google Scholar]
- Birrell MA, Hardaker E, Wong S, McCluskie K, Catley M, De Alba J, et al. Ikappa-B kinase-2 inhibitor blocks inflammation in human airway smooth muscle and a rat model of asthma. Am J Respir Crit Care Med. 2005;172:962–971. doi: 10.1164/rccm.200412-1647OC. [DOI] [PubMed] [Google Scholar]
- Brar H, Einarson A. Effects and treatment of inflammatory bowel disease during pregnancy. Can Fam Physician. 2008;54:981–983. [PMC free article] [PubMed] [Google Scholar]
- Buhimschi IA, Buhimschi CS, Weiner CP. Protective effect of N-acetylcysteine against fetal death and preterm labor induced by maternal inflammation. Am J Obstet Gynecol. 2003;188:203–208. doi: 10.1067/mob.2003.112. [DOI] [PubMed] [Google Scholar]
- Burke JR, Pattoli MA, Gregor KR, Brassil PJ, MacMaster JF, McIntyre KW, et al. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J Biol Chem. 2003;278:1450–1456. doi: 10.1074/jbc.M209677200. [DOI] [PubMed] [Google Scholar]
- Calzado MA, Bacher S, Schmitz ML. NF-kappaB inhibitors for the treatment of inflammatory diseases and cancer. Curr Med Chem. 2007;14:367–376. doi: 10.2174/092986707779941113. [DOI] [PubMed] [Google Scholar]
- Chariot A. The NF-kappaB-independent functions of IKK subunits in immunity and cancer. Trends Cell Biol. 2009;19:404–413. doi: 10.1016/j.tcb.2009.05.006. [DOI] [PubMed] [Google Scholar]
- Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods. 1986;89:271–277. doi: 10.1016/0022-1759(86)90368-6. [DOI] [PubMed] [Google Scholar]
- Descargues P, Sil AK, Sano Y, Korchynskyi O, Han G, Owens P, et al. IKKalpha is a critical coregulator of a Smad4-independent TGFbeta-Smad2/3 signaling pathway that controls keratinocyte differentiation. Proc Natl Acad Sci U S A. 2008;105:2487–2492. doi: 10.1073/pnas.0712044105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiulio DB, Romero R, Amogan HP, Kusanovic JP, Bik EM, Gotsch F, et al. Microbial prevalence, diversity and abundance in amniotic fluid during preterm labor: a molecular and culture-based investigation. PLoS ONE. 2008;3:e3056. doi: 10.1371/journal.pone.0003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elovitz MA. Anti-inflammatory interventions in pregnancy: now and the future. Semin Fetal Neonatal Med. 2006;11:327–332. doi: 10.1016/j.siny.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Gotsch F, Romero R, Kusanovic JP, Mazaki-Tovi S, Pineles BL, Erez O, et al. The fetal inflammatory response syndrome. Clin Obstet Gynecol. 2007;50:652–683. doi: 10.1097/GRF.0b013e31811ebef6. [DOI] [PubMed] [Google Scholar]
- Greten FR, Arkan MC, Bollrath J, Hsu LC, Goode J, Miething C, et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell. 2007;130:918–931. doi: 10.1016/j.cell.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehner SP, Hofmann TG, Droge W, Schmitz ML. The antiinflammatory sesquiterpene lactone parthenolide inhibits NF-kappa B by targeting the I kappa B kinase complex. J Immunol. 1999;163:5617–5623. [PubMed] [Google Scholar]
- Holmgren C, Esplin MS, Hamblin S, Molenda M, Simonsen S, Silver R. Evaluation of the use of anti-TNF-alpha in an LPS-induced murine model. J Reprod Immunol. 2008;78:134–139. doi: 10.1016/j.jri.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Hou Y, Li F, Karin M, Ostrowski MC. Analysis of the IKKbeta/NF-kappaB signaling pathway during embryonic angiogenesis. Dev Dyn. 2008;237:2926–2935. doi: 10.1002/dvdy.21723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang D, Fischer NH, Jang BC, Tak H, Kim JK, Lee W. Inhibition of the expression of inducible cyclooxygenase and proinflammatory cytokines by sesquiterpene lactones in macrophages correlates with the inhibition of MAP kinases. Biochem Biophys Res Commun. 1996;226:810–818. doi: 10.1006/bbrc.1996.1433. [DOI] [PubMed] [Google Scholar]
