

Abstract
In a scientific career spanning from 1955–2000, my research focused on phosphoenolpyruvate carboxykinase and glucose-6-phosphatase. Grounded in basic enzymology, and initially pursuing the steady-state rate behavior of isolated preparations of these critically important gluconeogenic enzymes, our key findings were confirmed and extended by in situ enzyme rate experiments exploiting isolated liver perfusions. These efforts culminated in the discovery of the liver cytosolic isozyme of carboxykinase, known today as (GTP)PEPCK-C (EC4.1.1.32) and also revealed a biosynthetic function and multicomponent nature of glucose-6-phosphatase (EC3.1.3.9). Discovery that glucose-6-phosphatase possessed an intrinsically biosynthetic activity, now known as carbamyl-P:glucose phosphotransferase– along with a deeper consideration of the enzyme’s hydrolytic activity as well as the action of liver glucokinase resulted in the evolution of Tuning/Retuning Hypothesis for blood glucose homeostasis in health and disease. This THEN & NOW review shares with the reader the joy and exhilaration of major scientific discovery and also contrasts the methodologies and approaches on which I relied with those currently in use.
Introduction
Many thanks to the Editors of Life Sciences for providing me this opportunity to write this retrospective, which is especially intended for younger scientists, as it touches on the evolution — both conceptually and experimentally — of a scientific theory for blood glucose homeostasis.
I am now ten years retired from my position at the University of North Dakota Medical School, where in 1960 I earned my Ph.D. degree from the (then) Department of Biochemistry, working under the direction of Professor Herbert J. Fromm. My NIH-sponsored pre-doctoral fellowship training was devoted to learning basic enzymology techniques and to developing new kinetic strategies for investigating enzymes, mainly ribitol dehydrogenase (Nordlie and Fromm 1959). I had the great good fortune to be selected as an NIH post-doctoral fellow in the laboratory of Professor Henry A. Lardy at the Institute for Enzyme Research at the University of Wisconsin–Madison. From him, I learned to consider the physiologic and regulatory roles that enzymes play in mammalian tissues. While in Lardy’s laboratory, I made two seminal observations regarding critical gluconeogenic enzymes. I discovered the cytosolic isozyme of liver phosphoenolpyruvate carboxykinase (Nordlie and Lardy 1963). My investigations of hepatic inorganic pyrophosphatases (Nordlie and Lardy, 1961a) led to my identification of the potent biosynthetic function of liver microsomal glucose-6-phosphatase (Nordlie and Arion 1964a) in my first year as a faculty member at North Dakota. As will become apparent, it was this melding of Fromm’s physical chemical perspective and Lardy’s far-reaching physiologic biochemical perspective that guided my entire scientific career.
At the University of North Dakota School of Medicine, I was appointed as the first James J. Hill Research Professor, and until I retired at age 70 in 2000, that same institution remained my base for conducting my research on gluconeogenesis. I also held appointments as the Chester Fritz Distinguished Professor, the William Eugene Cornatzer Professor, and the Chair of Biochemistry and Molecular Biology, the latter for 17 years. I received research support from NIH and NSF throughout my career. The specific aims of one NIH grant, entitled Inorganic Pyrophosphate Phosphotransferase, which was funded continuously for over 35 years, morphed into my life-long investigation of multifunctional glucose-6-phosphatase. In my career, I trained twenty Ph.D. students, sixteen M.S. candidates, and fifteen postdoctoral associates and published more than 130 research papers and reviews dealing with eight enzymes: thetin-homocysteine transmethylase, ribitol dehydrogenase, inorganic pyrophosphatases, glucose-6-phosphate dehydrogenase, glucose dehydrogenase, phosphoenolpyruvate carboxykinase, and glucose-6-phosphatase. Thirty papers appeared in The Journal of Biological Chemistry.
Experimental Tools
At the beginning of my biochemistry research career some fifty-five years ago, the available experimental tools and techniques were quite limited by today’s standards. All high-precision pipetting was tediously accomplished with hand-calibrated Lang-Levy glass-bulb pipettes. For instrumental measurements, we relied on pH meters, Klett colorimeters (which were rather primitive spectrophotometers), preparative and analytical ultracentrifuges, and Warburg manometric respirometers. We also used 14C-, 32P-, and 35S-labeled metabolites. Our biological preparations included (a) partially and a few highly purified enzymes; (b) isolated hepatocytes that were either used intact or after permeabilization, (c) isolated mitochondria, nuclei, microsomes, and cytosol fractions from livers perfused in vivo, and (d) entire rats, mice, rabbits, chickens that had been fed, fasted, made experimentally diabetic, or hormonally deprived or injected. Our most important measured parameters were Km, Vmax, and Ki values obtained from initial reaction velocity measurements. We also relied on accurate measurements of protein concentration.
Enzyme studies
Here, I will first consider briefly, in a THEN and NOW format, my early work with phosphoenolpyruvate carboxykinase, with the remainder of this Review devoted to the most exciting work of my career, that involving multifunctional glucose-6-phosphatase and the Tuning/Retuning of blood glucose concentrations.
Phosphoenolpyruvate carboxykinase
THEN
One fateful day, Professor Lardy suggested I diversify my experimental efforts to include not only inorganic pyrophosphatases (Nordlie and Lardy 1961a) but also phosphoenolpruvate carboxykinase. The latter had been characterized from chicken liver mitochondria by Professor Merton Utter at Case Western Reserve University (Utter and Kurahashi 1953; Hanson 2009). Lardy believed the carboxykinase might be a regulated enzyme in the pathway for gluconeogenesis. I thus developed an assay system for carboxykinase activity and then tried to measure the enzyme in rat liver mitochondria. On several tries, I found no activity, and then decided to do a full sub-cellular distribution study of rat liver homogenates. On one Saturday, the thermostat on the Lourdes refrigerated centrifuge malfunctioned! The preparation was ruined. What to do?? I came back on Sunday, repeated the whole process, and discovered that the cytosol was the sole cellular location of phosphoenolpyruvate carboxykinase in rat liver (Nordlie and Lardy 1963). This is what is now generally termed the cytosolic carboxykinase isozyme, (GTP)PEPCK-C (Hanson 2009). (As a note to graduate students and new post-docs, I would say that persistence occasionally pays off. And it doesn’t hurt to work on Sundays, some times!) Subsequent work, initiated by me and carried forward after my departure by Dr. Earl Shrago and others in the Lardy laboratory, revealed the hormone-sensitivity of PEPCK-C (Shrago et al. 1963). We also published several papers on this topic from North Dakota (Holten and Nordlie 1965; Nordlie et al. 1965), but then decided we couldn’t compete with the world from our remote scientific outpost. Our experience with PEPCK-C, however, would subsequently prove extremely useful as we explored the potential physiologic roles of the biosynthetic activity of glucose-6-phophatase (see below).
NOW
An abundant literature on PEPCK-C has appeared since our initial discovery of the liver cytosolic isozyme, with 136 publications on this isozyme appearing in the interval from 2007 to 2009. Premier among them are the elegant studies of Richard Hanson and his group at Case Western Reserve University, who used (GTP)PEPCK-C in their landmark applications of the tools of molecular genetics and molecular biology to mammalian enzymes (Liu et al. 2008; Hanson 2009; Yang et al. 2009b; Yang et al. 2009a). In very recent and exciting work that successfully introduced (GTP)PEPCK-C into muscle (Hakimi et al. 2007; Hanson and Hakimi 2008), the Hanson group produced a Supermouse that could run for up to 5 km at 20 m/min without stopping, compared to control mice that typically can run for only 0.2 km at the same speed. This work and pioneering study regarding a role for carboxykinase in glyceroneogenesis (Hanson 2009) provided the basis for Dr. Hanson’s recent presentation of the annual R.C. Nordlie Lecture at the University of North Dakota School of Medicine and Health Sciences. Although the bulk of work on this enzyme NOW is in the molecular biology area, a number of excellent recent, largely classical enzymological studies came to our attention (see, e.g. Carlsen et al. 1988; Gallwitz et al. 1988; Wiese et al. 1991; Lambeth et al. 1992; Hanson 2009).
Glucose-6-phosphatase
THEN
As a consequence of my interest in inorganic pyrophosphate (PPi) metabolism in the Lardy laboratory, my group first backed into our studies of glucose-6-phosphatase. One of Lardy’s graduate students had reported an enhancement of the P/O ratio upon inclusion of PPi in incubation mixtures (presumably via PPi-ADP transphosphorylation), and Professor Lardy suggested I try to repeat these experiments. I could not; but I did identify several inorganic pyrophosphatases in rat liver (Nordlie and Lardy 1961a, 1961b). Gale Rafter at Johns Hopkins University reported on the enzymatic formation of glucose-6-P formation, suggesting the reaction: PPi + glucose ⇌ glucose-6-P + Pi. So I looked for this same transphosphorylation activity with my rat liver homogenate preparation, relying on a spectrophotometric assay made possible by the availability in the Lardy laboratory of crystalline glucose-6-P dehydrogenase prepared by Dr. Steve Kuby. I found plenty of PPi-glucose phosphotransferase activity in my rat liver homogenates!
