

Abstract
Hypoxia is important in both biomedical and environmental contexts and necessitates rapid adaptive changes in metabolic organization. Mammals, as air breathers, have a limited capacity to withstand sustained exposure to hypoxia. By contrast, some aquatic animals, such as certain fishes, are routinely exposed and resistant to severe environmental hypoxia. Understanding the changes in gene expression in fishes exposed to hypoxic stress could reveal novel mechanisms of tolerance that may shed new light on hypoxia and ischemia in higher vertebrates. Using cDNA microarrays, we have studied gene expression in a hypoxia-tolerant burrow-dwelling goby fish, Gillichthys mirabilis. We show that a coherent picture of a complex transcriptional response can be generated for a nonmodel organism for which sequence data were unavailable. We demonstrate that: (i) although certain shifts in gene expression mirror changes in mammals, novel genes are differentially expressed in fish; and (ii) tissue-specific patterns of expression reflect the different metabolic roles of tissues during hypoxia.
Limitation in availability of oxygen (hypoxia) is a stress important in both biomedical and environmental contexts (1). In humans and other air-breathing vertebrates, hypoxia leads to rapid adaptive changes in metabolic organization, for instance in activation of anaerobic ATP-generating pathways like glycolysis. In mammals, a central role for a specific transcription factor, the hypoxia-inducible factor (HIF)-1α has been demonstrated. HIF-1α mediates the expression of a series of genes involved in both cellular and systemic responses to hypoxia, leading to enhanced anaerobic metabolism and induced erythropoiesis and angiogenesis (2).
The patterns of differential gene expression associated with hypoxic stress in aquatic animals, for instance fishes, remain largely unknown. Understanding the tissue-specific and temporal changes in gene expression in fishes exposed to hypoxia could reveal new mechanisms of hypoxia tolerance and shed light on the evolution of this adaptive response in vertebrates. The long-jaw mudsucker Gillichthys mirabilis is a hypoxia-tolerant species that inhabits estuaries along the coastline of central and southern California, where it lives in burrows that characteristically have low levels of dissolved oxygen (3). Here, we exploit DNA microarray technology to investigate the response of this euryoxic fish to prolonged (up to 6 d) hypoxia. The application of DNA microarrays allows the expression of hundreds to many thousands of genes to be monitored simultaneously, providing a broad and integrated picture of the way an organism responds to a changing environment (4). To date, however, microarray analyses have been applied almost exclusively to model species for which gene sequence data are abundant. We show the utility of microarray approaches for the study of gene expression in a species for which, at the onset of our investigation, sequence data were unavailable. Our study thus provides a road map for exploiting DNA microarrays in studies of adaptation in nonmodel species and reveals the potential power of this experimental approach in comparative and evolutionary biology.
Materials and Methods
Animals and Hypoxia Exposures.
Gillichthys mirabilis were collected near Santa Barbara, CA, by using baited minnow traps, and Gillichthys seta were captured by hand netting in the marine intertidal from San Felipe, Baja California. Fish were maintained in aerated aquaria with flowing seawater at 15°C and fed trout pellets ad libitum. Their pCrit was estimated by sealing an individual fish into a 15°C water-jacketed respirometer and recording the animal's rate of oxygen consumption as the pO2 declined over time. Hypoxic time-course experiments were conducted at 15°C, by bubbling nitrogen gas into an aquarium. The desired pO2 was controlled by using an oxygen sensor (Point-Four Systems, Port Moody, BC, Canada) coupled to a solenoid valve that regulated the flow of N2. At the onset of the time course, the oxygen content of the tank was lowered from an aerated pO2 of 100% (≈8.0 mg per liter) down to 10% (0.8 mg per liter) over a 90-min period. At the 8-, 24-, 72-, and 144-h time points, fish were sampled, and total RNA was extracted from liver, brain, skeletal, and cardiac muscle by using published methods (5). Poly(A)+ mRNA was isolated with oligo-dT cellulose (Ambion) from total RNA pooled from five individual fish. For cardiac and brain tissue, double-stranded cDNA containing a T7 RNA polymerase-binding site was prepared from 2 μg of total RNA (6), and amplified RNA was transcribed in vitro (MAXIscript, Ambion).
For Northern analysis, pooled total RNA samples (10 μg) were denatured, separated on 1.1% agarose gels, and transferred to Zetaprobe membranes (Bio-Rad). DNA probes were prepared by random-primer labeling (Strip-Eze, Ambion) of PCR products by using α-32P-dATP. Hybridizations were performed over night in 7% SDS/0.25 M sodium phosphate, pH 7.2 at 65°C, then washed under high stringency 0.1× SSC, 0.1% SDS, and exposed to x-ray film.
Subtractive Hybridization.
