

Abstract
BACKGROUND AND PURPOSE
Cucurbitacin R, a natural anti-inflammatory product, has been shown to exhibit activity against both adjuvant-induced arthritis and delayed-type hypersensitivity reactions induced by various agents. Previous studies have demonstrated that the effects of cucurbitacin R stem from its inhibition of both cytokine production and lymphocyte proliferation.
EXPERIMENTAL APPROACHES
Effects of cucurbitacin R were investigated on lipopolysaccharide-stimulated RAW 264.7 cells. Cell cycle evolution was analysed by flow cytometry, detection of apoptosis by DNA ladder, Bcl-2, p21, p53, Bax, cleaved caspase-1 (p10), caspase-9, and caspase-3, cleaved caspase (p17) and interleukin-1β detection was followed by Western blot analysis and mRNA expression with quantitative real time reverse transcription-polymerase chain reaction (qRT-PCR).
KEY RESULTS
Cucurbitacin R was found to induce apoptosis in lipopolysaccharide-stimulated RAW 264.7 macrophages through the inhibition of Bcl-2 expression, which regulates pro-inflammatory caspase-1 activation and interleukin-1β release. Also, cucurbitacin R arrested the cell cycle in the G2/M phase and increased the subG0 population in lipopolysaccharide-stimulated RAW 264.7 macrophages. Moreover, it increased the expression of proteins p53 and p21, down-regulated the expression of Bcl-2, activated the activity of caspase-1 and augmented the production of interleukin-1β. Finally, the transfection of RAW 264.7 macrophages with a Bcl-2 expression plasmid produced the inhibition of apoptosis and caspase-1 activation/interleukin-1β release induced by cucurbitacin R in RAW 264.7 cells.
CONCLUSIONS AND IMPLICATIONS
Taken together, these results point to a new apoptotic process in which interleukin-1β release is directly regulated by Bcl-2 status; this contributes to the evidence that apoptotic processes do not induce inflammation.
Keywords: apoptosis, cucurbitacin R, interleukin-1β, Bcl-2, caspase-1, RAW 264.7 macrophages
Introduction
Cucurbitacins are a group of natural products with a triterpene skeleton that are present in different plant species, especially those of the Cucurbitaceae and Cruciferae, which have been used as folk medicines for centuries in countries such as China and Brazil. However, only a small number of their pharmacological activities have been reported to date, notably their role as antagonists of ecdysteroids and their influence on the growth and development of insects (Ríos et al., 2005). Several cucurbitacins have also been shown to have anti-inflammatory and cytotoxic effects (Ríos et al., 2005). In previous papers, we have described the anti-inflammatory (Recio et al., 2004) and anti-arthritic (Escandell et al., 2007a) properties of cucurbitacin R. Indeed, in the latter study Escandell et al. (2007a) affirmed that the anti-arthritic effect of cucurbitacin R was due to its inhibition of signal transducer and activator of transcription-3 (STAT3) activation and the reduction of tumour necrosis factor-α (TNF-α). In parallel, the induction of nitric oxide synthase was inhibited along with nitric oxide production. However, while prostaglandin E2 production decreased, cyclooxygenase-2 induction did not. Taken together, these results suggest that cucurbitacin R could be a potential anti-inflammatory agent that acts through a mechanism in which the inhibition of TNF-α in lymphocytes is implicated, as is the activation of Janus kinase-2 and STAT3, as previously demonstrated by Sun et al. (2005) for other closely related compounds.
Recently, Escandell et al. (2008) demonstrated that cucurbitacins induced dramatic changes in the cytoskeleton of the cell, inhibiting cell proliferation in the G2/M phase and inducing apoptosis of both HCT116 and Hke-3 cells. However, the presence of oncogenic kRas clearly decreased the sensitivity of the cells to the three cucurbitacins tested, cucurbitacin R, dihydrocucurbitacin B and cucurbitacin I. Cucurbitacins also induced the expression of p53 and p21 in HCT116 cells harbouring mutant Ras. Using HCT116 cells with targeted deletion of p53 or p21, we confirmed that p53 and p21 actually protect cells from cucurbitacin-induced apoptosis.
Apoptosis is a physiological process critical for the development, homeostasis and normal functioning of the immune system. It involves the removal of cells from tissues in a deliberate and systematic manner, supposedly without an inflammatory reaction. Different proteins and enzymes are involved in the initiation, amplification and suppression of apoptosis, but it is generally caspases that mediate the final process of cell death (Wu and Shi, 2007). Caspases are intracellular proteases that cleave the substrates involved in both apoptosis and inflammation. They are expressed as inactive precursors or procaspases and are triggered into action as a result of their proteolytic processing at conserved aspartic acid residues to give rise to the active form (Wu and Shi, 2007). Although cell death can occur through caspase-independent, non-apoptotic mechanisms, the morphological characteristics that define the process of apoptosis still depend on caspases (Bleackley and Heibein, 2001).
Interleukin-1β, one of the major caspase-1 targets, is a multifunctional cytokine that is involved in a host of immune and pro-inflammatory responses (Dinarello, 1998). It is produced primarily by activated monocytes and macrophages, and signals through various adaptor proteins and kinases that lead to the activation of numerous downstream targets (Cao et al., 1996a,b;). Human interleukin-1β is synthesized as a 35 kDa precursor. To gain activity, the precursor must be cleaved by caspase-1 between Asp116 and Ala117 to yield a 17 kDa mature form (Cerretti et al., 1992; Thornberry et al., 1992). Detection of the 17 kDa mature form of interleukin-1β is a good indicator of the activity of the interleukin-1β converting enzyme, better known as caspase-1, which is required for this cleavage (Cerretti et al., 1992; Thornberry et al., 1992; Alnemri et al., 1996). Various agents, including lipopolysaccharide, are known to strongly induce the activation of caspase-1.
Recently, Bruey et al. (2007) identified a new mechanism for controlling caspase activation. They demonstrated that the anti-apoptotic proteins Bcl-2 and Bcl-XL interact with NALP1 (a nucleotide-dependent activator of cytokine-processing protease caspase-1), suppressing the NALP1-mediated activation of pro-inflammatory caspases and reducing interleukin-1β production. In addition, Greten et al. (2007) demonstrated that the inhibitor protein of NF-κB (IκB) kinase-β (IKKβ) inhibition produced an increase in both caspase-1 activation and interleukin-1β production.
In the present study, we have focused on the apoptosis induced by cucurbitacin R in RAW 264.7 macrophages to determine how these apoptotic processes can affect not only the release of different inflammatory mediators such as interleukin-1β but also their possible regulation by Bcl-2.
Methods
Chemicals and cell lines
Cucurbitacin R (Figure 1) was previously obtained from tayuya roots (Recio et al., 2004). RAW 264.7 macrophages were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and antibiotics, and propagated by scrapping. All biochemicals and chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA). Specific antibodies for β-actin were also obtained from Sigma-Aldrich, and antibodies recognizing caspase-3, p53, Bcl-2, Bax and p21 were purchased from Santa Cruz Biotechnologies (Santa Cruz, CA, USA).