- Karin M. The IkappaB kinase – a bridge between inflammation and cancer. Cell Res. 2008;18:334–342. doi: 10.1038/cr.2008.30. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Keelan JA, Mitchell MD. The effects of a ceramide analogue, N-acetylsphingosine, on basal and tumor necrosis factor-alpha-stimulated production of prostaglandins and cytokines in gestational tissues. Prostaglandins Other Lipid Mediat. 1998;55:141–158. [Google Scholar]
- Keelan JA, Blumenstein M, Helliwell RJ, Sato TA, Marvin KW, Mitchell MD. Cytokines, prostaglandins and parturition – a review. Placenta. 2003;24(Suppl. A):S33–S46. doi: 10.1053/plac.2002.0948. [DOI] [PubMed] [Google Scholar]
- Keelan JA, Khan S, Yosaatmadja F, Mitchell MD. Prevention of inflammatory activation of human gestational membranes in an ex-vivo model using a pharmacological NF-kappaB inhibitor. J Immunology. 2009;183:5270–5278. doi: 10.4049/jimmunol.0802660. [DOI] [PubMed] [Google Scholar]
- Kishore N, Sommers C, Mathialagan S, Guzova J, Yao M, Hauser S, et al. A selective IKK-2 inhibitor blocks NF-kappa B-dependent gene expression in interleukin-1 beta-stimulated synovial fibroblasts. J Biol Chem. 2003;278:32861–32871. doi: 10.1074/jbc.M211439200. [DOI] [PubMed] [Google Scholar]
- Kobori M, Yang Z, Gong D, Heissmeyer V, Zhu H, Jung YK, et al. Wedelolactone suppresses LPS-induced caspase-11 expression by directly inhibiting the IKK complex. Cell Death Differ. 2004;11:123–130. doi: 10.1038/sj.cdd.4401325. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Fukuda K, Adachi T, Nishida T. Inhibition by a selective IkappaB kinase-2 inhibitor of interleukin-1-induced collagen degradation by corneal fibroblasts in three-dimensional culture. Invest Ophthalmol Vis Sci. 2008;49:4850–4857. doi: 10.1167/iovs.08-1897. [DOI] [PubMed] [Google Scholar]
- Lappas M, Permezel M, Georgiou HM, Rice GE. Nuclear factor kappa B regulation of proinflammatory cytokines in human gestational tissues in vitro. Biol Reprod. 2002;67:668–673. doi: 10.1095/biolreprod67.2.668. [DOI] [PubMed] [Google Scholar]
- Lappas M, Permezel M, Rice GE. N-Acetyl-cysteine inhibits phospholipid metabolism, proinflammatory cytokine release, protease activity, and nuclear factor-kappaB deoxyribonucleic acid-binding activity in human fetal membranes in vitro. J Clin Endocrinol Metab. 2003;88:1723–1729. doi: 10.1210/jc.2002-021677. [DOI] [PubMed] [Google Scholar]
- Lappas M, Permezel M, Ho PW, Moseley JM, Wlodek ME, Rice GE. Effect of nuclear factor-kappa B inhibitors and peroxisome proliferator-activated receptor-gamma ligands on PTHrP release from human fetal membranes. Placenta. 2004;25:699–704. doi: 10.1016/j.placenta.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Lappas M, Yee K, Permezel M, Rice GE. Sulfasalazine and BAY 11-7082 interfere with the nuclear factor-kappa B and I kappa B kinase pathway to regulate the release of proinflammatory cytokines from human adipose tissue and skeletal muscle in vitro. Endocrinology. 2005;146:1491–1497. doi: 10.1210/en.2004-0809. [DOI] [PubMed] [Google Scholar]
