

Abstract
Introduction
In disease states such as heart failure, myocardial infarction and hypertrophy, changes in the expression and location of Connexin43 (Cx43) occur (Cx43 remodeling), and may predispose to arrhythmias. Stretch may be an important stimulus to Cx43 remodeling; however, it has only been investigated in neonatal cell cultures, which have different physiological properties to adult myocytes. We hypothesized that localized stretch in vivo causes Cx43 remodeling, with associated changes in conduction, mediated by the renin/angiotensin system (RAS).
Methods and Results
In an open-chest canine model a device was used to stretch part of the right ventricle (RV) by 22% for 6 hours. Activation mapping using a 312-electrode array was performed before and after stretch. Regional stretch did not change longitudinal conduction velocity (post-stretch vs. baseline: 51.5±5.2 vs. 55.3±8.1cm/s p=0.24, n=11), but significantly reduced transverse conduction velocity (28.7±2.5 vs. 35.4±5.4cm/s, p<0.01). It also reduced total Cx43 expression, by Western blotting, compared to a nonstretched area RV of the same animal (86.1±32.2 vs. 100±19.4%, p<0.02, n=11). Cx43 labeling redistributed to the lateral cell borders. Stretch caused a small but significant increase in the proportion of the dephosphorylated form of Cx43 (stretch 9.95±1.4% vs. control 8.74±1.2%, p<0.05).
Olmesartan, an angiotensin-II blocker, prevented the stretch induced changes in Cx43 levels, localization and conduction.
Conclusion
Myocardial stretch in vivo has opposite effects to that in neonatal myocytes in vitro. Stretch in vivo causes conduction changes associated with Cx43 remodeling that are likely caused by local stretch-induced activation of the RAS.
Keywords: Conduction, stretch, gap junctions
Introduction
Changes in wall stress and stretch of the ventricular myocardial cells occur physiologically, with increases in muscle length and width during diastolic filling and during systole from the resistance of the afterload. Greater than normal physiological stretch occurs pathologically in states of volume and pressure loading and triggers changes in muscle force development, cell size and electrical activity, attributed to activation of signaling pathways and sarcolemmal ion channels through mechanotransduction processes(1).
Gap junction remodeling has been found to occur in diseased myocardium in a variety of pathologies(2-6), all of which are associated with changes in myocardial wall stress, contractility, metabolic state, activation pattern, and other potentially common intermediaries or stimuli of the remodeling process. In investigating these processes, interventions that cause pressure and/or volume overload either directly or by inducing a myopathic process are generally used in vivo as the stimulus for increasing wall stress and stretch(1;7). Cell culture studies have also provided important insight into the role of stretch in modulating gap-junctional coupling, including the rapidity of profound changes within minutes of altered stretch, consistent with the short biological half life of the principal ventricular gap-junctional protein, connexin43 (Cx43)(8). However, these studies have mainly used neonatal cardiac cells that have very different two-dimensional topology of cell arrangement and cell-cell connectivity and a fundamentally different pattern of organization of gap junctions that is characteristic of neonatal myocytes(8;9) We have previously described a method for regionally loading the myocardium by applying local stretch in vivo(10). In addressing the hypothesis that localized myocardial stretch in vivo causes Cx43 remodeling with associated changes in conduction, mediated by the renin/angiotensin system (RAS), we used this method to study processes in the locally affected (stretched) region, avoiding the generalized systemic effects of pressure and volume overload.
Methods
Surgical preparation
Mongrel dogs weighing 20-30 kg were anesthetized with intravenous sodium pentobarbital (30-40 mg/kg). Maintenance of anesthesia was with boluses of sodium pentobarbital (10 mg/kg) as required. Positive pressure ventilation was through an endotracheal tube by an animal respirator. A femoral arterial line was used to measure blood pressure and a femoral venous line to give 0.9% sodium chloride solution and drugs. Through a median sternotomy, the heart was exposed and suspended in a pericardial cradle.
The dogs were divided into 4 groups; Group (1) had the stretch device sutured in place and a region of the right ventricle subjected to stretch for 6 hours; group (2) had the stretch device sutured in place but stretch was not applied for the 6-hour observation period; group (3) did not have the stretch device placed on the RV; and group (4) had Olmesartan administered prior to and during the period of stretch. Olmesartan (Sankyo) is a highly specific angiotensin-II receptor (AT1 subtype) antagonist that distributes primarily to the extracellular fluid compartment(11). An Olmesartan bolus (1 mg/kg iv) was given at baseline and again after 3 hours of stretch.
Stretch device
A custom built stretch device similar to one we have previously described(10) was sutured on the free wall of the right ventricle in Groups 1, 2 and 4. An area of the RV free wall approximately 1cm lateral to the interventricular groove and 1cm apical to the tricuspid annulus was selected for application of the stretch device. A small pneumatically driven piston pivoted the proximal ends of 4 levers around appropriately positioned fulcrums. Each of the levers was connected to a foot and the feet arranged to form the four corners of a square. The feet were sutured to the right ventricular epicardium. After 5 minutes of equilibration, the legs were splayed apart using the piston in groups 1 and 4. The diagonal distance between the legs was increased from 32 mm to 39 mm, an increase of 22%, which approximates the degree of stretch at end diastole during exercise, thus producing a significant change in muscle length but one that was within physiological limits and previously validated(10).