Figure 1.
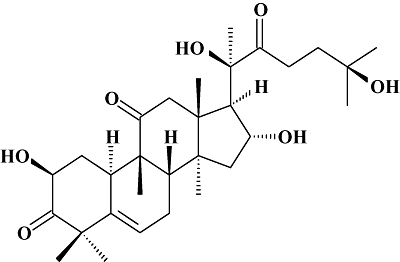
Chemical structure of cucurbitacin R.
Detection of apoptosis by DNA ladder in RAW 264.7 macrophages
Their viability was assessed with the aid of the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. First, cells were placed into each well of a 24-well flat-bottomed plate (Nunc, Raskilde, Denmark) with or without 1 µg·mL−1 lipopolysaccharide. Cucurbitacin R (100–12.5 µM) was added to the cells and the plates were incubated in 5% CO2-air humidified atmosphere at 37°C for different time periods. The cells were collected by means of centrifugation, and the pellet was resuspended in lysis buffer (100 mM EDTA, 10 mM Tris-HCl). Then, 10% SDS and 100 µg·mL−1 proteinase K were added to the homogenate, which was subsequently incubated for 1 h at 50°C. Proteins were precipitated through addition of 5 M NaCl, and the samples were centrifuged at 20 000 g for 15 min. The supernatant was mixed with an equal volume of phenol-chloroform-isoamylalcohol (25:24:1) and centrifuged at 20 000 g for 15 min. The uppermost layer was collected in a new vial, mixed with an equal volume of chloroform-isoamylalcohol (24:1) and centrifuged at 20 000 g for 5 min. Finally, the newly formed uppermost layer was mixed with 2-propanol, 10 mM MgCl2, and 10% glycogen (1 µL) and placed on ice for 15 min. DNA fragments were collected from this layer. To these fragments, 70% ethanol was added and the mixture was centrifuged at 20 000 g for 10 min. The desiccated pellet of DNA fragments was then dissolved in 25 µL of Tris-EDTA buffer (10 mM Tris-HCl, 1 mM EDTA, pH 7.4). Contaminating RNA was removed by incubating the samples with RNAase A (1 mg·mL−1) at 37°C for 20 min. Purified DNA (1 µg) was submitted to electrophoresis on a 2% agarose gel. The gel was stained with 0.5 µg·mL−1 ethidium bromide solution (Sigma-Aldrich), viewed on a UV transilluminator (LAS 3000 mini, Fuji, Tokyo, Japan) and photographed.
Cell cycle analysis
In order to analyse the cell cycle and apoptosis in RAW 264.7 macrophages, cells were first adjusted to a final concentration of 1 × 106 cells·mL−1 before use and then 1 mL of cell suspension was placed into each well of a six-well flat-bottomed plate (Nunc) with or without lipopolysaccharide (1 µg·mL−1) after treatment with the products. First, various concentrations of cucurbitacin R (100–12.5 µM) were added to the cells and the plates were incubated in 5% CO2-air-humidified atmosphere at 37°C for different time periods up to 24 h. In this experiment, we were looking for the subG0 peak as an index of apoptosis, and then for the G1, G2 and S peaks. The cells were harvested by means of centrifugation, washed in phosphate-buffered saline (pH 7.2) and then fixed in 70% ethanol for 30 min at −20°C. After the cells had been washed once with phosphate-buffered saline, the DNA was stained with propidium iodide (4 µg·mL−1, containing 100 µg·mL−1 of ribonuclease A; Sigma-Aldrich). Flow cytometry was carried out on a Beckman Coulter EPICS XCL (Fullerton, CA, USA); 104 cells were counted for each test sample.
Cell cycle histograms were analysed with the aid of the Cylchred program (University of Wales, College of Medicine, Cardiff, UK).
Annexin V apoptosis assay and determination of mitochondrial membrane potential
The detection of phosphatidylserine on the cell surface of the macrophages was performed with an Annexin V-fluorescein isothiocyanate apoptosis detection kit (BD PharMingen, San Diego, CA, USA), in accordance with the manufacturer's instructions. Briefly, unstimulated and lipopolysaccharide-stimulated cells (1 × 106 cells·mL−1) were cultured in 24-well flat-bottomed plates and subsequently exposed (or not) to cucurbitacin R (100-12.5 µM) for different time periods. The cells were collected by scrapping, washed with cold phosphate-buffered saline and resuspended in binding buffer. The cells were then stained with Annexin V-fluorescein isothiocyanate and propidium iodide. After a 30-min incubation period, the cells were analysed with the aid of flow cytometry.
Mitochondrial membrane potential was determined by means of flow cytometry through double staining with dihydrorhodamine 123 and propidium iodide. Lipopolysaccharide-stimulated cells were treated at different concentrations of cucurbitacin R for 18 h and stained with 1 µM of dihydrorhodamine 123 and propidium iodide for 1 h at 37°C, washed with phosphate-buffered saline, and analysed by the use of fluorescence techniques in the FL1 and FL3 channels of a flow cytometry Epics XL-MCL device (Beckman).
Analysis of caspase-3 and caspase-1 activity
The activity of caspase-3 was determined by the use of the cell-permeable fluorogenic substrate (EnzoLyte™ AMC Capase-3 Assay Kit, Anaspec, San Jose, CA, USA), which was used according to the manufacturer's instructions. The activity of caspase-1 was also determined with a cell-permeable fluorogenic specific substrate, also used according to the manufacturer's instructions. The activity was quantified by measuring fluorescence intensity upon caspase-3 or caspase-1 cleavage at Ex/Em = 354 nm/442 nm. Macrophages (1 × 106 cells·mL−1) were cultured in a 96-well black tissue culture microplate and treated with either cucurbitacin R (100 µM) or Ac-DEVD-CHO (10 µM). They were then stimulated with lipopolysaccharide for 24 h. Caspase-3 (peptide sequence = Ac-Asp-Glu-Val-Asp-AMC) substrate or caspase-1 (Ac-YVAD-AMC) was added to the cells, and the fluorescence was measured with a Victor Fluorescence microplate reader (Perkin Elmer, Wellesley, MA, USA).
Transient and stable transfections
Cells were transfected with a pool of siRNA specific for caspase-1 or non-specific siRNA (Dharmacon, Chicago, IL, USA) with the aid of lipofectamine 2000 (Invitrogen, Langley, OK, USA). Bcl-2 and PAI-2 expression plasmids or empty vector were also transfected with lipofectamine 2000 and selected by G418.
Quantitative reverse transcription-polymerase chain reaction (RT-PCR)
RAW 264.7 macrophages (1 × 106 cells·mL−1) were treated with cucurbitacin R at 100 µM for 8 h, after which the RNA was extracted with the aid of RNeasy mini spin (50) columns (Qiagen, Hilden, Germany). Real time RT-PCR was performed using a one-step qRT-PCR kit (Invitrogen, Paisley, UK) following the manufacturer's instructions. Applied biosystems (Foster City, CA, USA) 7500 qPCR platform was used. Results are expressed in time fold expression of interleukin-1β using GAPDH (R&D Systems, Minneapolis, MN, USA) as a housekeeping gene.