- Lee SE, Romero R, Park CW, Jun JK, Yoon BH. The frequency and significance of intraamniotic inflammation in patients with cervical insufficiency. Am J Obstet Gynecol. 2008;198:633.e1–633.e8. doi: 10.1016/j.ajog.2007.11.047. [DOI] [PubMed] [Google Scholar]
- Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, et al. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J. Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem. 1995;270:14255–14258. doi: 10.1074/jbc.270.24.14255. [DOI] [PubMed] [Google Scholar]
- Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42:145–151. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Luppi P. How immune mechanisms are affected by pregnancy. Vaccine. 2003;21:3352–3357. doi: 10.1016/s0264-410x(03)00331-1. [DOI] [PubMed] [Google Scholar]
- Moutsopoulos NM, Madianos PN. Low-grade inflammation in chronic infectious diseases: paradigm of periodontal infections. Ann N Y Acad Sci. 2006;1088:251–264. doi: 10.1196/annals.1366.032. [DOI] [PubMed] [Google Scholar]
- Murthy V, Kennea NL. Antenatal infection/inflammation and fetal tissue injury. Best Pract Res Clin Obstet Gynaecol. 2007;21:479–489. doi: 10.1016/j.bpobgyn.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Norgard B, Pedersen L, Christensen LA, Sorensen HT. Therapeutic drug use in women with Crohn's disease and birth outcomes: a Danish nationwide cohort study. Am J Gastroenterol. 2007;102:1406–1413. doi: 10.1111/j.1572-0241.2007.01216.x. [DOI] [PubMed] [Google Scholar]
- Paintlia MK, Paintlia AS, Singh AK, Singh I. Attenuation of lipopolysaccharide induced inflammatory response and phospholipids metabolism at the feto-maternal interface by N-Acetyl-cysteine. Pediatr Res. 2008;164:334–339. doi: 10.1203/PDR.0b013e318181e07c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande V, Ramos MJ. NF-kappaB in human disease: current inhibitors and prospects for de novo structure based design of inhibitors. Curr Med Chem. 2005;12:357–374. doi: 10.2174/0929867053363180. [DOI] [PubMed] [Google Scholar]
- Pandey MK, Sandur SK, Sung B, Sethi G, Kunnumakkara AB, Aggarwal BB. Butein, a tetrahydroxychalcone, inhibits nuclear factor (NF)-kappaB and NF-kappaB-regulated gene expression through direct inhibition of IkappaBalpha kinase beta on cysteine 179 residue. J Biol Chem. 2007;282:17340–17350. doi: 10.1074/jbc.M700890200. [DOI] [PubMed] [Google Scholar]
- Rahimi R, Nikfar S, Rezaie A, Abdollahi M. Pregnancy outcome in women with inflammatory bowel disease following exposure to 5-aminosalicylic acid drugs: a meta-analysis. Reprod Toxicol. 2008;25:271–275. doi: 10.1016/j.reprotox.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Redinbaugh MG, Turley RB. Adaption of the bicinchoninic acid protein assay for use with microtiter plates and sucrose gradient fractions. Anal Biochem. 1986;153:267–261. doi: 10.1016/0003-2697(86)90091-6. [DOI] [PubMed] [Google Scholar]
- Romero R, Espinoza J, Goncalves LF, Kusanovic JP, Friel LA, Nien JK. Inflammation in preterm and term labour and delivery. Semin Fetal Neonatal Med. 2006;11:317–326. doi: 10.1016/j.siny.2006.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero R, Espinoza J, Goncalves LF, Kusanovic JP, Friel L, Hassan S. The role of inflammation and infection in preterm birth. Semin Reprod Med. 2007a;25:21–39. doi: 10.1055/s-2006-956773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero R, Gotsch F, Pineles B, Kusanovic JP. Inflammation in pregnancy: its roles in reproductive physiology, obstetrical complications, and fetal injury. Nutr Rev. 2007b;65:S194–S202. doi: 10.1111/j.1753-4887.2007.tb00362.x. [DOI] [PubMed] [Google Scholar]