At that juncture, I moved my family and my operations to North Dakota, where on a fateful day, I decided purely on intuition to check rat liver microsomal preparations for PPi-glucose phosphotransferase activity. Hepatic endoplasmic reticulum, from which microsomes are generated, was known to contain glucose-6-phosphatase. Stimulated by earlier observations of others that nonspecific acid and alkaline phosphatases not only can hydrolyze phosphate esters but can also synthesize them (in limited fashion) via transphosphorylation, I examined microsomes for phosphotransferase activities. Low and behold, microsomes were loaded with PPi:glucose phosphotransferase and inorganic pyrophosphatase as well as glucose-6-phosphatase activity (Nordlie and Gehring 1963; Nordlie and Arion 1964a).
This was one of the truly thrilling days of my life as a young experimentalist! A series of ensuing studies confirmed that these hydrolytic and biosynthetic activities were in fact due to a single enzyme — glucose-6-phosphatase! Again, excitement! A telephone call to Professor Lardy settled me down. He wisely suggested that I submit a complete article to The Journal of Biological Chemistry, but only when the work was ready for publication. I nonetheless sent an abstract to FASEB for presentation at their Chicago meetings in 1964, a strategy that proved most prudent, as Dr. Marjorie Stetten had submitted an almost identical abstract for these same meetings. I can thus claim a 15-minute priority regarding this discovery, because our presentation (Nordlie and Arion 1964b) preceded Dr. Stetten’s (Stetten 1964a)! I could later claim a six-month priority, because our full manuscript, then already in press, subsequently appeared in The Journal of Biological Chemistry (Nordlie and Arion 1964a) a full six months before Stetten (1964b). Those using the textbook Principles of Biochemistry by White, Handler, Smith, and Dewitt Stetten told me I was now in good company. Young researchers, take notice!
Some characteristics of the glucose-6-phosphatase system
We have previously reviewed, frequently and in detail, characteristics of this enzyme system, and I refer the interested reader to a selected few (Nordlie 1971, 1974, 1976b; Nordlie and Sukalski 1982; Sukalski and Nordlie 1989; Nordlie et al. 1993b; Foster et al. 1997; Nordlie et al. 1999; Foster and Nordlie 2002). I will therefore zero in on two aspects, dealing with kinetics (ala Fromm) and control (ala Lardy), which truly reflect the major focus of my professional life.
Multiple catalytic activities
In addition to activities with glucose-6-P, PPi, and glucose described above, subsequent studies demonstrated high phosphotransferase activity with carbamyl-P at physiologic pH (Lueck and Nordlie 1970) as well as lesser activities with nucleoside di- and tri-phosphates (Nordlie and Arion 1965; Nordlie et al. 1968; Hanson et al. 1970; Nordlie et al. 1971). Some of these activities are listed in Fig. 1.
Fig. 1.
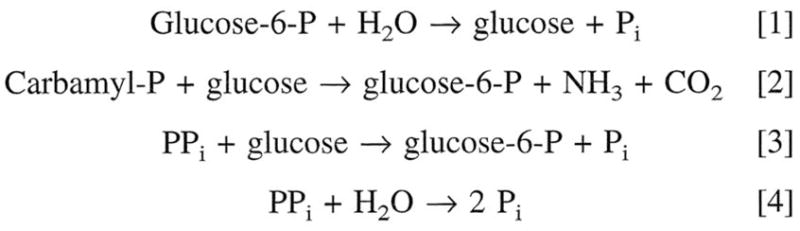
Some hydrolytic and biosynthetic reactions catalyzed by glucose-6-phosphatase (Nordlie and Arion 1964a; Lueck and Nordlie 1970).
Kinetic mechanism
A catalytic mechanism (Arion and Nordlie 1964; Lueck et al. 1972; Nordlie 1974) encompassing all the characterized hydrolytic and phosphotransferase activities of what is now termed the catalytic unit of the glucose-6-phosphatase system is presented in Fig. 2.
Fig. 2.

Kinetic mechanism of multifunctional glucose-6-phosphatase-phosphotransferase. RP is generalized phosphoryl substrate, e.g. PPi or carbamyl-P; E represents enzyme. Further details are given in the text. (From Nordlie 1974 by permission of Academic Press, New York).
Multiple components
The existence of multiple components for the microsomal glucose-6-phosphase system originally developed from observations in our laboratory (Arion and Nordlie 1967) indicating that the microsomal enzyme possessed latency (i.e., activity brought out by disruption of biomembranes (the microsomes) with detergent). Further, the degree of latency was not only dependant on the hormonal or dietary state of the experimental animal, but was uniquely specific to each of the several activities which were measured (Arion and Nordlie 1965, 1967).
Two models have been proposed (Sukalski and Nordlie 1989) to explain the differential, discriminant latency — the substrate translocase-catalytic unit model first proposed by Arion (Arion et al. 1975), and the combined conformational flexibility substrate transport model of Alfred Berteloot and Gerald van de Werve (Berteloot et al. 1995). The former model (Fig. 3) includes the multiplicity of activities inherent in the glucose-6-phosphatase system, and has support from molecular genetics studies (Waddell et al. 1988; Pan et al. 1999). The interested reader is referred also to the intriguing combined conformational flexibility-substrate transport model which is described in detail elsewhere (Sukalski and Nordlie 1989; Berteloot et al. 1995).
Fig. 3.

Structure-function relationships of the glucose-6-phosphatase system. The substrate translocase-catalytic unit model presented here (Foster et al. 1991) is based on earlier concepts of Arion et al. (1975). Depicted is a cross section of the endoplasmic reticulum (E.R. MEMBRANE); SP, stabilizing protein; T1, T2, and T3, substrate/product transporters with the indicated specificity; Catalytic unit, glucose-6-phosphatase (EC 3.1.3.9) enzyme. Two forms of T2 with differing specificity, termed T2α and T2β, have been proposed (Nordlie et al. 1992). (From Foster et al. 1991 by permission of Elsevier Science BV).
Glucose-6-phosphatase and the Tuning/Retuning Hypothesis
In our most recent review, in 1999 (Nordlie et al. 1999), I defined my use of the terminology Tuning/Retuning as follows: The ‘tuning’ of the liver refers to the establishment of the functional levels of the hepatic enzymes glucokinase, Glc-6-P phosphohydrolase, and biosynthetic activities of Glc-6-phosphatase which poise the liver to play a central role (along with other physiologic factors) in the maintenance of blood glucose concentrations at normal ambient values. ‘Retuning’ is used to refer to hormonally or otherwise induced changes in functional levels of these enzymes to repoise or reposition the liver for its still-central role in blood glucose homeostasis, but at an altered post-absorptive glucose concentration as, e.g., in the hyperglycemia of diabetes (Nordlie et al. 1999).
Historical role for glucose-6-phosphatase
In their landmark paper, Cahill et al. (1959) outlined fundamentals of the roles of the liver in regulating blood glucose concentration. A controllable balance between glucose-6-P hydrolysis and glucose phosphorylation was a critical element. Sir Hans Krebs (Krebs 1954; Nordlie 1974) proposed that two enzymes likely were involved. Subsequent studies led to the identification of those enzymes as glucose-6-phosphatase and glucokinase, the latter for glucose phosphorylation and the former for glucose-6-P hydrolysis (see Fig. 4).
Fig. 4.

Classic role of hepatic glucose-6-phosphatase in the regulation of blood glucose concentrations. The charged glucose-6-P molecule is impermeable to the plasma membrane. Net rate and direction of flux of glucose between blood and liver is postulated to be determined by the relative rates of glucose phosphorylation (B) and glucose-6-P hydrolysis (A), as first suggested by Cahill et al. (1959), and more recently elaborated upon by others (see Nordlie 1974). (From Nordlie 1980; by permission from University Park Press, Baltimore).