Western blot analysis
Cellular lysates from RAW 264.7 macrophages incubated for different periods of time with cucurbitacin R (100 µM) and lipopolysaccharide were obtained as described previously by Escandell et al. (2007a). For Bcl-2, p21, p53, Bax, cleaved caspase-1 (p10), caspase-3, cleaved caspase-3 (p17) (Cell Signalling, Danvers, MA, USA), caspase-9 (Cell Signalling), NALP1 (Cell Signalling) and interleukin-1β (R&D Systems) the membranes were incubated with the respective polyclonal antibodies (1:200 dilution, Santa Cruz). For β-actin, the membranes were incubated with monoclonal antibody (1:10 000 dilution, Sigma-Aldrich).
For Western blot of interleukin-1β in supernatants, 50 µL of media were collected at different time points from the LPS- or cucurbitacin R (100 µM)-treated RAW macrophages, and Western blot was carried out as described before.
Interleukin-1β production in RAW 264.7 macrophages
Macrophages (RAW 264.7) at 1 × 106 cells·mL−1 were co-incubated in a 96-well culture plate (200 µL) with either lipopolysaccharide (1 µg·mL−1) alone or with lipopolysaccharide (1 µg·mL−1) and cucurbitacin R (10–100 µM) for 24 h. The cells were lysed with a specific lysis buffer, and the resulting cell lysis was then collected and assayed for interleukin-1β by use of a specific enzyme immunoassay kit (eBioscience, San Diego, CA, USA), used according to the manufacturer's instructions.
Hoechst staining
Cells were grown on chamber slides and were either left untreated or were treated with cucurbitacin R as indicated. The cells were then fixed in a solution of methanol-acetic acid (95:5) for 20 min at −20°C. The slides were washed with phosphate-buffered saline and incubated with Hoechst 3324 at 37°C to stain the DNA. Samples were then examined with a fluorescent microscope; the images were acquired with a SPOT CCD camera (Nikon, Tokyo, Japan).
Results
Cucurbitacin R produced apoptosis in RAW 264.7 cells
To evaluate the effect of the anti-inflammatory drug cucurbitacin R (Figure 1) in RAW 264.7 macrophages, the cells were treated with the test compound (100 µM) and cells were collected by means of cell cycle analysis at different times after lipopolysaccharide stimulation. Not only did cucurbitacin R arrest the cell cycle in the G2/M phase starting at 12 h, but it consequently produced an increase in the subG0 population corresponding to apoptotic cells at 18 h (Figure 2A). These effects were observed only when the cells were treated with both lipopolysaccharide + cucurbitacin R; macrophages treated only with cucurbitacin R (100 µM) underwent neither apoptosis nor cell cycle arrest. The apoptotic process was confirmed by the DNA fragmentation assay. We found that only after the co-treatment of cells with lipopolysaccharide + cucurbitacin R was apoptosis visible at 18 and 24 h later (Figure 2B). To corroborate these data and evaluate how cucurbitacin R produced the cell cycle arrest, we tested the levels of cyclin A1, cyclin B1, cyclin D2, and cyclin E. While all the cyclin levels diminished at 18 and 24 h after treatment, the expression of cyclin D2 increased at 12 h (data not shown). These effects correlate with the reduction of cyclin levels in the apoptotic process.
Figure 2.
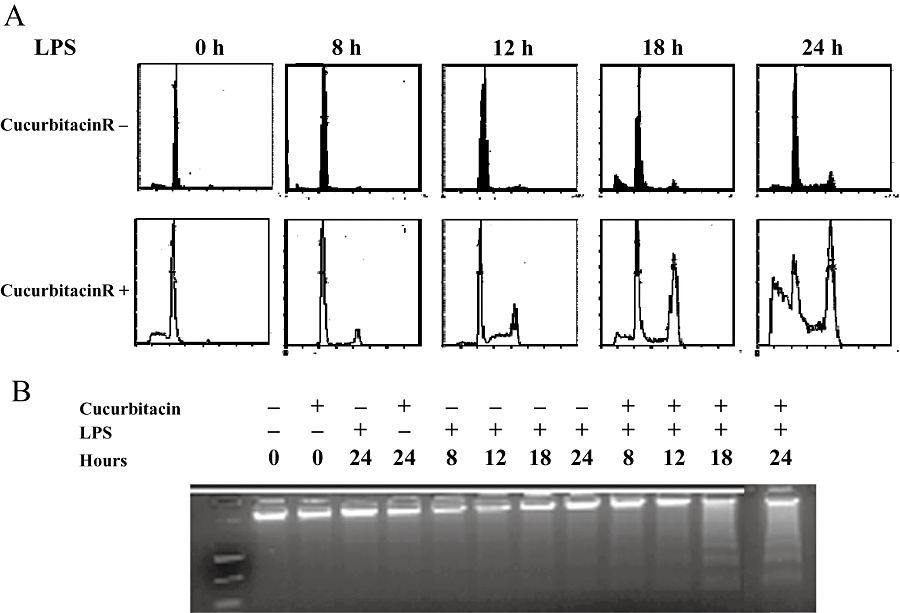
Cell cycle evolution after lipopolysaccharide (LPS) stimulation. (A) Inhibition of G0/G1 phase, G2/M arrest and apoptosis in cucurbitacin R-treated cells. Plots were obtained with propidium iodide staining at different times after cucurbitacin R (100 µM) treatment with or without LPS stimulation on RAW 264.7 macrophages. (B) DNA was obtained as described in the Methods section and the isolated DNA run on agarose gel (1.5%). DNA fragmentation was observed 18 and 24 h after cucurbitacin R treatment with LPS. Figures are representative of three experiments performed with similar results.
In the annexin V assay corresponding to phosphatidylserine externalization, cucurbitacin R at 100 µM led to 51% of the cells being apoptotic-necrotic cells (Figure 3A). This value was reduced to 19% when cells were treated with cucurbitacin R at 12.5 µM (Table 1). The concentration-dependent apoptotic effect of cucurbitacin R was confirmed by the DNA fragmentation assay and propidium iodide staining (Figure 3C,D), in which the major apoptotic effect occurred at concentrations of 50 and 100 µM; at the latter concentration, the apoptotic bodies were actually visible at 24 h (Figure 4).
Figure 3.