- Sadowsky DW, Adams KM, Gravett MG, Witkin SS, Novy MJ. Preterm labor is induced by intraamniotic infusions of interleukin-1beta and tumor necrosis factor-alpha but not by interleukin-6 or interleukin-8 in a nonhuman primate model. Am J Obstet Gynecol. 2006;195:1578–1589. doi: 10.1016/j.ajog.2006.06.072. [DOI] [PubMed] [Google Scholar]
- Sato TA, Keelan JA, Mitchell MD. Critical paracrine interactions between TNF-alpha and IL-10 regulate lipopolysaccharide-stimulated human choriodecidual cytokine and prostaglandin E2 production. J Immunol. 2003;170:158–166. doi: 10.4049/jimmunol.170.1.158. [DOI] [PubMed] [Google Scholar]
- Schmid JA, Birbach A. IkappaB kinase beta (IKKbeta/IKK2/IKBKB) – a key molecule in signaling to the transcription factor NF-kappaB. Cytokine Growth Factor Rev. 2008;19:157–165. doi: 10.1016/j.cytogfr.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Shahin AY, Hassanin IM, Ismail AM, Kruessel JS, Hirchenhain J. Effect of oral N-acetyl cysteine on recurrent preterm labor following treatment for bacterial vaginosis. Int J Gynaecol Obstet. 2009;104:44–48. doi: 10.1016/j.ijgo.2008.08.026. [DOI] [PubMed] [Google Scholar]
- Smolinski AT, Pestka JJ. Modulation of lipopolysaccharide-induced proinflammatory cytokine production in vitro and in vivo by the herbal constituents apigenin (chamomile), ginsenoside Rb(1) (ginseng) and parthenolide (feverfew) Food Chem Toxicol. 2003;41:1381–1390. doi: 10.1016/s0278-6915(03)00146-7. [DOI] [PubMed] [Google Scholar]
- Smolinski AT, Pestka JJ. Comparative effects of the herbal constituent parthenolide (Feverfew) on lipopolysaccharide-induced inflammatory gene expression in murine spleen and liver. J Inflamm. 2005;2:6. doi: 10.1186/1476-9255-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strnad J, Burke JR. IkappaB kinase inhibitors for treating autoimmune and inflammatory disorders: potential and challenges. Trends Pharmacol Sci. 2007;28:142–148. doi: 10.1016/j.tips.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Sun SC, Ley SC. New insights into NF-kappaB regulation and function. Trends Immunol. 2008;29:469–478. doi: 10.1016/j.it.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada Y, Kobayashi Y, Aggarwal BB. Evodiamine abolishes constitutive and inducible NF-kappaB activation by inhibiting IkappaBalpha kinase activation, thereby suppressing NF-kappaB-regulated antiapoptotic and metastatic gene expression, up-regulating apoptosis, and inhibiting invasion. J Biol Chem. 2005;280:17203–17212. doi: 10.1074/jbc.M500077200. [DOI] [PubMed] [Google Scholar]
- Verstrepen L, Bekaert T, Chau TL, Tavernier J, Chariot A, Beyaert R. TLR-4, IL-1R and TNF-R signaling to NF-kappaB: variations on a common theme. Cell Mol Life Sci. 2008;65:2964–2978. doi: 10.1007/s00018-008-8064-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu DX, Chen YH, Wang H, Zhao L, Wang JP, Wei W. Effect of N-acetylcysteine on lipopolysaccharide-induced intra-uterine fetal death and intra-uterine growth retardation in mice. Toxicol Sci. 2005;88:525–533. doi: 10.1093/toxsci/kfi300. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Gaynor RB. Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. J Clin Invest. 2001;107:135–142. doi: 10.1172/JCI11914. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.