Suppression subtractive hybridization (SSH) (7) was used to generate libraries of cDNA fragments enriched for hypoxia-regulated genes. Double-stranded cDNA was prepared from 1 μg of total RNA by reverse transcription–PCR amplification (SMART, CLONTECH). The cDNA was digested with RsaI and subjected to SSH as described (8). Both forward- and reverse-subtracted libraries were prepared by using RNA samples isolated from different tissues (liver, skeletal muscle, and brain) at different time points to increase the representation of temporally distinct and tissue-specific differentially expressed genes. The PCR-amplified cDNA fragments generated by SSH were then ligated into the plasmid pGEM-T Easy (Promega).
Full-Length cDNA Library Construction.
We devised a cDNA capture method to construct full-length cDNA libraries enriched for hypoxia-induced genes. In this method, a hypoxia-enriched library of cDNA fragments generated by SSH is biotinylated and then hybridized to a full-length cDNA library, allowing the respective full-length cDNA cognates to be captured by hybridization and then cloned. Briefly, four SSH libraries, identified as containing a high proportion of differentially expressed genes, were chosen as templates for the preparation of biotinylated probes. Single-stranded biotinylated probes were prepared in a linear PCR reaction identical to that used as the secondary PCR amplification in the SSH procedure (8), except that one of the nested primers was omitted, and the other contained a 5′ biotin group. Typically, 30–35 cycles of PCR were used. As a target for the capture reaction, double-stranded cDNA was amplified by PCR by using the SMART III cDNA library construction kit (CLONTECH) with the same RNA sample that had served as the tester sample for the respective SSH reaction. Then 10 ng of biotin-labeled probe was mixed with 4 μg of the double-stranded full-length cDNA in 10 μl of a hybridization buffer (0.5 M NaCl/50 mM Hepes, pH 8.3/1 mM EDTA), denatured at 95°C for 2 min, and hybridized at 65°C for 18 h in a thermal cycler. The hybridizing cDNAs were captured on streptavidin-coated magnetic beads (PerSeptive BioSystems) and washed twice in 2 ml 0.1× SSC/0.1% SDS. A small aliquot of the washed beads was added to a PCR mixture, and the captured full-length cDNAs were amplified by long-distance PCR (Advantage cDNA polymerase, CLONTECH), cleaved with SfiI, size fractionated over sephacryl S-400 and directionally cloned into pTriplEx2 (CLONTECH). During the preparation of this manuscript, a similar method to our cDNA capture-cloning procedure was published (9).
As an additional source of cloned cDNAs, we prepared a G. mirabilis normalized liver full-length cDNA library in pTriplEx2 by using an established protocol (method 4 of ref. 10). The source mRNA from which the normalized library was constructed was isolated from hypoxia-exposed fish.
Microarray Construction.
The Gillichthys microarray was constructed by arraying PCR-amplified cDNA clones at high density on glass microscope slides. cDNA clones were randomly picked from the collection of cDNA plasmid libraries prepared as described above, and their cDNA inserts amplified by PCR. For the data presented here, a 5,376-element array was constructed; 2,688 (half) of the clones on the array were cDNA fragments generated by SSH, 560 clones were from the liver-normalized library, and the remaining 2,128 clones were selected from the full-length captured cDNA libraries. The array consisted of 3,840 clones from liver, 1,152 from skeletal muscle, and 384 from brain. The arrays were prepared by using standard methods (see www.microarrays.org). Briefly, the plasmid libraries, prepared as described, were transformed into Escherichia coli, and bacterial colonies were picked at random into 384-well microtiter plates and grown at 37°C overnight. cDNA inserts were amplified by PCR in 96-well format by using <1 μl of bacterial culture in 50 μl of a standard PCR reaction and vector-specific primers. The PCR products were recovered by ethanol precipitation, washed once with 70% ethanol and resuspended in 50 μl 3× SSC, 0.0025% Nonidet P-40. The PCR products were printed robotically onto lysine-coated glass slides by using established protocols (11).
Gene Expression Analysis.
Fluorescently labeled first-strand cDNA probes were prepared by reverse transcription of 1 μg poly(A)+ RNA (liver and muscle), or 4 μg amplified RNA (heart and brain), prepared from control and hypoxia-treated fish in the presence of Cyanine 3 and 5 dUTP analogs (APBiotech, Uppsala, Sweden), respectively, and hybridized to microarrays by using established protocols (11). As blocking agents, 5 μg polydA and 10 μg yeast tRNA (APBiotech) were included in the hybridization. After hybridization, the slides were washed and dried (11), and Cy3 and Cy5 fluorescence was measured by using a confocal laser-scanning device (ScanArray 3000) and quantified with scanalyze software (12). The images were visually inspected, and spots from low-quality areas of the array were flagged and excluded from further analyses. Spots were also excluded from analyses if both the combined fluorescent intensity for both channels was less than 1.4 times that of the local background, and the spot's pixel-by-pixel correlation coefficient was less than 0.4. The flagged Cy5/Cy3 fluorescence ratios were log2 transformed, and then a normalization factor was applied so that the median fluorescence ratio of the well-measured spots on each array was 0.
