

Abstract
Purpose
Melanoma is a common and deadly tumor that upon metastasis to the central nervous system (CNS) has a median survival duration of less than 6 months. Activation of the signal transducer and activator of transcription 3 (STAT3) has been identified as a key mediator that drives the fundamental components of melanoma malignancy, including immune suppression in melanoma patients. We hypothesized that WP1193, a novel inhibitor of STAT3 signaling, would enhance the anti-tumor activity of IFN-α against metastatic melanoma.
Experimental Design
Combinational therapy of STAT3 blockade agents with IFN-α was investigated in a metastatic and an established syngeneic intracerebral murine tumor model of melanoma. The immunological in vivo mechanisms of efficacy were investigated by T cell and NK cell cytotoxic assays.
Results
IFN-α immunotherapy was synergistic with WP1193 demonstrating marked in vivo efficacy against metastatic and established intracerebral melanoma. At autopsy, it was noted that there was a decreased trend in mice with melanoma developing leptomeningeal disease (LMD) treated with combinational therapy. The combinational approach enhanced both NK and T cell-mediated anti-tumor cytotoxicity.
Conclusions
The immune modulatory effects of STAT3 blockade can enhance the therapeutic efficacy of IFN-α immunotherapy by enhancing both innate and adaptive cytotoxic T cell activities. This combination therapy has the potential in the treatment of metastatic melanoma that is typically refractory to this type of immune therapeutic approach.
Keywords: Melanoma, IFN-α, STAT3, leptomeningeal disease, CD8+ T cell
Introduction
Patients with stage IV melanoma, especially upon metastasis to the brain, have a median survival duration of less than 6 months (1) despite multi-modality therapy and the prognosis is even more dire in patients who develop leptomeningeal disease (LMD), who have a median survival of less than 2 months. Several large, cooperative-group adjuvant trials have documented a 24–38% reduction in the relative relapse risk with the use of high-dose IFN-α for stage II and III melanoma with overall survival prolongation (2). In contrast, no large cooperative-group trials have shown significant prolongation of survival with IFN-α for inoperable stage IV melanoma and especially for those with the central nervous system (CNS) melanoma and metastasis. IFN-α is known to have powerful effects on immune cells, including enhancing NK cell tumor cytotoxicity (3), dendritic cell maturation, Th1 skewing, enhancement of T cell survival, inducing immunological memory (4), and inhibiting the invasive ability of cancer cells (5). Therapeutic strategies using IFN-α in melanoma patients with LMD have included the direct intraventricular administration of IFN-α, which resulted in the clearance of malignant cells in the cerebrospinal fluid CSF (6) but was confounded by significant and sustained neurotoxicity (7). Thus, there is a clear clinical need to identify treatment modalities that exert therapeutic effects in melanoma patients with CNS disease.
A key transcription factor that drives the fundamental components of melanoma tumorigenesis and metastasis has been identified – the signal transducer and activator of transcription (STAT)-3 (8). Growth factors and cytokines, including interleukin (IL)-6, activate Janus kinase 2 (Jak2), and activate STAT3 by phosphorylation of the tyrosine residue (Tyr705) in the STAT3 transactivation domain (p-STAT3) resulting in translocation into the nucleus and the expression of a variety of target genes. STAT3 is frequently over activated in most cancers, including melanoma, and propagates tumorigenesis by preventing apoptosis (by increasing survivin, BCL-XL, and MCL1 expression) and enhancing proliferation (by increasing c-Myc and cyclin D1/D2 expression), angiogenesis (by increasing vascular endothelial growth factor [VEGF] and hypoxia-inducible factor [HIF]-1α expression), invasion (by increasing matrix metalloproteinase [MMP]-2 and MMP-9 expression), and metastasis (9, 10). The induction of the p-STAT3 pathway within the CNS may be particularly relevant because reactive astrocytes are a major inducible source of IL-6 (11) and these cells are often seen in direct contact with tumor metastasis (12). Furthermore, tissue microarray studies of human melanoma brain metastases have demonstrated higher levels of p-STAT3 in brain metastasis specimens compared with parenchymal tumors (8). Thus, the p-STAT3 pathway is a relevant therapeutic target for CNS melanoma metastasis.
In addition to the direct tumorigenic properties of the STAT3 pathway, STAT3 is also a key regulator of immunosuppression in patients with cancer (13). The signaling of STAT3 is up-regulated within the various immune cell populations upon becoming associated with the cancer microenvironment (13). In mice that had ablation of STAT3 in only the hematopoietic cells there was marked anti-tumor clearance by the immune system (14). Induced p-STAT3 has divergent functions within the various immune cell populations but the overall result is to shift the balance of cytokine production from IL-12, which activates T cells and natural killer (NK) cells, to IL-23, which activates regulatory T cells (Tregs)(13, 15–17). For example, STAT3 activity in dendritic cells reduces the expression of MHC II, CD80, CD86, and IL-12 in these cells, rendering them unable to stimulate T cells and generate antitumor immunity (14). Additionally, the activation of STAT3 in macrophages, CNS microglia, and NK cells suppresses their activation and function (14, 18–21). However, within immune suppressive cells, induced p-STAT3 enhances their functional activity. Specifically, STAT3 has been shown to be required for both TGF-β and IL-10 production by CD4+ T cells (22), factors necessary for the generation of tumor-associated Tregs and STAT3 binds to the first intron of the FoxP3 gene (23). We have previously shown that the number and functional activity of these FoxP3+ Tregs can be inhibited with WP1066, the small molecule inhibitor of the p-STAT3 pathway (24). Furthermore, the Th17 immune subset, which is induced by the IL-6/STAT3 pathway, has been shown to promote melanoma growth (25). Thus, STAT3 seems to be a key molecular hub for inhibiting immune surveillance and clearance of malignancy. Our hypothesis was that the addition of a STAT3 inhibitor would enhance the therapeutic efficacy of IFN-α for advanced melanoma, including within the typically treatment refractory CNS, and this would be secondary to enhanced NK and cytotoxic T cell-mediated tumor cytotoxicity.
Materials and Methods
Tumor cell lines and murine models
The B16/F10 murine melanoma cell line was derived from a spontaneous melanoma in the C57BL/6J mouse of the H-2B background and was provided by Dr. Isaiah Fidler (The University of Texas M. D. Anderson Cancer Center [M. D. Anderson], Houston, TX). The B16 model system is known for its propensity to develop LMD (26). The B16 cells were maintained in RPMI 1640 medium supplemented with 10% FBS at 37° C in a humidified atmosphere of 5% CO2 and 95% air. All cell lines were grown in antibiotic-free medium and were free of Mycoplasma contamination (27).
