

Abstract
Obstructive sleep apnea (OSA) is a complex disorder that consists of upper airway obstruction, chronic intermittent hypoxia and sleep fragmentation. OSA is well known to be associated with hypoxia, insulin resistance and glucose intolerance, and these factors can occur in the presence or absence of obesity and metabolic syndrome. Although it is well established that insulin resistance, glucose intolerance and obesity occur frequently with non-alcoholic fatty liver disease (NAFLD), it is now becoming apparent that hypoxia might also be important in the development of NAFLD, and it is recognized that there is increased risk of NAFLD with OSA. This review discusses the association between OSA, NAFLD and cardiovascular disease, and describes the potential role of hypoxia in the development of NAFLD with OSA.
Keywords: Sleep apnea syndrome, Hyperlipidemia, Non-alcoholic fatty liver disease, Insulin resistance
INTRODUCTION
Obstructive sleep apnea (OSA) is a condition that affects 1%-4% of the general population and 25%-35% of obese individuals. OSA is more common in men than women and is characterized by loud and frequent snoring, periods of apnea during sleep and excessive day somnolence[1]. Initially, OSA was thought to be due to failure to maintain small upper airway tone, which causes airway collapse and apnea, but recently, unstable ventilatory control and changes in lung volume have been implicated. An OSA disorder is generally defined as five or more apnea-hypopnea episodes per hour of sleep [i.e. the apnea-hypopnea index (AHI)][1,2]. OSA is associated with insulin resistance and hyperlipidemia and both conditions are associated with non-alcoholic fatty liver disease (NAFLD)[3,4]. Importantly, and potentially relevant to OSA, hypoxia is now considered as one of the aggravating factors for development of NAFLD[4], and interestingly, OSA is also regarded as one of the factors that accelerate the progression of NAFLD to non-alcoholic steatohepatitis (NASH)[5].
NAFLD is emerging as an important public health problem across the globe[6]. NAFLD refers to a wide spectrum of liver damage, which ranges from simple steatosis to steatohepatitis, advanced fibrosis, and cirrhosis. NAFLD is strongly associated with insulin resistance and is defined by accumulation of liver fat > 5% per liver weight, in the presence of < 10 g daily alcohol consumption[7]. The diagnosis of NAFLD can be established by ultrasound and can be confirmed by liver biopsy. The characteristic histology of NAFLD resembles that of alcohol-induced liver injury, but occurs in people who consume minimal or no alcohol. NAFLD is regarded as the most common cause of increased liver enzyme concentrations and is associated with type 2 diabetes, obesity and hyperlipidemia[8]. The reported prevalence of obesity with NAFLD varies between 30% and 100%, whereas the prevalence of NAFLD with type 2 diabetes varies between 10% and 75%[7]. In routine clinical practice, most cases of fatty liver disease are attributable to alcohol excess; however, fatty liver disease can also occur in association with a wide range of toxins, drugs, and diseases, such as morbid obesity, cachexia, type 2 diabetes, hyperlipidemia, and after jejuno-ileal bypass surgery. As important risk factors for NAFLD such as obesity and type 2 diabetes are increasing in prevalence this could explain the marked increase in numbers of individuals with NAFLD[9].
NAFLD can progress silently to cirrhosis, portal hypertension, and liver-related death in early adulthood. Importantly, NAFLD is also associated with an increased risk of all-cause death and predicts future cardiovascular disease (CVD) events, independently of age, sex, low-density lipoprotein (LDL)-cholesterol, smoking and the cluster of features of the metabolic syndrome[9]. Currently, there are no sensitive and specific biochemical markers for NAFLD. An increase (or decrease) in alanine aminotransferase (ALT) is often used as a biochemical marker to monitor progression (or amelioration) of NAFLD, despite the fact that ALT concentrations can be misleading and do not reflect the severity or outcome. Mass screening for significant liver injury in patients with NAFLD will be an important medical challenge in the years to come because of the epidemics of obesity and diabetes[10].
We have previously summarized the studies that have shown that NAFLD is associated with an increase in incidence of CVD[7]. Importantly, considerable numbers of studies have shown an increase in incidence of CVD with OSA[11-13]. The subsequent discussion focuses on the association of OSA with hypoxia, insulin resistance and hyperlipidemia, and how ultimately this can lead to NAFLD.