A hierarchical clustering algorithm (12) was applied to those genes that were unflagged on at least 75% of the arrays and whose log2 ratio was at least ±1.322 (equivalent to 2.5-fold differential expression) on at least 2 arrays. The average linkage clustered data were visualized by using treeview (12). For the differentially expressed clones, a complete sequence or a 5′ sequence tag was generated for the SSH fragments or full-length cDNA clones, respectively (GenBank accession nos. AF266165–266244, AW777099–777149, and AF268077). Clone redundancy was determined by sequence alignment by using sequencher software (Gene Codes, Ann Arbor, MI). A putative identification was given to those clones that yielded homologies with e-values ≤10−6 from BLASTX searches. The sequences of 119 other cDNAs whose expression did not change significantly were determined (EST accession nos. AW783808–783927) and confirmed that a diverse set of genes was present on the array.
Results and Discussion
For the purposes of our study, it was first necessary to define the environmental pO2 that would induce a hypoxic response in Gillichthys. Thus, we determined the critical oxygen tension (pCrit), defined as the oxygen tension (pO2) below which respiration rate falls as pO2 decreases. Below pCrit, O2 uptake may fail to meet metabolic demands, and adaptations to hypoxia must be induced. The pCrit for G. mirabilis and the congeneric species G. seta was ≈1.2 mg per liter O2 at 15°C. We chose 0.8 mg per liter O2 (≈10% of ambient pO2), a level of hypoxic stress well tolerated by Gillichthys, as the experimental hypoxic pO2.
For model species, the abundance of DNA sequence data has allowed the construction of microarrays that are tailored to suit a particular biological question (13). On the other hand, sequences for nonmodel organisms are poorly represented in the public database, and the cDNAs to be arrayed must be independently curated and then identified. We constructed a 5,376-element G. mirabilis microarray by using cDNA clones that were randomly picked from a collection of subtracted and normalized cDNA libraries that had been enriched for hypoxia-regulated genes. Our preliminary pilot microarrays were constructed with short cDNA fragments generated by suppression subtractive hybridization; however, these clones proved difficult to identify by sequence homology analysis. Thus, we printed a second array supplemented with full-length clones that were isolated by using a variation of cDNA capture. We discovered that many of the unidentifiable cDNA fragments from the preliminary arrays could be identified by homology when their full-length homologues, discovered to be present on the second array, were sequenced. For nonmodel organisms, the application of full-length cDNA microarrays may be advantageous, because the presence of conserved protein-coding regions aids gene identification and may permit the array to be used in hybridization analyses that cross species boundaries.
We then analyzed gene expression patterns in liver, brain, skeletal, and cardiac muscle tissues during a 6-d hypoxia time-course experiment, taking samples at 0, 8, 24, 72, and 144 h. RNA taken from control untreated fish (t = 0 h) was used as the Cy3-labeled reference sample (represented as green) to which each Cy5-labeled sample (represented as red) from the later time points was compared. To control for interindividual variation, pooled RNA from five fishes was assessed at each time point. We also used a selection criterion such that only those genes that were well detected on at least 75% of the arrays were selected for further analysis. For liver, 2,196 cDNA spots passed this criterion, and for muscle-type tissues, 1,291 spots passed. Those cDNAs that were 2.5-fold or greater differentially expressed on any 2 arrays in either the liver or muscle-type tissue expression dataset were then sequenced.