For the in vivo experiments, we used 4- to 6-week-old female C57BL/6J mice maintained in the M. D. Anderson Cancer Center Isolation Facility in accordance with Laboratory Animal Resources Commission standards and conducted according to an Institutional Animal Care and Use Committee-approved protocol, 08-06-11831. A mouse was euthanized when it became unable to reach food or water. To induce intracerebral tumors in C57BL/6J mice, B16 cells were collected in logarithmic growth phase, washed twice with PBS, mixed with an equal volume of 10% methyl cellulose and RPMI 1640 medium and loaded into a 250-μl syringe (Hamilton, Reno, NV) with an attached 25-gauge needle. The needle was positioned 2 mm to the right of bregma and 4 mm below the surface of the skull at the coronal suture using a stereotactic frame (Kopf Instruments, Tujunga, CA). The intracerebral tumorigenic dose for the B16 cells was 5 × 102 in a total volume of 5 μl. To induce metastatic disease, 1 × 105 cells in a total volume of 100 μl were injected into the tail vein.
Cell proliferation/survival assay
For cell proliferation assays, B16 cells were seeded at a density of 1,000 cells per well in 96-well culture plates and were treated with IFN-α (0 to 1250 U/ml) (PBL Interferon Source, Piscataway, NJ) with or without WP1193 (0.5 or 1.0 μM). After 72 h of treatment, 25 ml of 5 mg/ml dimethyl thiazolyl diphenyl tetrazolium salt (MTT, Sigma-Aldrich, St. Louis, MO) solution were added to each well, and the cells were cultured for 3 h at 37° C in a humidified atmosphere of 5% CO2 and 95% air. The cells were lysed with 100 μl/well of lysing buffer (50% dimethylformamide, 20% SDS, pH 5.6) and incubated at room temperature overnight. Cell proliferation and viability were evaluated by reading the O.D. at 570 nm.
Immunoblotting analysis
Murine melanoma B16 cells and splenocytes were used for protein isolation and immunoblotting analysis as described below. B16 cells were seeded at a density of 2 × 106 cells/well in 6-well culture plates and incubated at 37°C, in an atmosphere containing 5% CO2, with the RPMI 1640 medium overnight. Afterward, B16 cells were cultured in the absence or presence of WP1193 (5 μM, 10 μM). After 3.5 hours, the B16 cells were further cultured in the absence or presence of 2000 U/ml of IFN-α for 30 minutes. For the splenocyte preparations, spleens from two 4- to 6-week-old female mice were harvested and disassociated into a single cell suspension. After erythrocytes were lysed with 1× RBC lysis buffer (eBioscience, San Diego, CA), splenocytes were washed once with RPMI 1640 medium and were seeded at a density of 10 × 106 cells/well in 6-well culture plates and incubated at 37°C, in an atmosphere containing 5% CO2, with the RPMI 1640 medium in absence or presence of WP1193 (5 μM, 10 μM). After 1.5 hours, the splenocytes were further cultured in the absence or presence of 2000 U/ml of IFN-α for 30 minutes. Afterwards, all the above B16 cells and splenocytes were pelleted and rinsed with ice-cold PBS at 1500 rpm for 5 minutes. The cells were lysed for 30 minutes in ice-cold lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA) containing 1% Triton-X-100 and phosphatase and protease inhibitors (Sigma-Aldrich). The lysates were centrifuged at 14,000 rpm for 10 minutes at 4°C. The supernatants were collected and quantified for protein content. Equal amounts of proteins (65 μg) were electrophoretically fractionated in 8% sodium dodecyl sulfate (SDS)-polyacrylamide gels, transferred to nitrocellulose membranes, and subjected to immunoblot analysis with specific antibodies against p-STAT3 (Tyr705), STAT3 (Cell Signaling Technology, Inc., Danvers, MA), and β-actin (Sigma-Aldrich). Autoradiography of the membranes was performed using Amersham ECL Western blotting detection reagents (Amersham Biosciences). The densities of the protein bands compared with the β-actin protein control were measured with the Image J program provided by the NIH.1
Determination of inhibition of Tregs in vivo
To ascertain the inhibition of Tregs within the bone marrow, lymph node, spleen, thymus and peripheral blood compartment, non-tumor bearing mice were treated with IFN-α or “empty” plasmid control, WP1193, or IFN-α in combination with WP1193 for 9 and 16 days as described in the above schema. Single-cell suspensions were prepared from bone marrows, lymph nodes, spleens, thymuses and peripheral blood of mice. Single cells were surface-stained by FITC-conjugated anti-CD4 (L3T4), PerCP-conjugated anti-CD8 (53-6.7) and APC-conjugated anti-CD25 (PC61), and the cells were further subjected to intracellular staining with PE-conjugated mAbs to mouse FoxP3 (clone FJK-16s; eBioscience, San Diego, CA) using staining buffers and conditions specified by the manufacturer.
Ex vivo splenocytes cytotoxicity assays
Spleens from 4- to 6-week-old female mice in the above treatment schema were harvested and disassociated into a single cell suspension. After erythrocytes were lysed with 1× RBC lysis buffer (eBioscience), splenocytes were washed once with RPMI 1640 medium and were ready as effector cells for the standard cytotoxicity assay (25). For target cells, B16 cells in RPMI 1640 medium were cultured for 3 days, trypsinized, pelleted, and resuspended in FACS buffer at room temperature to achieve a concentration of 106 cells/ml. Carboxy-fluorescein diacetate succinimidyl ester (CFSE) stock solution (CellTrace CFSE Cell Proliferation Kit; Invitrogen, Eugene, OR) was added to achieve a final concentration of 4 μM. The mixture was incubated at 37°C for 10 minutes, and then the staining reaction was quenched by the addition of five volumes of ice-cold PBS for 5 minutes. The B16 cells were washed three times in RPMI 1640 medium and plated for the cytotoxicity assay. The ratios of splenocyte effector cells to B16 target cells were 30:1 and 100:1. After 48 h of incubation, the CFSE-labeled B16 melanoma cells were removed from the plates with trypsin-EDTA (0.05%) and analyzed by FACS. The B16 cells were stained with propidium iodide (PI; BD Biosciences) to distinguish viable cells from nonviable cells. B16 cells that were stained with CFSE and PI were considered nonviable. Flow cytometric acquisition of the B16 target cells was performed with a FACSCalibur flow cytometer (BD Biosciences), and data analysis was performed using FlowJo software (TreeStar, Ashland, OR).