Conduction mapping
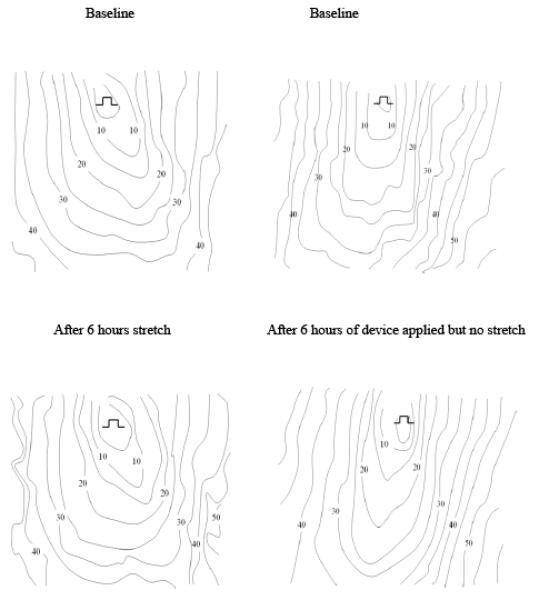
Activation mapping was performed using a 312-bipolar electrode plaque array, with a distance between bipoles of 1.25 mm × 1.25mm. Mapping was performed in the region that was designated for application of the stretch device, before the stretch device was placed on the right ventricle and after removal following 6 hours of stretch (groups 1 and 4) or no stretch (group 2). Sutures on the surface of the right ventricle were used as markers so that the mapping was conducted in the identical place both before and after stretch. Mapping of activation was also done at similar time periods and at a similar location in group 3, which did not have a stretch device applied.
A computerized mapping system recorded signals from all electrode pairs simultaneously during pacing at 300ms cycle length, at twice threshold from the centre of the array(12). Local activation times were assigned to each electrode manually using our previously published criteria and isochrones were constructed at 5 msec intervals(13). A custom written program was then used to calculate local conduction velocities from the gradient of activation times in myocardial surface areas of 1.25 × 1.25 mm2 (14). The gradient of activation times at each local area were calculated from the activation times (time resolution 1 ms) at the 4 electrode sites that delimited the local area. From the local activation time gradient the direction (of propagation) and magnitude of the local vector velocity was calculated. Local vector velocities having a direction between −90° and −60° were averaged to obtain a global value of conduction velocity in the area of the array in that direction of propagation. The direction in which the global value of the velocity was the fastest was defined as the longitudinal conduction velocity (LCV); and the slowest was defined as the transverse conduction velocity (TCV) (14). They corresponded with the long and short axes of the elliptical activation pattern and the anatomical long and short axis of the myocardial fibers on the epicardial surface of the right ventricle, confirmed by visual inspection.
Tissue processing
At the end of the experiment, the animals in each group were euthanized after inducing ventricular fibrillation by electrical stimulation. The hearts were then rapidly excised and plunged into ice-cold saline. Areas of the right ventricle, stretch and control, as well as from the LV were removed. The area of the right ventricle, between the four feet of the stretch device (stretched in group 1 and 4 or not stretched in group 2), was excised. The regions directly underneath where the feet had been applied were excluded from further analysis. The control area of the right ventricle was apical to and a minimum distance of 2cm away from where the stretch device had been applied. The LV tissue was from the free wall approximately 2cm from the interventricular groove midway between the base and the apex. The LV sample was divided into epicardial, mid-myocardial and endocardial sections. . After the samples had been snap frozen in liquid nitrogen, they were stored at −80°C. The frozen tissue then underwent quantitative Cx43 Western blotting and immunohistochemistry.
Immunohistochemistry
The ventricular myocardium was sectioned longitudinally (myocyte long axis) and transverse to the myocardial long axis on a cryostat and 10μm thick sections were collected onto poly-l-lysine slides. Sections were stained with hematoxylin and eosin stains (following standard histological procedures) and examined by standard light microscopy to confirm tissue preservation, cell structure and orientation. Tissue samples were orientated and re-orientated to ensure precisely orientated transversely- and longitudinally sectioned tissue from the respective blocks.
For immunolabeling of total Cx43 the sections were fixed in methanol at −20°C for 5 minutes. Sections were blocked in 1% Bovine Serum Albumin (BSA) solution (Sigma) for 30 minutes at room temperature. The primary antibody was a specific anti-Cx43 monoclonal antibody raised in mice against a synthetic peptide corresponding to positions 252-270 of the native rat connexin (Chemicon International Inc). The sections were incubated with this antibody (1:1000 dilution in 1% BSA) for 2 hours at room temperature. The secondary was an anti-mouse antibody tagged with a Cy3 fluorochrome (Chemicon International Inc). After washing, the sections were incubated with the secondary (1:500 dilution in 1% BSA) for 45 minutes at room temperature. Immunolabeled sections were examined using a Leica TCS 4D confocal microscope using the green (TRITC) high density filter.
For specific labeling of the dephosphorylated fraction of Cx43 labeling the primary antibody was a monoclonal antibody (Zymed Catalogue No. 13-8300) that recognizes Cx43 only if certain residues are not phosphorylated. The antibody was incubated at 1/500 dilution for 2 hours at room temperature. The secondary antibody was as above.
In order to assess the degree of lateralization of Cx43 for each specimen, a previously published, blinded, semiquantitative visual method was used(15). A simple, arbitrary score was derived based on the degree of clustering of the immunolabeled gap junctions in the following manner. After confirming longitudinal orientation by standard light microscopy of an adjacent section, immunolabeled sections were examined using phase-contrast transmission microscopy coupled with fluorescence microscope settings that maintained cell outline visualization to confirm longitudinal cell orientation throughout and assess the pattern of distribution of the immunolabeled gap junctions. Three randomly selected optical fields (~ 250,000 mm2) of at least three separate tissue sections of each specimen were examined by a single experienced immunofluorescence microscopist blinded to the origin of the specimens. For each specimen a simple, arbitrary score based on the visually assessed degree of clustering of the immunolabeled gap junctions in each tissue section was assigned by a blinded experienced assessor. The scoring system used assesses Cx43 label distribution, ranging from being confined exclusively to the normal, transversely orientated clusters at cell abutments (score 1), to a distribution of labeling within longitudinal arrays along the length of the myocyte, with markedly diminished labeling at the end-on abutments (score 5). Although only semiquantitative, this technique has proven a useful classification(15) and identifies changes that are relatively gross and of likely biologic relevance.