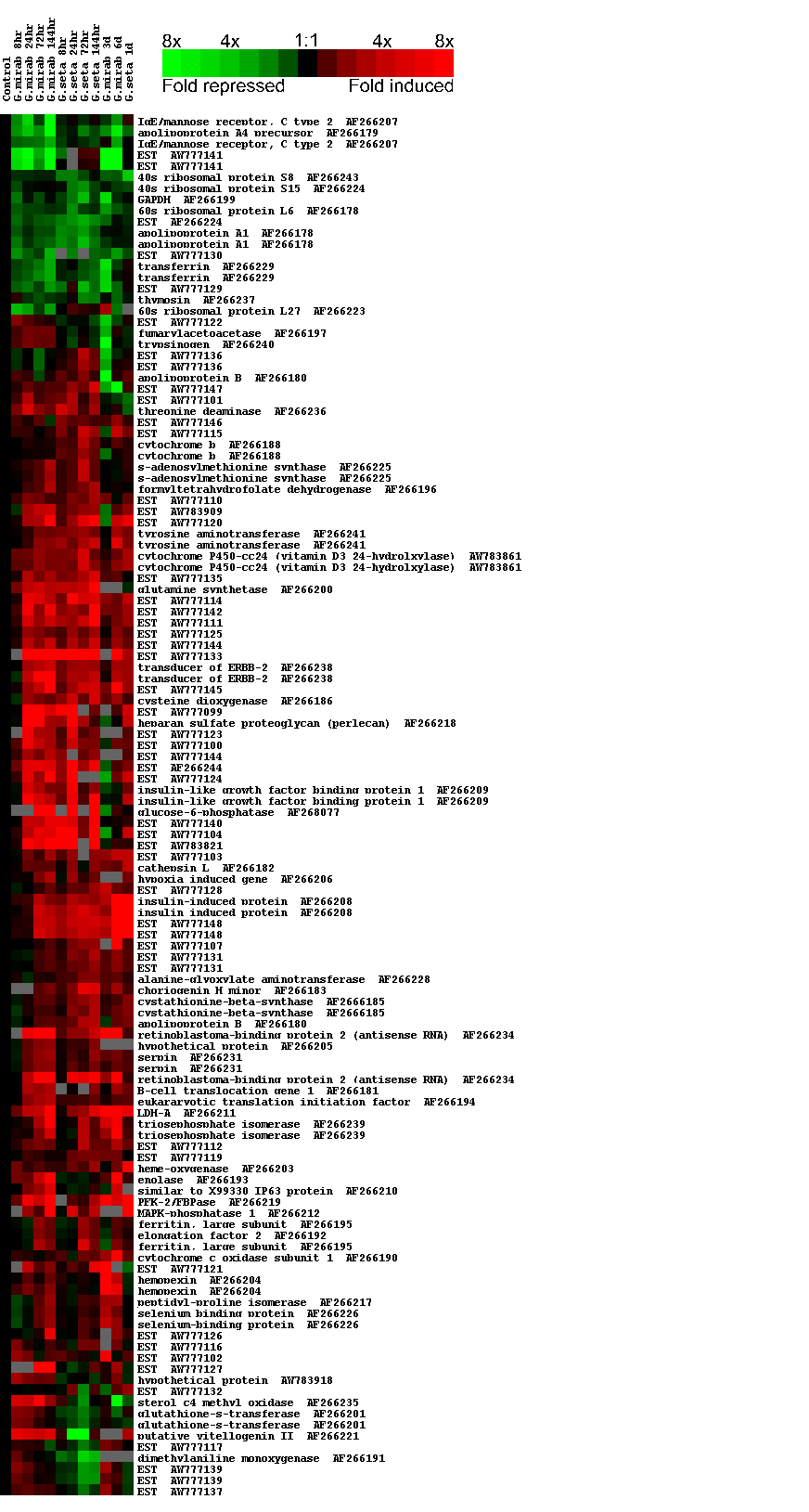
To interpret and organize the data, differentially expressed cDNAs were sequenced and grouped according to their temporal expression pattern by using a hierarchical clustering algorithm (12). Fig. 1 reveals that the expression levels of many genes changed progressively over the time course of hypoxia, and discrete tissue-specific patterns were evident. A total of 126 distinct hypoxia-regulated cDNAs were found, of which 36 were differentially expressed in both liver and muscle, 56 were liver specific, and 34 were muscle specific. By sequence analysis, 75 of the cDNAs could be identified by homology, and the remaining 51 were designated as novel expressed sequence tags (ESTs). We recognize that some of the 51 unidentified ESTs may represent untranslated regions of some of the already identified known genes or may represent multiple distinct fragments of unknown genes. Only four genes were differentially expressed in brain (data not presented). Generally, redundant cDNAs shared similar expression patterns and clustered together. The differential expression of a representative subset of genes was confirmed by Northern analysis (Fig. 2h). As a further confirmation of the repeatability of the results, we generated a liver expression profile for the congeneric species G. seta over an identical hypoxia time course (Fig. 1). This profile was in broad agreement with the data for G. mirabilis.
Figure 1.

Cluster images of the hypoxic expression profile in (a) liver and (b) muscle-type tissues. Supplemental expression data for liver are also presented: a repeat hypoxia time course on G. mirabilis (72 and 144 h, experiment 2) and acute exposure of G. seta to 5% pO2 (24 h acute). The expression data for muscle-type tissues are for G. mirabilis. For clarity, just two measurements are shown for genes that were represented by more than one array element. The quantitative changes in gene expression are represented in color: red indicates induced genes, and green indicates repressed genes. Missing data points are represented as gray bars. Expanded annotated figures showing the gene names and associated accession numbers are published as supplemental data on the PNAS web site, www.pnas.org.