Genesis of the Tuning/Retuning hypothesis
The concept that I have outlined below first struck me like lightning as I listened to Dr. Sidney Weinhouse, a world-renowned expert on glucokinase, in a guest lecture around 1970 at the University of North Dakota Medical School. I became aware of the insulin-dependence of glucokinase biosynthesis at that time. Later, preparing my lectures for the Medical students, I noted the statement in Professor Albert Lehninger’s classic textbook (Lehninger 1970) that, because of its somewhat high Km for glucose (~10 mM), glucokinase should be especially active in diabetes where hyperglycemia persists. I wrote to Professor Lehninger pointing out the absence of glucokinase in insulin deficiency, and informing him that we had a high-Km,glucose enzymatic activity, a biosynthetic activity of glucose-6-phosphatase, that increased in untreated diabetes. Professor Lehninger subsequently recommended me to serve on the prestigious NIH Biochemistry Study Section. Again, a message for younger biochemists– share your ideas with those you trust and admire, and they will look after your interests as well.
This inadequacy of glucokinase, at least under hyperglycemic, diabetic conditions, along with my feeling that the multiple activities and multiple components of the glucose-6-phosphatase system must be there for some physiologic purpose, prompted me to formulate what I term the Tuning/Retuning Hypothesis (Nordlie 1974, 1976b, 1976a).
Development and publication of the Tuning/Retuning model
The initial genesis of the Tuning/Retuning concept was as above. I then spent many hours of leisure time cogitating thereon, and finally for the first time published the model and supportive data in a volume of Current Topics in Cellular Regulation (Nordlie 1974). I was invited to submit the article by Prof. Bernard Horecker, whom I had gotten to know on his visit to North Dakota as a guest lecturer. I later organized and chaired a session on Carbohydrate Metabolism and It’s Regulation for a national meeting of the American Chemical Society in New York City and invited Prof. Horecker (and others) to present. My own presentation was on glucose-6-phosphatase and contained, for the first time, elements of the Tuning/Retuning concept. An outgrowth of all this was the initial publication of these at-the-time somewhat radical concepts in a first-rate scientific series (Nordlie 1974). I subsequently further developed the concept in a landmark compendium – Hanson and Mehlman’s Gluconeogenesis (Nordlie 1976a)
The Tuning/Retuning model itself
Our working model (Fig. 5) features the incorporation of the biosynthetic function of glucose-6-phosphatase along with its hydrolytic function and glucokinase (and minor contribution by hexokinase) in the regulation of blood glucose by the liver. Incorporation of this activity here seemed to us appropriate because, unlike non-specific phosphatases, the Vmax for the biosynthetic activity of glucose-6-phosphatase catalytic unit exceeds that for glucose-6-P hydrolysis (see Lueck et al. 1972). Consideration of carbamyl-P as a potential active substrate was first prompted by the work of Dr. James Lueck, a Post-doc in my laboratory. He had worked on nitrogen metabolism with Prof. Leon Miller at the University of Rochester as a graduate student, and thought carbamyl-P might be an effective substrate at physiologic pH with the microsomal glucose-6-phosphatase phosphotransferase system. It was (Lueck and Nordlie 1970)! Another great moment in biochemistry!
Fig. 5.

Schematic depiction of interrelationships among liver, blood, and peripheral tissues with respect to glucose flow and utilization. Net rate and direction of flux of glucose between liver and blood is determined by the relative rates of glucose phosphorylation (B) and glucose-6-P hydrolysis (A), as generally postulated by Cahill et al. (1959). The latter of these two processes (A) is catalyzed by hepatic glucose-6-phosphatase, while the former (B) involves hexokinase, glucokinase, and, we propose, transferase activity of glucose-6-phosphatase-phosphotransferase as well. (From Nordlie, 1974; by permission from Academic Press, New York).
Quoting again from our most recent review (Nordlie et al. 1999): Simply stated (Nordlie 1974, 1985; Nordlie et al. 1993a), the Tuning/Retuning hypothesis depends on the differences in Km,glc (Michaelis constant for glucose), for glucokinase (approximately 10 mM) and for phosphotransferase activity of glucose-6-phosphatase (approximately 40 mM at low carbamyl-P concentrations) [as well as on relative levels of activities of the two enzymes]. It follows from kinetic analysis that an increase in the ratio of phosphotransferase activity of glucose-6-phosphatase/glucokinase will contribute to an increase in the post-absorptive steady state blood glucose level. Because glucokinase requires insulin as an inducer and glucose-6-phosphatase increases in diabetes, the ambient level of blood glucose, which the liver is metabolically adjusted to maintain increases progressively with the severity of diabetes.
We believe the concept is best explained here by comparing enzyme activity data applicable with the normal, control rat (Fig. 6A) with that for the diabetic rat (Fig. 6B). Activity levels were calculated for varied, indicated concentrations of glucose for later reference. These two critical illustrations are taken from our original Current Topics in Cellular Regulation article (Nordlie 1974).
Fig. 6.

Activities of several hepatic enzymes, calculated as a function of varied glucose concentrations on the basis of presumed physiologic concentrations of other substrates and further adjusted for inhibition by estimated physiologic concentrations of various inhibitors. These inhibitors include ATP, Pi, HCO3−, and glucose-6-P for carbamyl-P:glucose phosphotransferase; and ATP, Pi, HCO3− for glucose-6-P phosphohydrolase activity. Hexokinase has been adjusted for inhibition by glucose and glucose-6-P. Details of calculation are given elsewhere (Nordlie 1971). Abbreviations employed are: G-6-P HY., glucose-6-P phosphohydrolase; CPGT, cabamyl-P:glucose phosphotransferase; GK, glucokinase; and HK, hexokinase. Vertical arrows designate crossover points, that is, those concentrations of glucose at which rates of glucose-6-P hydrolysis and glucose phosphorylation by the indicated combinations of enzymes, are equal. (From Nordlie 1974; by permission from Academic Press, New York).
In the normal [fed] rat, glucokinase may be the predominant activity for hepatic glucose phosphorylation, with carbamyl-P:glucose phosphotransferase (see Fig. 6A) of lesser importance. Under these conditions, the kinetics of overall hepatic glucose phosphorylation reflect principally the characteristics of glucokinase, the rectangular hyperbola describing such activity considered as a function of varied glucose concentration (see open triangles, Fig. 6A, i.e.) rising fairly rapidly with increasing levels of the hexose substrate.
Consequently such plots intersect with those depicting glucose-6-P hydrolysis at a point corresponding to relatively low levels of glucose (80–90 mg/100 ml in the example presented; see vertical arrows in Fig. 6A). In contrast with the situation for normal rats, as diabetes develops (see Fig. 6B) insulin-dependent glucokinase levels drop precipitously, and glucose-6-P phosphohydrolase and phosphotransferase levels rise considerably. With the latter [now] the predominant mechanism of hepatic glucose phosphorylation, the rectangular hyperbola depicting total rate of hepatic glucose phosphorylation as a function of varied glucose levels (indicated by a star or half shaded circle in Fig. 6B) is flattened [i.e., the radius of curvature is increased] relative to that in the normal situation (compare to Fig. 6A). And as a consequence of this ‘flattening’ and the increase in glucose-6-P phosphohydrolase, the plots for rates of total glucose phosphorylation and glucose-6-P hydrolysis intersect (see vertical arrows in Fig. 6B) at a point corresponding to a glucose concentration that is considerably higher in the diabetic (Fig. 6B) as compared with the normal (Fig. 6A) animal. Thus, control of glucose release, at the hepatic level, is still maintained through the elegant mechanism initially … outline by Cahill et al. (Cahill et al. 1959), but with the substitution of phosphotransferase activity of glucose-6-phophatase in place of insulin-dependent glucokinase as the major glucose phosphorylative activity (Nordlie 1974).
Applicability of the concept
The hypothesis is applicable to rationalize variations [from normal] in blood glucose levels in experimental diabetes, NIDDM, maturity-onset diabetes in the young, von Gierke patients, hyperglycemia of aging, and marked normal variations in blood glucose among various species (e.g., birds, frogs, humans) (Nordlie et al. 1999). Inherent in the hypothesized Tuning/Retuning mechanism are several metabolic regulatory advantages (Nordlie et al. 1993b). It provides for great sensitivity to even small variations in blood glucose levels over a broad range. Rather than being an either/or, on/off mechanism, it provides for a gradation of response of blood glucose concentration. Extent of response of ambient blood glucose concentration is correlated closely with the degree of insulin insufficiency or insulin insensitivity (Nordlie et al. 1999).
Experimental examination of the hypothesis
Some experimental observations that prompted the formulation of the Tuning/Retuning hypothesis have been described above. Further experimental analysis was in order, however. Remember, Albert Einstein formulated his theory of relativity exclusively on hypothetical grounds. It remained for others, who had to wait for a total solar eclipse to experimentally validate this theory that space bends back upon itself. So we set out as a major thrust of our scientific career to make use of available experimental technology (see above) to examine the validity of our Tuning/Retuning hypothesis. This involved a combination of isolated rat liver perfusions, isolated hepatocyte studies, and basic enzyme kinetics. As emphasized below, we believe we, and others, have made many experimental observations supportive of its validity.
Carbamyl-P as a potential hepatic glucose-phosphorylating agent
PPi was the initial credible phosphate substrate for biosynthetic activity of the glucose-6-phosphatase system (see above). However, its reaction was characterized by a rather low pH optimum, so we searched for other substrates that were more reactive near physiologic pH (Lueck and Nordlie 1970). We mentioned above Dr. James Lueck’s success with carbamyl-P as a potential substrate for phosphotransferase activity of the glucose-6-phosphatase system (Lueck and Nordlie 1970; Nordlie et al. 1971). We then tried to demonstrate glucose-6-P synthesis at pH 7.4 with a coupled, reconstituted system including buffer, mitochondria (source of carbamyl-P synthetase I), microsomes (source of glucose-6-phosphatase phosphotransferase), along with glucose and the substrates and activators for carbamyl-P synthesis (Lueck and Nordlie 1972). The system vigorously synthesized glucose-6-P in a manner totally dependent upon carbamyl-P formation. Compounds that inhibited either carbamyl-P synthetase or glucose-6-P phosphotransferase also inhibited glucose phosphorylation. The combined system worked in situ; maybe it might work in vivo as well? More excitement!
Hepatic glucose utilization initiated by a high-Km enzyme other than glucokinase
In 1977, I and Post-doc Fred Alvares (Alvares and Nordlie 1977) initiated studies with isolated, perfused rat livers which continued throughout my career. In this initial, landmark study (see Fig. 7, A and B) we measured glucose utilization as a function of perfusate glucose concentration. We also measured glucokinase and hexokinase activities in homogenates derived from these perforate livers, and with the use of kinetic parameters, calculated the levels of these activities applicable at various glucose concentrations. As shown in Fig. 7B, net glucose utilization rates exceeded those for combined glucokinase + hexokinase rates at and above 30 mM glucose. This disparity between observed net glucose utilization rates and combined kinase rates was even greater with livers from 48-hour fasted rats where glucokinase was much diminished (Fig. 7A). The point of inequality shifted downward to ~3 mM. The inadequacy of glucokinase alone to account for net glucose utilization in perfused livers from rats in various hormonal and metabolic states is elaborated upon by Nordlie and Sukalski (1982).
Fig. 7.

Net glucose uptake rates in livers from 48-hr fasted (A) and fed (B) rats perfused with various concentrations of D-glucose. Initial perfusate glucose concentrations are indicated on the axes of abscissa either as millimolar concentrations (II) or 10−2 × mg/100 ml (I). Closed circles indicate net glucose uptake values ± 1 S.D. (vertical bars). Dashed lines indicate summation of glucokinase plus hexokinase activitiy values calculated for the perfused glucose loads. Further details are given in Alvares and Nordlie 1977 . (Reprinted by permission of the American Society for Biological Chemistry and Molecular Biology).
Uptake of glucose by perfused livers from diabetic rats
Glucokinase essentially disappears in diabetes, insulin being required for induction of glucokinase protein synthesis. None-the-less, isolated perfused livers from experimentally–induced diabetic rats displayed pronounced net glucose uptake, even at glucose concentrations as low as 7 mM, when 3-mercaptopicolinate was included in perfusates (see Fig. 8). At that time, 3-mercaptopicolinate, an established inhibitor of (GTP)PEPCK-C, was included in perfusates to block glucose production via endogenous gluconeogenesis (Di Tullio et al. 1974; Jomain-Baum et al. 1976). More recently (Foster et al. 1994), 3-mercaptopicolinate was demonstrated in our laboratory to inhibit, in situ, the glucose-6-P phosphohydrolase, but not the carbamyl-P:glucose phosphotransferase, activity of glucose-6-phosphatase. Its presence would then facilitate net glucose uptake, as seen, by blocking its reformation via glucose-6-P hydrolysis (Nordlie et al. 1982), as well as inhibiting the indirect pathway (see Nordlie et al. 1982; Katz et al. 1986; Foster et al. 1997).
Fig. 8.
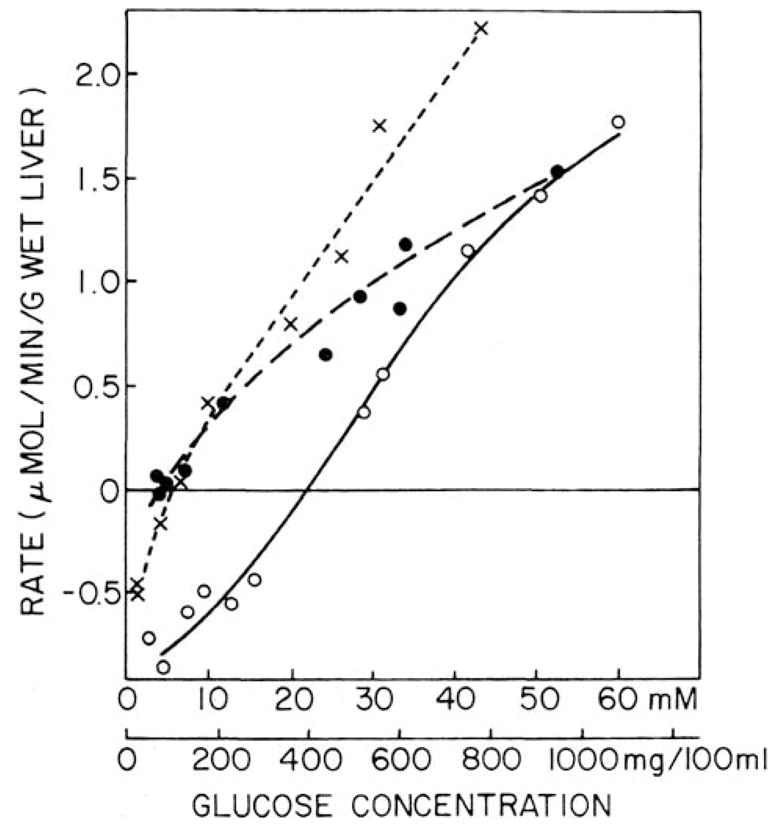
Effects of varied glucose concentrations on net uptake rates in perfused livers from fed control (X) and glucagon-treated alloxan-diabetic rat livers perfused in the absence (O) and presence (□) of 4 mM 3-mercaptopicolinate. Data for diabetic livers are from Tables I and III of Nordlie et al. 1982. Those for controls, included for reference, are from Alvares and Nordlie 1977. Cross-over points from net glucose production to net glucose utilization were observed at 6 mM, 22 mM, and 4 mM glucose with livers from control, diabetic, and mercatopicolinate-supplemented diabetic preparations, respectively. (Reprinted from Nordlie et al. 1982; by permission from Elsevier SV).
Phosphotransferase activities of glucose-6-phosphatase as an hepatic glucose-phosphorylation system?
The above studies establish the need for, and existence of, such an auxiliary high- Km,glucose enzyme for initiating glucose utilization in liver. They do not, however, in themselves establish the identity of such a system. Accordingly, we initiated studies of the glucose-6-phosphatase-phosphotransferase system to examine its possible suitability for this metabolic role.
We first undertook kinetic studies with the multifunctional glucose-6-phosphatase phosphotransferase system to establish if concurrent activities with substrates glucose-6- P and carbamyl-P could occur without serious competition between these phosphate substrates. We then followed these studies, which proved supportive of our hypothesis, with a series of isolated perfused liver studies designed to manipulate selectively, carbamyl-P production, glucokinase activity, individual activities of the glucose-6-phosphatase system, net glucose uptake, glycogenesis from glucose, and ureagenesis.
Carbamyl-P as a credible substrate, along with glucose-6-P, with the glucose-6-phosphatase system
With a multisubstrate enzyme system, such as the glucose-6-phosphatase phosphotransferase, it initially seemed reasonable to suspect that generalized Substrate A (say glucose-6-P) and Substrate B (say carbamyl-P) might be mutually competitive inhibitors, one versus the other (Hue and Hers 1974). On the other hand, especially because of the complexity of the glucose-6-phosphatase phosphotransferase system, this might not be true. The existence (see Fig. 3) of substrate translocases specific for glucose-6-P and carbamyl-P might in part resolve any potential dilemma here.
Because glucose-6-P and carbamyl-P may compete for a common portion of the enzyme catalytic unit’s active site (see Figs 2 and 3), we broached experimentally (Jorgenson and Nordlie 1980; Nordlie and Jorgenson 1981) possible mutual competitive inhibition there, using permeabilized isolated hepatocytes as the enzyme source (Nordlie et al 1984). While glucose-6-P and carbamyl-P do compete at the catalytic unit active site (Jorgenson and Nordlie 1980), the unique kinetics of the system indicate that this is not a problem, from the point of physiologic, metabolic impact. This is because here, where Km values are ≫ physiologic concentrations of substrate, mutual competitive inhibition between these substrate metabolites near physiologic concentration is VERY small, and of little or no metabolic relevance (Nordlie et al. 1984). This is illustrated in Table I. There, with a presumed physiologic concentration of substrate glucose-6-P (0.10 mM) and inhibitor carbamyl-P (0.06 mM), inhibition was only 2.5%. When the substrate concentration was raised to 3.0 or 26 mM and the ratio of [glucose-6-P]/[carbamyl-P] was maintained at 1.67, as before, inhibition increased to 28% and 39%, respectively. Comparable observations were made when carbamyl-P was presumed the substrate and glucose-6-P was the inhibitor (Nordlie et al. 1984).
Table 1.
Inhibition of the glucose-6-P phosphohydrolase reaction by various concentrations of carbamyl-P with the [glucose-6-P]/[carbamyl-P] ratio constant
| Substrate and inhibitor concentrations | Enzymatic activity (μmol/min/g liver) | Inhibition by carbamyl-P (%) |
|---|---|---|
| 0.10 mM Glucose-6-P | ||
| w/o cabamyl-P | 0.681 | |
| Plus 0.06 mM cabamyl-P | 0.664 | 2.5 |
| 3.0 mM Glucose-6-P | ||
| w/o carbamyl-P | 9.87 | |
| plus 1.8 mM carbamyl-P | 7.14 | 27.5 |
| 26.0 mM Glucose-6-P | ||
| w/o carbamyl-P | 16.7 | |
| Plus 15.6 mM carbamyl-P | 10.2 | 39.3 |
Activities, in the absence and presence of inhibitor, were calculated by classical inhibitiion kinetics (Dixon and Webb, 1979). Km for glucose-6-P was 2.6 mM; Ki, for carbamyl-P was 2.2 mM; Vmax for glucose-6-P phosphohydrolase was 18.4 μmol/min/g liver; and the [glucose-6-P]/[carbamyl-P] ratio was maintained constant at 1.67. All kinetic paramenters are from Nordlie and Jorgenson, 1980 . Physiologic concentrations of glucose-6-P (0.10 mM) and carbamyl-P (0.06 mM) are from the literature (see Nordlie and Sukalski, 1983 ). Abstracted from Nordlie et al. 1984.
Are there roles for carbamyl-P:glucose phosphotransferase in hepatic glucose phosphorylation?
Because the above study showed carbamyl-P to be a credible substrate, kinetically, a series of perfusion studies with isolated rat livers was initiated in our laboratory by Dr. Anne Bode and crew to explore carbamyl-P as a credible, potential phosphoryl donor in hepatic glucose metabolism. The strategy here was, by the use of inhibitors, each specific for one or another of the enzymes of carbohydrate metabolism, or for ureagenesis, or both, to gain insight regarding the behavior and function(s) or these enzyme activities in isolated, perfusing livers.
In a series of studies carried out in our laboratory by Dr. Bode and associates, a reciprocal correlation was demonstrated between glycogenesis from glucose, and ureagenesis, in isolated, perfused livers from 48-hour fasted rats. This brought into focus the relative availability of carbamyl-P for the two processes. In one study (Bode and Nordlie 1993), glutamine, a precursor of BOTH ammonium ions and the urea cycle intermediate aspartic acid, was more ureagenic than was proline. This is because the latter provides, via glutamate, EITHER an ammonium ion for carbamyl-P synthesis, OR an aspartate molecule for the urea cycle. Ammonium ion, a necessary precursor of carbamyl-P in liver (Lueck and Nordlie 1972), stimulated both ureagenesis and glycogenesis from glucose (Bode et al. 1994). Norvaline, which inhibits ornithine transcarbamylase, an enzyme of the urea cycle, reduced ureagenesis and increased glycogenesis from glucose (Bode et al. 1994). The carbonic anhydrase inhibitor ethoxyzolamide inhibited bicarbonate production and hence carbamyl-P synthesis. As a consequence, BOTH ureogenesis and glycogenesis from glucose were depressed (Bode et al. 1994). Hepatic glucose uptake and glycogenesis from glucose was lowered by ornithine, while this substrate for ornithine transcarbamylase, a urea-cycle enzyme, increased hepatic ureagenesis (Bode et al. 1992; Bode and Nordlie 1993). This series of critical studies terminated with Post-doc Dr. Bode’s departure for a permanent faculty position. Our feeling, then and now, is that these studies constitute some of the strongest experimental evidence yet to appear supporting the involvement of carbamyl-P:glucose phosphotransferase in hepatic glucose phosphorylation.
Glucose as an inducer of glucose-6-phosphatase catalytic unit protein biosynthesis
Our perfusion work came to a conclusion in 1993. We were excited in 1996 by the findings from two laboratories (Massillon et al. 1996; Argaud et al. 1997) that the rat liver glucose-6-phosphatase gene is up-regulated by glucose. Rossetti (Massillon et al. 1996) points out that …rather than the predicted decrease in glucose-6-phosphatase gene expression, instead prolonged hyperglycemia (as in diabetes) may result in overproduction of glucose via increased expression of the glucose-6-phosphatase protein. We believe that these critical observations provide strong support for our Tuning/Retuning hypothesis. Were glucose-6-P hydrolysis the only function of glucose-6-phophatase, the process of glucose liberation would become autocatalytic. Increased blood glucose would beget the release of still higher quantities of glucose via glucose-6-P phosphohydrolase. Instead, we propose that glucose phosphorylation via the biosynthetic function of the glucose-6-phosphatase system would also become increasingly more active in vivo as blood glucose concentration increased (Foster et al. 1997). Thus, while an increase in blood glucose concentration is ensured when it is most needed (e.g., in diabetes), the progressive replacement of insulin-dependent glucokinase with phosphotransferase activity of the glucose-6-phosphtase system would place an upper cap on the level of glucose that may be reached (Foster et al. 1997).
NOW
Glucose-6-phosphatase continues as an active area of research. We believe that our own work, outlined briefly above, has served as a stimulus. Many general concepts generated out of our observations with this enzyme system remain credible today. Multiple activities; differential latencies of these activities; varied responses of these activities to hormonal manipulations leading to the characterization of the multicomponent system — all serve as bases for ongoing investigations. It is worth noting that the multicomponent nature of glucose-6-phosphatase was hypothesized based on extensive kinetic analysis of the system – a methodology that has lost popularity in recent years but remains a very powerful research tool. Many of the multicomponent aspects of the glucose-6-phosphatase system have been confirmed using the contemporary technique of genetic analysis. Several groups, both industrial (Grempler et al. 2007; Ota et al. 2009; Samuel et al. 2009) and academic/federal (Chou and Mansfield 2007; Wang et al. 2007), are targeting individual components of the glucose-6-phosphatase system as foci for pharmaceutically regulating blood glucose concentration.
The existence of multiple forms of the enzyme’s catalytic unit, termed G6PC, G6PC2, and G6PC3, has been report by Dr. Richard O’Brien and his group in Nashville (Martin et al. 2008; Wang et al. 2008; Hutton and O’Brien 2009). These observations have been extended to include multiple tissue sites and variant functions of the enzyme therein (Martin et al. 2008; Wang et al. 2008; Hutton and O’Brien 2009; Chou et al. 2010). As we proposed in 1997 (Foster et al. 1997), the presence of and metabolic roles for glucose-6-phosphatase in organs other than liver, have now been well documented (Wang et al. 2007; Hutton and O’Brien 2009; Pillot et al. 2009).
The complete structures of both the hepatic catalytic unit and glucose-6-P transporter T1 have been characterized by modern molecular biology techniques by Dr. Janice Chou and associates at NIH (Pan et al. 1999; Shieh et al. 2002). They have applied their techniques to characterize in detail multiple genetic defects in glycogenosis types Ia, Ib, and Ic (Chen et al. 2008; Chou and Mansfield 2008). The possible role of Glycogenosis Type Ia in Sudden Infant Death Syndrome has been suggested (Forsyth et al. 2007). The molecular mechanism underlying the induction by glucose of glucose-6-phosphatase protein has been described (Pedersen et al. 2007).
The above are just a few selected, illustrative examples of ongoing work, much of it involving modern molecular genetics. Developments in recent years are much too extensive for me to review here. However, I am personally delighted, at age 80, with the veritable explosion in the glucose-6-phosphatase field. There is great potential there for clinical applications. My fervent hope is that some young, emergent metabolic group will recognize the potential inherent in the Tuning/Retuning concept described here, and will seize upon it as a research focus.
Epilogue
Some of the great cerebral experiences of my professional life have occurred while I was sitting in my back yard or watching a sunset from my cabin deck at Island Lake, contemplating nature, letting my mind wander, and coming up with some new insight which, remarkably, proved valid on experimental examination in the laboratory (Fig. 9). I still, in the same settings, occasionally intellectually ramble (reminisce), recalling some of those scientific Golden Moments, as I have done here. And life is good (Fig. 10)!
Fig. 9.

The author THEN – 1969
Fig. 10.

The author NOW at Island Lake – August 2009
Acknowledgments
My thanks to John Nordlie for his careful proofreading. Studies from our laboratory cited herein were financed by 35 years of continuous support by NIH grant AM/DK07141, and by smaller grants from NSF, the American and North Dakota Diabetes Associations, the North Dakota VFW Auxiliary, and the Dakota and Minnesota Aeries of Eagles. The senior author expresses his sincere thanks and appreciation to all the many graduate students, Post-docs and lab technicians who made this magical ride possible.
Footnotes
Dedication
This Retro-review is dedicated to my mentors, Professors Herbert J. Fromm and Henry A. Lardy, for nurturing me as a fledgling scientist; to Cathrine Snyder who edited many of my manuscripts; to Sally Nordlie who raised our wonderful family while I did science, and to the late Dr. William E. Cornatzer who gave me the opportunity for scientific self-realization.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alvares FL, Nordlie RC. Quantitative correlation of glucose uptake and phosphorylation with the activities of glucose-phosphorylating enzymes in perfused livers of fasted and fed rats. Journal of Biological Chemistry. 1977;252:8404–8414. [PubMed] [Google Scholar]
- Argaud D, Kirby LT, Newgard CB, Lange AJ. Stimulation of glucose-6-phosphatase gene expression by glucose and fructose-2,6-bisphosphate. Journal of Biological Chemistry. 1997;272 (19):12854–12861. doi: 10.1074/jbc.272.19.12854. [DOI] [PubMed] [Google Scholar]
- Arion WJ, Nordlie RC. Liver microsomal glucose 6-phosphatase, inorganic pyrophosphatase, and pyrophosphate-glucose phosphotransferase ii. Kinetic studies. Journal of Biological Chemistry. 1964;239:2752–2757. [PubMed] [Google Scholar]
- Arion WJ, Nordlie RC. Liver glucose-6-phosphatase and pyrophosphate-glucose phosphotransferase: Effects of fasting. Biochemical and Biophysical Research Communications. 1965;20:606–610. doi: 10.1016/0006-291x(65)90442-0. [DOI] [PubMed] [Google Scholar]
- Arion WJ, Nordlie RC. Biological regulation of inorganic pyrophosphate-glucose phosphotransferase and glucose 6-phosphate; activation by triamcinolone, in vivo, in the presence of actinomycin d. Journal of Biological Chemistry. 1967;242:2207–2210. [PubMed] [Google Scholar]
- Arion WJ, Wallin BK, Lange AJ, Ballas LM. On the involvement of a glucose 6-phosphate transport system in the function of microsomal glucose 6-phosphatase. Molecular and Cellular Biochemistry. 1975;6:75–83. doi: 10.1007/BF01732001. [DOI] [PubMed] [Google Scholar]
- Berteloot A, St-Denis JF, Van de Werve G. Evidence for a membrane exchangeable glucose pool in the functioning of rat liver glucose-6-phosphatase. Journal of Biological Chemistry. 1995;270:21098–21102. [PubMed] [Google Scholar]
- Bode AM, Nordlie RC. Reciprocal effects of proline and glutamine on glycogenesis from glucose and ureagenesis in isolated, perfused rat livers. Journal of Biological Chemistry. 1993;268:16298–16301. [PubMed] [Google Scholar]
- Bode AM, Foster JD, Nordlie RC. Effects of glutamine, proline, and ornithine on hepatic glycogenesis in isolated perfused rat livers. FASEB Journal. 1992;6:A189. [Google Scholar]
- Bode AM, Foster JD, Nordlie RC. Glycogenesis from glucose and ureagenesis in isolated perfused rat livers influence of ammonium ion, norvaline, and ethoxyzolamide. Journal of Biological Chemistry. 1994;269:7879–7886. [PubMed] [Google Scholar]
- Cahill GF, Jr, Ashmore J, Renold AE, Hastings AB. Blood glucose and the liver. American Journal of Medicine. 1959 Feb;:264–282. doi: 10.1016/0002-9343(59)90316-x. [DOI] [PubMed] [Google Scholar]
- Carlsen BD, Lambeth DO, Ray PD. Synthesis of malate from phosphoenolpyruvate by rabbit liver mitochondria: Implications for lipogenesis. Biochimica et Biophysica Acta. 1988;965 (1):1–8. doi: 10.1016/0304-4165(88)90143-2. [DOI] [PubMed] [Google Scholar]
- Chen SY, Pan CJ, Nandigama K, Mansfield BC, Ambudkar SV, Chou JY. The glucose-6-phosphate transporter is a phosphate-linked antiporter deficient in glycogen storage disease type ib and ic. FASEB Journal. 2008;22 (7):2206–2213. doi: 10.1096/fj.07-104851. [DOI] [PubMed] [Google Scholar]
- Chou JY, Mansfield BC. Gene therapy for type i glycogen storage diseases. Current Gene Therapy. 2007;7 (2):79–88. doi: 10.2174/156652307780363152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou JY, Mansfield BC. Mutations in the glucose-6-phosphatase-alpha (g6pc gene that cause type ia glycogen strorage disease. Hum Mutation. 2008;29 (7):921–930. doi: 10.1002/humu.20772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou JY, Jun HS, Mansfield BC. Neutropenia in type Ib glycogen storage disease. Current Opinion in Hematology. 2010;17 (1):36–42. doi: 10.1097/MOH.0b013e328331df85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Tullio NW, Berkoff CE, Blank B, Kostos V, Stack EJ, Saunders HL. 3-mercaptopicolinic acid, an inhibitor of gluconeogenesis. Biochemical Journal. 1974;138:387–394. doi: 10.1042/bj1380387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsyth L, Scott HM, Howatson A, Busuttil A, Burchell A. Genetic variation in hepatic glucose-6-phosphatase system genes in cases of sudden infant death syndrome. Journal of Pathology. 2007;212 (1):112–120. doi: 10.1002/path.2147. [DOI] [PubMed] [Google Scholar]
- Foster JD, Nordlie RC. The biochemistry and molecular biology of the glucose-6-phosphatase system. Experimental Biology and Medicine. 2002;227 (8):601–608. doi: 10.1177/153537020222700807. [DOI] [PubMed] [Google Scholar]
- Foster JD, Bode AM, Nordlie RC. Time-dependent inhibition of glucose-6-phosphatase by 3-mercaptopicolinic acid. Biochimica et Biophysica Acta. 1994;1208(2):222–228. doi: 10.1016/0167-4838(94)90107-4. [DOI] [PubMed] [Google Scholar]
- Foster JD, Pederson BA, Nordlie RC. Glucose-6-phosphatase structure, regulation, and function: An update. Experimental Biology and Medicine. 1997;215:314–332. doi: 10.3181/00379727-215-44142. [DOI] [PubMed] [Google Scholar]
- Foster JD, Nelson KL, Sukalski KA, Lucius RW, Nordlie RC. Hysteretic behavior of the hepatic microsomal glucose-6-phosphatase system. Biochimica et Biophysica Acta. 1991;1118:91–98. doi: 10.1016/0167-4838(91)90445-6. [DOI] [PubMed] [Google Scholar]
- Gallwitz WE, Jacoby GH, Ray PD, Lambeth DO. Purification and characterization of the isozymes of phosphoenolpyruvate carboxykinase from rabbit liver. Biochimica et Biophysica Acta. 1988;964 (1):36–45. doi: 10.1016/0304-4165(88)90064-5. [DOI] [PubMed] [Google Scholar]
- Grempler R, Zibrova D, Schoelch C, van Marle A, Rippmann JF, Redemann N. Normalization of prandial blood glucose and improvement of glucose tolerance by liver-specific inhibition of sh2 domain containing inositol phosphatase 2 (ship2) in diabetic kkay mice: Ship2 inhibition causes insulin-mimetic effects on glycogen metabolism, gluconeogenesis, and glycolysis. Diabetes. 2007;56 (9):2235–2241. doi: 10.2337/db06-1660. [DOI] [PubMed] [Google Scholar]
- Hakimi P, Yang J, Casadesus G, Massillon D, Tolentino-Silva F, Nye CK, Cabrera ME, Hagen DR, Utter CB, Baghdy Y, Johnson DH, Wilson DL, Kirwan JP, Kalhan SC, Hanson RW. Overexpression of the cytosolic form of phosphoenolpyruvate carboxykinase (gtp) in skeletal muscle repatterns energy metabolism in the mouse. Journal of Biological Chemistry. 2007;282 (45):32844–32855. doi: 10.1074/jbc.M706127200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson RW. Thematic minireview series: A perspective on the biology of phosphoenolpyruvate carboxykinase 55 years after its discovery. Journal of Biological Chemistry. 2009;284 (40):27021–27023. doi: 10.1074/jbc.R109.040519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson RW, Hakimi P. Born to run; the story of the pepck-cmus mouse. Biochimie. 2008;90 (6):838–842. doi: 10.1016/j.biochi.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson TL, Lueck JD, Horne RN, Nordlie RC. Adenosine 5′-triphosphate-glucose and phosphoenolpyruvate-glucose phosphotransferase activities of liver microsomal glucose 6-phosphatase. Journal of Biological Chemistry. 1970;245:6078–6084. [PubMed] [Google Scholar]
- Holten DD, Nordlie RC. Comparative studies of catalytic properties of guinea pig liver intra-and extra-mitochondrial phosphoenolpyruvate carboxykinases. Biochemistry. 1965;4:723–731. doi: 10.1021/bi00880a018. [DOI] [PubMed] [Google Scholar]
- Hue L, Hers HG. Utile and futile cycles in the liver. Biochemical and Biophysical Research Communications. 1974;58:540–548. doi: 10.1016/s0006-291x(74)80454-7. [DOI] [PubMed] [Google Scholar]
- Hutton JC, O’Brien RM. Glucose-6-phosphatase catalytic subunit gene family. Journal of Biological Chemistry. 2009;284 (43):29241–29245. doi: 10.1074/jbc.R109.025544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jomain-Baum M, Schramm VL, Hanson RW. Mechanism of 3-mercaptopicolinic acid inhibition of hepatic phosphoenolpyruvate carboxykinase (gtp) Journal of Biological Chemistry. 1976;251:37–44. [PubMed] [Google Scholar]
- Jorgenson RA, Nordlie RC. Multifunctional glucose-6-phosphatase studied in permeable isolated hepatocytes. Journal of Biological Chemistry. 1980;255 (12):5907–5915. [PubMed] [Google Scholar]
- Katz J, Kuwajima M, Foster DW, McGarry JD. The glucose paradox: New perspectives on hepatic carbohydrate metabolism. Trends in Biochemical Sciences. 1986;11(3):136–140. [Google Scholar]
- Krebs HA. Bulletin of the Johns Hopkins Hospital. 1954;95:19–33. [PubMed] [Google Scholar]
- Lambeth DO, Muhonen WW, Jacoby GH, Ray PD. Factors affecting the manganese and iron activation of the phosphoenolpyruvate carboxykinase isozymes from rabbit. Biochimica et Biophysica Acta. 1992;1156 (1):85–91. doi: 10.1016/0304-4165(92)90100-9. [DOI] [PubMed] [Google Scholar]
- Lehninger AL. Biochemistry: The molecular basis of cell structure and function. New York: Worth Publishers; 1970. [Google Scholar]
- Liu GE, Weirauch MT, Van Tassell CP, Li RW, Sonstegard TS, Matukumalli LK, Hanson RW, Yang J. Identification of conserved regulatory elements in mammalian promoter regions: A case study using the pck1 promoter. Journal of Genomics Proteomics and Bioinformatics. 2008;6 (3–4):129–143. doi: 10.1016/S1672-0229(09)60001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lueck JD, Nordlie RC. Carbamyl phosphate: Glucose phophotransferase activity of hepatic microsomal glucose 6-phosphatase at physiological ph. Biochemical and Biophysical Research Communications. 1970;39:190–196. doi: 10.1016/0006-291x(70)90776-x. [DOI] [PubMed] [Google Scholar]
- Lueck JD, Nordlie RC. The utilization of intramitochondrially generated carbamyl phosphate for microsomal glucose 6-phosphate biosynthesis. FEBS Letters. 1972;20:195–198. doi: 10.1016/0014-5793(72)80792-0. [DOI] [PubMed] [Google Scholar]
- Lueck JD, Herrman JL, Nordlie RC. General kinetic mechanism of microsomal carbamyl phosphate: Glucose phosphotransferase, glucose 6-phosphatase, and other associated activities. Biochemistry. 1972;11:2792–2799. doi: 10.1021/bi00765a010. [DOI] [PubMed] [Google Scholar]
- Martin CC, Flemming BP, Wang Y, Oeser JK, O’Brien RM. Foxa2 and mafa regulate islet-specific glucose-6-phosphatase catalytic subunit-related protein gene expression. Journal of Molecular Endocrinolology. 2008;41 (5):315–328. doi: 10.1677/JME-08-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massillon D, Barzalai N, Chen W, Hu M, Rossetti L. Glucose regulates in vivo glucose-6-phosphatase gene expression in the liver of diabetic rats. Journal of Biolological Chemistry. 1996;271 (17):9871–9874. doi: 10.1074/jbc.271.17.9871. [DOI] [PubMed] [Google Scholar]
- Nordlie RC. The enzymes. Vol. 4. New York: Academic Press; 1971. Glucose-6-phosphatase, hydrolytic and synthetic activities; pp. 543–609. [Google Scholar]
- Nordlie RC. Metabolic regulation by multifunctional glucose-6-phosphatase. Current Topics in Cellular Regulation. 1974;8:33–117. doi: 10.1016/b978-0-12-152808-9.50009-2. [DOI] [PubMed] [Google Scholar]
- Nordlie RC. Multifunctional hepatic glucose-6-phosphatase and the ‘tuning” of blood glucose levels. Trends in Biochemical Sciences. 1976a;1:199–202. [Google Scholar]
- Nordlie RC. Gluconeogenesis. New York: John Wiley & Sons, Inc; 1976b. Glucose-6-phosphatase-phosphotransferase: Roles and regulation in relation to gluconeogenesis; pp. 93–152. [Google Scholar]
- Nordlie RC. Metabolic regulation. Amsterdam: Elsevier Science Publishers; 1985. Fine tuning of blood glucose concentrations; pp. 60–69. [Google Scholar]
- Nordlie RC, Fromm HJ. Ribitol dehydrogenase; ii. Studies on the reaction mechanism. Journal of Biological Chemistry. 1959;234:2523–2531. [PubMed] [Google Scholar]
- Nordlie RC, Lardy HA. Sub-cellular distribution of rat-liver inorganic pyrophosphatase activity. Biochimica et Biophysica Acta. 1961a;50:189–191. doi: 10.1016/0006-3002(61)91083-6. [DOI] [PubMed] [Google Scholar]
- Nordlie RC, Lardy HA. Adenine nucleotide inhibition of mitochondrial inorganic pyrophosphatase activity. Biochimica et Biophysica Acta. 1961b;53:309–323. doi: 10.1016/0006-3002(61)90443-7. [DOI] [PubMed] [Google Scholar]
- Nordlie RC, Gehring AW. Inorganic pyrophosphate hydrolysis by rat-liver microsomes. Biochimica et Biophysica Acta. 1963;77:100–107. doi: 10.1016/0006-3002(63)90472-4. [DOI] [PubMed] [Google Scholar]
- Nordlie RC, Lardy HA. Mammalian liver phosphoenolpyruvate carboxykinase activities. Journal of Biological Chemistry. 1963;238:2259–2263. [PubMed] [Google Scholar]
- Nordlie RC, Arion WJ. Evidence for the common identity of glucose 6-phosphatase, inorganic pyrophosphatase, and pyrophosphate-glucose phosphotransferase. Journal of Biological Chemistry. 1964a;239:1680–1685. [PubMed] [Google Scholar]
- Nordlie RC, Arion WJ. The common identity of inorganic pyrophosphatase, glucose-6-phosphatase, and pyrophosphate-glucose phosphotransferase. Federation Proceedings. 1964b;23:534. [PubMed] [Google Scholar]
- Nordlie RC, Arion WJ. Liver microsomal glucose 6-phosphatase, inorganic pyrophosphatase, and pyrophosphate-glucose phosphotransferase iii. Associated nucleoside triphosphate- and nucleoside diphosphate-glucose phosphotransferase activities. Journal of Biological Chemistry. 1965;240:2155–2164. [PubMed] [Google Scholar]
- Nordlie RC, Jorgenson RA. Latency and inhibitability by metabolites of glucose-6-phosphatase of permeable hepatocytes from fasted and fed rats. Journal of Biological Chemistry. 1981;256:4768–4771. [PubMed] [Google Scholar]
- Nordlie RC, Sukalski KA. Multifunctional glucose-6-phosphatase and regulation of metabolic flux through the glucose = glucose-6-phosphate cycle in liver. In: Lennon D, Stratman FW, Zahlten RN, editors. Biochemistry of metabolic processes. New York: Elsevier; 1982. pp. 125–138. [Google Scholar]
- Nordlie RC, Varricchio FE, Holten DD. Effects of altered hormonal states and fasting on rat-liver mitochondrial phosphoenolpyruvate carboxykinase levels. Biochimica et Biophysica Acta. 1965;97:214–221. doi: 10.1016/0304-4165(65)90085-1. [DOI] [PubMed] [Google Scholar]
- Nordlie RC, Alvares FL, Sukalski KA. Stimulation by 3-mercaptopicolinate of net glucose uptake by perfused livers from diabetic rats. Biochimica et Biophysica Acta. 1982;719:244–250. doi: 10.1016/0304-4165(82)90095-2. [DOI] [PubMed] [Google Scholar]
- Nordlie RC, Sukalski KA, Robbins BL. Some unique kinetic aspects of multifunctional glucose-6-phosphatase. Federation Proceedings. 1984;43:1960. [Google Scholar]
- Nordlie RC, Bode AM, Foster JD. Recent advances in hepatic glucose 6-phosphatase regulation and function. Experimental Biology and Medicine. 1993a;203:274–285. doi: 10.3181/00379727-203-43600. [DOI] [PubMed] [Google Scholar]
- Nordlie RC, Bode AM, Foster JD. Recent advances in hepatic glucose-6-phosphatase regulation and function. Experimental Biology and Medicine. 1993b;203:274–285. doi: 10.3181/00379727-203-43600. [DOI] [PubMed] [Google Scholar]
- Nordlie RC, Foster JD, Lange AJ. Regulation of glucose production by the liver. Annual Review of Nutrition. 1999;19:379–406. doi: 10.1146/annurev.nutr.19.1.379. [DOI] [PubMed] [Google Scholar]
- Nordlie RC, Hanson TL, Johns PT, Lygre DG. Inhibition by nucleotides of liver microsomal glucose-6-phosphatase. Proceedings of the National Academy of Science (USA) 1968;60:590–597. doi: 10.1073/pnas.60.2.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordlie RC, Lueck JD, Hanson TL, Johns PT. The nature of pH discriminant differences in the behavior of various phosphoanhydrides, mixed phosphate anhydrides, and phosphate esters as substrates and inhibitors with microsomal glucose 6-phosphatase. Journal of Biological Chemistry. 1971;246:4807–4812. [PubMed] [Google Scholar]
- Nordlie RC, Scott HM, Waddell ID, Hume R, Burchell A. Analysis of human hepatic microsomal glucose-6-phosphatase in clinical conditions where the T2 pyrophosphate/phosphate transport protein is absent. Biochemical Journal. 1992;281:859–863. doi: 10.1042/bj2810859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota S, Horigome K, Ishii T, Nakai M, Hayashi K, Kawamura T, Kishino A, Taiji M, Kimura T. Metformin suppresses glucose-6-phosphatase expression by a complex i inhibition and ampk activation-independent mechanism. Biochemical and Biophysical Research Communications. 2009;388 (2):311–316. doi: 10.1016/j.bbrc.2009.07.164. [DOI] [PubMed] [Google Scholar]
- Pan CJ, Lin B, Chou JY. Transmembrane topology of human glucose 6-phosphate transporter. Journal of Biological Chemistry. 1999;274:13865–13869. doi: 10.1074/jbc.274.20.13865. [DOI] [PubMed] [Google Scholar]
- Pedersen KB, Zhang P, Doumen C, Charbonnet M, Lu D, Newgard CB, Haycock JW, Lange AJ, Scott DK. The promoter for the gene encoding the catalytic subunit of rat glucose-6-phosphatase contains two distinct glucose-responsive regions. American Journal of Physiology - Endocrinology and Metabolism. 2007;292 (3):E788–801. doi: 10.1152/ajpendo.00510.2006. [DOI] [PubMed] [Google Scholar]
- Pillot B, Soty M, Gautier-Stein A, Zitoun C, Mithieux G. Protein feeding promotes redistribution of endogenous glucose production to the kidney and potentiates its suppression by insulin. Endocrinology. 2009;150 (2):616–624. doi: 10.1210/en.2008-0601. [DOI] [PubMed] [Google Scholar]
- Rafter GW. A pyrophosphatase activity associated with mouse liver particles. Journal of Biological Chemistry. 1958;230:643–648. [PubMed] [Google Scholar]
- Samuel VT, Beddow SA, Iwatsuki T, Zhang XM, Chu X, Still CD, Gerhard GS, Shulman GI. Fasting hyperglycemia is not associated with increased expression of pepck or g6pc in patients with type 2 diabetes. Proceedings of the National Academy of Science (USA) 2009;106 (29):12121–12126. doi: 10.1073/pnas.0812547106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh JJ, Terzioglu M, Hiraiwa H, Marsh J, Pan CJ, Chen LY, Chou JY. The molecular basis of glycogen storage disease type 1a: Structure and function analysis of mutations in glucose-6-phosphatase. Journal of Biological Chemistry. 2002;277:5047–5053. doi: 10.1074/jbc.M110486200. [DOI] [PubMed] [Google Scholar]
- Shrago E, Lardy HA, Nordlie RC, Foster DO. Metabolic and hormonal control of phosphoenolpyruvate carboxykinase and malic enzyme in rat liver. Journal of Biological Chemistry. 1963;238:3188–3192. [PubMed] [Google Scholar]
- Stetten MR. Federation Proceedings. 1964a;23:535. [Google Scholar]
- Stetten MR. Metabolism of inorganic pyrophosphate i. Microsomal inorganic pyrophosphate phosphotransferase of rat liver. Journal of Biological Chemistry. 1964b;239:3576–3583. [PubMed] [Google Scholar]
- Sukalski KA, Nordlie RC. Glucose-6-phosphatase: Two concepts of membrane-function relationship. Advances in Enzymology and Related Areas of Molecular Biology. 1989:93–117. doi: 10.1002/9780470123089.ch3. [DOI] [PubMed] [Google Scholar]
- Utter MF, Kurahashi K. Journal of the American Chemical Society. 1953;75:758. [Google Scholar]
- Waddell ID, Lindsay JG, Burchell A. The identification of T2; the phosphate/pyrophosphate transport protein of the hepatic microsomal glucose-6-phosphatase system. FEBS Letters. 1988;229:179–182. doi: 10.1016/0014-5793(88)80822-6. [DOI] [PubMed] [Google Scholar]
- Wang Y, Martin CC, Oeser JK, Sarkar S, McGuinness OP, Hutton JC, O’Brien RM. Deletion of the gene encoding the islet-specific glucose-6-phosphatase catalytic subunit-related protein autoantigen results in a mild metabolic phenotype. Diabetologia. 2007;50 (4):774–778. doi: 10.1007/s00125-006-0564-1. [DOI] [PubMed] [Google Scholar]
- Wang Y, Flemming BP, Martin CC, Allen SR, Walters J, Oeser JK, Hutton JC, O’Brien RM. Long-range enhancers are required to maintain expression of the autoantigen islet-specific glucose-6-phosphatase catalytic subunit-related protein in adult mouse islets in vivo. Diabetes. 2008;57 (1):133–141. doi: 10.2337/db07-0092. [DOI] [PubMed] [Google Scholar]
- Wiese TJ, Lambeth DO, Ray PD. The intracellular distribution and activities of phosphoenolpyruvate carboxykinase isozymes in various tissues of several mammals and birds. Comparative Biochemistry and Physiology Part B. 1991;100 (2):297–302. doi: 10.1016/0305-0491(91)90378-q. [DOI] [PubMed] [Google Scholar]
- Yang J, Reshef L, Cassuto H, Aleman G, Hanson RW. Aspects of the control of phosphoenolpyruvate carboxykinase gene transcription. Journal of Biological Chemistry. 2009a;284 (40):27031–27035. doi: 10.1074/jbc.R109.040535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Kong X, Martins-Santos ME, Aleman G, Chaco E, Liu GE, Wu SY, Samois D, Hakimi P, Chiang CM, Hanson RW. Activation of sirt1 by resveratrol represses transcription of the gene for the cytosolic form of phosphoenolpyruvate carboxykinase (gtp) by deacetylating hepatic nuclear factor 4alpha. Journal of Biological Chemistry. 2009b;284 (40):27042–27053. doi: 10.1074/jbc.M109.047340. [DOI] [PMC free article] [PubMed] [Google Scholar]