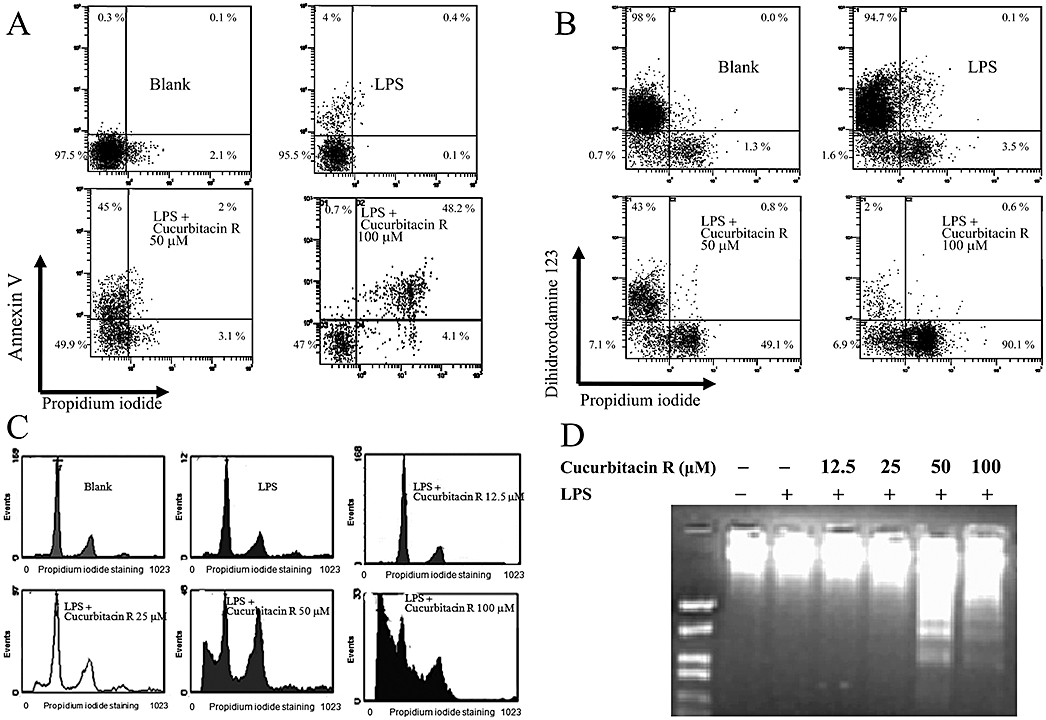
Cucurbitacin R induced apoptosis in RAW 264.7 cells in a concentration-dependent manner. Cells were treated for 24 h with cucurbitacin R (12.5–100 µM) after LPS stimulation. (A) Annexin V apoptosis assay. (B) Mitochondrial membrane potential assay. Cells were treated with cucurbitacin R at various concentrations for 18 h and apoptotic cells, apoptotic-necrotic cells, and necrotic cells were examined with annexin V or dihidrorodamine 123 and propidium iodide binding as described in the Methods section. (C) Plots were obtained with propidium iodide staining after cucurbitacin R treatment at different concentrations (12.5–100 µM). (D) DNA fragmentation assay in RAW 264.7 cells. Figures are representative of three experiments performed with similar results.
Table 1.
Annexin V staining showed that cucurbitacin R produced apoptosis in RAW 264.7 cells in a concentration-dependent manner
| Viable cells % | Apoptotic cells % | Apoptotic-necrotic cells % | Necrotic cells % | |
|---|---|---|---|---|
| Blank | 95 ± 3 | 0 ± 2 | 0 ± 3 | 4 ± 1 |
| LPS | 94 ± 5 | 5 ± 4 | 0 ± 1 | 1 ± 1 |
| LPS + cucurbitacin R 100 µM | 40 ± 10 | 0 ± 12 | 51 ± 1 | 9 ± 3 |
| LPS + cucurbitacin R 50 µM | 56 ± 8 | 41 ± 4 | 3 ± 2 | 3 ± 2 |
| LPS + cucurbitacin R 25 µM | 69 ± 3 | 28 ± 3 | 1 ± 1 | 3 ± 2 |
| LPS + cucurbitacin R 12.5 µM | 79 ± 10 | 19 ± 3 | 3 ± 2 | 1 ± 2 |
Cells were treated for 24 h with cucurbitacin R (12.5–100 µM) after LPS stimulation. Cells were collected and processed as described in Methods. Data are an average of three independent experiments.
LPS, lipopolysaccharide.
Figure 4.
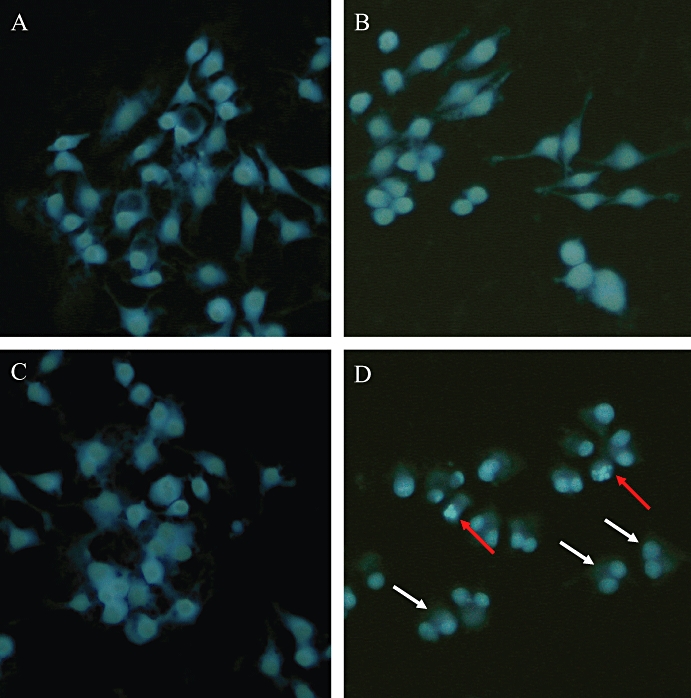
Apoptotic effect of cucurbitacin R in RAW 264.7 cells. Cells were treated for 24 h with cucurbitacin R (100 µM). Cells were fixed with methanol-acetic acid (95:5) and stained with Hoechst 3324. White arrows indicate that the cell cycle stopped in the G2/M phase; red arrows indicate apoptotic cells. (A) Blank group, without stimulation or cucurbitacin R treatment; (B) cucurbitacin R-blank group, treated with cucurbitacin but without stimulation with lipopolysaccharide (LPS); (C) control group, LPS-stimulated cells without cucurbitacin R treatment; (D) cells stimulated with LPS and treated with cucurbitacin R. White arrows indicate cells that were arrested in the G2/M phase. Red arrows indicate apoptotic cells. Figures are representative of two experiments performed with similar results.
To determine how the apoptotic pathway is regulated, we treated RAW macrophages with cucurbitacin R at 100 µM and collected cell extracts at different time points. The apoptotic death produced by cucurbitacin R induced the expression of p53 (Figure 5), which is mainly responsible for the arrest of the cell cycle and regulator apoptosis. In addition, cucurbitacin R also induced the production of the apoptotic protein p21, which is also involved in cell cycle arrest. Interestingly, whereas Bax was left unregulated, Bcl-2 was down-regulated when the RAW 264.7 macrophages were treated with cucurbitacin R (Figure 5), producing a destabilization of the mitochondrial membrane potential, as measured by dihydrorhodamine 123. This dye is the reduced form of rhodamine 123, which is a commonly used fluorescent mitochondrial dye. Dihydrorhodamine 123 itself is non-fluorescent, but it readily enters most of the cells and is oxidized by cellular redox systems to the fluorescent rhodamine 123 that accumulates in mitochondrial membranes. When the mitochondrial membrane potential is compromised, the cell is not able to oxidize this compound. As shown in Figure 3B, the fluorescence levels of dihydrorhodamine 123 were reduced in the cucurbitacin R-treated cells. These reduced values are accompanied by higher levels of propipium iodide staining, corresponding to apoptotic-necrotic cells.