For immunohistochemical quantification of phosphorylation state, the area of immunolabeling was measured using confocal microscopy. Single slice images were taken and the immunolabeled area of dephosphorylated Cx43 was expressed as a percentage of the total field area for 6 randomly selected fields. Measurements were made using PC Image Analysis (Foster-Findlay Associates, UK) software.
Quantitative immunoblotting
Total tissue homogenates were prepared and a 0.5μg/μl solution of homogenates was made up in sample buffer. 2.5μg of total protein from each sample was resolved by polyacrylamide gel electrophoresis (BioRad, Hercules, CA, USA) on a 12.5% gel (with a 4.5% stacker).The gel was electrophoretically transferred onto Polyvinyldene fluoride (PVDF) membrane (Immobilon-P-Transfer membrane), at 30volts constant voltage overnight at room temperature, using the Mini Trans-Blot® Electrophoretic Transfer Cell [BIORAD]. Transfer of proteins was assessed by Ponceau S [Sigma] staining, and the membrane was blocked with TBS (0.2mM Tris/HCl pH 7.6, 0.15M NaCl [Sigma]) /0.1% Tween [Merck]/ 1% blot qualified Bovine Serum Albumin (BSA) [Sigma], at room temperature. The membrane was then incubated with the same primary antibody examining total Cx43 as used for immunohistochemistry, anti-Cx43 (Chemicon) at the 1:1000 for 2 hours. The membrane was then washed (in TBS/0.1% Tween/1% BSA) and incubated with the secondary alkaline phosphatase-conjugated antibody. The membrane was then thoroughly washed, and the enzyme activity was revealed by incubation with freshly prepared NBT (Nitro Blue Tetrazolium)/BCIP (%-Bromo-4-Chloro-3-Indoyl-Phosphate) substrate solution [Promega Corporation], made up in alkaline phosphatase buffer (0.1M Tris/HCl pH 9.5, 0.1M NaCl, 5mM MgCl2).
After densitometric quantification of band intensity all values were corrected for protein loading by expressing Cx43 as a ratio to the actin band on a coomassie stained gel run in parallel.
Statistical Methods
Values for conduction velocity and Cx43 quantity were normally distributed. Paired t-tests were therefore used to analyze the changes in conduction velocity before and after stretch, and to compare Cx43 values of stretched areas with control nonstretched areas. Where multiple measurements of a variable were made in a single animal/tissue preparation, the mean of these values was used as the value for that preparation (n=1) for statistical analysis. Significant difference between groups was defined as p<0.05.
The authors had full access to the data and take responsibility for its integrity. All authors have read and agree to the manuscript as written.
Results
Conduction velocity
The preparation was stable over the 6-hour period of the experiment (Table 1); with either no stretch (Group 2) or no device (Group 3) there was no significant change in transverse (TCV) or longitudinal conduction velocity (LCV).
Table 1.
Conduction velocities in the right ventricle (mean±S.D.) at baseline and after the 6 hour intervention period
| Conduction velocity cm/s at Baseline |
Conduction velocity cm/s after 6 hours of Stretch |
Ratio of Conduction Velocity Post-Stretch /Baseline |
||||
|---|---|---|---|---|---|---|
| Longitudinal | Transverse | Longitudinal | Transverse | Longitudinal | Transverse | |
| Group 1 | 55.3±8.1 | 35.4±5.4 | 51.5±5.2 | 28.7±2.5 | 0.949±0.15 | 0.829±0.15 |
| Stretch (n=11) |
p=NS | p<0.01 | p=NS | p<0.01 | ||
| Group 2 | 53.6±6.00 | 31.9±1.17 | 47.8±5.98 | 33.5±2.28 | 0.895±0.09 | 1.056±0.15 |
| Device on no stretch (n=6) |
p=NS | p= NS | p=NS | p=NS | ||
| Group 3 | 46.8±4.99 | 29.8±2.5 | 46.3±1.08 | 29.8±3.35 | 0.996±0.08 | 0.999±0.06 |
| No Device (n=4) |
p=NS | p= NS | p=NS | p=NS | ||
| Group 4 | 51.3±5.4, | 32.3±4.6 | 50.3±7.3 | 34.2±5.1 | 0.984±0.14 | 1.063±0.08 |
| Stretch + Olmesartan (n=6) |
p=NS | p=NS | p=NS | p=NS | ||
A 6 hour regional stretch of the right ventricle did not change LCV (55.3±8.1 cm/s to 51.5±5.2 cm/s, p=NS-baseline vs. post-stretch). However, it produced an 18.8% reduction in TCV (35.4±5.4 cm/s to 28.7±2.5 (p<0.01 –baseline vs. post-stretch). Olmesartan (Group 4) abolished this change.
