

Abstract

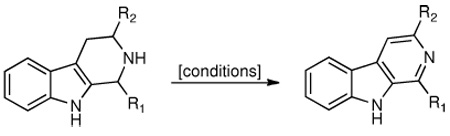
2-Iodoxybenzoic acid (IBX) is a convenient reagent for the dehydrogenation of tetrahydro-β-carbolines to their aromatic forms under mild conditions. The utility of the method was demonstrated in a total synthesis of the marine indole alkaloid eudistomin U.
The aromatic β-carboline moiety is found in numerous natural products and synthetic congeners.1 Compounds bearing this ring system display a diverse range of biological properties including antimalarial,2 antitumor,3 and anti-HIV activities.4 Others show potent binding affinities toward benzodiazepine receptors in the central nervous system, thereby acting as diazepam antagonists.5 In light of these pharmacological properties, mild synthetic methods for the construction of the β-carboline unit are desirable. One strategy for its preparation centers on the formal dehydrogenation of a suitable tetrahydro-β-carboline precursor. Transformations of this type have been previously conducted by heating the substrate with palladium on carbon6 or sulfur7 in refluxing cumene or xylenes over extended periods of time. Other oxidizing agents such SeO28 and MnO29 also require high temperatures and must often be used in excess. Organic-based reagents capable of effecting the dehydrogenation are limited to quinone-derived reagents such as chloranil10 and DDQ11 and yields are often unsatisfactory. Trichloroisocyanuric acid (TCCA) has recently been identified as an additional oxidant.12
We were prompted to seek a mild set of conditions for the aromatization of tetrahydro-β-carbolines during our studies in alkaloid synthesis. Aiming to improve upon existing methodologies, we desired a process that would employ an inexpensive oxidant and proceed smoothly at ambient temperature. In this Letter, we describe a new method to achieve this transformation and demonstrate its utility in a total synthesis of the marine indole alkaloid eudistomin U.
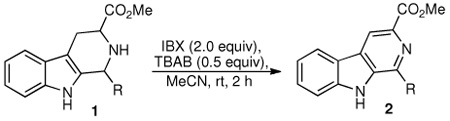
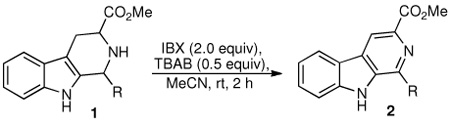
Drawn to examples by Nicolaou and co-workers of iodine(V)-mediated syntheses of pyridines from N-heterocyclic precursors,13 our attention turned to hypervalent iodine reagents such as the Dess–Martin periodinane and 2-iodoxybenzoic acid (IBX).14 A survey of conditions revealed that each could effect the dehydrogenation, though a large excess of the former reagent was required to achieve comparable yields (Table 1, entries 1 and 2). Placement of an ester group at C(3) led to shorter reaction times (cf. Table 1, entries 3 and 4). In each case, however, amounts of partially oxidized 3,4-dihydro-β-carboline intermediates (~30%) were observed by 1H HMR analysis of the crude reaction mixtures. Gratifyingly, addition of tetrabutylammonium bromide (TBAB)15 with acetonitrile as the solvent led to enhanced conversions (Table 1, entries 5 and 6). Under these conditions, the desired products could be obtained in good yields within two hours. Overall, the presence of the ester function at C(3) was important for good conversions; substrates without this group either did not oxidize completely (entry 7) or were mostly inert (entry 8) toward the reaction conditions.
Table 1.
Optimization studies on the IBX-mediated aromatization of tetrahydro-β-carbolines
 | |||
|---|---|---|---|
| entry | R1/R2 | reaction conditionsa | yield (%)b |
| 1 | Ph/H | DMP (5.5 equiv), CH2Cl2, rt, 13 h |
70% |
| 2 | Ph/H | IBX (2.5 equiv), DMSO, rt, 24 h |
60% |
| 3 | Ph/H | IBX (2.5 equiv), DMSO, 45 °C, 9 h |
50% |
| 4 | Ph/CO2Me | IBX (2.0 equiv), DMSO, rt, 2.5 h |
55% |
| 5 | Ph/CO2Me | IBX (2.0 equiv), TBAB (0.5 equiv), MeCN, rt, 2 h |
77% |
| 6 | H/CO2Me | IBX (2.0 equiv), TBAB (0.5 equiv), MeCN, rt, 2 h |
89% |
| 7 | Ph/H | IBX (2.0 equiv), TBAB (0.5 equiv), MeCN, rt, 2 h |
53%c |
| 8 | H/H | IBX (2.0 equiv), TBAB (0.5 equiv), MeCN, rt, 2 h |
<5%d |
DMP = Dess–Martin periodinane, IBX = 2-iodoxybenzoic acid, TBAB = tetrabutylammonium bromide.
Yield determined by 1H NMR analysis of crude reaction mixture.
Product accompanied by 47% of 3,4-dihydro-β-carboline intermediate.
Product accompanied by 30% of 3,4-dihydro-β-carboline.
Following the optimized procedure, the generality of the method was explored. A number of tetrahydro-β-carbolines, obtained by Pictet–Spengler condensation of tryptophan methyl ester with the appropriate aldehyde, were examined (Table 2). Yields were generally good and appeared to be dependent on the electronic characteristics of the substituent at C(1); substrates bearing electron-donating groups (Table 2, entries 2 and 3) affording higher yields than those with electron-withdrawing groups (Table 2, entries 4 and 5). Overall, the conditions proved to be tolerant of aromatic functional groups and did not require protection of the indole nitrogen. We note that these mild conditions offer some advantages over those previously described for the dehydrogenation of a few of these substrates, particularly in regard to reaction time and temperatures. For example, aromatization of 1b to β-carboline 2b (Table 2, entry 2) was previously reported to occur in 73% yield after exposure to sulfur in refluxing xylenes for 48 h,16 while a 77% yield was obtained using selenium dioxide in refluxing dioxane.17 Similarly, β-carboline 2a (Table 2, entry 1) has been previously obtained from 1a in 75% yield upon treatment with sulfur in xylenes for 6 h,17 and in 80% yield employing Pd/C in xylenes after 43 h.7a
Table 2.
IBX-mediated oxidation of tetrahydro-β-carbolinesa
 | |||
|---|---|---|---|
| entry | substrate | product | yield (%)b |
| 1 | 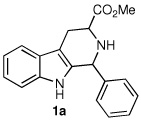 |
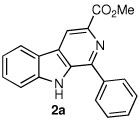 |
77% |
| 2 | 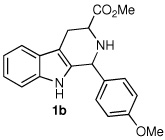 |
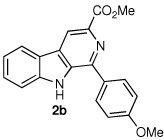 |
86% |
| 3 | 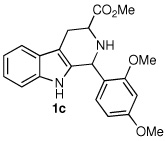 |
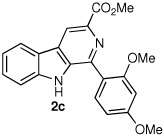 |
82% |
| 4 | 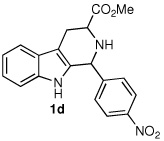 |
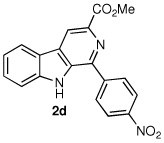 |
58% |
| 5 | 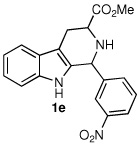 |
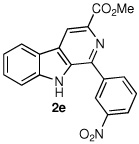 |
61% |
Reaction conditions: substrate (0.5 mmol), IBX (1.0 mmol), TBAB (0.25 mmol), MeCN (5 mL), rt, 2 h.
Isolated, purified yields after flash column chromatography.
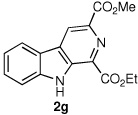
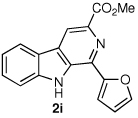
We next turned our attention to substrates bearing aliphatic or heteroaromatic substituents at C(1), as these systems would serve to further extend the scope of the methodology and allow access to a variety of interesting β-carboline scaffolds. Oxidation of 1f proceeded in good yield, as did 1g having two ester moieties (Table 3, entries 1 and 2). Oxidation of 1h, a known ligand for the benzodiazepine receptor bearing a 3-pyridinyl moiety,7a provided β-carboline 2h in 91% yield (Table 3, entry 3) and represents an improvement over the 77% yield reportedly obtained using selenium dioxide (9.4 equiv) in refluxing acetic acid.8b Aromatization of 1i, a structural analogue of the anti-HIV agent flazin,4a,18 was equally facile (Table 3, entry 4). When bis-indole 1j was subjected to the reaction conditions, β-carboline 2j was obtained in 71% yield (Table 3, entry 5).
Table 3.
Mild aromatization of tetrahydro-β-carboline systemsa
 | |||
|---|---|---|---|
| entry | substrate | product | yield (%)b |
| 1 | 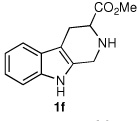 |
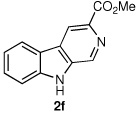 |
89% |
| 2 | 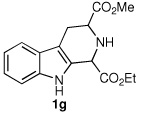 |
 |
80% |
| 3 | 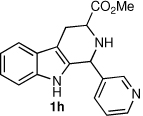 |
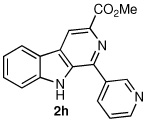 |
91% |
| 4 | 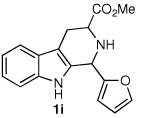 |
 |
80% |
| 5 | 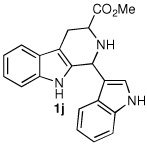 |
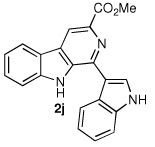 |
71% |
Reaction conditions: substrate (0.5 mmol), IBX (1.0 mmol), TBAB (0.25 mmol), MeCN (5 mL), rt, 2 h.
Isolated, purified yields after flash column chromatography.
The facility by which bis-indole 1j underwent dehydrogenation, together with the structural similarity of β-carboline 2j to the naturally occurring indole alkaloid eudistomin U, prompted us to undertake a total synthesis of the natural product. Isolated from the marine ascidian Lissoclinum fragile, eudistomin U was found to display DNA-binding activity as well as strong antimicrobial properties.19 Application of the IBX-mediated oxidation would provide a direct entry to its unique framework.20 Pictet–Spengler condensation of N-acetyl indole-3-carboxaldehyde21 (3, Scheme 1) and tryptophan methyl ester provided tetrahydro-β-carboline 4 as diastereomers (76% yield).22 Dehydrogenation of 4 under the optimized conditions proceeded smoothly, affording β-carboline 5 in 75% yield. Saponification of the ester moiety was accompanied by loss of the N-acetyl group to provide carboxylic acid 6. Cu/quinoline-mediated decarboxylation of 6 furnished eudistomin U (7) whose spectroscopic data was identical to that of natural material.19
Scheme 1.

Application of the IBX-Mediated Aromatization in the Total Synthesis of Eudistomin U
In summary, we have developed a convenient protocol for the synthesis of aromatic β-carbolines via IBX-mediated dehydrogenation of tetrahydro-β-carboline precursors. The method provides a milder alternative to traditional metal-based reagents and should prove useful in instances when forcing conditions must be avoided. The procedure is especially suited for projected SAR studies and should enable ready access to a diverse array of β-carboline scaffolds for biological evaluation. Finally, the utility of the process was highlighted in a four-step total synthesis of the indole alkaloid eudistomin U. Progress is underway in applying the methodology toward the total synthesis of additional biologically active alkaloid natural products.
Supplementary Material
Acknowledgement
We gratefully acknowledge the University of Vermont for financial support. This work was also generously supported by the Vermont Genetics Network through Grant Number P20 RR16462 from the INBRE Program of the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH). We thank Dr. John Greaves, Director of the Mass Spectrometry Facility at the University of California, Irvine, for obtaining high-resolution mass spectra.
Footnotes
Supporting Information Available: Experimental procedures, characterization data, and NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews on the chemistry and biology of β-carbolines, see: Love BE. Org. Prep. Proceed. Int. 1996;28:3–64. Cao R, Peng W, Wang Z, Xu A. Curr. Med. Chem. 2007;14:479–500. doi: 10.2174/092986707779940998.