Figure 5.
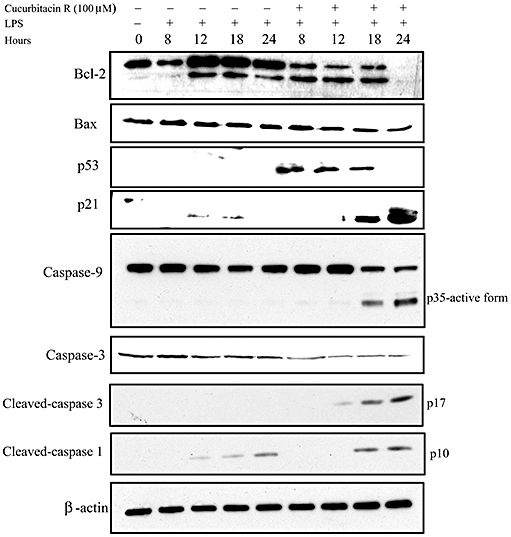
Cucurbitacin R modulates the expression of proteins implicated in apoptosis. RAW macrophages were treated with cucurbitacin R (100 µM) at different times and then expression of p53, p21, Bcl-2, Bax, cleaved-caspase-1 p10 subunit, caspase-9, cleaved caspase-3 p17 subunit and caspase-3 were determined with the aid of Western blot techniques. β-Actin was used as a control. Figures are representative of two experiments performed with similar results. LPS, lipopolyysaccharide.
The intrinsic apoptotic pathway was confirmed by caspase-9 Western blot. Upon apoptotic stimulation, the mitochondrial membrane potential is destabilized and the cytochrome c released from mitochondria associates with the 47 kDa procaspase-9/Apaf 1 (Li et al., 1997). This complex processes procaspase-9 into a large active fragment (35 or 17 kDa). As expected, we were able to detect the active form at 35 kDa of caspase-9 at 18 and 24 h after lipopolysaccharide stimulation in the presence of cucurbitacin R 100 µM. We checked for different effector caspases and the active fragments, like caspase-3, cleaved caspase-3 fragment p17 and cleaved caspase-1 fragment p10. As shown in Figure 5, we detected the active fragment of caspase-3 at the later time points after cucurbitacin R treatment. Surprisingly, we could detect an increase in the levels of active caspase-1 (p10), and this correlated with lower levels of Bcl-2.
Cucurbitacin R-activated caspase-1 and interleukin-1β release
As the cleavage of interleukin-1β is a good measure of caspase-1 activation, we measured the levels of this cytokine by different techniques. Firstly, we performed Western blot on the extracts and supernatants of cucurbitacin-treated cells. Whereas in lipopolysaccharide-treated cells, we detected the inactive form of interleukin-1β and only low levels of the cleaved form 17 kDa, in the cucurbitacin-treated cells, we detected mainly the cleaved form of interleukin-1β, at the different time points after activation of the apoptotic pathway (Figure 6A). We also subjected the cell culture supernatants to Western blot analysis of interleukin-1β. As expected, we detected higher levels of interleukin-1β in the cell supernatants treated with cucurbitacin R.
Figure 6.
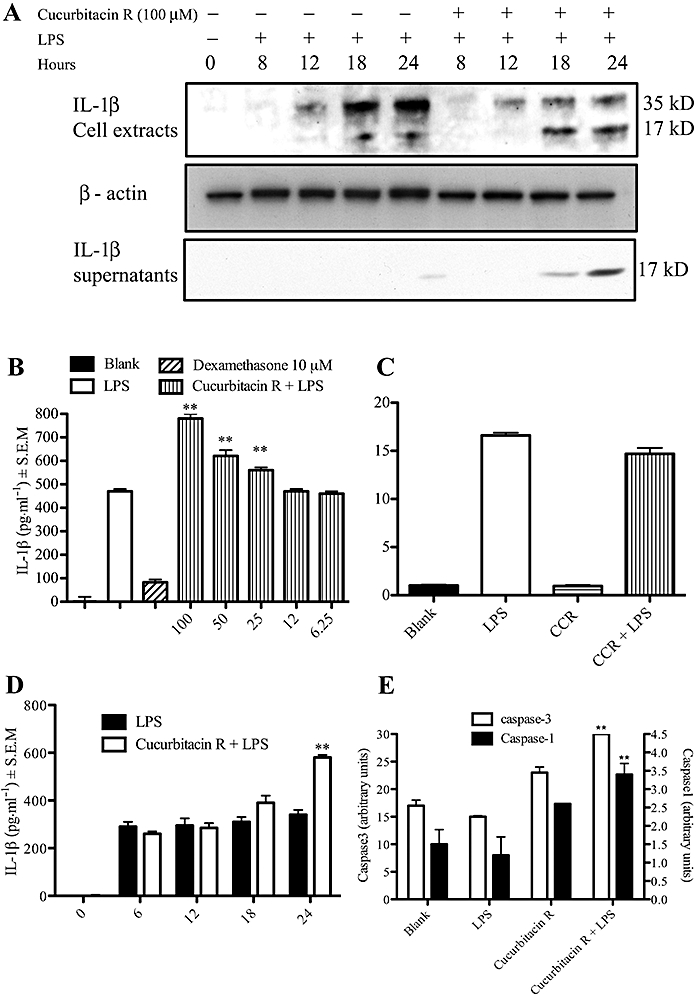
Cucurbitacin R treatment produced an increase in the release of interleukin-1β (IL-1β) in RAW 264.7 macrophages due to an increase in caspase-1 activity. (A) Western blot analysis on RAW macrophages cell extracts and cells supernatants of IL-1β. Cells were treated with cucurbitacin R (100 µM) and lipopolyysaccharide (LPS) for different times and IL-1β levels were determined in cell lysates or supernatants as described in the Methods section. (B) Cells were treated with cucurbitacin R (6.25–100 µM) or dexamethasone (10 µM) for 24 h and IL-1β levels were determined in cell lysates as described in the Methods section. (C) Real time reverse transcription-polymerase chain reaction (RT-PCR). RAW macrophages were treated with cucurbitacin R (100 µM) and LPS. Cells were collected after 8 h of LPS stimulation and real time RT-PCR were carried out as described in Methods. (D) Time course effect of cucurbitacin R in LPS stimulated RAW 264.7 cells. Columns represent cytokine production in pg mL−1 in the presence or absence of test compounds with SEM and statistical significance: blank (untreated cells), control (LPS-stimulated cells). (E) Caspase activity in cucurbitacin R-treated cells. Cells were treated with cucurbitacin R 100 µM with or without LPS for 24 h and cell lysates were incubated with respective caspase-1 and caspase-3 fluorometric substrates for 2 h. The vertical lines indicate the SEM. Statistically significant difference with respect to the control is expressed as **P < 0.01 (Dunnett's t-test). Figures are representative of three experiments performed with similar results.