Figure 2.

Genes selected from data presented in Fig. 1 and grouped into categories on the basis of their probable biological role. (a) ATP metabolism; (b) locomotion and contraction; (c) protein translation; (d) iron metabolism; (e) antigrowth and proliferation; (f) amino acid metabolism; and (g) cryptic role. Note that only a small fraction of the unknown genes discovered in this work are shown. (h) Northern blot analysis confirming the expression of a number of hypoxia-regulated genes in liver of G. mirabilis. The expression of β-actin, shown for reference, does not appear to significantly change in liver.
One measure of the reliability of our procedure was that we detected the differential expression of a number of genes known to be regulated by hypoxia in mammals, such as those associated with glycolysis (Fig. 2a), the chief anaerobic ATP-generating pathway (14). The mRNA levels for the glycolytic enzymes lactate dehydrogenase A, enolase (ENO), and triosephosphate isomerase were elevated in liver and increased with time of hypoxia exposure. In muscle tissue, in contrast to liver, mRNA levels of ENO and glyceraldehyde-3-phosphate dehydrogenase decreased, especially in heart. A possible explanation for this is that muscle-type tissues have an already high glycolytic capacity that may be more than adequate to match the reduced metabolic demands of hypoxia.
Many of the differentially expressed genes shared roles in a common physiological process (Fig. 2). For example, three genes involved in successive steps of iron/heme catabolism were regulated in the liver in response to hypoxia (Fig. 2d). Hemopexin, a serum protein that binds and transports heme to the liver for breakdown, heme oxygenase-1, which cleaves the heme ring to yield iron, and ferritin, an intracellular molecule that stores iron in a soluble nontoxic form, were all induced. Conversely, transferrin, an iron transport protein, was down-regulated in liver, despite being a known hypoxia-inducible gene in mammals (15). Intracellular iron can stimulate cell growth and proliferation (16), thus declining transferrin mRNA levels may be linked to the suppression of growth during hypoxia (see below). Changes in the metabolism and sequestration of iron could be linked to hypoxia-induced erythropoiesis (2) and increased demand for iron for hemoglobin synthesis.
In liver, the expression of several genes encoding enzymes of amino acid catabolism increased during hypoxia (Fig. 2f). For example, S-adenosylmethionine synthase and cystathione synthase, which catalyze steps in methionine degradation, were up-regulated. Several aminotransferases were also induced. Consistent with the induction of aminotransferases was the coexpression of glutamine synthetase, which catalyzes the major liver ammonia detoxification reaction through the synthesis of glutamine from glutamate. Catabolism of gluconeogenic amino acids, such as tyrosine and serine, yields either pyruvate or an intermediate of the trichloroacetic acid cycle, metabolites that can serve as carbon skeletons for gluconeogenesis. Further evidence linking amino acid catabolism with hypoxia-induced gluconeogenesis is that the expression of glucose-6-phosphatase (G-6-Pase) was strongly induced. G-6-Pase catalyzes the dephosphorylation of glucose-6-phosphate to glucose, which can be transported in the circulation to other tissues to fuel glycolysis. The time course of the induction of G-6-Pase does not appear to be associated with the mobilization of stored glycogen. In a parallel time-course study, liver glycogen levels were essentially depleted within 24 h of hypoxia exposure, falling from 6.87 ± 1.5 to 0.21 ± 0.1 mg/g (data not shown), yet peak G-6-Pase mRNA levels were not detected until 72 h in the present study. Significantly, expression of the bifunctional enzyme, phosphofructokinase-2/fructose bisphosphate-2, which sets the cellular concentration of the allosteric modulator, fructose-2,6-bisphosphate (F-2,6-BP), was up-regulated in both liver and muscle (Fig. 2a). Changes in F-2,6-BP levels have been linked to hypoxic stress (17). A possible role for the transcriptional activation of this gene would be to increase the capacity for allosteric regulation of the glycolytic and gluconeogenic pathways during hypoxia. Thus, for G. mirabilis, amino acid catabolism coupled with gluconeogenesis in the liver may represent a mechanism to maintain blood glucose levels during hypoxia and may perhaps contribute to continued brain function. The shuttling of metabolic substrates between tissues for either direct oxidation or gluconeogenesis also appears to have been adopted by frogs subjected to prolonged hypoxia (18).