OSA AND FATTY LIVER DISEASE
Experimental studies have shown that OSA can lead to an increase in insulin resistance and an alteration in lipid metabolism and can precipitate NAFLD[14-16]. Savransky et al[14] have exposed lean C57BL/6J mice (n = 15) on a regular chow diet to chronic intermittent hypoxia (CIH) for 12 wk and compared these mice with pair-fed mice, exposed to intermittent air (IA, n = 15). CIH caused liver injury with an increase in serum ALT (224 ± 39 U/ L vs 118 ± 22 U/L in the IA group, P < 0.05). CIH also induced hyperglycemia, lipid peroxidation of liver tissue, and increased activity of nuclear factor (NF)-κB but not an inflammatory response [as tumor necrosis factor (TNF)-α was not detectable], which suggests that CIH induces oxidative stress in the liver. Liver histology shows swelling of hepatocytes, with marked accumulation of glycogen in hepatocytes, but no evidence of hepatic steatosis. CIH greatly exacerbates acetaminophen-induced liver toxicity, which causes fulminant hepatocellular injury[14]. It is therefore likely that in the absence of factors that induce obesity as a primary stressor on the liver, IH per se leads to mild liver injury. The same authors have repeated the same experiment in C57BL/6J mice on a high-fat, high-cholesterol diet, exposed to CIH for 6 mo. CIH caused liver injury with an increase in serum ALT (461 ± 58 U/L vs 103 ± 16 U/L in the control group, P < 0.01) and aspartate aminotransferase (AST) (637 ± 37 U/L vs 175 ± 13 U/L in the control group, P < 0.001). Histology revealed evidence of inflammation and fibrosis in the liver, which was not evident in the control mice. CIH caused marked increases in lipid peroxidation in serum and liver tissue; marked increases in hepatic levels of myeloperoxidase, pro-inflammatory cytokines interleukin (IL)-1β, IL-6, the chemokine macrophage inflammatory protein-2; a trend towards an increase in TNF-α; and an increase in α1 (I)-collagen mRNA[15]. Thus, it is plausible that a high-fat diet that occurs in the presence of hypoxia with OSA promotes NAFLD. Furthermore, in a rat model of NAFLD (a choline-deficient high-fat diet) IH has been shown to induce NASH[16]. The metabolic disorders that predispose patients to NASH include insulin resistance and obesity but the mechanism by which repeated hypoxic events, such as occur in OSA, can lead to the progression of liver disease is unclear. It has been shown that hypoxia decreases insulin sensitivity in mice and might ultimately increase expression of the lipogenic genes sterol-regulatory-element-binding protein-1c (SREBP-1c), peroxisome-proliferator-activated receptor-γ (PPAR-γ), acetyl-CoA carboxylase 1 (ACC1) and acetyl-CoA carboxylase 2 (ACC2). Furthermore, hypoxia also decreases expression of genes that regulate mitochondrial β oxidation [e.g. PPAR-α and carnitine palmitoyltransferase-1 (CPT-1)][17], which suggests that fat oxidation is also inhibited. Therefore, hypoxia can increase lipogenesis and inhibit fat oxidation; both factors that promote fat accumulation and development of NAFLD.
Human studies have shown that OSA is associated with an increase in liver enzymes, and treatment of OSA has been shown to decrease liver enzymes. For example, Chin et al[18] have shown that OSA is associated with an increase in liver enzyme concentrations in 14 of 44 (35%) obese individuals. Furthermore, continuous positive airway pressure (CPAP) therapy decreases concentrations of liver enzymes (ALT and AST). In contrast, in a randomized controlled trial, administration of CPAP for 4 wk had no effect on liver enzymes[19]. In a cohort of morbidly obese patients who required bariatric surgery, OSA was found to be a risk factor for increased liver enzyme concentrations but not for NASH[20]. However, Kallwitz et al[21] have shown that, in obese patients with NAFLD, OSA is associated with elevated ALT levels and a trend toward histological evidence of progressive liver disease. This finding has been endorsed by Mishara et al[22], who have shown that, in 101 patients awaiting bariatric surgery, OSA was a risk factor for progression of NAFLD to NASH. Histopathological evidence from 20 obese individuals has shown that OSA is associated with NASH and insulin resistance[23]. Importantly, in 109 patients with OSA, serum aminotransferase levels were better predicted by markers of oxygen desaturation than by factors traditionally associated with the metabolic syndrome[24]. Markers of hypoxia were correlated significantly with AST and ALT levels, whereas the AHI, body mass index (BMI), blood pressure, fasting glucose, triglyceride, and cholesterol levels did not[24]. Importantly, in obese children, OSA has also been shown to be associated with hepatic steatosis and insulin resistance[25], which suggests that early exposure to relative hypoxia also has a deleterious impact on the liver (Figure 1).
Figure 1.

Obstructive sleep apnea can induce non-alcoholic fatty liver disease through increasing insulin resistance, dyslipidemia and inflammation. The presence of metabolic syndrome and obesity with obstructive sleep apnea (OSA) can aggravate non-alcoholic fatty liver disease (NAFLD). OSA might aggravate NAFLD in the absence of obesity and metabolic syndrome.
OSA AND INSULIN RESISTANCE
There is a strong link between insulin resistance and excessive deposition of triglyceride in hepatocytes, which is the hallmark of NAFLD[26]. Although clinical and experimental studies have shown an association between OSA and insulin resistance, whether CPAP therapy improves insulin resistance remains a controversial issue[27]. In 14 obese individuals with OSA, there were marked increases in leptin, insulin resistance, TNF-α and IL-6, compared with non-apneic obese men. The sleep apnea patients had a significantly greater amount of visceral fat compared to obese controls (P < 0.05) and indexes of sleep disordered breathing were positively correlated with visceral fat, but not with BMI, or total or subcutaneous fat. Furthermore, a greater degree of insulin resistance was observed in the group of apnea patients than in BMI-matched non-apneic controls[28]. This finding suggests that OSA is not only associated with insulin resistance but also with inflammation.
Punjabi et al[29] have shown that the prevalence of sleep-disordered breathing in 150 healthy mildly obese men, without diabetes, ranged from 40% to 60%, and impairment in glucose tolerance was related to severity of oxygen desaturation. For a 4% decrease in oxygen saturation, the associated OR for worsening glucose tolerance was 1.99 (95% CI: 1.11-3.56) after adjusting for percent body fat, BMI, and AHI. Multivariate linear regression analyses revealed that increasing OSA was associated with worsening insulin resistance independent of obesity[29]. Ip et al[30] also have shown that OSA is independently associated with insulin resistance. Furthermore, Meslier et al[31] have carried out a cross-sectional study in 494 patients with OSA and have found that the prevalence of type 2 diabetes was 30% and impaired glucose tolerance was 20%, and importantly, diabetes and BMI were independent predictors of OSA[32]. Mallon et al[33] have shown in a 12-year follow-up study that difficulty maintaining sleep, or short sleep duration (≤ 5 h), was associated with an increased incidence of diabetes in men; whereas in contrast, the Finnish type 2 diabetes survey (FIN-D2D) (a large population study in Finland) has shown that sleep duration of ≤ 6 h, or ≥ 8 h, was independently associated with type 2 diabetes in middle-aged women, but not in men[34]. Taken together, these data suggest that an alteration in the normal sleep pattern increases risk of diabetes in men and women.