NK cell and T cell cytotoxicity assay against melanoma cells
Splenocytes were prepared as described above. NK1.1+CD3− NK effector cells or CD3+CD8+ T effector cells were sorted from splenocytes on a FACSAria Cell Sorter (BD Biosciences) with FITC-conjugated anti-mouse NK1.1 (eBioscience), PE-conjugated anti-CD3, and allophycocyanin (APC)-conjugated anti-CD8 antibodies (Miltenyi Biotec, Auburn, CA). B16 target cells were prepared as described above. The ratio of NK1.1+CD3− NK effector cells or CD8+ T effector cells to B16 target cells was 10:1 and 5:1, respectively. We were limited to minimizing the titration ratios of effector to targets secondary to limitations of purification and the logistics of the assay set up. In the presence of NK1.1+CD3− NK cells or CD8+ T cells, treatment groups consisted of B16 target cells alone; B16 cells with 2 μM of WP1193; B16 cells with 2000 U/ml of IFN-α; B16 cells with 2 μM of WP1193 and 2000 U/ml of IFN-α. After 48 hours of incubation, the CFSE-labeled B16 melanoma cells were removed from the plates with trypsin and analyzed by FACS. Then, B16 cells were stained with PI (BD Biosciences) to distinguish viable cells from nonviable cells. B16 cells that were stained with CFSE and PI were considered nonviable. Flow cytometric acquisition of the B16 target cells was performed with a FACSCalibur flow cytometer (BD Biosciences), and data analysis was performed using FlowJo software (TreeStar).
Detection of NK cell receptors
Spleens from 4- to 6-week-old female mice were harvested and disassociated into a single-cell suspension as described above. Splenocytes were seeded at a density of 4 × 106 cells/well in 24-well culture plates and cultured with the RPMI 1640 medium in absence or presence of WP1193 (2 μM) and/or INF-α (2000 U/ml) for 24 hours. Afterwards, the cells were harvested and washed twice in PBS with 5% FCS, resuspended in staining buffer and labeled with FITC- or PE-conjugated anti-mouse NK1.1 (eBioscience, San Diego, CA) to identify the NK population. To stain in duplicate, 106 cells were transferred to a 96-well plates and Fc staining was blocked with rat anti-mouse CD16/CD32 serum (BD Biosciences) and the cells were incubated for 15 minutes at room temperature. Secondary staining was performed with FITC-conjugated rat anti-mouse CD94 (KLRD1) mAb (Lifespan Biosciences, Seattle, WA), biotin-conjugated rat anti-mouse NKG2C mAb (AbD Serotec; Raleigh, NC), PE-conjugated rat anti-mouse NKG2D (CD314) mAb (Biolegend, San Diego, CA), FITC-conjugated rat anti-mouse NKp46 (NCR1) mAb (R&D Systems, Minneapolis, MN), or Alexa Fluor 647-conjugated rat anti-mouse CD226 (DNAM-1) mAb (Biolegend). Negative control wells were stained with the corresponding isotypes. Following incubation, the cells were washed twice with FACS buffer and then analyzed with a BD FACSCalibur with gates set for viable splenocytes.
Detection of MHC class I (MHC I), MHC class II (MHC II), and NK cell ligands on B16 melanoma cells
To ascertain the MHC I, MHC II, and NK ligands expressed on melanoma cells, B16 cells were seeded at a density of 2 × 106 cells/well in 24-well culture plates and cultured with the RPMI 1640 medium in the absence or presence of WP1193 (2 μM) and/or INF-α (2000 U/ml) for 24 hours. Afterwards, the cells were harvested and washed twice, and 106 cells in duplicate were Fc blocked with purified rat anti-mouse CD16/CD32 (BD Biosciences) for 15 minutes at room temperature. The B16 cells were washed and then stained for approximately 30 minutes at 4°C with FITC-conjugated rat anti-mouse MHC I mAb (Abcam, Cambridge, MA), PE- conjugated rat anti-mouse MHC Class II mAb (Abcam), FITC-conjugated rat anti-mouse Rae-1 mAb (R&D Systems), APC-conjugated rat anti-mouse H60 mAb (R&D Systems) or PE-conjugated rat anti-mouse CD155 mAb (Biolegend). Negative control cells were stained with the corresponding isotypes. Following incubation, the cells were washed twice with FACS buffer and then analyzed with a BD FACSCalibur with gates set for viable cells.
Immune therapeutics
pORF.IFN-α (IFN-α) plasmid was obtained from Invivogen. Hydrodynamic gene transfer (HGT) consisted of a single intravenous (i.v.) injection of 3 μg endotoxin-free pORF plasmid encoding murine IFN-α or pORF control plasmid DNA (InvivoGen, San Diego, CA) in 2 ml of saline as previously described(28). This results in the sustained in vivo expression of IFN-α at serum levels of 900 pg/ml for more than 30 days after administration (29). The STAT3 inhibitor, WP1193, was synthesized and supplied by Dr. Priebe (M. D. Anderson). WP1193, a third-generation analogue inhibitor of the p-STAT3 pathway, was dissolved in a mixture of 20 parts dimethylsulfoxide (DMSO) to 80 parts polyethylene glycol (PEG) 300 (Sigma-Aldrich, St Louis, MO) at titered concentrations and delivered in a final volume of 100 μL. Prior to use, WP1193 was stored as a lyophilized powder at 4°C.
Treatment for the established tumors was with WP1193 (starting on day 3) and/or IFN-α (starting on day 5) after B16 tumor cell challenge. Mice were injected intravenously (i.v.) with 2 ml (3 μg) of IFN-α plasmid in saline once. Mice were treated with a sub-therapeutic dose of 30 mg/kg of WP1193 by oral gavage (o.g.) in a vehicle of DMSO/PEG300 (20 parts/80 parts) on Monday, Wednesdays and Fridays, on a q.i.d. schedule (5 days on, 2 days off) for 5 treatments (the metastatic model) and 9 treatments (the intracerebral model), respectively. For the metastatic model, the mice were euthanized on day 14 after the B16 cell intravenous injection. Lungs were dissected and B16 nodules in the lungs were counted. For the intracerebral model, survival was observed. When mice were treated in the therapeutic range of 40 mg/kg, >80% of animals survived long-term (more than 70 days) and synergy with IFN-α could not be assessed (24). Ten mice per experimental group were used, including treatment with the DMSO/PEG300 vehicle alone in the control group.
Statistics
Kaplan-Meier product-limit survival probability estimates of overall survival were calculated (30) and log-rank tests (31) were performed to compare overall survival between treatment groups and the control arm. Ex-vivo or in vitro data are presented as means ± standard errors (SEs) of three repeated experiments. Student’s t test was performed. A P value below 0.05 was considered statistically significant.
Results
WP1193, an inhibitor of p-STAT3
Using molecular modeling and medicinal chemistry approaches, we designed and developed a panel of unique small molecule inhibitors (32) that block STAT3 phosphorylation (33, 34), in vitro and in vivo, based on the caffeic acid benzyl ester/AG490 scaffold. WP1193 is a third-generation analogue that has an additional aromatic ring on the benzyl amine moiety (Fig. 1A) and was selected for these studies based on its potential to be a potent immune modulator. Both IFN-α and WP1193 have direct cytotoxic effects on B16, with the combination therapy exerting an additive effect (Fig. 1B). WP1193 can inhibit p-STAT3 in B16 cells and in immune cells (Fig. 1C and D, respectively).