- 2.(a) Shilabin AG, Kasanah N, Tekwani BL, Hamann MT. J. Nat. Prod. 2008;71:1218–1221. doi: 10.1021/np800163u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Winkler JD, Londregan AT, Hamann MT. Org. Lett. 2006;8:2591–2594. doi: 10.1021/ol060848d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Boursereau Y, Coldham I. Bioorg. Med. Chem. Lett. 2004;14:5841–5844. doi: 10.1016/j.bmcl.2004.09.036. [DOI] [PubMed] [Google Scholar]
- 3.(a) Guan H, Chen H, Peng W, Ma Y, Cao R, Liu X, Xu A. Eur. J. Med. Chem. 2006;41:1167–1179. doi: 10.1016/j.ejmech.2006.05.004. [DOI] [PubMed] [Google Scholar]; (b) Rashid MA, Gustafson KR, Boyd MR. J. Nat. Prod. 2001;64:1454–1456. doi: 10.1021/np010214+. [DOI] [PubMed] [Google Scholar]; (c) Prinsep MR, Blunt JW, Munro MHG. J. Nat. Prod. 1991;54:1068–1076. doi: 10.1021/np50076a023. [DOI] [PubMed] [Google Scholar]
- 4.(a) Tang JG, Wang YH, Wang RR, Dong ZJ, Yang LM, Zheng YT, Liu JK. Chem. Biodiversity. 2008;5:447–460. doi: 10.1002/cbdv.200890044. [DOI] [PubMed] [Google Scholar]; (b) Wang YH, Tang JG, Wang RR, Yang LM, Dong ZJ, Du L, Shen X, Liu JK, Zheng YT. Biochem. Biophys. Res. Commun. 2007;355:1091–1095. doi: 10.1016/j.bbrc.2007.02.081. [DOI] [PubMed] [Google Scholar]; (c) Yu X, Lin W, Li J, Yang M. Bioorg. Med. Chem. Lett. 2004;14:3127–3130. doi: 10.1016/j.bmcl.2004.04.022. [DOI] [PubMed] [Google Scholar]
- 5.(a) Hagen TJ, Skolnick P, Cook JM. J. Med. Chem. 1987;30:750–753. doi: 10.1021/jm00387a033. [DOI] [PubMed] [Google Scholar]; (b) Hagen TJ, Guzman F, Schultz C, Cook JM, Skolnick P, Shannon HE. Heterocycles. 1986;10:2845–2855. [Google Scholar]; (c) Müller WE, Fehske KJ, Borbe HO, Wollert U, Nanz C, Rommelspacher H. Pharmacol. Biochem. Behav. 1981;14:693–699. doi: 10.1016/0091-3057(81)90133-7. [DOI] [PubMed] [Google Scholar]
- 6.(a) Soerens D, Sandrin J, Ungemach F, Mokry P, Wu GS, Yamanaka E, Hutchins L, DiPierro M, Cook JM. J. Org. Chem. 1979;44:535–545. [Google Scholar]; (b) Hibino S, Miko O, Masataka I, Kohichi S, Takashi I. Heterocycles. 1985;23:261–264. [Google Scholar]; (c) Coutts RT, Micetich RG, Baker GB, Benderly A, Dewhurst T, Hall TW, Locock AR, Pyrozko J. Heterocycles. 1984;22:131–142. [Google Scholar]
- 7.(a) Cain M, Weber RW, Guzman F, Cook JM, Barker SA, Rice KC, Crawley JN, Paul SM, Skolnick P. J. Med. Chem. 1982;25:1081–1091. doi: 10.1021/jm00351a015. [DOI] [PubMed] [Google Scholar]; (b) Still IJW, McNulty J. Heterocycles. 1989;29:2057–2059. [Google Scholar]; (c) Qifeng W, Rihui C, Manxiu F, Xiangdong G, Chunming M, Jinbing L, Huacan S, Wenlie P.Eur. J. Med. Chem 200944533–540.18462839 [Google Scholar]
- 8.(a) Gatta F, Misiti D. J. Heterocycl. Chem. 1987;24:1183–1187. [Google Scholar]; (b) Cain M, Campos O, Guzman F, Cook JM. J. Am. Chem. Soc. 1983;105:907–913. [Google Scholar]; (c) Campos O, DiPierro M, Cain M, Mantei R, Gawish A, Cook JM. Heterocycles. 1980;14:975–984. [Google Scholar]
- 9.(a) Yin W, Srirama Sarma PVV, Ma J, Han D, Chen JL, Cook JM. Tetrahedron Lett. 2005;46:6363–6368. [Google Scholar]; (b) Dantale SB, Söderberg BCG. Tetrahedron. 2003;59:5507–5514. [Google Scholar]
- 10.(a) Lippke KP, Schunack WG, Wenning W, Müller WE. J. Med. Chem. 1983;26:499–503. doi: 10.1021/jm00358a008. [DOI] [PubMed] [Google Scholar]; (b) Snyder HR, Hansch CH, Katz L, Parmerter SM, Spaeth EC. J. Am. Chem. Soc. 1948;70:219–221. doi: 10.1021/ja01181a063. [DOI] [PubMed] [Google Scholar]
- 11.(a) Yeun-Mun C, Hamann MC. Heterocycles. 2007;71:245–252. [Google Scholar]; (b) Kobayashi J, Cheng J, Ohta T, Nozoe S, Ohizumi Y, Sasaki T. J. Org. Chem. 1990;55:3666–3670. [Google Scholar]
- 12.(a) Haffer G, Nickisch K, Tilstam U. Heterocycles. 1998;48:993–998. [Google Scholar]; (b) Tilstam U, Weinmann H. Org. Process Res. Dev. 2002;6:384–393. [Google Scholar]
- 13.(a) Nicolaou KC, Mathison CJN, Montagnon T. Angew. Chem. Int. Ed. 2003;42:4077–4082. doi: 10.1002/anie.200352076. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC, Mathison CJN, Montagnon T. J. Am. Chem. Soc. 2004;126:5192–5201. doi: 10.1021/ja0400382. [DOI] [PubMed] [Google Scholar]
- 14.For reviews, see: Varvoglis A. Hypervalent Iodine in Organic Synthesis. London: Academic Press; 1997. Zhdankin VV, Stang PJ. Chem. Rev. 2002;102:2523–2584. doi: 10.1021/cr010003+. Zhdankin VV. Curr. Org. Synth. 2005;2:121–145. Wirth T. Angew. Chem. Int. Ed. 2005;44:3656–3665. doi: 10.1002/anie.200500115.
- 15.For discussions on the beneficial role of quaternary ammonium salts in other IBX-mediated transformations, see: Fontaine P, Chiaroni A, Masson G, Zhu J. Org. Lett. 2008;10:1509–1512. doi: 10.1021/ol800199b. Drouet F, Fontaine P, Masson G, Zhu J. Synthesis. 2009:1370–1374. Shukla VG, Salgaonkar PD, Akamanchi KG. J. Org. Chem. 2003;68:5422–5425. doi: 10.1021/jo034483b. Bhalerao DS, Mahajan US, Chaudhari KH, Akamanchi KG. J. Org. Chem. 2007;72:662–665. doi: 10.1021/jo0619074.
- 16.Nazari Formagio AS, Santos PR, Zanoli K, Ueda-Nakamura KT, Düsman Tonin LT, Nakamura CV, Sarragiotto MH. Eur. J. Med. Chem. 2009;44:4695–4701. doi: 10.1016/j.ejmech.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 17.Cao R, Peng W, Chen H, Hou X, Guan H, Chen Q, Ma Y, Xu A. Eur. J. Med. Chem. 2005;40:249–258. doi: 10.1016/j.ejmech.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 18.Nakatsuka SI, Feng BN, Goto T, Kihara K. Tetrahedron Lett. 1986;27:3399–3402. [Google Scholar]
- 19.Badre A, Boulanger A, Abou-Mansour E, Banaigs B, Combaut G, Francisco C. J. Nat. Prod. 1994;57:528–533. doi: 10.1021/np50106a016. [DOI] [PubMed] [Google Scholar]
- 20.For previous syntheses, see: Rocca P, Marsais F, Godard A, Quéguiner G, Adams L, Alo B. Tetrahedron Lett. 1995;36:7085–7088. Molina P, Fresneda PM, García-Zafra S. Tetrahedron Lett. 1995;36:3581–3582.
- 21.Ottoni O, Cruz R, Alves R. Tetrahedron. 1998;54:13915–13928. [Google Scholar]
- 22.The formyl group of indole-3-carboxaldehyde is known to be weakly electrophilic as the compound tends to behave as a vinylogous amide. Use of its N-acetyl congener suppressed this behavior and facilitated the Pictet–Spengler reaction.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.