Data from experimental animal models have shown that IH is also associated with insulin resistance. In obese mice, short-term IH led to a decrease in blood glucose levels accompanied by a marked increase in serum insulin levels, and intriguingly, this effect was completely abolished by prior leptin infusion. Obese mice exposed to IH for 12 wk developed a time-dependent increase in fasting serum insulin levels (from 3.6 ± 1.1 ng/mL at baseline to 9.8 ± 1.8 ng/mL at wk 12, P < 0.001) and worsening glucose tolerance, consistent with an increase in insulin resistance[35]. However, in lean C57BL/6J mice, exposure to IH for 5 d did not induce the same metabolic changes seen in obese mice[35]. Furthermore, in lean C57BL/6J mice IH induced insulin resistance. This effect was seen during the time of exposure to IH[36]. These data suggest that the presence of obesity or metabolic syndrome (as a first insult) in association with OSA (a second insult) might lead to NAFLD and ultimately NASH. Therefore, it is plausible to suggest that OSA in association with insulin resistance increases risk of type 2 diabetes. Several mechanisms are thought to contribute to the development of insulin resistance with OSA (Figure 2).
Figure 2.

The complex relationship between non-alcoholic fatty liver disease, obstructive sleep apnea and insulin resistance. TNF: Tumor necrosis factor; NAFLD: Non-alcoholic fatty liver disease; PPAR: Peroxisome-proliferator-activated receptor; SREBP-1c: Sterol-regulatory-element-binding protein-1c.
Death of adipose tissue and associated excess release of free fatty acid
Adipose tissue hypoxia (ATH) is a new concept in understanding the pathogenesis of insulin resistance and inflammation in OSA. The concept suggests that inhibition of adipogenesis and triglyceride synthesis by hypoxia might be a mechanism for the increased free fatty acid (FFA) concentrations in obesity that occurs with insulin resistance. Gross obesity might be associated with ATH and adipocyte death. However, the exact cause of adipocyte death in obesity is not known. It has been suggested that gross obesity per se is associated with a reduction in blood flow to adipocytes due to diminished angiogenesis or vasoconstriction[37]. Yin et al [38] have shown that hypoxia might inhibit insulin-induced glucose uptake by reducing concentrations of the insulin-signaling molecules insulin receptor β and insulin receptor substrate-1 in mice. Hypoxia might also stimulate lipolysis and inhibit uptake of FFA in adipocytes, which leads to FFA elevation in the plasma of obese subjects, because the increase in FFA might occur as a result of the inhibitory effect of hypoxia on the fatty acid transporters (FATP1, CD36) and the transcription factor (PPAR-γ)[38]. It is tempting to speculate that OSA might act in synergism with gross obesity to accelerate the process of adipocyte death that could ultimately aggravate the course of insulin resistance.
Inflammation
Numerous studies have shown that OSA is a trigger for inflammation. This might explain the associated increase in insulin resistance, dyslipidemia and hypertension in OSA[36]. NF-κB is the transcription factor that is involved in inflammatory pathways and might be involved in modulating insulin sensitivity[39]. Importantly, NF-κB is also increased not only with OSA but also with obesity and metabolic syndrome. NF-κB is the master regulator of inflammatory process and its activation with hypoxia also leads to activation of TNF-α, IL-1, IL-6, monocyte chemoattractant protein-1, macrophage migration-inhibition factor, inducible nitric oxide synthase and matrix metalloproteinase 9. Some of these mediators are also activated by the hypoxia inducible factor (HIF)[40-43]. TNF-α and IL-1 are known to be increased not only with OSA, but also with obesity and metabolic syndrome[40], and the increase in TNF-α and IL-1 is known to be associated with an increase in insulin resistance[44].
Modulation of transcription factors
The modulation of transcription factors has a crucial role in the development of insulin resistance. Evidence is now emerging that hypoxia stimulates SREBP-1c, which is a positive transcription factor for activity of ACC and fatty acid synthase genes, both genes whose activity can promote development of fatty liver[45]. Hypoxia-induced fatty liver has been shown to be associated with an increase in the expression of SREBP-1c[45]. Insulin-resistant ob/ob mice have increased concentrations of SREBP-1c and also develop spontaneous fatty liver[45]. We have presented evidence that suggests strongly that abnormalities of SREBP-1c function play an important pathogenetic role in contributing to the NAFLD phenotype[46]. PPAR-γ is required for maintenance of insulin sensitivity and lipid metabolism[47,48]. Importantly, hypoxia and the associated increase of NF-κB, TNF-α, IL-1 and IL-6 are all known to inhibit PPAR-γ[49]. Overexpression of hepatic PPAR-γ leads to lipid accumulation and it is suggested as a mechanism for hypoxia-induced fatty liver. Furthermore, PPAR-α is also reduced by hypoxia. PPAR-α is highly expressed in the liver, and animal models deficient in PPAR-α develop NAFLD and insulin resistance[49]. In addition, hypoxia also decreases the expression of mitochondrial fatty acid transporter CPT-1[17], which might decrease fat oxidation and promote lipid accumulation. Hypoxia might also modulate AMP-activated protein kinase through mitochondrial respiration or oxidative stress, and ultimately, this might enhance insulin resistance[50].