Western blot analysis of Cx43 in stretched myocardium
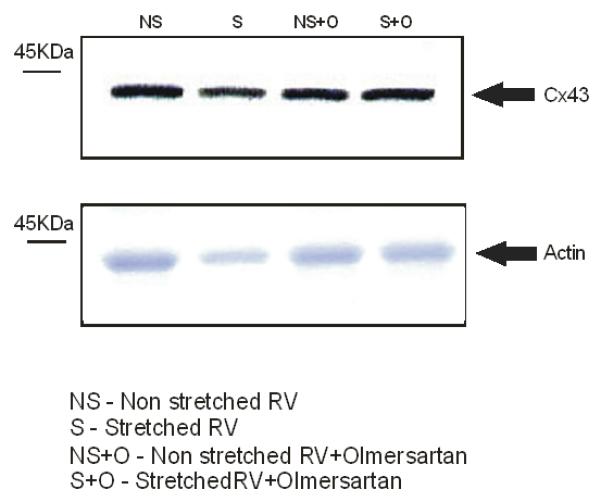
Total Cx43 in stretched RV (Group 1) was reduced compared to the nonstretched control region of the RV in the same hearts (86.1±32.2% vs 100±19.4%, p<0.02, n=11: Fig 1), a reduction that was not seen in the corresponding area in either group 2 (device applied to RV but no stretch) or group 3 (sham operation with no device) (Figure 1), indicating that stretch alone causes a decrease in Cx43 by 13.9% (Figure 1). No change in Cx43 in the LV occurred in any experimental group. In contrast, examination of Cx43 in animals treated with Olmesartan (Group 4) showed no significant change in Cx43 expression when comparing RV stretch area to the RV control area (stretch 93.6±16.7 vs. nonstretched 100.0± 16.9%, p=0.21, n=6) (Figure 1) indicating that blockade of the angiotensin-II receptor inhibited the stretch-induced loss of Cx43 in the RV.
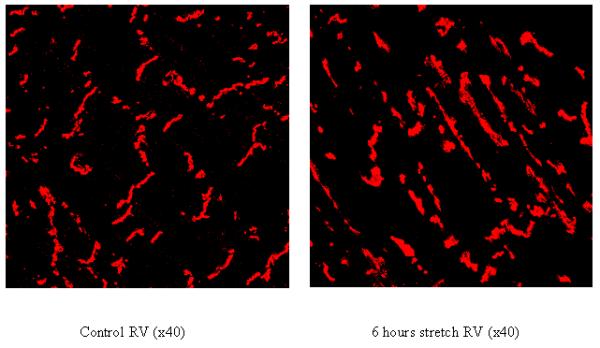
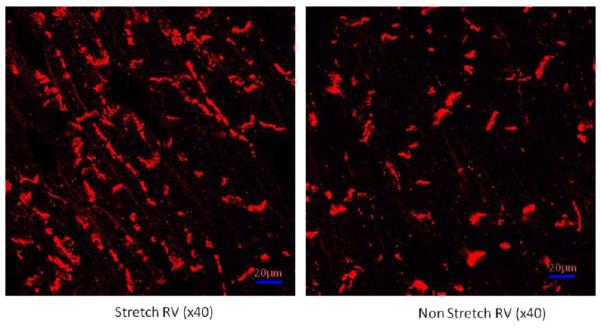
Determination of Cx43 localization following myocardial stretch
Using the semiquantitative score, derived from immunolabeling, described above we analyzed the ability of myocardial stretch to alter Cx43 localization away from the intercalated disk. Distribution scoring (Table 2) shows that there is a significant increase in Cx43 labeling on the lateral membranes in the stretched myocardium compared to nonstretched RV in group 1 and in comparable regions of nonstretched myocardium in groups 2 and 3 and in the Olmesartan treated animals in group 4.
Table 2.
Redistribution scores from right ventricle. The scores are semi-quantitative scores derived from the clustering of gap junctions at the lateral membranes the greater the lateralization the higher the scores (range 1-5). Figures are mean±S.D.
| Redistribution score from RV area intervention |
Redistribution score from RV area control |
|
|---|---|---|
| Stretch (n=11) | 3.1±1.02 | 1.73±0.5 p< 0.002 |
| Stretch + Olmesartan (n=6) |
1.88±1.03 | 1.5±0.71 p= NS |
| Device on no stretch (n=6) |
2.5±1.5 | 1.33±0.58 p= NS |
Figure 2 shows that while control myocardium has Cx43 primarily at the intercalated disk region, stretched myocardium has increased Cx43 on the lateral membranes of myocytes. This lateralization of Cx43 is inhibited by pretreatment with Olmesartan. Both normal and sham treated myocardium show no changes in Cx43 localization and there were no differences in standard tissue histology between any of the myocardial areas of any of the experimental groups (data not shown).
Alterations in Cx43 phosphorylation following myocardial stretch
Because dephosphorylation of Cx43 is associated with a loss of Cx43 function, the phosphorylation status of Cx43 following myocardial stretch was examined by quantitative immunolabeling with an antibody that is specific for the dephosphorylated form of Cx43. In the ventricular myocardium of all groups the majority of the Cx43 was in the phosphorylated form (Figure 3), with stretch causing a small but significant increase in the dephosphorylated form of Cx43 (Figure 3) (9.95±1.4%, vs. 8.74±1.2%, p<0.04). Olmesartan inhibited this dephosphorylation (8.64±2.9%).
Discussion
Results of Our Experiments
The effects of stretch alone on adult myocardium in vivo have not been previously reported. The results of our experiments show that localized stretch of adult ventricular myocytes in vivo causes a reduction of Cx43, a redistribution of Cx43 to lateral sarcolemmal membranes and a slowing of conduction in the transverse direction. These effects were completely prevented by blocking the angiotensin II receptor (subtype AT1) with olmesartan.
The method that we used to investigate the effects of stretching has the following advantages(10). It provides a measured stimulus to a segment of ventricular myocardium and allows correlation of that stimulus with local structural and functional measurements in the stretched myocardium compared to control nonstretched myocardium adjacent to the stretched region. This method eliminates and controls for the multiple reflex vascular and neurohormonal responses to models of pressure/volume overload.
How Does Stretch Remodel Cx43?
The decrease in Cx43 quantity caused by stretch might result from a decrease in synthesis, an increase in breakdown or a decrease in translocation to the membrane triggered by the stretch. Since the turnover of Cx43 in adult myocardium is relatively rapid (half life of ~2h )(16) any of these mechanisms or a combination might explain the findings of the present study, but our experiments do not distinguish between mechanisms. Mechanotransduction processes have been described in ventricular myocardium in vitro, whereby a mechanical stimulus can result in changes of gene expression(17) and activation of signaling pathways(1), and localized stretch of adult myocardium in vivo can result in the upregulation of genes such as c-fos and c-myc which may be involved in the hypertrophic response(10). The effects of stretch on pathways involved in formation, breakdown or translocation of Cx43 in adult myocardium are unknown. In disease states, such as human heart failure(18), and in corresponding animal models, there is evidence indicating reduced formation of Cx43(18;19) and increased breakdown(20). There is evidence that changes in phosphorylation may also play a role in regulating Cx43 turnover(21).
That the remodeling of Cx43 was prevented by olmesartan, implicates angiotensin II in the stretch induced changes. In vivo studies have demonstrated that mRNA expression of cardiac angiotensinogen(22;23), angiotensin converting enzyme (ACE) (24;25) and Ang II type 1 and type 2 receptors (AT1 and AT2, respectively)(26;27) are upregulated in response to pressure overload or after myocardial infarction in various animal species.
These in vivo studies have been unable to distinguish between systemic and potential paracrine effects(28). The present study indicates that local (paracrine) release of angiotensin II can cause Cx43 remodeling, but the mechanism by which angiotensin II is related to Cx43 remodeling in our study is unknown.
How Does Stretched Induced Remodeling of Cx43 Influence Conduction Velocity
We found a decrease in transverse but not longitudinal conduction velocity in our experiments. The decrease in Cx43 in our experiments (13%) was modest and might not be expected to cause a decrease in velocity based on prior modeling studies. However, there are unlikely to be other changes that decrease velocity specifically in the transverse direction. For example, if there were a decrease in sodium current caused by stretch, a decrease in longitudinal velocity would also be expected. The significant increase in Cx43 on lateral membranes might be expected to increase conduction velocity through improved transverse coupling. This assumption is based on the supposition that the Cx43 in the lateral membranes forms functional gap junctions. However, in a canine model of lateralization of Cx43 in the epicardial border zone of myocardial infarcts in which transverse conduction is also slowed(13) a corresponding increase in lateral gap junctions on electron microscopic examination was not seen, leading to the conclusion that the lateral Cx43 did not form morphologically distinct intact gap junctions(29;30), and that the decrease in transverse velocity was attributed to the decrease in total Cx43. Modelling studies have indicated that modest decreases in myocardial gap-junctional coupling here are likely to have a greater affect on transverse than longitudinal propagation(31). This differential effect on transverse conduction is confirmed by studies with gap junctional blockers(32) and in animal models with selectively reduced Cx43 expression, particularly it seems in the right ventricle(33). The same authors found that in senescent mice with reduced Cx43 and increased fibrosis there was an effect only on transverse conduction velocity in the right ventricle(34). They speculated whether this may have been secondary to the transmural nature of fibrosis in the right ventricle combined with the reduction in Cx43.
Stretch Effects on Neonatal Myocardium; Differences from Adult Myocardium
Our results are different from those described in studies on neonatal myocytes in tissue culture, where there are marked differences from the adult myocyte genotype and phenotype.
Upregulation of Cx43 protein determined by Western blotting was reported after 4 hours of 20% pulsatile stretch of neonatal rat myocytes(35). Both pulsatile and nonpulsatile stretch (1-6 hours) caused an increase in Cx43 immunoreactive signal assumed to represent gap junction formation, and an increase in conduction velocity (8).
The underlying mechanisms for these effects of stretch in neonatal myocytes in culture have been studied in detail, including the effects of stretch on patterns of gene expression, and signaling pathways. Multiple MAPK family members, including p44/p42 MAPKs, stress-activated protein kinase (p38 MAPK), and focal adhesion kinase [p125(FAK)], are all activated by pulsatile stretch(36) . The effects of stretch to increase Cx43 were mimicked by exposure of the cultures to angiotensin(37). Angiotensin is released by stretch of neonatal myocytes in culture(28) and the concept of a paracrine effect on Cx43 levels of this locally released angiotensin is further supported by the observation that the angiotensin II receptor blocker losartan attenuates the increased Cx43 expression, with a similar weight of evidence to that supporting interpretation of the present study, but with opposite direction of change(38). Neonatal myocytes might be expected to respond to stretch differently than adult myocytes. Differences between neonatal and adult myocytes in mechanisms for transduction of mechanical stimuli to activation of signaling pathways, factors controlling protein synthesis, and effects of angiotensin II on protein synthesis have been described(39). There is significant active synthesis of Cx43; Cx43 mRNA levels accumulate progressively during early neonatal stages accompanied by a parallel but temporally delayed accumulation of Cx43 protein. Transcriptional regulation is likely to be the major determinant of Cx43 abundance, a possible difference from adult myocytes(40).
In addition, the differences in the physical characteristics and multicellular architecture between neonatal myocytes in culture and adult myocytes in intact myocardium might influence the response to stretch. Neonatal myocytes are more rounded with no anisotropy of structure, unlike the elongated adult myocytes alignment in anisotropic tissue. Cx43 in cultured neonatal myocytes (as in intact neonatal ventricular myocardium) is distributed around the entire cell perimeter rather than being located at the poles of the cells in intercalated disks as in adult myocytes(9;41;42). Therefore, lateralization as part of the remodeling process cannot be investigated. Further, conductance of gap junctions in neonatal myocytes is lower than in adults(43), resulting in slower conduction and confirming an absence of the typical anisotropic conduction patterns of adult myocardium(44). A progressive increase in cellular coupling accompanies postnatal cardiac development that is related both to the increase in Cx43 and to eventual restriction of the Cx43 to the intercalated disk(9).
Significance of Effects of Stretch on Cx43 Remodeling in Cardiac Pathology
Changes in wall stress and stretch of the ventricular myocardial cells occur naturally during beating of the heart and cause increases in muscle length and width during diastole from the filling of the chambers and during systole from the resistance of the afterload. Numerous studies of failing human myocardium have demonstrated downregulation of Cx43 and remodeling of gap junctions(45-47). Mechanical stretch is thought to play an important role in the remodeling of the cardiac phenotype in both myocardial hypertrophy and heart failure. However, a relationship between stretch and Cx43 remodeling in adult myocardium has not previously been shown. Our results are consistent with studies on experimental and clinical cardiac failure where the amount of Cx43 has been shown to be decreased and lateralized and cell to cell coupling is decreased(46;48).
Conclusion
Stretch of the ventricular myocardium is an independent stimulus for Cx43 gap junction remodeling mediated by the RAS. The remodeling resembles that which occurs in heart failure and myocardial infarction. Therefore, stretch is likely to be one of a group of stimuli occurring in multiple cardiac pathologies that contribute to gap junction remodeling.
Acknowledgments
Supported by the British Heart Foundation RG/05/009, by NIHR Biomedical Research Centre (UK) funding, by grants HL 066140 and HL083205 from the Heart, Lung and Blood Institutes, National Institutes of Health and by a grant from Sankyo Pharmaceuticals.
Footnotes
No disclosures.
Reference List
- 1.Sadoshima J, Izumo S. The cellular and molecular response of cardiac myocytes to mechanical stress. Annu Rev Physiol. 1997;59:551–571. doi: 10.1146/annurev.physiol.59.1.551. [DOI] [PubMed] [Google Scholar]
- 2.Uzzaman M, Honjo H, Takagishi Y, Emdad L, Magee AI, Severs NJ, Kodama I. Remodeling of gap junctional coupling in hypertrophied right ventricles of rats with monocrotaline-induced pulmonary hypertension. Circ Res. 2000;86:871–878. doi: 10.1161/01.res.86.8.871. [DOI] [PubMed] [Google Scholar]
- 3.Akar FG, Nass RD, Hahn S, Cingolani E, Shah M, Hesketh GG, DiSilvestre D, Tunin RS, Kass DA, Tomaselli GF. Dynamic changes in conduction velocity and gap junction properties during development of pacing-induced heart failure. Am J Physiol Heart Circ Physiol. 2007;293:H1223–H1230. doi: 10.1152/ajpheart.00079.2007. [DOI] [PubMed] [Google Scholar]
- 4.Poelzing S, Rosenbaum DS. Altered connexin43 expression produces arrhythmia substrate in heart failure. Am J Physiol Heart Circ Physiol. 2004;287:H1762–H1770. doi: 10.1152/ajpheart.00346.2004. [DOI] [PubMed] [Google Scholar]
- 5.Hall DG, Morley GE, Vaidya D, Ard M, Kimball TR, Witt SA, Colbert MC. Early onset heart failure in transgenic mice with dilated cardiomyopathy. Pediatr Res. 2000;48:36–42. doi: 10.1203/00006450-200007000-00009. [DOI] [PubMed] [Google Scholar]
- 6.Sato T, Ohkusa T, Honjo H, Suzuki S, Yoshida MA, Ishiguro YS, Nakagawa H, Yamazaki M, Yano M, Kodama I, Matsuzaki M. Altered expression of connexin43 contributes to the arrhythmogenic substrate during the development of heart failure in cardiomyopathic hamster. Am J Physiol Heart Circ Physiol. 2008;294:H1164–H1173. doi: 10.1152/ajpheart.00960.2007. [DOI] [PubMed] [Google Scholar]
- 7.Qu J, Volpicelli FM, Garcia LI, Sandeep N, Zhang J, Marquez-Rosado L, Lampe PD, Fishman GI. Gap junction remodeling and spironolactone-dependent reverse remodeling in the hypertrophied heart. Circ Res. 2009;104:365–371. doi: 10.1161/CIRCRESAHA.108.184044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhuang J, Yamada KA, Saffitz JE, Kleber AG. Pulsatile stretch remodels cell-to-cell communication in cultured myocytes. Circ Res. 2000;87:316–322. doi: 10.1161/01.res.87.4.316. [DOI] [PubMed] [Google Scholar]
- 9.Peters NS, Severs NJ, Rothery SM, Lincoln C, Yacoub MH, Green CR. Spatiotemporal relation between gap junctions and fascia adherens junctions during postnatal development of human ventricular myocardium. Circulation. 1994;90:713–725. doi: 10.1161/01.cir.90.2.713. [DOI] [PubMed] [Google Scholar]
- 10.Meghji P, Nazir SA, Dick DJ, Bailey ME, Johnson KJ, Lab MJ. Regional workload induced changes in electrophysiology and immediate early gene expression in intact in situ porcine heart. J Mol Cell Cardiol. 1997;29:3147–3155. doi: 10.1006/jmcc.1997.0545. [DOI] [PubMed] [Google Scholar]
- 11.Brousil JA, Burke JM. Olmesartan medoxomil: an angiotensin II-receptor blocker. Clin Ther. 2003;25:1041–1055. doi: 10.1016/s0149-2918(03)80066-8. [DOI] [PubMed] [Google Scholar]
- 12.Ciaccio EJ, Saltman AE, Hernandez OM, Bornholdt RJ, Coromilas J. Multichannel data acquisition system for mapping the electrical activity of the heart. Pacing Clin Electrophysiol. 2005;28:826–838. doi: 10.1111/j.1540-8159.2005.00167.x. [DOI] [PubMed] [Google Scholar]
- 13.Dillon SM, Allessie MA, Ursell PC, Wit AL. Influences of anisotropic tissue structure on reentrant circuits in the epicardial border zone of subacute canine infarcts. Circ Res. 1988;63:182–206. doi: 10.1161/01.res.63.1.182. [DOI] [PubMed] [Google Scholar]
- 14.Coromilas J, Saltman AE, Waldecker B, Dillon SM, Wit AL. Electrophysiological effects of flecainide on anisotropic conduction and reentry in infarcted canine hearts. Circulation. 1995;91:2245–2263. doi: 10.1161/01.cir.91.8.2245. [DOI] [PubMed] [Google Scholar]
- 15.Patel PM, Plotnikov A, Kanagaratnam P, Shvilkin A, Sheehan CT, Xiong W, Danilo P, Rosen MR, Peters NS. Altering ventricular activation remodels gap junction distribution in canine heart. J Cardiovasc Electrophysiol. 2001;12:570–577. doi: 10.1046/j.1540-8167.2001.00570.x. [DOI] [PubMed] [Google Scholar]
- 16.Laing JG, Tadros PN, Green K, Saffitz JE, Beyer EC. Proteolysis of connexin43-containing gap junctions in normal and heat-stressed cardiac myocytes. Cardiovasc Res. 1998;38:711–718. doi: 10.1016/s0008-6363(98)00060-1. [DOI] [PubMed] [Google Scholar]
- 17.Komuro I, Kaida T, Shibazaki Y, Kurabayashi M, Katoh Y, Hoh E, Takaku F, Yazaki Y. Stretching cardiac myocytes stimulates protooncogene expression. J Biol Chem. 1990;265:3595–3598. [PubMed] [Google Scholar]
- 18.Dupont E, Matsushita T, Kaba RA, Vozzi C, Coppen SR, Khan N, Kaprielian R, Yacoub MH, Severs NJ. Altered connexin expression in human congestive heart failure. J Mol Cell Cardiol. 2001;33:359–371. doi: 10.1006/jmcc.2000.1308. [DOI] [PubMed] [Google Scholar]
- 19.Itoh M, Takeishi Y, Nakada S, Miyamoto T, Tsunoda Y, Takahashi H, Kubota I, Tomoike H. Long-term treatment with angiotensin II type 1 receptor antagonist, CV-11974, restores beta-catenin mRNA expression in volume-overloaded rabbit hearts. Heart Vessels. 2002;17:36–41. doi: 10.1007/s003800200040. [DOI] [PubMed] [Google Scholar]
- 20.Jongsma HJ, Wilders R. Gap junctions in cardiovascular disease. Circ Res. 2000;86:1193–1197. doi: 10.1161/01.res.86.12.1193. [DOI] [PubMed] [Google Scholar]
- 21.Solan JL, Lampe PD. Key connexin 43 phosphorylation events regulate the gap junction life cycle. J Membr Biol. 2007;217:35–41. doi: 10.1007/s00232-007-9035-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baker KM, Chernin MI, Wixson SK, Aceto JF. Renin-angiotensin system involvement in pressure-overload cardiac hypertrophy in rats. Am J Physiol. 1990;259:H324–H332. doi: 10.1152/ajpheart.1990.259.2.H324. [DOI] [PubMed] [Google Scholar]
- 23.Lindpaintner K, Lu W, Neidermajer N, Schieffer B, Just H, Ganten D, Drexler H. Selective activation of cardiac angiotensinogen gene expression in post-infarction ventricular remodeling in the rat. J Mol Cell Cardiol. 1993;25:133–143. doi: 10.1006/jmcc.1993.1017. [DOI] [PubMed] [Google Scholar]
- 24.Schunkert H, Dzau VJ, Tang SS, Hirsch AT, Apstein CS, Lorell BH. Increased rat cardiac angiotensin converting enzyme activity and mRNA expression in pressure overload left ventricular hypertrophy. Effects on coronary resistance, contractility, and relaxation. J Clin Invest. 1990;86:1913–1920. doi: 10.1172/JCI114924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirsch AT, Talsness CE, Schunkert H, Paul M, Dzau VJ. Tissue-specific activation of cardiac angiotensin converting enzyme in experimental heart failure. Circ Res. 1991;69:475–482. doi: 10.1161/01.res.69.2.475. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki J, Matsubara H, Urakami M, Inada M. Rat angiotensin II (type 1A) receptor mRNA regulation and subtype expression in myocardial growth and hypertrophy. Circ Res. 1993;73:439–447. doi: 10.1161/01.res.73.3.439. [DOI] [PubMed] [Google Scholar]
- 27.Nio Y, Matsubara H, Murasawa S, Kanasaki M, Inada M. Regulation of gene transcription of angiotensin II receptor subtypes in myocardial infarction. J Clin Invest. 1995;95:46–54. doi: 10.1172/JCI117675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sadoshima J, Xu Y, Slayter HS, Izumo S. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell. 1993;75:977–984. doi: 10.1016/0092-8674(93)90541-w. [DOI] [PubMed] [Google Scholar]
- 29.Peters NS, Coromilas J, Severs NJ, Wit AL. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation. 1997;95:988–996. doi: 10.1161/01.cir.95.4.988. [DOI] [PubMed] [Google Scholar]
- 30.Peters NS. Myocardial gap junction organization in ischemia and infarction. Microsc Res Tech. 1995;31:375–386. doi: 10.1002/jemt.1070310507. [DOI] [PubMed] [Google Scholar]
- 31.Spach MS, Heidlage JF, Dolber PC, Barr RC. Electrophysiological effects of remodeling cardiac gap junctions and cell size: experimental and model studies of normal cardiac growth. Circ Res. 2000;86:302–311. doi: 10.1161/01.res.86.3.302. [DOI] [PubMed] [Google Scholar]
- 32.Dhein S, Hammerath SB. Aspects of the intercellular communication in aged hearts: effects of the gap junction uncoupler palmitoleic acid. Naunyn Schmiedebergs Arch Pharmacol. 2001;364:397–408. doi: 10.1007/s002100100462. [DOI] [PubMed] [Google Scholar]
- 33.van Rijen HV, Eckardt D, Degen J, Theis M, Ott T, Willecke K, Jongsma HJ, Opthof T, de Bakker JM. Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation. 2004;109:1048–1055. doi: 10.1161/01.CIR.0000117402.70689.75. [DOI] [PubMed] [Google Scholar]
- 34.Noorman M, van Rijen HV, van Veen TA, de Bakker JM, Stein M. Differences in distribution of fibrosis in the ventricles underlie dominant arrhythmia vulnerability of the right ventricle in senescent mice. Neth Heart J. 2008;16:356–358. doi: 10.1007/BF03086178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang TL, Tseng YZ, Chang H. Regulation of connexin 43 gene expression by cyclical mechanical stretch in neonatal rat cardiomyocytes. Biochem Biophys Res Commun. 2000;267:551–557. doi: 10.1006/bbrc.1999.1988. [DOI] [PubMed] [Google Scholar]
- 36.Seko Y, Takahashi N, Tobe K, Kadowaki T, Yazaki Y. Pulsatile stretch activates mitogen-activated protein kinase (MAPK) family members and focal adhesion kinase (p125(FAK)) in cultured rat cardiac myocytes. Biochem Biophys Res Commun. 1999;259:8–14. doi: 10.1006/bbrc.1999.0720. [DOI] [PubMed] [Google Scholar]
- 37.Dodge SM, Beardslee MA, Darrow BJ, Green KG, Beyer EC, Saffitz JE. Effects of angiotensin II on expression of the gap junction channel protein connexin43 in neonatal rat ventricular myocytes. J Am Coll Cardiol. 1998;32:800–807. doi: 10.1016/s0735-1097(98)00317-9. [DOI] [PubMed] [Google Scholar]
- 38.Shyu KG, Chen CC, Wang BW, Kuan P. Angiotensin II receptor antagonist blocks the expression of connexin43 induced by cyclical mechanical stretch in cultured neonatal rat cardiac myocytes. J Mol Cell Cardiol. 2001;33:691–698. doi: 10.1006/jmcc.2000.1333. [DOI] [PubMed] [Google Scholar]
- 39.Schluter KD, Piper HM. Regulation of growth in the adult cardiomyocytes. FASEB J. 1999;13(Suppl):S17–S22. doi: 10.1096/fasebj.13.9001.s17. [DOI] [PubMed] [Google Scholar]
- 40.Fishman GI, Hertzberg EL, Spray DC, Leinwand LA. Expression of connexin43 in the developing rat heart. Circ Res. 1991;68:782–787. doi: 10.1161/01.res.68.3.782. [DOI] [PubMed] [Google Scholar]
- 41.Thomas SP, Bircher-Lehmann L, Thomas SA, Zhuang J, Saffitz JE, Kleber AG. Synthetic strands of neonatal mouse cardiac myocytes: structural and electrophysiological properties. Circ Res. 2000;87:467–473. doi: 10.1161/01.res.87.6.467. [DOI] [PubMed] [Google Scholar]
- 42.Angst BD, Khan LU, Severs NJ, Whitely K, Rothery S, Thompson RP, Magee AI, Gourdie RG. Dissociated spatial patterning of gap junctions and cell adhesion junctions during postnatal differentiation of ventricular myocardium. Circ Res. 1997;80:88–94. doi: 10.1161/01.res.80.1.88. [DOI] [PubMed] [Google Scholar]
- 43.Wittenberg BA, White RL, Ginzberg RD, Spray DC. Effect of calcium on the dissociation of the mature rat heart into individual and paired myocytes: electrical properties of cell pairs. Circ Res. 1986;59:143–150. doi: 10.1161/01.res.59.2.143. [DOI] [PubMed] [Google Scholar]
- 44.Litchenberg WH, Norman LW, Holwell AK, Martin KL, Hewett KW, Gourdie RG. The rate and anisotropy of impulse propagation in the postnatal terminal crest are correlated with remodeling of Cx43 gap junction pattern. Cardiovasc Res. 2000;45:379–387. doi: 10.1016/s0008-6363(99)00363-6. [DOI] [PubMed] [Google Scholar]
- 45.Kostin S, Dammer S, Hein S, Klovekorn WP, Bauer EP, Schaper J. Connexin 43 expression and distribution in compensated and decompensated cardiac hypertrophy in patients with aortic stenosis. Cardiovasc Res. 2004;62:426–436. doi: 10.1016/j.cardiores.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 46.Peters NS, Green CR, Poole-Wilson PA, Severs NJ. Reduced content of connexin43 gap junctions in ventricular myocardium from hypertrophied and ischemic human hearts. Circulation. 1993;88:864–875. doi: 10.1161/01.cir.88.3.864. [DOI] [PubMed] [Google Scholar]
- 47.Kaprielian RR, Gunning M, Dupont E, Sheppard MN, Rothery SM, Underwood R, Pennell DJ, Fox K, Pepper J, Poole-Wilson PA, Severs NJ. Downregulation of immunodetectable connexin43 and decreased gap junction size in the pathogenesis of chronic hibernation in the human left ventricle. Circulation. 1998;97:651–660. doi: 10.1161/01.cir.97.7.651. [DOI] [PubMed] [Google Scholar]
- 48.Cooklin M, Wallis WR, Sheridan DJ, Fry CH. Changes in cell-to-cell electrical coupling associated with left ventricular hypertrophy. Circ Res. 1997;80:765–771. doi: 10.1161/01.res.80.6.765. [DOI] [PubMed] [Google Scholar]