Secondly, we performed ELISA of interleukin-1β. Thus, various concentrations of cucurbitacin R (100–12.5 µM) were incubated with cells for 1 h and then stimulated for 24 h. When cucurbitacin R was used at apoptotic concentrations, the levels of interleukin-1β were higher than in the control (lipopolysaccharide-stimulated cells) (Figure 6B). We went on to determine the interleukin-1β levels in macrophages treated with cucurbitacin R (100 µM) at different times. An increase in interleukin-1β levels occurred at both 18 and 24 h, times at which cucurbitacin R 100 µM was found to induce apoptosis in RAW 264.7 macrophages (Figure 6D). This correlation between interleukin-1β levels and apoptosis was also found in human lymphocytes (data not shown).
To confirm the stimulatant effect of cucurbitacin R on interleukin-1β levels and to determine whether this effect occurs at the transcriptional level, qRT-PCR analyses were performed. The total RNA obtained from RAW 264.7 macrophages was used to estimate the mRNA level by use of qRT-PCR. Whereas LPS treatment increased the levels of interleukin-1β mRNA, the cucurbitacin R (100 µM) treatment in lipopolysaccharide-treated cells did not change the mRNA levels of interleukin-1β, which indicates that the over-release of interleukin-1β did not occur at the transcriptional level. Consequently, the maturation of this protein was over-activated.
We confirmed the effect of cucurbitacin R on the activity of caspase-1 as well as on that of the effector caspase-3 by use of fluorogenic substrates. Although cucurbitacin R alone, at a concentration of 100 µM, stimulated the activity of both caspase-1 and caspase-3, when combined with lipopolysaccharide I produced even higher levels of activity (Figure 6E).
Over-expression of Bcl-2 inhibited cucurbitacin R-induced apoptosis and interleukin-1β levels
To determine whether Bcl-2 and the plasminogen activator (PAI)-2 regulated cucurbitacin R-induced apoptosis and the over-expression of interleukin-1β, we produced two stably transfected cell lines with the Bcl-2 and PAI-2 expression plasmid. In addition, we silenced caspase-1 and NALP1 with specific siRNA (Figure 7C) to examine their role in both the apoptotic process and interleukin-1β release. Whereas altering PAI-2 expression or silencing NALP1 or caspase-1 did not change the apoptotic levels in cells treated with cucurbitacin R, RAW-Bcl-2 was found to be resistant to cucurbitacin R-induced apoptosis (Figure 7A). In addition, the levels of interleukin-1β in RAW-Bcl-2 were clearly diminished in both the lipopolysaccharide-treated cells and cucurbitacin R plus lipopolysaccharide-treated cells (Figure 7B). As expected, caspase-1 silencing reduced the levels of interleukin-1β in the cells as this caspase is required for the processing of interleukin-1β. In addition, PAI-2 expression did not affect either the pro-apoptotic properties or the interleukin-1β levels in cucurbitacin R-induced apoptosis. Moreover, cucurbitacin R did not affect NF-κB activity (data not shown; Escandell et al., 2006).
Figure 7.
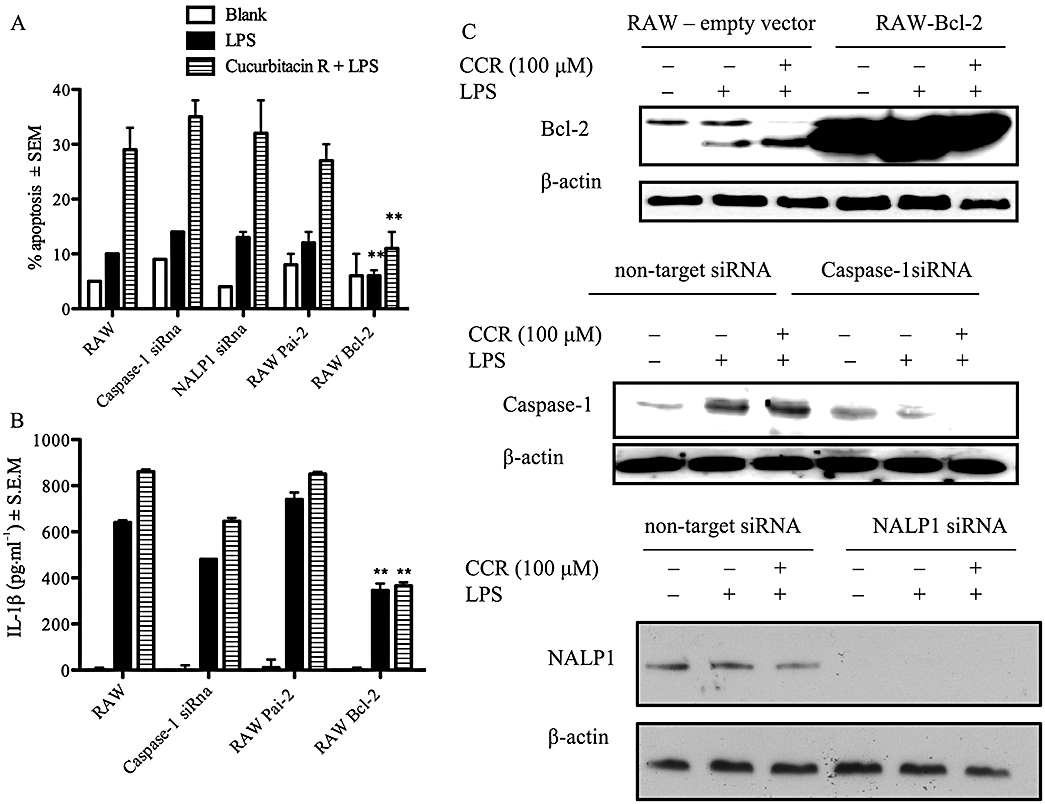
Effect of Bcl-2, PAI-2, NALP1 and caspase-1 on cucurbitacin R-induced apoptosis. (A) RAW 264.7 cells, RAW- Bcl-2 cells, NALP1 silenced cells, RAW-PAI-2 cells and caspase-1 silenced cells were treated with cucurbitacin R (100 µM) and lipopolyysaccharide (LPS) for 24 h and the levels of apoptosis were measured by propidium iodide staining. (B) RAW 264.7 cells, RAW- Bcl-2 cells, RAW-PAI-2 cells, and caspase-1 silenced cells were treated with cucurbitacin R (100 µM) and LPS for 24 h and the IL-1β levels were measured. (C) Western blots corresponding to caspase-1 silenced cells, RAW-Bcl-2 cells and NALP1 silenced cells. Statistically significant difference with respect to the control is expressed as **P < 0.01 (Dunnett's t-test). Figures are representative of three experiments performed with similar results.