Adiponectin
Adiponectin is a cytokine that is produced by adipocytes. Serum levels of adiponectin correlate with systemic insulin sensitivity[51]. A reduction in adiponectin contributes to insulin resistance in obesity. However, it is still not clear why adiponectin concentrations are decreased in obesity[52]. Decreased adiponectin is known to be associated with NAFLD[53] and studies have now shown that hypoxia reduces adiponectin expression in adipocytes[54,55]. In adipose tissue, the inhibitory effect of hypoxia on adiponectin might result in increased expression of inflammatory cytokines[56]. Furthermore, TNF-α has been shown to inhibit adiponectin in adipocytes[56]. Thus, these data suggest that hypoxia might directly inhibit adiponectin expression, directly or indirectly, through TNF-α, although whether the decrease in adiponectin causes NAFLD is still uncertain.
Leptin
Leptin is a hormone that is secreted by adipose tissue and increases with obesity. The main role of leptin is to reduce appetite[57]. OSA is known to be associated with an increase in leptin plasma levels, and the increase in leptin occurs in proportion to the severity of OSA[57]. Therefore, it is likely that adipose tissue hypoxia might in part contribute to the increase in plasma leptin level. HIF-1α is associated with increased leptin level[58]. Despite the increase in plasma leptin in the majority of obese individuals with OSA, there is no improvement in appetite due to leptin resistance associated with excess fats[59-61]. CPAP has been shown to be associated with a decrease in leptin[59-61], which suggests that hypoxia might modulate insulin sensitivity at least in part via changes in leptin concentrations. In contrast, other studies have suggested that hypoxia is associated with a decrease in leptin level[62-64]. Yasumasu et al[62] have shown that hypoxia is associated with a decrease in leptin secretion in cultured rat adipocytes[62]. Furthermore, short-term hypoxia does not affect leptin in humans. Hypoxia for 8 wk in a neonatal animal model was not associated with marked changes in plasma leptin levels[64]. Therefore, further research is needed to establish the impact of hypoxia on leptin.
Mitochondrial dysfunction and endoplasmic reticulum stress
Hypoxia is known to inhibit biogenesis and respiration of the mitochondria[65]. Furthermore, hypoxia might also gradually decrease the number and function of the mitochondria and this could lead to insulin resistance[65]. We have shown that alteration in mitochondrial function is associated with NAFLD[66]. Hypoxia is known to induce endoplasmic reticulum stress and inhibition of this has been found to protect mice against insulin resistance. OSA is thought to induce endoplasmic reticulum stress in obesity[67-69]. An increase in endoplasmic reticulum stress is also associated with NAFLD[70].
OSA AND METABOLIC SYNDROME
NAFLD is regarded as the hepatic component of the metabolic syndrome[71]. In October 2009, a joint interim statement from the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and the International Association for the Study of Obesity was published that defined diagnostic criteria for identifying the presence of the metabolic syndrome, without having to resort to measurements that require sophisticated equipment[72]. Metabolic syndrome was defined by the presence of three of five criteria, including: increased waist circumference, elevated triglycerides, reduced high-density lipoprotein (HDL)-cholesterol levels, elevated blood pressure, and elevated fasting-glucose levels. In this new definition, waist circumference is not an obligate requirement for defining the syndrome and is one of five criteria that physicians can use when diagnosing the metabolic syndrome[72]. Furthermore, Vgontzas et al[73] have shown that sleep apnea patients have a significantly greater amount of visceral fat and insulin resistance compared to obese controls (P < 0.05), and indexes of sleep disordered breathing are positively correlated with visceral fat, but not with BMI or total or subcutaneous fat. This finding has led to the suggestion that OSA should be considered as part of the metabolic syndrome[73]. Coughlin et al[74] have shown that subjects with OSA are more obese, have higher blood pressure and fasting insulin concentrations, are more insulin resistant, and have lower HDL-cholesterol concentrations, which provides evidence that there is increased prevalence of metabolic syndrome (87% vs 35%, P < 0.0001). A regression analysis adjusted for age, BMI, smoking and alcohol consumption, has demonstrated that OSA is independently associated with increased systolic and diastolic blood pressure, higher fasting insulin and triglyceride concentrations, decreased HDL-cholesterol, increased cholesterol:HDL ratios, and a trend towards higher homeostasis model assessment values. Importantly, the authors have concluded that, in individuals with OSA, the prevalence of metabolic syndrome was 9.1 times higher (95% CI: 2.6-31.2, P < 0.0001) than in the general population. These data suggest that OSA is independently associated with an increase in the cardiovascular risk factors, and are supported by the work of Tkacova et al[75] who have shown that severe OSA is associated with an increased incidence of CVD independent of insulin resistance and obesity.
There could be a differing contribution of risk factors for OSA between ethnic groups. In 819 Japanese patients with OSA (719 men and 100 women) and 89 control subjects without OSA, metabolic syndrome was significantly more common in patients with OSA than in the controls (49.5% vs 22.0% for men, P < 0.01; 32.0% vs 6.7% for women, P < 0.01)[70]. Men and women with moderate and severe OSA have a higher risk of metabolic syndrome compared with controls. In men, age, BMI and OSA are significantly associated with metabolic syndrome, whereas, in women, BMI is the only risk factor for metabolic syndrome[76].