Fig 1.

A novel small molecule, WP1193, inhibits STAT3 activity. A. Chemical structure of WP1193. B. The combination of IFN-α and WP1193 exerts direct cytotoxic effects on B16 cells. C. and D. WP1193 inhibits the phosphorylation of p-STAT3 in both B16 cells (C) and in splenocytes (D). B16 cells and splenocytes isolated from C57BL/6J mice and were incubated with either the medium, medium supplemented with IFN-α (2000 U/ml), or medium supplemented with both IFN-α and WP1193 (5 or 10 μM). After 2 hours (splenocytes) or 4 hours (B16 cells), cells were lysed, electrophoretically fractionated in 8% SDS-polyacrylamide gels, transferred to nitrocellulose membranes, and immunoblotted with antibodies to p-STAT3, total STAT3 and β-actin. Semi-quantitative densitometry was used to determine the relative levels of p-STAT3 to STAT3 and β-actin.
In vivo treatment of mice with IFN-α and WP1193 inhibits bone marrow-derived Tregs but combinational therapy is not synergistic
To ascertain the in vivo effects of IFN-α and WP1193, and in combination on Tregs, non-tumor bearing mice were treated for 16 days. Both WP1193 and IFN-α significantly inhibited the number of Tregs (CD4+Foxp3+) in the bone marrow by 31% and 78%, respectively, compared with the control (P< 0.05 and P< 0.01; Fig. 2). WP1193 and IFN-α also significantly inhibited the number of Tregs (CD4+Foxp3+) in the peripheral blood by 20% and 46%, respectively, compared with the control (P< 0.05; data not shown). However, the combination of IFN-α and WP1193 was not additive or synergistic for inhibiting the number of Tregs in either the bone marrow or the blood. Furthermore, WP1193 or IFN-α alone or in combination did not inhibit the number of Foxp3+ Tregs in the thymus, lymph nodes, or spleen (data not shown). This suggested to us that an additive inhibition of Tregs would not be a mechanism of efficacy with combinational therapy.
Fig 2.

In vivo inhibition of Tregs in the bone marrow of mice treated with WP1193, IFN-α, or both. Both WP1193 and IFN-α inhibited Tregs by 31% (P<0.05) and 78% (P<0.01), respectively, compared with the control; inhibition in the peripheral blood was 20% (P<0.05) and 46% (P<0.05), respectively. However, the combination of IFN-α and WP1193 was not synergistic for Treg inhibition in either the bone marrow or the blood. The experiment was been reproduced twice at day 11 and 14 with similar findings.
In vivo treatment of mice with IFN-α or WP1193 enhances melanoma immune-mediated cytotoxicity
To ascertain the immunological mechanisms of combinational therapy, we examined splenocytes cytotoxic responses directed against B16 melanoma cells. Splenocytes from control mice and mice treated with WP1193, IFN-α, or IFN-α + WP1193 were isolated and co-cultured with CFSE-labeled B16 target cells for 48 hours to assess splenocyte cytotoxicity against B16 cells. In both ratios of splenocyte effector cells to B16 target cells (30:1 and 100:1), the splenocytes from the WP1193 or IFN-α-treated mice had significantly increased cytotoxic clearance of the B16 target cells compared with control mice (P< 0.05; Fig. 3A). Furthermore, in mice treated with both IFN-α and WP1193, there was additive enhanced cytotoxic clearance of the B16 target cells compared with mice that were treated with either WP1193 or IFN-α alone (P< 0.01; Fig. 3A).
Fig 3.


Cytotoxicity of B16 in vitro produced by splenocytes from mice treated with WP1193, IFN-α, or both. A. The splenocyte effector cells from mice that were treated with either WP1193 or IFN-α induced modest tumor lysis. However, splenocyte effector cells from mice that were treated with the combination of WP1193 and IFN-α potently enhanced B16-specific lysis (P< 0.01). B. Cytotoxicity of B16 in vitro produced by NK and CD8+ T cells treated with WP1193, IFN-α, or both. The NK and CD8+ immune effector cell populations were sorted as described and were treated with either WP1193, IFN-α or both. Similar to the data in Fig 3A, either treatment induced modest tumor lysis. However, both effector immune cell populations treated with the combination of WP1193 and IFN-α potently enhanced B16-specific lysis (P<0.01). This experiment was reproduced twice in its entirety with identical findings.
To further ascertain the underlying immunological mechanisms, we performed cytotoxic responses directed against B16 melanoma cells by NK cells and CD8+ cells. The NK1.1+CD3− (NK) cells and CD3+CD8+ T cells from spleens of 4- to 6-week-old mice were isolated, co-cultured with CFSE-labeled B16 target cells and treated with RPMI 1640 medium (control), WP1193 (2 μM), IFN-α (2000 U/ml), or IFN-α (2000 U/ml) + WP1193 (2 μM) for 48 h to assess NK or T cell cytotoxicity against B16 cells. Both the NK and CD8+ T cell populations from the WP1193 (P =0.01 and P =0.02, respectively) or IFN-α-treated group (P =0.0002 and P =0.0006, respectively) increased cytotoxic clearance of the B16 target cells compared with the control group (Fig. 3B). Furthermore, there was enhanced cytotoxic clearance of the B16 target cells in the NK cells or CD8+ T cells treated with both IFN-α and WP1193 compared with the NK cells or CD8+ T cells treated with either WP1193 or IFN-α alone (P< 0.01; Fig. 3B).
MHC I and NK activating ligands are expressed on melanoma cells but are not further enhanced by combination therapy
Because we observed NK and CD8+ T cell- mediated anti-tumor cytotoxicity were enhanced with combinational therapy, to ascertain if either IFN-α, WP1193 or both were augmenting the expression of MHC or NK activating receptors or their ligands, splenocytes and B16 cells were treated with RPMI 1640 medium (control), WP1193 (2 μM), IFN-α (2000 U/ml), or IFN-α (2000 U/ml) + WP1193 (2 μM) for 24 hours. The NK-activating ligands (Rae-1, H60 and CD155) and MHC (I and II) on B16 cells and the NK-activating receptors (NKG2D, KLRD1, NKp46 and DNAM-1) on NK1.1+ NK cells were analyzed by flow cytometric analysis. MHC I but not MHC II was expressed on B16. IFN-α enhanced MHC I expression but this was not further enhanced with WP1193 (Fig. 4A). Additionally, B16 expressed H60, Rae-1 and CD155; however, neither IFN-α nor the WP1193 treatment altered the mean fluorescent intensity on the surface indicating that these treatments do not alter the receptor density of the NK ligands (Fig. 4B). Furthermore, NKG2D, KLRD1, NKp46 and DNAM-1 were expressed on the NK cells but also did not appear to be up-regulated by either WP1193 or IFN-α indicating that these treatments do not alter the receptor density of the NK receptors (Fig. 4C).
Fig 4.




Regulation of MHC and NK-activating receptors and their respective ligands by WP1193 and IFN-α. Splenocytes or B16 cells were treated with WP1193, IFN-α, or both and MHC, the NK-activating receptors and ligands were subsequently analyzed by flow cytometric analysis. The isotype control is shown by the dashed black line and the respective target antigen by a solid black line. A. B16 cells stained for surface expression of MHC I and II after exposure to WP1193, IFN-α or the combination of WP1193 and IFN-α. B. B16 cells stained for surface expression of the NK-activating receptor ligands H60, Rae-1 and CD155 after exposure to WP1193, IFN-α or the combination of WP1193 and IFN-α. C. NK cells labeled with anti-NK1.1+ antibody from murine splenocytes stained for surface expression of the NK activating receptors NKG2D, KLRD1, NKp46, and DNAM-1 after exposure to WP1193, IFN-α or the combination of WP1193 and IFN-α.
STAT3 blockade enhances the efficacy IFN-α against metastatic melanoma
To determine whether the IFN-α and STAT3 blockade combination therapy yielded a synergistic efficacy against the process of metastasis, IFN-α (i.v.) or WP1193 (o.g.) was administrated alone or in conjunction with each other in C57BL/6J mice with systemic, metastatic melanoma. In order to observe the synergistic effect of STAT3 blockade and IFN-α, the sub-therapeutic dose of WP1193, 30 mg/kg was used in this study. The number of pulmonary metastasis was quantified 14 days after tumor inoculation. The number of metastasis for tumor-bearing mice without further intervention was 28 ± 12. Neither sub-therapeutic WP1193 (34 ± 17, P=0.31 compared with control) nor IFN-α alone (28 ± 11, P=0.48 compared with control) inhibited B16 pulmonary metastasis (Fig. 5A). However, the number of pulmonary metastasis was significantly reduced for WP1193 + IFN-α combinatorial therapy (9 ± 5) compared to control (P<0.05), WP1193 alone (P<0.05), or IFN-α alone (P<0.05) (Fig. 5A).
Fig 5.
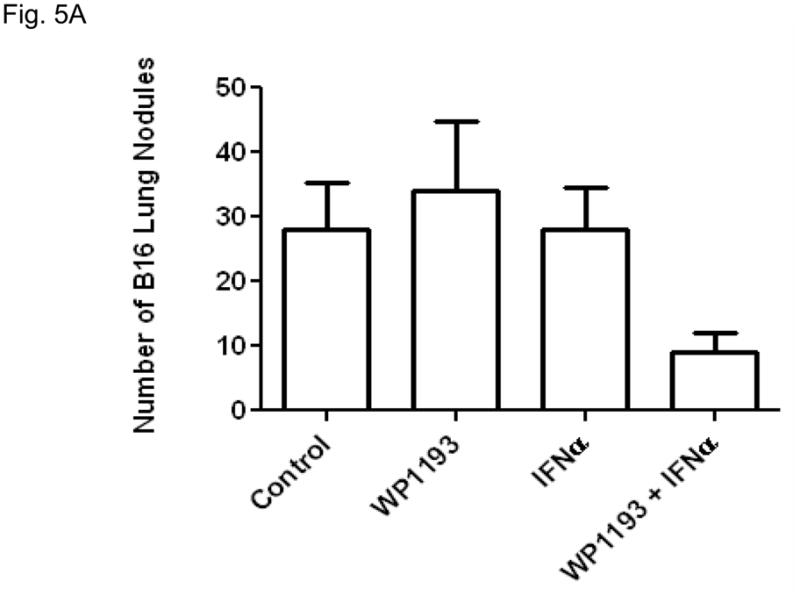

STAT3 blockade enhances the efficacy IFN-α against metastatic melanoma. A. The number of pulmonary metastasis was quantified 14 days after tumor inoculation. The number of metastasis for tumor-bearing mice without further intervention was 28 ± 12. Neither sub-therapeutic WP1193 (34 ± 17, P=0.31 compared with control) nor IFN-α alone (28 ± 11, P=0.48 compared with control) inhibited B16 pulmonary metastasis (Fig. 5A). However, the number of pulmonary metastasis was significantly reduced for WP1193 + IFN-α combinatorial therapy (9 ± 5) compared to control (P<0.05), WP1193 alone (P<0.05), or IFN-α alone (P<0.05). B. Survival data from C57BL/6J mice treated with WP1193, IFN-α, or both after B16 cells were established in the brain. Median overall survival for mice with intracerebral tumors without further intervention (n=11) was 17 days. C57BL/6J mice with established intracerebral B16 cells treated with a sub-therapeutic dose of WP1193 via oral gavage (n=12) showed a 9% increase in their median survival time to 18.5 days (P<0.04 compared with control). In mice with established tumor treated with IFN-α alone (n=8), there was a 62% increased in median survival to 27.5 days (P< 0.01 compared with control). In those mice with established tumors treated with the combination of IFN-α and WP1193 (n=11), there was a 135% increase in median survival to 40 days that was significantly longer compared with IFN-α alone (P<0.02). In mice that survived long-term subsequent re-challenge by injection of B16 cells into the contralateral hemisphere indicated that minimal immunological memory was induced. This experiment was repeated in its entirety with similar results.
Treatment of established intracerebral melanoma with both IFN-α and STAT3 blockade is efficacious
To determine whether the IFN-α and STAT3 blockade combination therapy yielded a synergistic efficacy against established CNS tumors, IFN-α (i.v.) or WP1193 (o.g.) was administrated alone or in conjunction with each other to C57BL/6J mice in which intracerebral melanoma had been established via intracranial injection of log-phase B16 tumor cells. Kaplan-Meier survival curves were plotted for those mice. Upon death, the etiology was confirmed to be tumor progression. Median overall survival for tumor-bearing mice without further intervention was 17 days (95% CI, 16, NA; n=11) and was significantly enhanced by all therapies, including with either sub-therapeutic WP1193 (18.5 days; 95% CI, 17, NA; P<0.04 compared with control; n=12) or IFN-α alone (27.5 days; 95% CI, 21, NA; P< 0.01 compared with control; n=8) (Fig. 5B). Moreover, median overall survival (40 days; 95% CI, 31, NA; P<0.001; n=11) was significantly longer for WP1193 + IFN-α combinatorial therapy than for IFN-α alone (P<0.02). For the mice treated with the combinatorial therapy of WP1193 and IFN-α, 27% survived long term (>84 days) (P<0.001 compared with the control group), and there was at least a 135% increase in median survival time when the experiment was terminated to perform the tumor rechallenge experiments.
To determine whether mice with intracerebral tumors treated with both WP1193 and IFN-α were able to generate long-lasting protective immune memory, mice that survived for 84 days after the initial tumor cell implantation were re-inoculated with B16 cells in the contralateral hemisphere. Upon rechallenge, in the animal group that had received the WP1193 and IFN-α, the median survival time was 16 days, which did not differ significantly from the median survival time (17 days) of naïve, control mice. There were no long-term survivors in the rechallenged group (data not shown), indicating that long-lasting immune memory was not induced by the combination therapy.
IFN-α inhibits in vivo death secondary to LMD
Because IFN-α has previously been demonstrated to inhibit invasion and melanoma LMD in clinical trials, we assessed at the time of treatment failure the etiology of the animal’s death whose bodies could be recovered for autopsy. Both the control group (n=8) and in those treated with a sub-therapeutic dose of WP1193 (n=9) had macroscopic evidence of LMD, 75% and 67%, respectively, (Fig. 6A). In contrast, in those mice treated with IFN-α (n=6) or the combination of IFN-α and WP1193 (n=4), only 17% in the former and none of the animals treated with the combination died of LMD (Fig. 6B). s would be expected, in a sub-analysis within each treatment group, mice that developed LMD died sooner than did those that died of tumor. Specifically, in the control group, median survival was 16.7 ± 0.7 days in those mice that developed LMD and 18.5 ± 1.5 days in those that died of tumor. Furthermore, in the sub-therapeutic WP1193 group, median survival was 17.7 ± 0.3 days in mice that developed LMD and 20 ± 2.1 days in those that died of tumor. In contrast, within the IFN-α treatment group, the median survival of mice with progressive tumor was 25.8 ± 2.5 days, which was further increased to 29.7 ± 5.5 days in the combination treatment group. Within individual treatment groups there was insufficient power to draw statistically meaningful conclusions; however when we assessed all mice who died of LMD, the median survival was 16.9 ± 0.4 days compared with 24.7 ± 2 days for mice dying of tumor progression (P=0.0006), indicating this model system conforms to the negative prognostic influence of LMD observed in human patients.
Fig 6.

The CNS and survival data of C57/BL6 mice with intracerebral melanoma treated with WP1193, IFN-α, or both. A. The neuro-axis of mice at the time of death (treatment failure) was formalin fixed, paraffin embedded, and then stained with hematoxylin and eosin. The top panel demonstrates a whole mount axial section of the CNS from a mouse with death secondary to progression of LMD (shown by the black arrow). The asterisk (*) denotes the brainstem, and # denotes the hippocampus at 2.5× magnification. The bottom panel demonstrates a coronal section of the CNS from a mouse with death secondary to progression of intraparenchymal tumor. The white arrow shows the tumor-brain interface at 2.5× magnification. B. The C57/BL6 mice died of LMD or tumor depending on the treatment. Both the control and the sub-therapeutically WP1193 group died of progressive LMD. In contrast, in those C57/BL6 mice treated with IFN-α or the combination of IFN-α and WP1193, treatment failure-related deaths were secondary to tumor progression rather than LMD.
Discussion
In this report, we demonstrated that the combination of IFN-α and p-STAT3 blockade can exert efficacy against metastatic melanoma including intracerebral established CNS melanoma. Patients with CNS melanoma, especially those with LMD, are typically refractory to currently available standard therapies and our preclinical data would suggest that this combination might have clinical utility. This is notable considering that immunotherapeutic approaches for melanoma have been disappointing (35). The cytokine IFN-α is currently FDA-approved for patients with surgically resected stage III melanoma without evidence of radiographic measurable disease (36) and is not particularly efficacious against CNS disease. In our experimental model systems of metastatic and CNS melanoma, IFN-α induced a modest therapeutic response. However, when IFN-α was used in combination with WP1193, marked therapeutic efficacy was seen and there appeared to be a trend to a diminished propensity for the development of LMD. This warrants further investigation in murine models of established LMD with larger cohorts of animals. Previous immune therapeutic approaches to treat melanoma LMD have included the intrathecal administration of IL-2 (37) and IFN-α (6). IL-2 enhances NK cell activity, activates cytotoxic T cells, stimulates IFN-γ release and activates macrophages and IFN-α has direct anti-proliferative effects on tumor cells, activates NK cells and cytotoxic T cells, and enhances antigen presentation and MHC expression. Clinical trials of IL-2 in melanoma patients with LMD demonstrated a high rate of tumor clearance from the CSF (38–40); however treatment resulted in meningeal irritation, fever, brain edema, seizures, stupor and one death (38–40). Similarly, IFN-α in clinical trials also demonstrated clearance of malignancy within the CSF (6); however treatment was confounded by profound neuro-toxicity (7). Our data would suggest that systemic IFN-α in combination with WP1193 might be a novel approach to also inhibit the development of LMD.
The combination of IFN-α and WP1193 enhanced tumor cytotoxicity mediated by both the NK and CD8+ T cell populations. It was not surprising to observe an enhancement of CD8+ T cell tumor cytotoxic activity with the combination of STAT3 blockade and IFN-α, because this was consistent with our previously published data of p-STAT3 inhibitors (21) and other investigators work that demonstrated that IFN-α enhances CD8+ cytotoxic responses (41). Because melanomas have been shown to evade immune detection by down-modulating MHC (42–44), immunological responses that are antigen-MHC independent such as NK-mediated cytotoxicity are appealing for overcoming at least one mechanism of immune therapeutic resistance. To determine whether the enhancement of NK-mediated cytotoxic function was related to up-regulation of NK-activating receptors or ligands by treatment with the combination of IFN-α and WP1193, we assessed the expression levels on NK cells and on B16, respectively. The B16 cells expressed the MHC I, Rae1, H60 and CD155, indicating that they would be capable of triggering NK cytotoxic responses, resulting in tumor clearance similar to findings in a previous report (45), but treatment did not alter the expression of the ligands. Furthermore, we did not find changes in the NK-activating receptor expression levels of NKG2D, KLRD1, NKp46 or DNAM-1. It is possible that there was a transient increase in the NK-activating receptors that was not identified under the current experimental conditions.
We have shown that the p-STAT3 inhibitors inhibit Tregs in murine models of melanoma and from melanoma patients (24, 46). IFN-α has been shown to augment IL-10 production and IFN-α-treated dendritic cells induce IL-10-producing regulatory T cells (47). Additionally, in studies of human melanoma patients treated with high-dose IFN-α, Wang et al., demonstrated an enhancement of Tregs in the lymph nodes but the Treg population was not analyzed in the bone marrow and blood (48). Furthermore, we found that IFN-α induces the immune suppressive p-STAT3 and others have shown that p-STAT3 is a promoter of FoxP3 expression in Tregs. Thus, we hypothesized that the p-STAT3 inhibitors would enhance the therapeutic efficacy of IFN-α by inhibiting the induced Tregs. Although there is inhibition of the number of Tregs in both the bone marrow and blood with IFN-α and WP1193, there was no additive effect on inhibiting the number of Tregs. Interestingly, IFN-α demonstrated inhibition of the number of Tregs in bone marrow and the peripheral blood and slight enhancement of the numbers of Tregs in the lymph nodes and so a paradox arises as to the mechanism of IFN-α in inhibiting Tregs that we observed in vivo. Within a few days after hydrodynamic gene transfer of IFN-γ, the total bone marrow cellularity drops with the CD4 T cell population being the most affected which is consistent with other reports of IFN-α (49). Within the CD4 fraction, the FoxP3+ Tregs numbers are even more suppressed compared with non-Treg CD4+ T cells, thus the reason why in the IFN-γ treatment group the Treg numbers were most dramatically inhibited within the bone marrow. Using sorted Tregs from a FoxP3-GFP reporter mouse from IFN-α-treated or control mice we did not see a decrease in the suppressive activity (data not shown). Thus, IFN-α inhibits the relative number of Tregs but not their suppressive activity, whereas WP1193 only modestly inhibits the number of Tregs in the bone marrow compared with IFN-α but does suppress their functional activity as we have previously demonstrated (24, 46). Alternatively, we may not have appreciated an additive effect on Treg inhibition since these studies were conducted in non-tumor bearing animals in which Treg induction may have been minimal.
In conclusion, the combination approach of WP1193 and IFN-α enhances both NK and CD8+ cytotoxicity and appears to be a promising potential treatment modality for melanoma patients with CNS disease who currently have very few therapeutic options available and who are typically excluded from clinical trials.
Statement of Translational Relevance.
Patients who have stage IV melanoma with central nervous system (CNS) metastasis have dismal survival despite multi-modality treatment and are typically excluded from clinical trials. Systemic interferon (IFN)-α is FDA approved for the adjuvant treatment of surgically resected melanoma patients that are at high risk for recurrence and death. However, IFN-α adjuvant therapy is not particularly effective in melanoma patients with CNS metastasis including leptomeningeal disease (LMD). Clinical trials of melanoma patients with LMD using direct intraventricular administration of IFN-α were confounded by severe neurotoxicity. This study demonstrates that the efficacy of systemic IFN-α can be significantly enhanced with an inhibitor of the signal transducer and activator of transcription (STAT) 3 in the treatment of metastatic melanoma and established intracerebral syngeneic murine melanoma. Therapeutic efficacy was secondary to enhanced CD8+ T cell and NK tumor cytotoxicity. Thus, the STAT3 inhibitor, WP1193, and IFN-α appear to be a promising potential treatment modality for melanoma patients with CNS disease.
Acknowledgments
We thank Adelina “Keats” Fuentes and Michael Worley for editorial assistance. This work was supported by funding from The National Institutes of Health (CA120813-03) (A177225-01) (ABH); P50 CA093459 (WP).
Abbreviations used
- CNS
Central nervous system
- LMD
leptomeningeal disease
- IFN-α
interferon
- STAT3
transducer and activator of transcription 3
Footnotes
References
- 1.Sampson JH, Carter JH, Jr, Friedman AH, Seigler HF. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg. 1998;88:11–20. doi: 10.3171/jns.1998.88.1.0011. [DOI] [PubMed] [Google Scholar]
- 2.Kirkwood JM, Manola J, Ibrahim J, et al. A pooled analysis of eastern cooperative oncology group and intergroup trials of adjuvant high-dose interferon for melanoma. Clin Cancer Res. 2004;10:1670–7. doi: 10.1158/1078-0432.ccr-1103-3. [DOI] [PubMed] [Google Scholar]
- 3.Liang S, Wei H, Sun R, Tian Z. IFNα regulates NK cell cytotoxicity through STAT1 pathway. Cytokine. 2003;23:190–9. doi: 10.1016/s1043-4666(03)00226-6. [DOI] [PubMed] [Google Scholar]
- 4.Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202:637–50. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ravine TJ, Ledinko N. Treatment with human recombinant leukocyte interferons inhibits in vitro invasive ability of human lung carcinoma cells. Clin Exp Metastasis. 1986;4:191–203. doi: 10.1007/BF00117932. [DOI] [PubMed] [Google Scholar]
- 6.Obbens EA, Feun LG, Leavens ME, et al. Phase I clinical trial of intralesional or intraventricular leukocyte interferon for intracranial malignancies. J Neurooncol. 1985;3:61–7. doi: 10.1007/BF00165173. [DOI] [PubMed] [Google Scholar]
- 7.Meyers CA, Obbens EA, Scheibel RS, Moser RP. Neurotoxicity of intraventricularly administered alpha-interferon for leptomeningeal disease. Cancer. 1991;68:88–92. doi: 10.1002/1097-0142(19910701)68:1<88::aid-cncr2820680118>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 8.Xie TX, Huang FJ, Aldape KD, et al. Activation of stat3 in human melanoma promotes brain metastasis. Cancer Res. 2006;66:3188–96. doi: 10.1158/0008-5472.CAN-05-2674. [DOI] [PubMed] [Google Scholar]
- 9.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 10.Huang S. Regulation of metastases by signal transducer and activator of transcription 3 signaling pathway: clinical implications. Clin Cancer Res. 2007;13:1362–6. doi: 10.1158/1078-0432.CCR-06-2313. [DOI] [PubMed] [Google Scholar]
- 11.Van Wagoner NJ, Oh JW, Repovic P, Benveniste EN. Interleukin-6 (IL-6) production by astrocytes: autocrine regulation by IL-6 and the soluble IL-6 receptor. J Neurosci. 1999;19:5236–44. doi: 10.1523/JNEUROSCI.19-13-05236.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fitzgerald DP, Palmieri D, Hua E, et al. Reactive glia are recruited by highly proliferative brain metastases of breast cancer and promote tumor cell colonization. Clin Exp Metastasis. 2008;25:799–810. doi: 10.1007/s10585-008-9193-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 14.Kortylewski M, Kujawski M, Wang T, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11:1314–21. doi: 10.1038/nm1325. [DOI] [PubMed] [Google Scholar]
- 15.Bollrath J, Phesse TJ, von Burstin VA, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91–102. doi: 10.1016/j.ccr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 16.Grivennikov S, Karin E, Terzic J, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–13. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kortylewski M, Xin H, Kujawski M, et al. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell. 2009;15:114–23. doi: 10.1016/j.ccr.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takeda K, Clausen BE, Kaisho T, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- 19.O’Farrell AM, Liu Y, Moore KW, Mui AL. IL-10 inhibits macrophage activation and proliferation by distinct signaling mechanisms: evidence for Stat3-dependent and -independent pathways. EMBO J. 1998;17:1006–18. doi: 10.1093/emboj/17.4.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002;169:2253–63. doi: 10.4049/jimmunol.169.5.2253. [DOI] [PubMed] [Google Scholar]
- 21.Hussain SF, Kong L-Y, Jordan J, et al. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007;67:9630–6. doi: 10.1158/0008-5472.CAN-07-1243. [DOI] [PubMed] [Google Scholar]
- 22.Kinjyo I, Inoue H, Hamano S, et al. Loss of SOCS3 in T helper cells resulted in reduced immune responses and hyperproduction of interleukin 10 and transforming growth factor-β1. J Exp Med. 2006;203:1021–31. doi: 10.1084/jem.20052333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zorn E, Nelson EA, Mohseni M, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood. 2006;108:1571–9. doi: 10.1182/blood-2006-02-004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kong LY, Abou-Ghazal MK, Wei J, et al. A novel inhibitor of signal transducers and activators of transcription 3 activation is efficacious against established central nervous system melanoma and inhibits regulatory T cells. Clin Cancer Res. 2008;14:5759–68. doi: 10.1158/1078-0432.CCR-08-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L, Yi T, Kortylewski M, et al. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. 2009;206:1457–64. doi: 10.1084/jem.20090207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alterman AL, Stackpole CW. B16 melanoma spontaneous brain metastasis: occurrence and development within leptomeninges blood vessels. Clin Exp Metastasis. 1989;7:15–23. doi: 10.1007/BF02057178. [DOI] [PubMed] [Google Scholar]
- 27.Coligan JE, Kruisbeck AM, Margulies DH, Shevach EM, Strober W. Current protocols in immunology. New York: Green & Wiley Interscience; 1994. [Google Scholar]
- 28.Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Therapy. 1999;6:1258–66. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- 29.Sikora AG, Jaffarzad N, Hailemichael Y, et al. IFN-a enhances peptide vaccine-induced CD8+ T cell numbers, effector function, and antitumor activity. J Immunol. 2009;182:7398–407. doi: 10.4049/jimmunol.0802982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–81. [Google Scholar]
- 31.Mantel N. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother Rep. 1966;50:163–70. [PubMed] [Google Scholar]
- 32.Priebe W, Donato N, Talpaz M, Fokt I, Szymanski S. Novel compounds for treatment of cell proliferative diseases. WO/104013214 US Patent Office. 2004;832
- 33.Priebe W, Fokt I, Szymanski S, et al. Design, synthesis and structure-activity relationships of novel Jak2/STAT3 signaling inhibitors. 97th Annual Meeting of the American Association for Cancer Research; 2006. p. LB-298. [Google Scholar]
- 34.Madden T, Kazerooni R, Myer J, et al. The preclinical pharmacology of WP1066, a potent small molecule inhibitor of the JAK2/STAT3 pathway. 97th Annual Meeting of the American Association for Cancer Research; Washington, DC. 2006. [Google Scholar]
- 35.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–15. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agarwala SS, Kirkwood JM. Interferons in melanoma. Curr Opin Oncol. 1996;8:167–74. doi: 10.1097/00001622-199603000-00015. [DOI] [PubMed] [Google Scholar]
- 37.List J, Moser RP, Steuer M, et al. Cytokine responses to intraventricular injection of interleukin 2 into patients with leptomeningeal carcinomatosis: rapid induction of tumor necrosis factor α, interleukin 1β, interleukin 6, γ-interferon, and soluble interleukin 2 receptor (Mr 55,000 protein) Cancer Res. 1992;52:1123–8. [PubMed] [Google Scholar]
- 38.Moser RP, Bruner JM, Grimm EA. Biologic therapy for brain tumors. Cancer Bull. 1991;43:117–26. [Google Scholar]
- 39.Papadopoulos NE, Moser RP, Grimm E, et al. Intrathecal use of recombinant interleukin-2 (rIL-2)in the treatment of leptomeningeal disease (LMD) from metastatic melanoma. Proc Annu Meet Am Soc Clin Oncol. 1995:14. [Google Scholar]
- 40.Heimans JJ, Wagstaff J, Schreuder WO, et al. Treatment of leptomeningeal carcinomatosis with continuous intraventricular infusion of recombinant interleukin-2. Surg Neurol. 1991;35:244–7. doi: 10.1016/0090-3019(91)90079-o. [DOI] [PubMed] [Google Scholar]
- 41.Halloran PF, Urmson J, Van der Meide PH, Autenried P. Regulation of MHC expression in vivo. II. IFN-α/β inducers and recombinant IFN-α modulate MHC antigen expression in mouse tissues. J Immunol. 1989;142:4241–7. [PubMed] [Google Scholar]
- 42.Garrido C, Algarra I, Maleno I, et al. Alterations of HLA class I expression in human melanoma xenografts in immunodeficient mice occur frequently and are associated with higher tumorigenicity. Cancer Immunol Immunother. 2009 doi: 10.1007/s00262-009-0716-5. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khong HT, Wang QJ, Rosenberg SA. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: tumor escape by antigen loss and loss of MHC expression. J Immunother. 2004;27:184–90. doi: 10.1097/00002371-200405000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferrone S, Marincola FM. Loss of HLA class I antigens by melanoma cells: molecular mechanisms, functional significance and clinical relevance. Immunol Today. 1995;16:487–94. doi: 10.1016/0167-5699(95)80033-6. [DOI] [PubMed] [Google Scholar]
- 45.Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 2001;413:165–71. doi: 10.1038/35093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kong L-K, Wei J, Sharma AK, et al. A novel phosphorylated STAT3 inhibitor enhances T cell cytotoxicity against melanoma through inhibition of regulatory T cells. Cancer Immunol Immunother. 2008;58:1023–32. doi: 10.1007/s00262-008-0618-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ito T, Amakawa R, Inaba M, et al. Differential regulation of human blood dendritic cell subsets by IFNs. J Immunol. 2001;166:2961–9. doi: 10.4049/jimmunol.166.5.2961. [DOI] [PubMed] [Google Scholar]
- 48.Wang W, Edington HD, Rao UN, et al. Effects of high-dose IFNα2b on regional lymph node metastases of human melanoma: modulation of STAT5, FOXP3, and IL-17. Clin Cancer Res. 2008;14:8314–20. doi: 10.1158/1078-0432.CCR-08-0705. [DOI] [PubMed] [Google Scholar]
- 49.Zoumbos NC, Gascon P, Djeu JY, Young NS. Interferon is a mediator of hematopoietic suppression in aplastic anemia in vitro and possibly in vivo. Proc Natl Acad Sci U S A. 1985;82:188–92. doi: 10.1073/pnas.82.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]