Discussion
Cucurbitacins are natural products known to have anti-inflammatory and anti-cancer properties. We have previously reported on the anti-arthritic and anti-inflammatory effects of two such compounds, cucurbitacin R and dihydrocucurbitacin B, along with their effect on the cell cycle of human lymphocytes. In previous experiments, however, under the experimental condition used, neither compound was found to induce apoptosis (Recio et al., 2004; Escandell et al., 2006; 2007a,b;).
Recently, however, we demonstrated the apoptotic effect of cucurbitacin R on colon cancer cells (Escandell et al., 2008). In the present study, we have examined the apoptotic effect of cucurbitacin R on murine macrophages RAW 264.7, its effect on the release of interleukin-1β and its possible regulation by the anti-apoptotic protein Bcl-2. Apoptosis is one of the mechanisms that living organisms have for removing cells from tissues in a deliberate and systematic manner. It is brought on by different cell signals and regulated by various pro- or anti-apoptotic proteins such as caspases and the family of Bcl proteins. Activation of inflammatory caspases occurs upon assembly of the inflammasome (Martinon and Tschopp, 2004) or of the molecular platform for the activation of pro-inflammatory caspases such as caspase-1 (Petrilli et al., 2005). The inflammasome system processes the cytoplasmatic pro- interleukin-1β, which is transcriptionally regulated by NF-κB. As a result, interleukin-1β is processed and released during apoptosis (Hogquist et al., 1991; Martinon and Tschopp, 2004). Because the anti-apoptotic proteins Bcl-2 and Bcl-XL interact with NALP1, one of the central proteins in the inflammasome complex, to suppress caspase-1 and interleukin-1β production (Petrilli et al., 2005; Bruey et al., 2007), we decided to study the effect of a pro-apoptotic compound, cucurbitacin R, on the expression of both the anti-apoptotic protein Bcl-2 and the pro-apoptotic protein Bax. We also examined its role in the activation of caspase-1 and the release of interleukin-1β. We found that when cucurbitacin R was applied to RAW 264.7 cells, the production of interleukin-1β clearly increased, as did apoptosis and cyclin D2 production. Other cyclins, in contrast, were reduced. Indeed, several studies have indicated that lipopolysaccharide induces the expression of cyclin D2 in macrophages (Vadiveloo et al., 2000) while other reports have implicated the over-expression of cyclin D2 in the arrest of the cell cycle and apoptosis (Banerji et al., 2001).
Cucurbitacin R down-regulated the expression of the anti-apoptotic protein Bcl-2, whereas the protein p53 was over-expressed 8, 12 and 18 h after treatment. This transcription factor plays a significant role in the regulation of the cell cycle to prevent mutations and cancer (Harms and Chen, 2005), as well as to control the functions of p21. It regulates the progression of the cell cycle, which was clearly increased at 18 and 24 h as a consequence of the increased p53 activity, in the S phase (Gartel and Radhakrishnan, 2005). In our experiments, the expression of the pro-apoptotic protein Bax was not modified. The intrinsic pathway for caspase activation in cells involves the participation of the mitochondria, with a change in the mitochondrial membrane potential releasing caspase-activating proteins such as cytochrome c into the cytosol (St-Louis and Archambault, 2007). In our experiments, when RAW 264.7 macrophages were treated with cucurbitacin R at various concentrations, the mitochondrial membrane potential was modified at 18 h, and caspase-9 and caspase-3 were activated. This result indicates that the mitochondrial apoptotic pathway is implicated in cucurbitacin R-induced apoptosis in RAW 264.7 macrophages.
In their work, Bruey et al. (2007) demonstrated the relationship between the Bcl-2 levels and the inhibition of interleukin-1β. This effect is due to the inhibition of the activation of NALP1, a protein that constitutes part of the inflammosome, which activates caspase-1. In addition, Park et al. (2005) showed that PAI-2 inhibited both apoptosis and interleukin-1β secretion in Toll-like receptor 4 stimulated cells. Using stably transfected cell lines which included Bcl-2 and PAI-2 expression plasmids and which had been silenced for caspase-1 activity, we found that neither PAI-2 expression nor caspase-1 silencing or NALP1 silencing produced a change in the apoptotic levels of cells that had been treated with cucurbitacin R. In contrast, the RAW-Bcl-2 cells were resistant to cucurbitacin R-induced apoptosis. This indicates that Bcl-2 plays an important role in the apoptotic mechanism of cucurbitacin R. In addition, the level of interleukin-1β in RAW-Bcl-2 cells was clearly lower in the treated cells. Moreover, neither caspase-1 silenced cells nor PAI-2 over-expressed cells were found to undergo any change in the apoptotic levels produced by cucurbitacin R. These results show that Bcl-2, but not PAI-2 or caspase-1, regulate both apoptosis and interleukin-1β secretion in cucurbitacin R-induced apoptosis and that there is a clear correlation between interleukin-1β levels and apoptosis.
As noted above, NF-κB regulates both interleukin-1β expression and interleukin-1β production during apoptosis (Martinon and Tschopp, 2004). One possibility that has been proposed for reducing NF-κB activation is that of blocking IKKβ, one of the two catalytic subunits of the IKK complex (Karin et al., 2004). IKKβ activity is required for the activation of a severe inflammatory reaction in response to ischaemia-reperfusion and for the protection of a large number of different cell types from apoptosis. This allows for the use of specific inhibitors as potential anti-inflammatory agents (Karin et al., 2004). Various drugs have been described as inhibitors of IKKβ kinase activity, including natural products such as flavonoids and resveratrol. Because IKKβ inhibition increases caspase-1 activation and interleukin-1β production, Greten et al., (2007) consider NF-κB to be a negative regulator of interleukin-1β secretion. However, in our study, the inhibition of IKKβ had no effect (data not shown; Escandell et al., 2006).
In conclusion, we have demonstrated that cucurbitacin R produces apoptosis in lipopolysaccharide-activated RAW 264.7 macrophages. This effect is regulated by Bcl-2 whereas Bax, caspase-1, NALP1 and PAI-2 do not seem to be involved. Moreover, there is a clear correlation between interleukin-1β levels produced by the cells and the apoptotic effects. In addition, Bcl-2 was found to act as a negative regulator of interleukin-1β secretion in cucurbitacin R-induced apoptosis. Taken together, these results allow us to describe a new apoptotic process in which interleukin-1β release is directly regulated by the Bcl-2 status, which contributes to the evidence that apoptotic processes do not induce inflammation.
Acknowledgments
This work was supported by the Spanish Government (SAF2006-06726) and EU (FEDER). J.M.E. is a fellowship of the Generalitat Valenciana (CTBPRB/2003/315).
We are indebted to the Centre de Transfusions de la Comunitat Valenciana (Valencia, Spain) for its generous supply of human blood. PAI-2 expression plasmid was kindly provided by Dr Karin (La Jolla, San Diego, USA) and Bcl-2 expression plasmid by Dr Klampfer (Albert Einstein Cancer Center, New York, USA).