In a small study in China, the independent determinants of OSA in men and women were age, sex, BMI and the metabolic syndrome[77]. In another study in the Japanese population with OSA, the concurrent presence of metabolic syndrome constituted an additional cardiovascular risk factor[78]. From the evidence mentioned above, it is possible to conclude that OSA is associated with an increase in risk of CVD in the presence, (or absence), of metabolic syndrome. In addition, when the metabolic syndrome (including NAFLD) occurs in association with OSA, there might be a further increase in risk of CVD. Therefore, the complex relationship between OSA, metabolic syndrome and NAFLD to increase risk of CVD suggests the importance of identifying and treating NAFLD in individuals with metabolic syndrome and OSA.
OSA, NAFLD AND HYPERLIPIDEMIA
NAFLD is not only associated with insulin resistance but also with dyslipidemia[79]. Importantly, in numerous studies, NAFLD has been shown to be associated with an increase in risk of CVD. NAFLD is associated with increased incidence of CVD in type 2 diabetes[80-87]. Furthermore, OSA is associated with significant cardiovascular morbidity and mortality[88]. There is increasing evidence that OSA is associated with dyslipidemia in animal models as well as human studies. Acute exposure to hypoxia increases LDL-cholesterol concentrations but does not influence the concentration of cholesterol and fatty acids in rats[89]. Repeated exposure to hypobaric hypoxia causes a significant increase in the concentration of cholesterol, fatty acids, chylomicron, LDL-cholesterol and very-low-density beta-lipoproteins (VLDL) in rats, whereas the level of HDL-cholesterol decreases[89]. Furthermore, Li et al[90] have shown that, in leptin-deficient obese C57BL/6J-Lep(ob) mice, exposure to IH increases fasting serum levels of total cholesterol, HDL-cholesterol, and triglycerides, as well as liver triglyceride content. These changes are not observed in obese mice, which have hyperlipidemia and fatty liver at baseline. In lean mice, IH increases SREBP-1c levels in the liver, increases mRNA and protein levels of stearoyl-coenzyme A desaturase 1 (SCD-1), an important enzyme that is involved in desaturation of fatty acids, controlled by SREBP-1, and increases monounsaturated fatty acid content in serum, which indicates augmented SCD-1 activity. In addition, in lean mice, IH decreases protein levels of scavenger receptor B1, which regulates uptake of cholesterol esters and HDL by the liver[91]. In mice with a conditional knockout of SREBP cleavage-activating protein (SCAP) in the liver, which exhibits low levels of an active nuclear isoform of SREBP-1c (nSREBP-1c), IH does not have any effect on serum and liver lipids, and expression of lipid metabolic genes is not altered[89]. In wild-type mice, IH increases fasting levels of serum total and HDL-cholesterol, serum triglycerides, serum and liver phospholipids, mRNA levels of SREBP-1c and mitochondrial glycerol-3-phosphate acyltransferase (mtGPAT), and protein levels of SCAP, nSREBP-1, and mtGPAT in the liver. These data suggest that hyperlipidemia in response to IH is mediated in part via the SREBP-1c pathway[91], and we have previously suggested that modulation of SREBP provides a potential treatment of NAFLD[46].
In contrast, Savransky et al[92] have shown that C57BL/6J mice exposed to CIH and a high-cholesterol diet develop dyslipidemia, aortic atherosclerosis, and upregulation of SCD-1. Therefore, inhibition of SCD-1 might have the potential to prevent dyslipidemia and atherosclerosis during OSA. In another study by Savransky et al[93] in mice and in obese humans, C57BL/6J mice were exposed to CIH or normoxia for 10 wk while being treated with SCD-1 or control antisense oligonucleotides. In mice, hypoxia increased hepatic SCD-1 and plasma VLDL levels and induced atherosclerotic lesions in the ascending aorta (the cross-section area of 156 514 ± 57 408 μm2, and descending aorta (7.0% ± 1.2% of the total aortic surface). In mice exposed to CIH and treated with SCD-1 antisense oligonucleotides, dyslipidemia and atherosclerosis in the ascending aorta were abolished, whereas there was a 56% decrease in lesions in the descending aorta. None of the mice exposed to normoxia developed atherosclerosis. Furthermore, Savransky et al[93] have studied obese human subjects who have undergone an intraoperative liver biopsy at the time of bariatric surgery for treatment of sleep apnea and obesity. In these patients, hepatic SCD mRNA levels correlated with the degree of nocturnal hypoxemia (r = 0.68, P = 0.001) and patients who showed oxyhemoglobin desaturation at night showed higher plasma triglyceride and LDL-cholesterol levels, compared to subjects without hypoxemia[93].
Modulation of HIF-1 activity could also be a precipitating factor for dyslipidemia with OSA. HIF-1 is a master transcriptional regulator of genes that are involved in physiological responses to hypoxia, including erythropoiesis, angiogenesis, and glucose metabolism. Li et al[94] have hypothesized that HIF-1 might be involved in dyslipidemia associated with OSA. They have performed a 5-d IH experiment using C57BL/6J (wild-type) or heterozygous Hif1α+/− mice (with partial HIF-1α deficiency). During IH, Hif1α+/− mice experienced blunted rises in serum triglycerides, liver triglycerides, light-phase fasting insulin, and glucose level, and attenuated transcription or translation of several liver lipid biosynthesis enzymes. HIF-1α deficiency diminished the rise of SREBP-1 and SCD-1 protein levels during IH without affecting serum cholesterol[94]. This suggests that, besides obesity, insulin resistance and a high intake of dietary cholesterol, modulation of HIF-1α could represent another factor that mediates hypoxia-induced dyslipidemia. In summary, hypoxia might lead to an increase in plasma and hepatic lipid profile through different factors and this could precipitate fatty liver.
Clinical studies have shown that CPAP is associated with a reduction in cholesterol and C-reactive protein. In two studies, CPAP was associated with a reduction in cholesterol, LDL-cholesterol, C-reactive protein and homocysteine[95,96]. In children with OSA, tonsillectomy improves parameters of the lipid profile such as LDL-cholesterol, apolipoprotein B and HDL-cholesterol[97].
OSA AND NAFLD AND MODULATION OF CVD RISK
Recently, Floras and Bradley have reviewed the association between OSA and CVD[98]. Their conclusion was that OSA is associated with an increased risk of CVD and this has been demonstrated in epidemiological, clinical and physiological studies. Epidemiological studies have shown a significant independent association between OSA and hypertension, coronary artery disease, arrhythmias, heart failure and stroke[99-105]. Although the association between NAFLD and OSA and CVD is not yet fully elucidated, from the evidence presented, it is tempting to postulate that the association between OSA and NAFLD accelerates atherosclerosis development. The complex interaction between OSA and NAFLD, and the fact that they share similar metabolic pathways that are well known to be associated with an increase in the incidence of CVD, suggest the need for clinical trials in this field (Figure 3).
Figure 3.
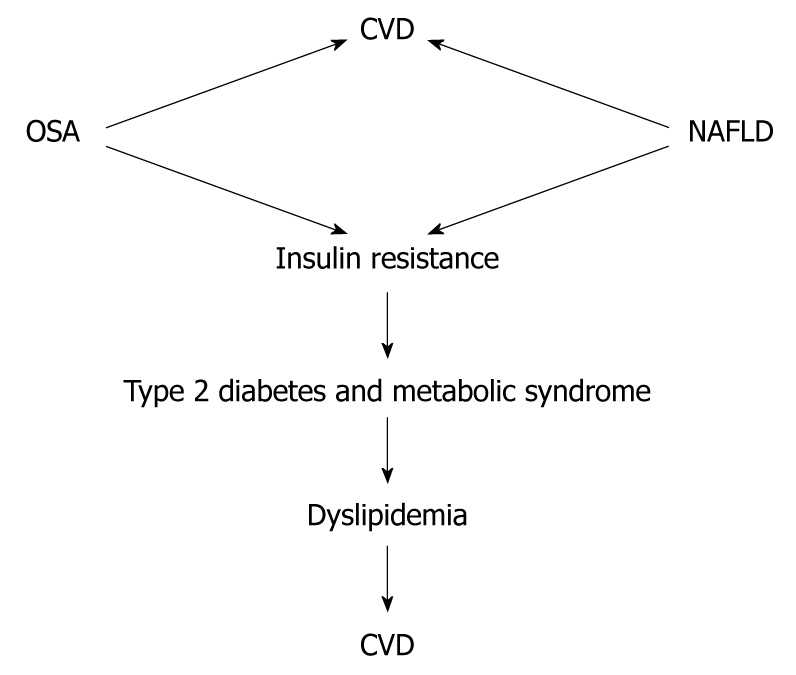
Association of non-alcoholic fatty liver disease and obstructive sleep apnea with cardiovascular disease. Whether the combination of non-alcoholic fatty liver disease (NAFLD) and obstructive sleep apnea (OSA) has a synergistic effect in the incidence of cardiovascular disease (CVD) needs to be demonstrated.
CONCLUSION
OSA is associated with NAFLD in experimental animals and in humans. Importantly, OSA can aggravate the development of NAFLD to NASH in obese individuals or those with metabolic syndrome. OSA might induce NAFLD in the absence of obesity and metabolic syndrome, and the link with hypoxia might be instrumental in precipitating fatty liver development. We suggest that the relationship between CVD, OSA and NAFLD requires further study to elucidate the precise nature of these relationships. Importantly, individuals with OSA require a full evaluation of their CVD risk, and clinicians should be aware that these individuals are also at increased risk of NAFLD.
Acknowledgments
The authors thank Lucinda England for help with the text of the manuscript.
Footnotes
Peer reviewer: Michael Torbenson, MD, Associate Professor of Pathology, Room B314, 1503 E Jefferson (Bond Street Building), The Johns Hopkins University School of Medicine, Baltimore, MD 21231, United States
S- Editor Wang YR L- Editor Kerr C E- Editor Ma WH
References
- 1.Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med. 1993;328:1230–1235. doi: 10.1056/NEJM199304293281704. [DOI] [PubMed] [Google Scholar]
- 2.Sleep-related breathing disorders in adults: recommendations for syndrome definition and measurement techniques in clinical research. The Report of an American Academy of Sleep Medicine Task Force. Sleep. 1999;22:667–689. [PubMed] [Google Scholar]
- 3.Botros N, Concato J, Mohsenin V, Selim B, Doctor K, Yaggi HK. Obstructive sleep apnea as a risk factor for type 2 diabetes. Am J Med. 2009;122:1122–1127. doi: 10.1016/j.amjmed.2009.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Minoguchi K, Yokoe T, Tanaka A, Ohta S, Hirano T, Yoshino G, O’Donnell CP, Adachi M. Association between lipid peroxidation and inflammation in obstructive sleep apnoea. Eur Respir J. 2006;28:378–385. doi: 10.1183/09031936.06.00084905. [DOI] [PubMed] [Google Scholar]
- 5.Byrne CD. Hypoxia and non-alcoholic fatty liver disease. Clin Sci (Lond) 2009;118:397–400. doi: 10.1042/CS20090565. [DOI] [PubMed] [Google Scholar]
- 6.Ahmed MH, Abu EO, Byrne CD. Non-Alcoholic Fatty Liver Disease (NAFLD): New challenge for general practitioners and important burden for health authorities? Prim Care Diabetes. 2010:Epub ahead of print. doi: 10.1016/j.pcd.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 7.Byrne CD, Olufadi R, Bruce KD, Cagampang FR, Ahmed MH. Metabolic disturbances in non-alcoholic fatty liver disease. Clin Sci (Lond) 2009;116:539–564. doi: 10.1042/CS20080253. [DOI] [PubMed] [Google Scholar]
- 8.Ahmed MH, Byrne CD. Non Alcoholic Steatohepatitis and Metabolic syndrome. In: Byrne C, Wild S, editors. Metabolic syndrome. Chichester, UK: John Wiley & Sons; 2005. pp. 279–305. [Google Scholar]
- 9.Ahmed MH, Byrne CD. Metabolic syndrome, diabetes & CHD risk. In: Packard CJ, editor. The Year in Lipid Disorders. Oxford, UK: Clinical Publishing; 2007. pp. 3–26. [Google Scholar]
- 10.Ahmed MH. Biochemical markers: the road map for the diagnosis of nonalcoholic fatty liver disease. Am J Clin Pathol. 2007;127:20–22. doi: 10.1309/JXWUM661T8VT1ETX. [DOI] [PubMed] [Google Scholar]
- 11.Nishibayashi M, Miyamoto M, Miyamoto T, Suzuki K, Hirata K. Correlation between severity of obstructive sleep apnea and prevalence of silent cerebrovascular lesions. J Clin Sleep Med. 2008;4:242–247. [PMC free article] [PubMed] [Google Scholar]
- 12.Dorkova Z, Petrasova D, Molcanyiova A, Popovnakova M, Tkacova R. Effects of continuous positive airway pressure on cardiovascular risk profile in patients with severe obstructive sleep apnea and metabolic syndrome. Chest. 2008;134:686–692. doi: 10.1378/chest.08-0556. [DOI] [PubMed] [Google Scholar]
- 13.Takama N, Kurabayashi M. Influence of untreated sleep-disordered breathing on the long-term prognosis of patients with cardiovascular disease. Am J Cardiol. 2009;103:730–734. doi: 10.1016/j.amjcard.2008.10.035. [DOI] [PubMed] [Google Scholar]
- 14.Savransky V, Nanayakkara A, Vivero A, Li J, Bevans S, Smith PL, Torbenson MS, Polotsky VY. Chronic intermittent hypoxia predisposes to liver injury. Hepatology. 2007;45:1007–1013. doi: 10.1002/hep.21593. [DOI] [PubMed] [Google Scholar]
- 15.Savransky V, Bevans S, Nanayakkara A, Li J, Smith PL, Torbenson MS, Polotsky VY. Chronic intermittent hypoxia causes hepatitis in a mouse model of diet-induced fatty liver. Am J Physiol Gastrointest Liver Physiol. 2007;293:G871–G877. doi: 10.1152/ajpgi.00145.2007. [DOI] [PubMed] [Google Scholar]
- 16.Takayama F, Egashira T, Kawasaki H, Mankura M, Nakamoto K, Okada S, Mori A. A Novel Animal Model of Nonalcoholic Steatohepatitis (NASH): Hypoxemia Enhances the Development of NASH. J Clin Biochem Nutr. 2009;45:335–340. doi: 10.3164/jcbn.09-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piguet AC, Stroka D, Zimmermann A, Dufour JF. Hypoxia aggravates non-alcoholic steatohepatitis in mice lacking hepatocellular PTEN. Clin Sci (Lond) 2009;118:401–410. doi: 10.1042/CS20090313. [DOI] [PubMed] [Google Scholar]
- 18.Chin K, Nakamura T, Takahashi K, Sumi K, Ogawa Y, Masuzaki H, Muro S, Hattori N, Matsumoto H, Niimi A, et al. Effects of obstructive sleep apnea syndrome on serum aminotransferase levels in obese patients. Am J Med. 2003;114:370–376. doi: 10.1016/s0002-9343(02)01570-x. [DOI] [PubMed] [Google Scholar]
- 19.Kohler M, Pepperell JC, Davies RJ, Stradling JR. Continuous positive airway pressure and liver enzymes in obstructive sleep apnoea: data from a randomized controlled trial. Respiration. 2009;78:141–146. doi: 10.1159/000170785. [DOI] [PubMed] [Google Scholar]
- 20.Jouët P, Sabaté JM, Maillard D, Msika S, Mechler C, Ledoux S, Harnois F, Coffin B. Relationship between obstructive sleep apnea and liver abnormalities in morbidly obese patients: a prospective study. Obes Surg. 2007;17:478–485. doi: 10.1007/s11695-007-9085-3. [DOI] [PubMed] [Google Scholar]
- 21.Kallwitz ER, Herdegen J, Madura J, Jakate S, Cotler SJ. Liver enzymes and histology in obese patients with obstructive sleep apnea. J Clin Gastroenterol. 2007;41:918–921. doi: 10.1097/01.mcg.0000225692.62121.55. [DOI] [PubMed] [Google Scholar]
- 22.Mishra P, Nugent C, Afendy A, Bai C, Bhatia P, Afendy M, Fang Y, Elariny H, Goodman Z, Younossi ZM. Apnoeic-hypopnoeic episodes during obstructive sleep apnoea are associated with histological nonalcoholic steatohepatitis. Liver Int. 2008;28:1080–1086. doi: 10.1111/j.1478-3231.2008.01822.x. [DOI] [PubMed] [Google Scholar]
- 23.Polotsky VY, Patil SP, Savransky V, Laffan A, Fonti S, Frame LA, Steele KE, Schweizter MA, Clark JM, Torbenson MS, et al. Obstructive sleep apnea, insulin resistance, and steatohepatitis in severe obesity. Am J Respir Crit Care Med. 2009;179:228–234. doi: 10.1164/rccm.200804-608OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Norman D, Bardwell WA, Arosemena F, Nelesen R, Mills PJ, Loredo JS, Lavine JE, Dimsdale JE. Serum aminotransferase levels are associated with markers of hypoxia in patients with obstructive sleep apnea. Sleep. 2008;31:121–126. doi: 10.1093/sleep/31.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kheirandish-Gozal L, Sans Capdevila O, Kheirandish E, Gozal D. Elevated serum aminotransferase levels in children at risk for obstructive sleep apnea. Chest. 2008;133:92–99. doi: 10.1378/chest.07-0773. [DOI] [PubMed] [Google Scholar]
- 26.Byrne CD. Fatty liver: role of inflammation and fatty acid nutrition. Prostaglandins Leukot Essent Fatty Acids. 2010;82:265–271. doi: 10.1016/j.plefa.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 27.Idris I, Hall AP, O’Reilly J, Barnett A, Allen M, Andrews R, Grunstein P, Lewis K, Goenka N, Wilding JP. Obstructive sleep apnoea in patients with type 2 diabetes: aetiology and implications for clinical care. Diabetes Obes Metab. 2009;11:733–741. doi: 10.1111/j.1463-1326.2009.01045.x. [DOI] [PubMed] [Google Scholar]
- 28.Vgontzas AN, Papanicolaou DA, Bixler EO, Hopper K, Lotsikas A, Lin HM, Kales A, Chrousos GP. Sleep apnea and daytime sleepiness and fatigue: relation to visceral obesity, insulin resistance, and hypercytokinemia. J Clin Endocrinol Metab. 2000;85:1151–1158. doi: 10.1210/jcem.85.3.6484. [DOI] [PubMed] [Google Scholar]
- 29.Punjabi NM, Sorkin JD, Katzel LI, Goldberg AP, Schwartz AR, Smith PL. Sleep-disordered breathing and insulin resistance in middle-aged and overweight men. Am J Respir Crit Care Med. 2002;165:677–682. doi: 10.1164/ajrccm.165.5.2104087. [DOI] [PubMed] [Google Scholar]
- 30.Ip MS, Lam B, Ng MM, Lam WK, Tsang KW, Lam KS. Obstructive sleep apnea is independently associated with insulin resistance. Am J Respir Crit Care Med. 2002;165:670–676. doi: 10.1164/ajrccm.165.5.2103001. [DOI] [PubMed] [Google Scholar]
- 31.Meslier N, Gagnadoux F, Giraud P, Person C, Ouksel H, Urban T, Racineux JL. Impaired glucose-insulin metabolism in males with obstructive sleep apnoea syndrome. Eur Respir J. 2003;22:156–160. doi: 10.1183/09031936.03.00089902. [DOI] [PubMed] [Google Scholar]
- 32.West SD, Nicoll DJ, Stradling JR. Prevalence of obstructive sleep apnoea in men with type 2 diabetes. Thorax. 2006;61:945–950. doi: 10.1136/thx.2005.057745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mallon L, Broman JE, Hetta J. High incidence of diabetes in men with sleep complaints or short sleep duration: a 12-year follow-up study of a middle-aged population. Diabetes Care. 2005;28:2762–2767. doi: 10.2337/diacare.28.11.2762. [DOI] [PubMed] [Google Scholar]
- 34.Tuomilehto H, Peltonen M, Partinen M, Seppä J, Saaristo T, Korpi-Hyövälti E, Oksa H, Puolijoki H, Saltevo J, Vanhala M, et al. Sleep duration is associated with an increased risk for the prevalence of type 2 diabetes in middle-aged women - The FIN-D2D survey. Sleep Med. 2008;9:221–227. doi: 10.1016/j.sleep.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 35.Polotsky VY, Li J, Punjabi NM, Rubin AE, Smith PL, Schwartz AR, O’Donnell CP. Intermittent hypoxia increases insulin resistance in genetically obese mice. J Physiol. 2003;552:253–264. doi: 10.1113/jphysiol.2003.048173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iiyori N, Alonso LC, Li J, Sanders MH, Garcia-Ocana A, O’Doherty RM, Polotsky VY, O’Donnell CP. Intermittent hypoxia causes insulin resistance in lean mice independent of autonomic activity. Am J Respir Crit Care Med. 2007;175:851–857. doi: 10.1164/rccm.200610-1527OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye J. Emerging role of adipose tissue hypoxia in obesity and insulin resistance. Int J Obes (Lond) 2009;33:54–66. doi: 10.1038/ijo.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yin J, Gao Z, He Q, Zhou D, Guo Z, Ye J. Role of hypoxia in obesity-induced disorders of glucose and lipid metabolism in adipose tissue. Am J Physiol Endocrinol Metab. 2009;296:E333–E342. doi: 10.1152/ajpendo.90760.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams A, Scharf SM. Obstructive sleep apnea, cardiovascular disease, and inflammation--is NF-kappaB the key? Sleep Breath. 2007;11:69–76. doi: 10.1007/s11325-007-0106-1. [DOI] [PubMed] [Google Scholar]
- 40.Htoo AK, Greenberg H, Tongia S, Chen G, Henderson T, Wilson D, Liu SF. Activation of nuclear factor kappaB in obstructive sleep apnea: a pathway leading to systemic inflammation. Sleep Breath. 2006;10:43–50. doi: 10.1007/s11325-005-0046-6. [DOI] [PubMed] [Google Scholar]
- 41.Greenberg H, Ye X, