

Abstract
Regulation of hepatocellular apoptosis is crucial for liver homeostasis. Increased sensitivity of hepatocytes towards apoptosis results in chronic liver injury, whereas apoptosis resistance is linked to hepatocarcinogenesis and non-responsiveness to therapy-induced cell death. Recently, we have demonstrated an essential role of the anti-apoptotic Bcl-2 family member Myeloid cell leukemia-1 (Mcl-1) in hepatocyte survival. In mice lacking Mcl-1 specifically in hepatocytes (Mcl-1Δhep) spontaneous apoptosis caused severe liver damage. Here, we demonstrate that chronically increased apoptosis of hepatocytes coincides with strong hepatocyte proliferation resulting in hepatocellular carcinoma (HCC). Liver cell tumor formation was observed in >50% of Mcl-1Δhep mice already by the age of 8 months, whereas 12 month-old wild-type and heterozygous Mcl-1flox/wt mice lacked tumors. Tumors revealed a heterogenous spectrum ranging from small dysplastic nodules to HCC. The neoplastic nature of the tumors was confirmed by histology, expression of the HCC marker glutamine synthetase and chromosomal aberrations. Liver carcinogenesis in Mcl-1Δhep mice was paralleled by markedly increased levels of survivin, an important regulator of mitosis which is selectively overexpressed in common human cancers.
Conclusion
The present study provides in vivo evidence that increased apoptosis of hepatocytes not only impairs liver homeostasis but is also accompanied by hepatocyte proliferation and hepatocarcinogenesis. Our findings might have implications for understanding apoptosis-related human liver diseases.
The survival of multicellular organisms depends on the maintenance of tissue homeostasis. Under physiological conditions apoptosis contributes to liver homeostasis by removing damaged hepatocytes. Proliferation, growth and programmed hepatocyte cell death are highly coordinated and tightly controlled events in the normal liver (1).
On the one hand, increased apoptosis sensitivity contributes to liver injury. On the other hand, defective apoptosis was demonstrated to lead to excessive hepatocellular survival and has emerged as a major mechanism by which pre-malignant hepatocytes obtain a competitive advantage over normal liver cells (2). Various molecular alterations have been characterized causing an imbalance in the regulation of apoptosis. Among these are alterations in p53 signalling, expression of death receptors, growth factors and mitochondrial integrity (3). Decreased activity of pro-apoptotic signalling as well as increased activity of anti-apoptotic events are associated with HCC development and progression (4).
Among the main cellular changes that trigger apoptosis of hepatocytes is the permeabilization of the outer mitochondrial membrane followed by the release of pro-apoptotic factors (5). The Bcl-2 protein family plays a pivotal role for mitochondrial integrity and the selective interactions between pro- and anti-apoptotic family members regulate mitochondrial activation (6). Bcl-2 family members are similar within the Bcl-2 homology regions (BH1-BH4) and can be divided in pro- and anti-apoptotic Bcl-2 proteins.
Pro-apoptotic Bcl-2 proteins comprise (1) multi-domain members, which lack the BH4 domain (e.g. Bax, Bak), and (2) BH3-only proteins, which lack BH1, 2 and 4 domains (e.g. Bid, Noxa, Puma). BH3-only proteins initiate the mitochondrial signalling cascade by sensing cellular damage (7). After activation, BH3-only proteins are released to neutralise anti-apoptotic Bcl-2 proteins. Subsequently, Bax and Bak trigger mitochondrial membrane leakage and the release of mitochondrial proteins, including cytochrome c, Smac/DIABLO (second mitochondria-derived activator of caspases/direct IAP-binding protein with low pI) and apoptosis-inducing factor (AIF). Smac/DIABLO proteins inactivate the IAP (inhibitors of apoptosis proteins) family, which consists of IAP1/2, BRUCE, NAIP, ILP2, ML-IAP, survivin and XIAP. XIAP is a direct caspase inhibitor. Other IAPs including survivin have several functions apart from caspase inhibition, eg, triggering of ubiquitination processes (8). Anti-apoptotic Bcl-2 family members (eg, Bcl-2, Bcl-xL and Mcl-1), interact with Bax and Bak to inhibit the activation of mitochondria (7).
Both Bcl-xL and Mcl-1 have been identified as major anti-apoptotic Bcl-2 proteins in the liver (9-11). Liver homeostasis is severely disturbed in Mcl-1Δhep mice (10, 11). Spontaneous hepatocyte apoptosis was observed in livers of Mcl-1Δhep mice in profound liver cell damage and increased susceptibility of hepatocytes towards pro-apoptotic stimuli (10). In addition, Mcl-1 has been shown to be highly expressed in a subset of human HCC, contributing to apoptosis resistance of cancer cells (12, 13). Thus, abrogation of the pro-survival function of Mcl-1 (1) either by diminishing its levels or (2) by inactivating its function, have shown promising results with regards to treatment of HCC (12, 13).
In this study, we show that liver-specific depletion of Mcl-1 increases hepatocyte apoptosis, induces hepatocellular proliferation and causes HCC in the absence of overt inflammation.
Keywords: liver, hepatocellular carcinoma, apoptosis, Bcl-2 proteins, survivin
Experimental Procedures
Mcl-1 knockout mice
Conditional liver-specific Mcl-1 knockout mice (homozygous: Mcl-1flox/flox-AlbCre, referred to as Mcl-1Δhep, heterozygous: Mcl-1flox/wt-AlbCre referred to as Mcl-1flox/wt, control littermates: Mcl-1wt/wt) were generated and genotyped as described (10). Animal experiments were performed as described elsewhere (10). All animals received humane care according to the criteria published by the National Institutes of Health (NIH publication 86-23 revised 1985).
Aminotransferase levels
Levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were determined as described (10).
TUNEL Assay
TUNEL assays were performed as described (10).
Caspase Assay
Caspase 3, 8 and 9 activity was measured as previously described (14).
Histology and immunohistochemistry
Histology and immunohistochemistry on a Ventana stainer from Roche were performed as described (10, 15). TGFβ1 was stained with a polyclonal antibody (IgG, species rabbit) from Santa Cruz Biotechnology Inc. (Code: sc-146, 1: 200), p53 was stained with a polyclonal antibody (IgG, species rabbit) from Imgenex (IMG-80442, 1: 200). A6 antibody was kindly provided by Valentina Factor.
DNA and RNA isolation, quantitative RT-PCR
DNA and RNA isolation as well as quantitative RT-PCR were performed as described (10). For quantitative RT-PCR, QuantiTect primers (Qiagen, Hilden, Germany) for murine Mcl-1 (refer to (10)), Bcl-xL (NM_009743, QT00149254), XIAP (QT01755649), Noxa (QT00142940), Puma (QT01657432), CD95/APO-1/Fas (QT00095333), CD95L (QT00104125), Ciapin1 (QT00118986), Bid (QT00145061), cFLIP (QT01776033), cIAP1 (forward: 5-cgaggaggaggagtcagatg-3; reverse: 5-gtgatggcccttgcacttag-3), ll1β (forward: 5-gcaactgttcctgaactcaact-3; reverse: 5-atcttttggggtccgtcaact-3) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, NM_008084, XM_001003314, XM_990238, QT01658692) were used.
Tissue lysis and immuno blotting
Immuno blot analyses were performed as described (10), using the following primary antibodies: Mcl-1 (Rockland, Gilbertsville, PA), Bcl-xL (H-62; Santa Cruz Biotechnology, Heidelberg, Germany), survivin (NB500-201; Novus Biologicals, Littleton, CO), lamin (2032, Cell Signaling, Danvers, MA) and α-tubulin (Sigma).
Detection of cell proliferation
Cell proliferation was assessed by BrdU and Ki67 staining as reported previously (10).
Array-based Comparative Genomic Hybridization (aCGH)
Agilent oligonucleotide array based CGH for Genomic DNA analysis for FFPE samples (Mouse Genome CGH Microarray 4×44K) was performed on paraffin embedded liver tissues according to the protocol provided by Agilent Technologies. Chromosomal copy number aberration in HCC samples of Mcl-1Δhep mice in relation to WT samples were investigated by aCGH (Agilent DNA Analytics 4.0 CGH Module User Guide). Log2-ratios of signal intensity values of WT (Cy5) versus signal intensity values of HCC (Cy3) samples were computed with Agilent Feature Extraction software Version 9.5.3.1. Log2 ratios were imported into the DNA Analytics Software 4.0.76 (Agilent Technologies, USA). Saturated and non-uniform data points were filtered out. Values of probes that occurred several times within one chip were combined and averaged. The aCGH data were then normalized in a linear way using the DNA Analytics centralization method. Aberrations were detected using the Aberration Detection Method Nr.1 (ADM-1) as implemented in the DNA Analytics software (Agilent DNA Analytics 4.0 CGH Module User Guide, Agilent Technologies, Inc. 2008) with standard settings. Those aberrations that were not covered by more than two probes, were filtered out. Single log2 ratio intensities, moving average of these ratios and aberration detection results were graphically displayed in the genome browser of the DNA Analytics software. Statistical significance of amplification and deletion patterns in aCGH for monoclonal tumors was calculated by applying a permutation test. The samples were compared pair-wise as follows using an in-house written program. First, the sequence overlap (o) of amplifications/deletions was calculated for the two samples. Then, the amplifications/deletions of one sample were kept but randomly distributed on the other sample and the new overlap (ri) calculated. This step was repeated n = 1 × 107 times and r = sum (ri > o) computed. Finally, the p-value for the pair-wise comparison was estimated as p = r/n.
Original data from aCGH on HCC tissues of Mcl-1Δhep mice as well as liver tissues of wild-type control mice are available in the Gene Expression Omnibus (GEO) database under accession number GSE16580.
Data analysis
All graphs represent at least three independent experiments. Histological images show representative results. Data were analyzed by Mann-Whitney U test using SPSS software, with P < 0.05 considered significant.
Results
Sustained chronic liver injury and increased hepatocyte proliferation in aged Mcl-1Δhep mice
We have previously shown that deletion of Mcl-1 in hepatocytes results in liver injury of mice <6 months of age, caused by spontaneous induction of hepatocellular apoptosis (10). In the present study, we examined the long-term consequences of liver specific deletion of Mcl-1 by investigating the liver phenotype of adult Mcl-1Δhep mice >6 months of age. First, we tested the ablation efficiency of Mcl-1 in livers of 12 month-old Mcl-1Δhep. Mcl-1 protein and mRNA expression were strongly reduced or virtually absent in whole-liver extracts of Mcl-1Δhep mice compared to age matched wild-type animals (Fig. 1A, B). The residual low expression levels of Mcl-1 were most likely attributed to non-parenchymal liver cells.
Fig. 1. Chronic liver injury in Mcl-1Δhep mice.
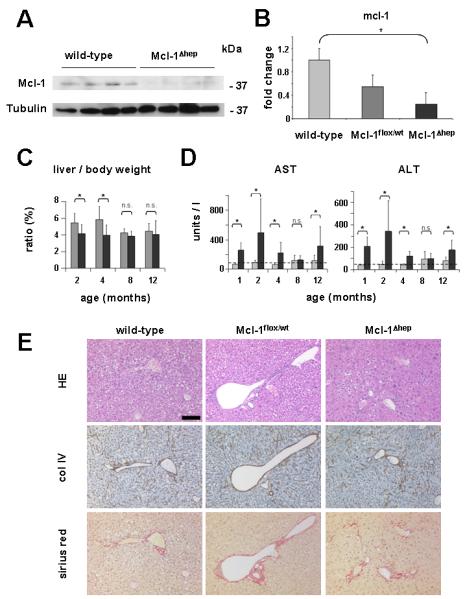
(A) Whole-liver extracts derived from 12 month-old wild-type and Mcl-1Δhep mice were analyzed by immuno blot analysis for the protein expression of Mcl-1 and Tubulin (loading control). kDa: kilo Dalton. (B) Whole-liver extracts from 12 month-old wild-type, heterozygous Mcl-1flox/wt and Mcl-1Δhep mice were analyzed for the mRNA expression levels of Mcl-1 by qRT-PCR. (C) Liver/body weight ratio of control (gray bars) and Mcl-1Δhep (black bars) mice was determined. Boxes represent the average. Standard deviation is indicated by error bars. * indicates a statistical significance of p<0.05. n.s.: statistically not significant. (D) Serum AST and ALT levels of control (gray bars) and Mcl-1Δhep (black bars) mice. Mean levels of control and Mcl-1Δhep mice are shown. Boxes represent the average. Standard deviation is indicated by error bars. * indicates a statistical significance of p<0.05. n.s.: statistically not significant. Transaminase levels of 1-4 month-old animals have been published previously (10) and are shown for completeness. (E) Liver paraffin sections of wild-type, heterozygous Mcl-1flox/wt and Mcl-1Δhep mice, all 12 month-old, were analyzed histologically. Consecutive paraffin sections were stained: hematoxylin/eosin (HE), collagen IV (col IV) and Sirius red. Scale bar: 100 μm.
Although no significant differences in body weight were detected between Mcl-1Δhep and control mice from 0-12 months of age, liver weight was significantly reduced (p<0.05) in 2 and 4 month-old Mcl-1Δhep animals (10). These differences decreased with age: 8 and 12 month-old Mcl-1Δhep animals did no longer show a significant reduction of liver to body weight ratio compared to age-matched controls (Fig. 1C). Furthermore, transaminase levels were determined as a surrogate marker for liver cell-damage. No significant elevation of AST and ALT levels was found in 8 month-old Mcl-1Δhep mice. This was in contrast to the significant differences (p<0.05) in transaminase levels observed in sera of Mcl-1Δhep mice at 2 and 4 months of life shown previously (Fig. 1D and (10). Interestingly, a significant rise in serum transaminase levels was again reproducibly detected in 12 month-old Mcl-1Δhep mice (p<0.05; Fig. 1D).
As already observed at the age of 4 months (10), 8 to 12 month-old Mcl-1Δhep mice still showed an abnormal liver morphology. Mcl-1Δhep livers displayed numerous pleomorphic and atypical hepatocytes and an altered, remarkably nodular liver structure (Fig. 1E, right panel), which was in contrast to livers of age-matched wild-type and heterozygous Mcl-1flox/wt mice. This was accompanied by a subtle, mostly pericellular fibrosis, not found in control littermates (Fig. 1E).
Similar to 4 month-old and younger mice (10) also 8 and 12 month-old Mcl-1Δhep mice histologically still showed an increased rate of liver cell apoptosis, highlighted by caspase 3 staining and TUNEL assay (Fig. 2A), which was not observed in wild-type and heterozygous Mcl-1flox/wt mice (data not shown). This was paralleled by an increased activity of caspase 3 and caspase 9 in liver homogenates of Mcl-1Δhep mice (Fig. 2B). Caspase 8 activity in livers of Mcl-1Δhep mice, however, was not significantly different compared to wild-type and Mcl-1flox/wt mice (Fig. 2B).
Fig. 2. Increased apoptosis in Mcl-1Δhep mice.
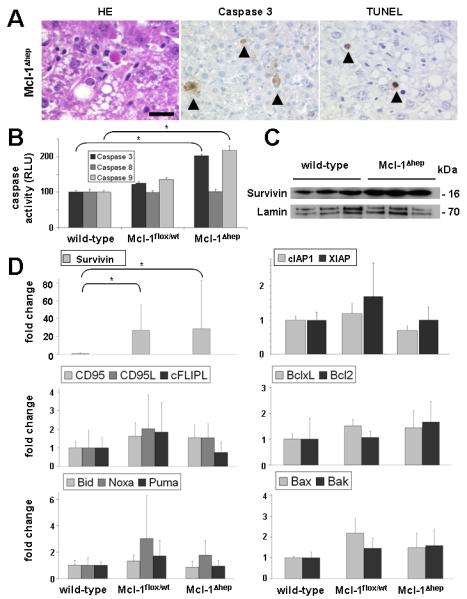
(A) Liver histology of 8 and 12 month-old Mcl-1Δhep mice revealed an increased basal rate of apoptosis that was not observed in livers of wild-type and heterozygous Mcl-1flox/wt (data not shown). Hematoxylin/eosin (HE), activated caspase 3 and TUNEL staining. Arrow heads indicate apoptotic hepatocytes. Scale bar: 20 μm. (B) Analysis for caspase 3, caspase 8, and caspase 9 activity in whole-liver lysates of wild-type, heterozygous Mcl-1flox/wt and Mcl-1Δhep mice. Boxes represent the average. Standard deviation is indicated by error bars. * indicates a statistical significance of p<0.05. (C) Nuclear extracts of livers from 12 month-old wild-type and Mcl-1Δhep mice were analyzed by immuno blot analysis for the expression of survivin and lamin. kDa: kilo Dalton. (D) mRNA expression analysis of several apoptosis-related factors (CD95, CD95L, cFLIP, BclxL, Bcl2, Bid, Noxa, Puma, Bax, Bak, Survivin, XIAP and cIAP1) by qRT-PCR. Boxes represent the average expression level of control and Mcl-1flox/wt, Mcl-1Δhep mice. All values are normalized to GAPDH mRNA expression. Standard deviation is indicated by error bars. * indicates a statistical significance of p<0.05.
To test for potential compensatory anti-apoptotic mechanisms in livers of Mcl-1Δhep mice, the expression of several apoptosis-related factors was analyzed. Remarkably, strongly elevated transcript levels of survivin, a protein of the IAP family associated with hepatocyte proliferation and carcinogenesis (16, 17), were detected in Mcl-1flox/wt and Mcl-1Δhep livers (Fig. 2D). On protein level, survivin expression was significantly higher in nuclear fractions of Mcl-1Δhep compared to wildtype livers (Fig. 2C). No differences in survivin protein expression were observed in the cytosolic fraction (data not shown). In contrast to survivin, XIAP and cIAP-1, other members of the IAP family, were not up-regulated (Fig. 2D). Previous reports suggested that proteins of the death inducing signalling complex (DISC), such as CD95/APO-1/Fas and cellular FLICE-inhibitory protein, long isoform (c-FLIP), can couple cell death and proliferation in hepatocytes (18). However, neither transcript levels of CD95 nor cFLIP, nor transcript levels of the ligand of CD95, CD95L, were significantly different comparing livers of Mcl-1Δhep mice to wild-type and Mcl-1flox/wt mice (Fig. 2D, middle panel). Furthermore, hepatic mRNA expression of the anti-apoptotic Bcl-2 proteins Bcl-xL, and Bcl-2 (Fig. 2D, middle panel, and Supplement Fig. 1), BH3-only proteins Bid, Puma and Noxa, as well as Bax and Bak (Fig. 2D, lower panel), respectively, was analyzed. No significantly different expression levels were found comparing livers of 12 month-old Mcl-1Δhep mice to age-matched wild-type and Mcl-1flox/wt mice. Chronic liver injury potentially causes an inflammatory reaction. Although no overt inflammatory infiltrates were detectable histologically (Fig. 1E and data not shown), the expression of several inflammatory mediators was studied. Increased levels of IL6, a trigger of survivin expression and an important regulator of proliferation in the liver (19, 20) were found in liver lysates of 12 months Mcl-1Δhep mice compared to wild-type and Mcl-1flox/wt mice (Fig. 3A). Albeit remarkable, these differences were not statistically different. Similarily, a slight up-regulation of TNFα mRNA (2-6 fold) was detected in Mcl-1Δhep mice compared to wild-type and Mcl-1flox/wt mice (data not shown). In contrast, mRNA levels of interleukin 1β (IL1β land IFNγ were not different (Fig. 3A). Remarkably, livers of Mcl-1Δhep mice revealed scattered cells immunoreactive for the cytokine TGFβ, an important inducer of carcinogenesis, which were not detectable in control mice (Fig. 3B).
Fig. 3. Increased hepatocyte proliferation in Mcl-1Δhep mice.
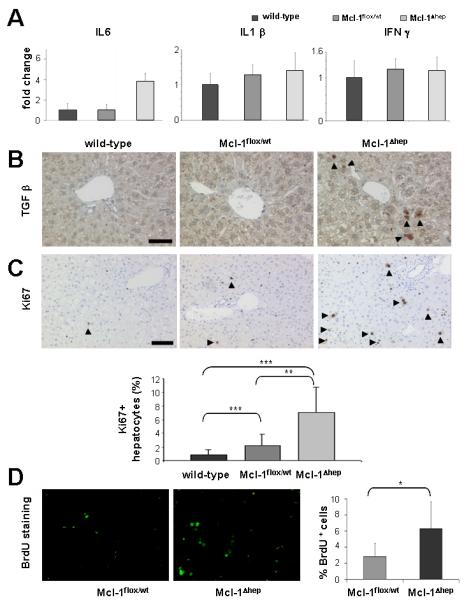
(A) mRNA expression analysis of whole liver homogenates derived from wild-type, Mcl-1flox/wt, and Mcl-1Δhep mice for the inflammatory cytokines IL6, IL1β, and IFNγ by qRT-PCR. Boxes represent the average expression level. Standard deviation is indicated by error bars. All values are normalized to GAPDH mRNA expression. (B) Livers of wild-type, heterozygous Mcl-1flox/wt and Mcl-1Δhep mice, all 12 month-old, were analyzed histologically for the expression of TGFβ. Arrow heads indicate positive cells, which morphologically appeared to be non-parenchymal cells. Scale bar: 50 μm. (C, upper row) To assess hepatocyte proliferation in wild-type, heterozygous Mcl-1flox/wt and Mcl-1Δhep mice, Ki67 staining was performed. Arrow heads indicate positive cells, which morphologically appeared to be hepatocytes. Scale bar: 100 μm. (C, lower row) 5 independent high power fields (400x) of 4 to 7 mice per genotype were quantified for Ki67-positive hepatocytes and statistically analyzed. *** and ** indicate a statistical significance of p<0.0001 and p<0.001, respectively. (D) Livers of 9 to 12 month-old Mcl-1flox/wt and Mcl-1Δhep mice were stained for BrdU incorporation (left panels) and the ratio of BrdU-positive nuclei to total liver cells was calculated (right panel). Boxes represent the average. Standard deviation is indicated by error bars. * indicates a statistical significance of p<0.05.
Next, we addressed whether the relative increase of liver weight in Mcl-1Δhep animals and the strong up-regulation of survivin might be linked to a higher hepatocyte proliferation rate, which we had observed previously in 4 month-old mice (10). Indeed, Mcl-1Δhep mice revealed a highly significant increase of Ki67+ hepatocytes compared to wild-type and heterozygous Mcl-1flox/wt mice at the age of 8 and 12 months (p<0.0001, and p<0.001, respectively, Fig. 3C). Remarkably, heterozygous Mcl-1flox/wt livers also displayed increased proliferation indices compared to wild-type livers. Quantification by BrdU incorporation corroborated this finding: Livers of 8 month-old Mcl-1Δhep mice still showed a significantly higher proliferation rate when compared to age-matched heterozygous Mcl-1flox/wt mice (p<0.05; Fig. 3D).
Development of liver cell tumors in Mcl-1Δhep mice
Wild-type and heterozygous Mcl-1flox/wt mice revealed macroscopically normal livers at the age of 8 and 12 months. This was in contrast to age matched Mcl-1Δhep livers which contained tumors in >50% of Mcl-1Δhep livers (Table 1). Liver tumors ranged from ~2 mm to 3 cm in diameter (Fig. 4A). In addition, non-tumorous parts of Mcl-1Δhep livers revealed a spectrum of findings ranging from a macroscopically unremarkable (some animals) to a strongly nodular structure (most animals) (Fig. 3A). Histology confirmed that ~50% of all Mcl-1Δhep mice (11/21) displayed liver tumors (Fig. 4B). Larger tumors showed cellular atypia, altered liver-architecture with broadening of liver cell cords (highlighted by collagen IV staining) and loss of reticulin fibres (shown by Gomori staining). In addition, proliferation rate again increased compared to non-tumorous areas, and a focal pattern of strong immunoreactivity for glutamine synthetase was observed (Fig. 4C). These findings support that the tumors histologically qualified as HCC.
Table 1. Development of liver tumors in Mcl-1Δhep mice.
At the age of 8 and 12 months, no tumors were detected in wild-type or heterozygous Mcl-1flox/wtmice. >50% of the Mcl-1Δhep mice revealed up to 14 macroscopically detectable tumor nodules / liver.
| Genotype | mice (n) | incidence of liver tumors |
number of tumor nodules (range) |
|---|---|---|---|
| wilde-type | 9 | 0/9 | 0 |
| Mcl-1flox/wt | 15 | 0/15 | 0 |
| Mcl-1Δhep | 21 | 11/21 | 1-14 |
Fig. 4. Development of liver cell tumors (HCC) in Mcl-1Δhep mice.
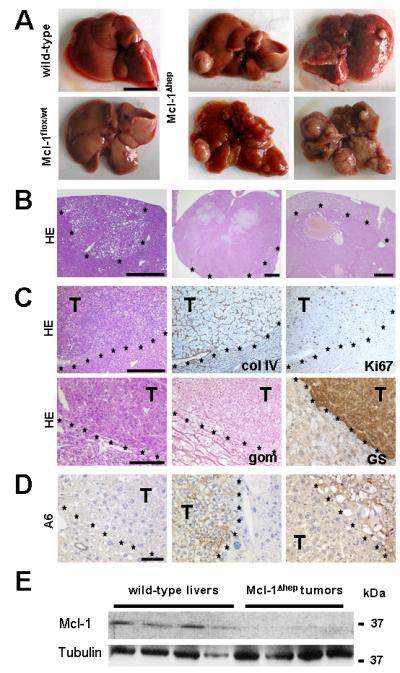
(A) Macroscopic view of livers from wild-type, Mcl-1flox/wt and Mcl-1Δhep mice are shown. The macroscopic spectrum of Mcl-1Δhep livers with tumors is shown. Scale bar: 1 cm. (B) Representative HE-stained liver sections confirm hepatocellular tumors ranging from small nodules to tumors of several centimetres in diameter. Scale bars: 500 μm (left), 1 mm (middle), 500 μm (right). Tumor borders are marked by asterisks. (C) Tumor sections were stained for collagen IV (col IV), reticulin fibres (Gomori staining, gom), Ki67 and glutamine synthetase (GS). Scale bars: 500 μm (upper panel), 100 μm (lower panel). (D) Presence of A6-positive cells was investigated in livers of Mcl-1Δhep mice containing HCC. T= tumor. Tumor borders are marked by asterisks. Scale bar: 50 μm. (E) Whole liver homogenates of 12 month-old wild-type mice and tumor nodules from Mcl-1Δhep mice were analyzed for Mcl-1 and Tubulin expression. kDa: kilo Dalton.
Mcl-1 deficient livers displayed different staining patterns for the oval cell marker A6. Mostly, tumors and non-tumorous tissues were A6−. However, in several instances A6+ tumors, surrounded by A6− liver tissue, were detected. Besides, we could also detect one A6− tumor surrounded by A6+ liver tissue (Fig. 4D).
Since tumors might have originated from a minute subset of Mcl-1 positive hepatocytes due to a lack of Cre-recombination, quantitative real-time RT-PCR and immuno blot analyses of tumor nodules in Mcl-1Δhep mice was performed. These analyses revealed only marginal Mcl-1 mRNA and protein expression compared to wild-type livers (Fig. 4E and data not shown). This strongly suggests that tumors did not originate from a conceivable subset of hepatocytes with a growth advantage due to leaky knockout of Mcl-1, but rather from Mcl-1 deficient hepatocytes.
Finally, we set out to investigate whether HCC nodules of Mcl-1Δhep mice contain chromosomal aberrations. Five HCC (ranging from 5 to 30 mm in diameter) derived from independent Mcl-1Δhep livers were analyzed by array-CGH (aCGH) analysis. This revealed numerous, chromosomal aberrations with amplifications and deletions on several chromosomal regions that were statistically significant (p<0.05; Fig. 5). No clearly mutual pattern of chromosomal aberrations was detected in Mcl-1Δhep HCC. These observations not only confirmed the neoplastic nature of the tumor nodules, but also indicated that HCC contained different chromosomal abberations. To further explore possible signalling mechanisms, which may contribute to hepatocarcinogenesis in the presented model, p53 expression was analyzed. No significantly different mRNA expression levels were detected comparing livers of Mcl-1Δhep mice to wild-type and Mcl-1flox/wt mice. In addition, no p53 accumulation was detectable by immunostaining, neither in tumor nor non-tumor tissues of Mcl-1Δhep mice (Suppl. Fig. 1B). Based on these findings, there was no evidence for p53 being a key play for HCC formation in the presented model.
Fig. 5. Chromosomal aberrations in HCC of Mcl-1Δhep mice shown by aCGH analysis.
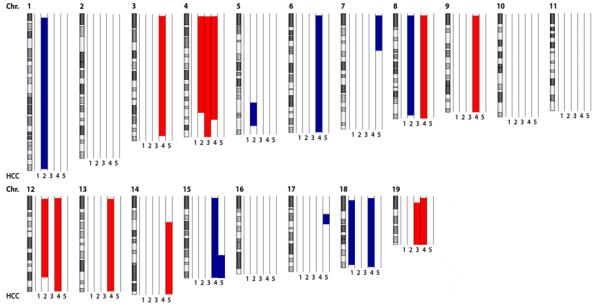
The q-arm of each chromosome is shown and chromosome numbers are indicated. Gray and black bars within the symbolized chromosomes represent G bands. Deletions according to the genomic segmentation workflow are indicated in blue, amplifications in red (see methods for details). HCC of 5 individual Mcl-1hep mice were hybridized and normalized to liver tissue of age-matched wild-type mice and analyzed by aCGH analysis. Columns next to each chromosome represent individual Mcl-1Δhep HCC (1, 2, 3, 4, 5).
Enhanced expression of the vascular endothelial growth factor-A (VEGF-A) has been discussed to be involved in hepatocarcinogenesis (21). Although few tumors revealed a slightly enhanced VEGF-A expression by immunhistochemistry, this was not a constant finding (Suppl. Fig. 1C).
Discussion
HCC is one of the most common cancers worldwide and frequently develops in the context of chronic liver disease and cirrhosis (22). However, the molecular mechanisms causing this sequence of events are still poorly understood. In this study, we describe HCC development in mice with hepatocyte-specific depletion of the anti-apoptotic Bcl-2 family member Mcl-1.
Apoptosis is generally considered as a tumor-preventing mechanism by removing unwanted or dangerous cells, e.g. those with oncogenic alterations. Conversely, evasion of apoptotic cell death is considered a basic cellular feature contributing to cancer (23). We have recently shown that Mcl-1 is a crucial anti-apoptotic factor in hepatocytes (10, 24). It is well known that liver cell death through apoptosis is a key pathogenic feature of acute and chronic liver diseases, including cholestasis, hepatitis C virus infection as well as alcoholic and non-alcoholic steatohepatitis (25). Since mitochondrial activation is a central event in the induction of hepatocellular apoptosis, Bcl-2 family members play a pivotal role for the apoptosis regulation of hepatocytes. To date, five anti-apoptotic Bcl-2 family members are known: Bcl-2, Bcl-w, Bfl-1, Bcl-xL and Mcl-1. However, only deletion of Bcl-xL or Mcl-1 resulted in severe liver phenotypes due to apoptosis induction (9-11).
Controlled hepatocyte apoptosis is essential for liver homeostasis. However, apoptosis can also induce compensatory proliferation of hepatocytes. Here, we show that increased apoptosis of hepatocytes - due to the lack of Mcl-1 - finally results in hepatocarcinogenesis. At first glance our data that increased hepatocyte apoptosis can cause liver cancer are seemingly incongruous with the known phenomenon of apoptosis resistance of pre-malignant and malignant hepatocytes. However, our results suggest a link between uncontrolled hepatocyte apoptosis, hepatocyte proliferation and hepatocarcinogenesis. In line with these findings, our study demonstrates chronic liver damage and aberrant liver architecture in 8 and 12 month-old Mcl-1Δhep mice. In addition, pericellular fibrosis triggered by chronic liver injury could be observed. Similar data were obtained in Bcl-xL deficient livers demonstrating a link between apoptosis induction and fibrogenesis (9, 11).
In a previous study, we reported that liver injury caused by apoptosis induction was accompanied by a decreased relative liver weight in 1-4 month-old Mcl-1Δhep mice (10). Here we demonstrate that relative liver weight was back to normal in 12 month-old mice Mcl-1Δhep mice. The transient increase in relative liver weight compared to 1-4 month-old Mcl-1Δhep mice is presumably caused by a compensatory hepatocellular proliferation: Indeed, we observed significantly increased proliferation rates of hepatocytes in Mcl-1Δhep mice. The observation we made in heterozygous Mcl-1flox/wt mice further supports this interpretation: These mice revealed an increased hepatocyte apoptosis rate compared to wild-type mice, but significantly lower than Mcl-1Δhep mice. This was paralleled by an increased hepatocyte proliferation rate in heterozygous Mcl-1flox/wt mice compared to wild-type mice which again was significantly lower compared to Mcl-1Δhep mice.
In this study, we found sustained caspase 3 and 9 activity in 8 and 12 months old Mcl-1Δhep mice. In contrast, caspase 8 activity was not different in Mcl-1Δhep hepatocytes, most likely due to the fact that caspase 8 activation mainly occurs upstream of mitochondrial activation. Caspases may not only trigger controlled cell death but also proliferation to preserve homeostasis after tissue damage (26). Distinct mechanisms of compensatory proliferation are well described in Drosophila melanogaster (27, 28). Our data do not prove a direct causality between apoptosis induction in the liver and hepatocyte proliferation. However, it is very likely that the chronic induction of apoptosis and chronically elevated caspase activities - observed in livers of Mcl-1Δhep mice - may not only cause hepatocyte apoptosis, but also induction of compensatory proliferation.
Further putative mechanisms contributing to the proliferative state of Mcl-1Δhep hepatocytes and HCC development might be linked to the strong up-regulation of the IAP family member survivin we observed. Recent studies have shown that survivin is involved in carcinogenesis, not only by its anti-apoptotic effects, but also by regulation of mitosis and angiogenesis (29).
In contrast to survivin, mRNA expression of other IAP family members (e.g. XIAP, cIAP-1) was not up-regulated in Mcl-1Δhep livers. Further, potentially interesting players of the apoptotic network were studied, but found to be of minor or no relevance for hepatocarcinogenesis in Mcl-1Δhep mice: (1) Absence of Mcl-1 was not compensated by enhanced expression of other anti-apoptotic Bcl-2 proteins (Bcl-xL, Bcl-2 or A1). (2) Moreover, the multi-domain pro-apoptotic Bcl-2 members Bax and Bak were not significantly changed, as described in hepatocytes deficient in NEMO/IKKγ (30). (3) Besides, no down-regulation of the BH3-only protein Bid, essential for death receptor-induced activation of mitochondria in hepatocytes (5), or other BH3-only proteins (Noxa, Puma) was observed in of Mcl-1Δhep livers. Persistent apoptosis of hepatocytes could lead to compensatory hepatocyte proliferation probably due to an increased activation of hepatic progenitor (oval) cells. Oval cells are located in the periphery of the biliary tract and represent a constant source to restore the pool of hepatocytes. The finding that most of the liver tumors from Mcl-1Δhep mice lacked A6+ cells argues against a prominent involvement of oval cells and liver tumor formation in Mcl-1Δhep mice. However, it is also possible that A6 positivity of HCC – demarcating oval cell origin - is lost in the environment of Mcl-1Δhep HCC and therefore a link between oval cell proliferation and HCC development can not be absolutely excluded.
Remarkably, HCC development in Mcl-1Δhep mice occurred independently of overt hepatitis. This is in contrast to recently published mouse models that link HCC formation to inflammation, e.g. deletion of NEMO/IKKγ (15, 30). In line with the observed absence of morphologically overt inflammation, we could also not detect an upregulation of IFNγ or IL1β in livers of Mcl-1Δhep mice when compared to wild-type controls. Only a slightly increased expression of the pro-inflammatory cytokine IL6 was found in livers of Mcl-1Δhep mice. Increased levels of IL6 in Mcl-1Δhep livers are very likely to be produced by activated Kupffer cells, the main source of cytokines in the liver (31). IL6 may also co-contribute to hepatocarcinogenesis in Mcl-1Δhep mice as described in other HCC models. For example, in dimethylnitrosamine-induced HCC in mice, triggering of IL6 production is considered as a key mechanism for chemically induced hepatocarcinogenesis (32).
Since chronic (viral) hepatitis is not only characterized by inflammation, but also by an increased rate of apoptosis, we believe that from our model also useful insight can be gained for understanding the impact of increased apoptosis on liver cancer development including those disease with concomitant severe chronic inflammation as well as those diseases without overt chronic inflammation (e.g. alcoholic liver disease without steatohepatitis). In our model, pro-inflammatory cytokines such as IL6, likely derived from Kupffer cells, as well as regulators of mitosis such as survivin may play a major role for hepatocarcinogenesis. Indeed, increased levels of survivin and IL6 have also been described in human HCC tissues and sera of patients at increased risk for HCC development, respectively (33, 34).
Hepatocellular proliferation in livers of Mcl-1Δhep mice preceded HCC development detected in 8 and 12 month-old Mcl-1Δhep animals. These regenerative responses may represent a risk factor for HCC formation by triggering genomic aberrations. In line with this hypothesis, we found genetic aberrations in tumor nodules of Mcl-1Δhep mice by aCGH. Various genomic alterations including amplifications and deletions were observed.
We have detected liver tumors of varying size from the age of 8 months in Mcl-1Δhep mice. Some liver tumors were small in size and did not (yet) meet the histological criteria of HCC. However, morphology on macroscopic and histological level of larger tumors, combined with the expression of glutamine synthetase and overt chromosomal aberrations confirmed them as HCC.
In summary, HCC were heterogenous as corroborated by different patterns of morphology, immunohistochemistry (GS; A6) as well as chromosomal aberrations. This argues against one particular molecular pathway involved in HCC formation in Mcl-1Δhep mice. In contrast it favours the notion that compensatory mechanisms underlie HCC formation in Mcl-1Δhep mice.
We show that HCC of Mcl-1Δhep mice revealed no significant expression of Mcl-1 emphasizing that hepatocarcinogenesis in Mcl-1Δhep mice occurs in the absence of the pro-survival protein Mcl-1. Remarkably, Mcl-1 was also found to be highly expressed in human malignancies, including HCC, and is discussed to contribute to apoptosis resistance of HCC cells (12, 13). Thus, depending on the context, Mcl-1 plays seemingly contrary roles in hepatocarcinogenesis. On the one hand, as observed in livers of Mcl-1Δhep mice, Mcl-1-deficiency can result in a hyper-apoptotic environment provoking compensatory up-regulation of anti-apoptotic pathways (e.g. survivin) and compensatory hyper-proliferation, finally resulting in the outgrowth of a malignant cell population. On the other hand, in tumors with Mcl-1-independent initiation, Mcl-1 over-expression can be acquired during tumor progression as an anti-apoptotic factor of cancer cells. Whether Mcl-1 up-regulation in human malignancies is causally linked to carcinogenesis or a correlative finding still has to be examined.
Taken together, we provide for the first time in vivo evidence that deletion of an anti-apoptotic protein in the liver not only induces hepatocyte apoptosis but is also accompanied by compensatory proliferation in the liver, resulting in HCC formation.
Supplementary Material
Supplemental Figure 1 (A) Whole-liver extracts derived from independent 12 month-old wild-type and Mcl-1Δhep mice were analyzed by immuno blot analysis for the expression of Bcl-xL and Tubulin (loading control). kDa: kilo Dalton. (B) Upper panel: mRNA expression analysis of whole liver homogenates derived from wild-type, Mcl-1flox/wt, and Mcl-1Δhep mice for p53 by qRT-PCR. Boxes represent the average expression level. Standard deviation is indicated by error bars. All values are normalized to GAPDH mRNA expression. Lower panel: Representative tumor section showing no p53 expression. Tumor borders are marked by asterisks. (C) Livers of Mcl-1Δhep mice with tumors were analyzed histologically for the expression of VEGF. Tumor borders are marked by asterisks. Scale bar: 100 μm.
Acknowledgements
We thank the Dana Farber Cancer Institute, Boston, MA, for providing Mcl-1flox/flox mice. In addition, we thank Sandra Heine, Silvia Behnke and Fian K. Mirea for excellent technical assistance.
Financial Support: This work was supported by grants of the Deutsche Forschungsgemeinschaft, DFG SCHU 1443/3-1, to HSB; Oncosuisse foundation, OCS 02113-08-2007, to MH and AW; the research foundation at the Medical Faculty Zurich (MH), the Prof. Dr. Max-Cloëtta foundation (MH) and the “Julius-Müller-Stiftung” (MH) and the “Kurt-Senta-Hermann”-Stiftung to MH and JH.
List of Abbreviations
- aCGH
Array-based Comparative Genomic Hybridization
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- HCC
hepatocellular carcinoma
- mRNA
messenger RNA
- Mcl-1
myeloid cell leukaemia-1
- RT-PCR
real-time polymerase chain reaction
- WT
wild-type
References
- 1.Galle PR. Apoptosis in liver disease. J Hepatol. 1997;27:405–412. doi: 10.1016/s0168-8278(97)80189-4. [DOI] [PubMed] [Google Scholar]
- 2.Schulte-Hermann R, Bursch W, Low-Baselli A, Wagner A, Grasl-Kraupp B. Apoptosis in the liver and its role in hepatocarcinogenesis. Cell Biol Toxicol. 1997;13:339–348. doi: 10.1023/a:1007495626864. [DOI] [PubMed] [Google Scholar]
- 3.Schattenberg JM, Galle PR, Schuchmann M. Apoptosis in liver disease. Liver Int. 2006;26:904–911. doi: 10.1111/j.1478-3231.2006.01324.x. [DOI] [PubMed] [Google Scholar]
- 4.Fabregat I, Roncero C, Fernandez M. Survival and apoptosis: a dysregulated balance in liver cancer. Liver Int. 2007;27:155–162. doi: 10.1111/j.1478-3231.2006.01409.x. [DOI] [PubMed] [Google Scholar]
- 5.Li S, Zhao Y, He X, Kim TH, Kuharsky DK, Rabinowich H, Chen J, et al. Relief of extrinsic pathway inhibition by the Bid-dependent mitochondrial release of Smac in Fas-mediated hepatocyte apoptosis. J Biol Chem. 2002;277:26912–26920. doi: 10.1074/jbc.M200726200. [DOI] [PubMed] [Google Scholar]
- 6.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 7.Huang DC, Strasser A. BH3-Only proteins-essential initiators of apoptotic cell death. Cell. 2000;103:839–842. doi: 10.1016/s0092-8674(00)00187-2. [DOI] [PubMed] [Google Scholar]
- 8.Eckelman BP, Salvesen GS, Scott FL. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep. 2006;7:988–994. doi: 10.1038/sj.embor.7400795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takehara T, Tatsumi T, Suzuki T, Rucker EB, 3rd, Hennighausen L, Jinushi M, Miyagi T, et al. Hepatocyte-specific disruption of Bcl-xL leads to continuous hepatocyte apoptosis and liver fibrotic responses. Gastroenterology. 2004;127:1189–1197. doi: 10.1053/j.gastro.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 10.Vick B, Weber A, Urbanik T, Maass T, Teufel A, Krammer PH, Opferman JT, et al. Knockout of myeloid cell leukemia-1 induces liver damage and increases apoptosis susceptibility of murine hepatocytes. Hepatology. 2009;49:627–636. doi: 10.1002/hep.22664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hikita H, Takehara T, Shimizu S, Kodama T, Li W, Miyagi T, Hosui A, et al. Mcl-1 and Bcl-xL cooperatively maintain integrity of hepatocytes in developing and adult murine liver. Hepatology. 2009 doi: 10.1002/hep.23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fleischer B, Schulze-Bergkamen H, Schuchmann M, Weber A, Biesterfeld S, Muller M, Krammer PH, et al. Mcl-1 is an anti-apoptotic factor for human hepatocellular carcinoma. Int J Oncol. 2006;28:25–32. [PubMed] [Google Scholar]
- 13.Sieghart W, Losert D, Strommer S, Cejka D, Schmid K, Rasoul-Rockenschaub S, Bodingbauer M, et al. Mcl-1 overexpression in hepatocellular carcinoma: a potential target for antisense therapy. J Hepatol. 2006;44:151–157. doi: 10.1016/j.jhep.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 14.Schulze-Bergkamen H, Fleischer B, Schuchmann M, Weber A, Weinmann A, Krammer PH, Galle PR. Suppression of Mcl-1 via RNA interference sensitizes human hepatocellular carcinoma cells towards apoptosis induction. BMC Cancer. 2006;6:232. doi: 10.1186/1471-2407-6-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haybaeck J, Zeller N, Wolf M, Weber A, Wagner U, Kurrer M, Bremer J, et al. A lymphotoxin-driven pathway to hepatocellular carcinoma. Cancer Cell. 2009 doi: 10.1016/j.ccr.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baba HA, Wohlschlaeger J, Schmitz KJ, Nadalin S, Lang H, Benesch A, Gu Y, et al. Survivin is upregulated during liver regeneration in rats and humans and is associated with hepatocyte proliferation. Liver Int. 2009;29:585–592. doi: 10.1111/j.1478-3231.2008.01911.x. [DOI] [PubMed] [Google Scholar]
- 17.Altieri DC. New wirings in the survivin networks. Oncogene. 2008;27:6276–6284. doi: 10.1038/onc.2008.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilot D, Serandour AL, Ilyin GP, Lagadic-Gossmann D, Loyer P, Corlu A, Coutant A, et al. A role for caspase-8 and c-FLIPL in proliferation and cell-cycle progression of primary hepatocytes. Carcinogenesis. 2005;26:2086–2094. doi: 10.1093/carcin/bgi187. [DOI] [PubMed] [Google Scholar]
- 19.Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol. 2004;5:836–847. doi: 10.1038/nrm1489. [DOI] [PubMed] [Google Scholar]
- 20.Gao B. Cytokines, STATs and liver disease. Cell Mol Immunol. 2005;2:92–100. [PubMed] [Google Scholar]
- 21.Yang JC, Teng CF, Wu HC, Tsai HW, Chuang HC, Tsai TF, Hsu YH, et al. Enhanced expression of vascular endothelial growth factor-A in ground glass hepatocytes and its implication in hepatitis B virus hepatocarcinogenesis. Hepatology. 2009;49:1962–1971. doi: 10.1002/hep.22889. [DOI] [PubMed] [Google Scholar]
- 22.Motola-Kuba D, Zamora-Valdes D, Uribe M, Mendez-Sanchez N. Hepatocellular carcinoma. An overview. Ann Hepatol. 2006;5:16–24. [PubMed] [Google Scholar]
- 23.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 24.Schulze-Bergkamen H, Brenner D, Krueger A, Suess D, Fas SC, Frey CR, Dax A, et al. Hepatocyte growth factor induces Mcl-1 in primary human hepatocytes and inhibits CD95-mediated apoptosis via Akt. Hepatology. 2004;39:645–654. doi: 10.1002/hep.20138. [DOI] [PubMed] [Google Scholar]
- 25.Guicciardi ME, Gores GJ. Apoptosis: a mechanism of acute and chronic liver injury. Gut. 2005;54:1024–1033. doi: 10.1136/gut.2004.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lamkanfi M, Festjens N, Declercq W, Vanden Berghe T, Vandenabeele P. Caspases in cell survival, proliferation and differentiation. Cell Death Differ. 2007;14:44–55. doi: 10.1038/sj.cdd.4402047. [DOI] [PubMed] [Google Scholar]
- 27.Fan Y, Bergmann A. Apoptosis-induced compensatory proliferation. The Cell is dead. Long live the Cell! Trends Cell Biol. 2008;18:467–473. doi: 10.1016/j.tcb.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huh JR, Guo M, Hay BA. Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Curr Biol. 2004;14:1262–1266. doi: 10.1016/j.cub.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 29.Mita AC, Mita MM, Nawrocki ST, Giles FJ. Survivin: key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin Cancer Res. 2008;14:5000–5005. doi: 10.1158/1078-0432.CCR-08-0746. [DOI] [PubMed] [Google Scholar]
- 30.Luedde T, Beraza N, Kotsikoris V, van Loo G, Nenci A, De Vos R, Roskams T, et al. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 11:119–132. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 31.Ramadori G, Armbrust T. Cytokines in the liver. Eur J Gastroenterol Hepatol. 2001;13:777–784. doi: 10.1097/00042737-200107000-00004. [DOI] [PubMed] [Google Scholar]
- 32.Sander LE, Trautwein C, Liedtke C. Is interleukin-6 a gender-specific risk factor for liver cancer? Hepatology. 2007;46:1304–1305. doi: 10.1002/hep.21982. [DOI] [PubMed] [Google Scholar]
- 33.Augello C, Caruso L, Maggioni M, Donadon M, Montorsi M, Santambrogio R, Torzilli G, et al. Inhibitors of apoptosis proteins (IAPs) expression and their prognostic significance in hepatocellular carcinoma. BMC Cancer. 2009;9:125. doi: 10.1186/1471-2407-9-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakagawa H, Maeda S, Yoshida H, Tateishi R, Masuzaki R, Ohki T, Hayakawa Y, et al. Serum IL-6 levels and the risk for hepatocarcinogenesis in chronic hepatitis C patients: An analysis based on gender differences. Int J Cancer. 2009 doi: 10.1002/ijc.24720. epub ahead of print. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1 (A) Whole-liver extracts derived from independent 12 month-old wild-type and Mcl-1Δhep mice were analyzed by immuno blot analysis for the expression of Bcl-xL and Tubulin (loading control). kDa: kilo Dalton. (B) Upper panel: mRNA expression analysis of whole liver homogenates derived from wild-type, Mcl-1flox/wt, and Mcl-1Δhep mice for p53 by qRT-PCR. Boxes represent the average expression level. Standard deviation is indicated by error bars. All values are normalized to GAPDH mRNA expression. Lower panel: Representative tumor section showing no p53 expression. Tumor borders are marked by asterisks. (C) Livers of Mcl-1Δhep mice with tumors were analyzed histologically for the expression of VEGF. Tumor borders are marked by asterisks. Scale bar: 100 μm.