Glossary
Abbreviations
- IκB
inhibitor protein of NF-κB
- IKK
IκB kinase
- JAK2
Janus kinase-2
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NALP1
(nucleotide-dependent activator of cytokine-processing protease caspase-1
- NF-κB
nuclear factor-κB
- PAI
plasminogen activator
- qRT-PCR
quantitative reverse transcription-polymerase chain reaction
- STAT3
signal transducer and activator of transcription-3
- TNF-α
tumour necrosis factor-α
Conflict of interest
The authors state no conflict of interest.
References
- Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, et al. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- Banerji L, Glassford J, Lea NC, Thomas NSB, Klaus GGB, Lam EWF. BCR signals target p27Kip1 and cyclin D2 via the PI3-K signalling pathway to mediate cell cycle arrest and apoptosis of WEHI 231 B cells. Oncogene. 2001;20:7352–7367. doi: 10.1038/sj.onc.1204951. [DOI] [PubMed] [Google Scholar]
- Bleackley RC, Heibein JA. Enzymatic control of apoptosis. Nat Prod Rep. 2001;18:431–440. doi: 10.1039/a909080k. [DOI] [PubMed] [Google Scholar]
- Bruey JM, Bruey-Sedano N, Luciano F, Zhai D, Balpai R, Xu C, et al. Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell. 2007;129:45–56. doi: 10.1016/j.cell.2007.01.045. [DOI] [PubMed] [Google Scholar]
- Cao Z, Henzel WJ, Gao X. IRAK: a kinase associated with the interleukin-1 receptor. Science. 1996a;271:1128–1131. doi: 10.1126/science.271.5252.1128. [DOI] [PubMed] [Google Scholar]
- Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel D. TRAF6 is a signal transducer for interleukin-1. Nature. 1996b;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- Cerretti EP, Kozolsky CJ, Mosley B, Nelson N, Van Ness K, Greenstreet TA, et al. Molecular cloning of the interleukin-1β converting enzyme. Science. 1992;256:97–100. doi: 10.1126/science.1373520. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. Interleukin-1β, interleukin-18, and the interleukin-1β converting enzyme. Ann N Y Acad Sci. 1998;856:1–11. doi: 10.1111/j.1749-6632.1998.tb08307.x. [DOI] [PubMed] [Google Scholar]
- Escandell JM, Recio MC, Máñez S, Giner RM, Cerdá-Nicolás M, Ríos JL. Dihydrocucurbitacin B, isolated from Cayaponia tayuya, reduces damage in adjuvant-induced arthritis. Eur J Pharmacol. 2006;532:145–154. doi: 10.1016/j.ejphar.2005.12.028. [DOI] [PubMed] [Google Scholar]
- Escandell JM, Recio MC, Giner RM, Máñez S, Cerdá-Nicolás M, Ríos JL. Cucurbitacin R reduces the inflammation and bone damage associated with adjuvant arthritis in Lewis rats by suppression of TNF-α in T lymphocytes and macrophages. J Pharmacol Exp Ther. 2007a;320:581–590. doi: 10.1124/jpet.106.107003. [DOI] [PubMed] [Google Scholar]
- Escandell JM, Recio MC, Máñez S, Giner RM, Cerdá-Nicolás M, Gil-Benso R, et al. Dihydrocucurbitacin B inhibits delayed-type hypersensitivity reaction by suppressing lymphocyte proliferation. J Pharmacol Exp Ther. 2007b;322:1261–1268. doi: 10.1124/jpet.107.122671. [DOI] [PubMed] [Google Scholar]
- Escandell JM, Kaler P, Recio MC, Sasazuki T, Shirasawa S, Augenlicht L, et al. Activated kRas protects colon cancer cells from cucurbitacin-induced apoptosis: the role of p53 and p21. Biochem Pharmacol. 2008;76:198–207. doi: 10.1016/j.bcp.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res. 2005;65:3980–3985. doi: 10.1158/0008-5472.CAN-04-3995. [DOI] [PubMed] [Google Scholar]
- Greten FR, Arkan MC, Bollrath J, Hsu LC, Goode J, Miething C, et al. NF-κB is a negative regulator of IL-1β secretion as revealed by genetic and pharmacological inhibition of IKKβ. Cell. 2007;130:918–931. doi: 10.1016/j.cell.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms KL, Chen X. The C terminus of p53 family proteins is a cell fate determinant. Mol Cell Biol. 2005;25:2014–2030. doi: 10.1128/MCB.25.5.2014-2030.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogquist KA, Nett MA, Unanue ER, Chaplin DD. Interleukin 1 is processed and released during apoptosis. Proc Natl Acad Sci USA. 1991;88:8485–8489. doi: 10.1073/pnas.88.19.8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Yamamoto Y, Wang QM. The IKK NF-κB system: a treasure trove for drug development. Nat Rev Drug Discov. 2004;3:17–26. doi: 10.1038/nrd1279. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase 9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561–574. doi: 10.1016/j.cell.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Park JM, Greten FR, Wong A, Westrick RJ, Arthur JSC, Otsu K, et al. Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis – CREB and NF-κB as key regulators. Immunity. 2005;23:319–329. doi: 10.1016/j.immuni.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Petrilli V, Papin S, Tschopp J. The inflammasome. Curr Biol. 2005;15:R581. doi: 10.1016/j.cub.2005.07.049. [DOI] [PubMed] [Google Scholar]
- Recio MC, Prieto M, Bonucelli M, Orsi C, Máñez S, Giner RM, et al. Anti-inflammatory activity of two cucurbitacins isolated from Cayaponia tayuya roots. Planta Med. 2004;70:414–420. doi: 10.1055/s-2004-818968. [DOI] [PubMed] [Google Scholar]
- Ríos JL, Escandell JM, Recio MC. New insight on the bioactivity of cucurbitacins. In: Atta-ur-Rahman, editor. Studies in Natural Products Chemistry: Bioactive Natural Products. Amsterdam: Elsevier; 2005. pp. 519–572. Vol. 32. [Google Scholar]
- St-Louis MC, Archambault D. The equine arteritis virus induces apoptosis via caspase-8 and mitochondria-dependent caspase-9 activation. Virology. 2007;367:147–155. doi: 10.1016/j.virol.2007.05.023. [DOI] [PubMed] [Google Scholar]
- Sun J, Blaskovich MA, Jove R, Livingston SK, Coppola D, Sebti SM. Cucurbitacin Q: a selective STAT3 activation inhibitor with potent antitumor activity. Oncogene. 2005;24:3236–3245. doi: 10.1038/sj.onc.1208470. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A novel heterodimeric cysteine protease is required for interleukin-1β processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- Vadiveloo PK, Christopoulos H, Novak U, Kola I, Hertzog PJ, Hamilton JA. Type I interferons mediate the lipopolysaccharide induction of macrophage cyclin D2. J Interferon Cytokine Res. 2000;20:355–359. doi: 10.1089/107999000312289. [DOI] [PubMed] [Google Scholar]
- Wu G, Shi Y. Apoptosis signaling pathways and lymphocyte homeostasis. Cell Res. 2007;17:759–771. doi: 10.1038/cr.2007.52. [DOI] [PubMed] [Google Scholar]