We detected the induction of a number of genes involved in the suppression of cell growth and proliferation (Fig. 2e). For example, elevated levels of mRNA for insulin-like growth factor-binding protein 1 (IGFBP-1), which regulates the availability of insulin-like growth factors in the circulation, were observed in liver. Elevated levels of IGFBP-1 have been reported in fetuses with long-term chronic hypoxia and are correlated with intrauterine growth restriction (19). A strong induction of the stress-inducible mitogen-activated protein (MAP) kinase phosphatase 1 (MKP-1) (20) was also observed in all tissues examined (data not shown for brain). MKP-1 attenuates the activity of the ERK group of MAP kinases, which are phosphorylated in response to the binding of growth factor to cell-surface receptors and activate a signaling cascade that stimulates cell growth (21). In addition, two structurally related antiproliferation genes, transducer of Erb-B2 (Tob) and B-cell translocation gene-1 (BTG-1), were induced in liver. Elevated levels of Tob were also detected in skeletal muscle at 72 and 144 h. Tob interacts with the oncogene Erb-B2 to suppress growth via inhibition of the cell cycle (22). We also identified an inducible pseudogene encoding an antisense mRNA for the 5′ end of retinoblastoma-binding protein 2 (RBP2) (GenBank accession no. AF266234). This antisense transcript was strongly up-regulated in liver and muscle after 48 h and shared a similar expression profile to Tob. The antisense RBP2 cDNA does not appear to be an artifact of cloning, because it was isolated from both a liver and skeletal cDNA library and was shown to bind to an equivalently sized hypoxia-induced RNA transcript by Northern analysis (Fig. 2h). RBP2 has been shown to bind and inhibit the retinoblastoma (pRB) gene product (23). pRB is a potent inhibitor of cell proliferation (24). Although the functional role of the putative antisense-RBP2 is undetermined, we conjecture that it may antagonize translation of RBP2 and the inhibition of pRB activity, thus allowing pRB-mediated repression of proliferation to persist during hypoxia. The expression of all these genes may serve as a complex layered control mechanism to suppress cell growth under hypoxia. IGFBP-1 reduces the level of circulating growth factor, and MKP-1 attenuates the effects of growth factor at the cellular level, whereas Tob and BTG-1 block the cell cycle, thus reducing the energetic demands on the animal from growth and proliferation. The putative adaptive significance of the expression of these genes in response to hypoxia may be to divert important energy resources away from growth toward those metabolic processes essential for survival. Tob, BTG-1, and antisense-RBP2 were not induced until 48 h of hypoxia exposure and thus may represent a longer-term adaptive strategy rather than a first line of defense to a hypoxic challenge.
The expression profile in muscle tissues was dominated by a strong coordinated down-regulation of genes that encode elements of the protein translation machinery, such as the ribosomal proteins and elongation factor 2 (Fig. 2c). Down-regulation of genes associated with translation are characteristic of stressed yeast (12) and cultured mammalian cells with low doubling times (25), suggesting that the repression of these genes in Gillichthys is related to decreased cell growth during hypoxic stress. A depression in protein synthesis rates in muscle has previously been observed in another fish, the crucian carp, during anoxia exposure (26). Curtailment of protein synthesis in skeletal muscle may also have freed amino acids for exploitation in gluconeogenic processes (see above). One unexpected finding was that hypoxia resulted in the repression of genes that code for the contractile proteins tropomyosin, myosin heavy and light chains, and actins (Fig. 2b). Reduced expression of these genes in skeletal muscle may be associated with the decreased locomotory activity Gillichthys exhibited during hypoxia (unpublished results) and in heart with hypoxia-induced bradycardia (27). Because contractile proteins are so abundant in muscle, the reduced expression of these genes may represent an especially important energy-saving strategy during hypoxia. Creatine kinase mRNA levels in muscle also declined during hypoxia (Fig. 2a), suggesting that the demand for phosphocreatine falls consistent with decreased muscle contraction and metabolism. Clustering revealed that the down-regulated translation and contractile genes in skeletal muscle shared similar expression profiles (Fig. 1), and it is tempting to speculate that their expression may be coordinated through a common transduction mechanism. Of the up-regulated muscle genes, a disproportionately large number were unknown, suggesting that these may be involved in an as-yet-uncharacterized aspect of oxygen-related physiology.
Examination of the temporal expression profile revealed that relatively few genes were up-regulated at 8 h of hypoxia, suggesting that the initial stress response is not mediated through gene induction. In contrast, down-regulation of many genes was observed by 8 h, for instance many of the genes for contractile proteins (Fig. 2b) and protein translation (Fig. 2c). This pattern suggests a reorganization of metabolism, such that the major energy-requiring processes like protein synthesis are shut down very rapidly after onset of hypoxia, even before increased expression of proteins needed for enhanced anaerobic ATP production takes place. Other studies have also implicated metabolic suppression as a key adaptive strategy in other hypoxia-tolerant organisms such as turtles (28). In particular, these studies have pointed to the importance of hypoxia-induced reductions in membrane permeability or “channel arrest” as a mechanism to reduce the energetic costs of ion-balancing ATPases (29). However, none of the differentially expressed genes identified in the present study appeared to be linked to this phenomenon.
The effects of hypoxia varied substantially among tissues and reflected the relative importance of the tissues to survival (Fig. 1). In liver, few genes were down-regulated compared with skeletal muscle, which likely reflects the large number of critical biochemical transformations that must be maintained in liver, regardless of environmental conditions. Reduced locomotion powered by skeletal muscle might pose a minimal threat to survival under extreme hypoxia when swimming activity will be minimized. Cardiac muscle, like liver, showed fewer instances of down-regulation compared with skeletal muscle, in keeping with the need for sustained activity of the heart. The differences in patterns of expression may also be related to the extent of the hypoxic insult that each tissue experiences. For example, the heart receives blood directly from the gills and thus will have a better oxygen supply than other tissues. Notably, very few genes exhibited differential activity in brain. During hypoxic stress, the brain is likely to receive a preferential supply of oxygen and glucose, so alterations in gene expression may be minimal relative to tissues like skeletal muscle that undergo substantial decreases in activity.
In mammals, the transcriptional response to hypoxia is commonly mediated by the activation of the transcription factor HIF-1α (2). To date, approximately 36 hypoxia-regulated genes have been reported in mammals (1), and the majority of these genes appear to be under HIF-1α control. HIF-1α activity was not measured in the present study, but the observed induction of a number of known HIF-1α-responsive genes in mammals, such as the glycolytic genes (2), heme oxygenase-1 (30), and insulin-like growth factor-binding protein 1 (31), offers indirect evidence for the conservation of a HIF-1α-mediated transcriptional mechanism in Gillichthys. However, the transcriptional control mechanism of a substantial proportion of the hypoxia-induced genes in Gillichthys remains unknown.
Summary
The utility of cDNA microarray technologies for studies of nonmodel organisms' responses to physiological stress is demonstrated by this study. The power of this approach to identify tissue-specific changes in gene expression and the temporal patterning of these changes is manifested by the finding that wide-scale reorganization of energy-consuming and ATP-generating pathways occurred in G. mirabilis when exposed to hypoxic stress. Our expression profile suggests the following hypoxia survival mechanism in Gillichthys. First, in skeletal muscle, which accounts for approximately one-half of the mass of the organism, the major energy-requiring processes like protein synthesis and locomotion are shut down very rapidly after onset of hypoxia. Then after 24 h, a strong induction in the liver of genes needed for enhanced anaerobic ATP production and for gluconeogenesis occurs. Concurrently, cell growth and proliferation are suppressed, which may serve to divert important energy resources away from growth toward those metabolic processes more essential for hypoxia survival. This microarray-based study represents an important first step toward understanding the mechanisms of hypoxia survival in fish. Nevertheless, further studies using more conventional biochemical and physiological approaches will be needed to verify the interpretation of the mRNA transcriptional data.
Supplementary Material
Acknowledgments
We thank M. Wilson for help with and advice on microarray printing and scanning and G. Schoolnik and P. Small for allowing access to the printing robot and scanning apparatus. This work was supported by National Science Foundation Grant IBN9727721 (to G.N.S.). This is contribution number 34 of the Partnership for Interdisciplinary Studies of Coastal Oceans (PISCO): A Long-Term Ecological Consortium, funded by the David and Lucile Packard Foundation.
Abbreviations
- SSH
suppression subtractive hybridization
- Tob
transducer of Erb-B2
- RBP2
retinoblastoma-binding protein 2
- HIF
hypoxia-inducible factor
Footnotes
References
- 1.Semenza G L. J Appl Physiol. 2000;88:1474–1480. doi: 10.1152/jappl.2000.88.4.1474. [DOI] [PubMed] [Google Scholar]
- 2.Semenza G L. Annu Rev Cell Dev Biol. 1999;15:551–578. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- 3.Barlow G W. Copeia. 1961;121:423–437. [Google Scholar]
- 4.Brown P O, Botstein D. Nat Genet. 1999;21:33–37. doi: 10.1038/4462. [DOI] [PubMed] [Google Scholar]
- 5.Chomzynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 6.Wodicka L, Dong H, Mittmann M, Ho M H, Lockhart D J. Nat Biotechnol. 1997;15:1359–1367. doi: 10.1038/nbt1297-1359. [DOI] [PubMed] [Google Scholar]
- 7.Diatchenko L, Lau Y F C, Campbell A P, Chenchik A, Moqadam F, Huang B, Lukyanov S, Lukyanov K, Gurskaya N, Sverdlov E D, Siebert P D. Proc Natl Acad Sci USA. 1996;93:6025–6030. doi: 10.1073/pnas.93.12.6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diatchenko L, Lukyanov S, Lau Y F C, Siebert P D. Methods Enzymol. 1999;303:349–380. doi: 10.1016/s0076-6879(99)03022-0. [DOI] [PubMed] [Google Scholar]
- 9.Ciavatta V, Cairney J. BioTechniques. 2000;29:444–450. doi: 10.2144/00293bm08. [DOI] [PubMed] [Google Scholar]
- 10.Bonaldo M D F, Lennon G, Soares M B. Genome Res. 1996;6:791–806. doi: 10.1101/gr.6.9.791. [DOI] [PubMed] [Google Scholar]
- 11.Eisen M B, Brown P O. Methods Enzymol. 1999;303:179–205. doi: 10.1016/s0076-6879(99)03014-1. [DOI] [PubMed] [Google Scholar]
- 12.Eisen M B, Spellman P T, Brown P O, Botstein D. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alizadeh A A, Eisen M B, Davis R E, Ma C, Lossos I S, Rosenwald A, Boldrick J C, Sabet H, Tran T, Yu X, et al. Nature (London) 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 14.Semenza G L, Roth P H, Fang H M, Wang G L. J Biol Chem. 1994;269:23757–23763. [PubMed] [Google Scholar]
- 15.Rolfs A, Kvietikova I, Gassmann M, Wenger R H. J Biol Chem. 1997;272:20055–20062. doi: 10.1074/jbc.272.32.20055. [DOI] [PubMed] [Google Scholar]
- 16.Li Z R, Hromchak R, Mudipalli A, Bloch A. Cancer Res. 1998;58:4282–4287. [PubMed] [Google Scholar]
- 17.Storey K B. Physiol Zool. 1987;60:601–607. [Google Scholar]
- 18.Donohoe P H, Boutilier R G. Respir Physiol. 1999;116:171–179. doi: 10.1016/s0034-5687(99)00036-5. [DOI] [PubMed] [Google Scholar]
- 19.Chard T. Growth Regul. 1994;4:91–100. [PubMed] [Google Scholar]
- 20.Keyse S M, Emslie E A. Nature (London) 1992;359:644–647. doi: 10.1038/359644a0. [DOI] [PubMed] [Google Scholar]
- 21.Whitmarsh A J, Davis R J. Nature (London) 2000;403:255–256. doi: 10.1038/35002220. [DOI] [PubMed] [Google Scholar]
- 22.Matsuda S, Kawamura-Tsuzuku J, Ohsugi M, Yoshida M, Emi M, Nakamura Y, Onda M, Yoshida Y, Nishiyama A, Yamamoto T. Oncogene. 1996;12:705–713. [PubMed] [Google Scholar]
- 23.Kim Y W, Otterson G A, Kratzke R A, Coxon A B, Kaye F J. Mol Cell Biol. 1994;14:7256–7264. doi: 10.1128/mcb.14.11.7256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaelin W G., Jr BioEssays. 1999;21:950–958. doi: 10.1002/(SICI)1521-1878(199911)21:11<950::AID-BIES7>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 25.Ross D T, Scherf U, Eisen M B, Perou C M, Rees C, Spellman P, Iyer V, Jeffrey S S, Van de Rijn M, Waltham M, et al. Nat Genet. 2000;24:227–235. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]
- 26.Smith R W, Houlihan D F, Nilsson G E, Brechin J G. Am J Physiol. 1996;271:R897–R904. doi: 10.1152/ajpregu.1996.271.4.R897. [DOI] [PubMed] [Google Scholar]
- 27.Aguilar N M. Ph.D. thesis. Univ. of California, San Diego: Scripps Oceanographic Institute; 2000. p. 201. [Google Scholar]
- 28.Hochachka P W, Buck L T, Doll C J, Land S C. Proc Natl Acad Sci USA. 1996;93:9493–9498. doi: 10.1073/pnas.93.18.9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boutilier R G, St-Pierre J. Comp Biochem Physiol A Mol Integr Physiol. 2000;126:481–490. doi: 10.1016/s1095-6433(00)00234-8. [DOI] [PubMed] [Google Scholar]
- 30.Lee P J, Jiang B H, Chin B Y, Iyer N V, Alam J, Semenza G L, Choi A M. J Biol Chem. 1997;272:5375–5381. [PubMed] [Google Scholar]
- 31.Tazuke S I, Mazure N M, Sugawara J, Carland G, Faessen G H, Suen L F, Irwin J C, Powell D R, Giaccia A J, Giudice L C. Proc Natl Acad Sci USA. 1998;95:10188–10193. doi: 10.1073/pnas.95.17.10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}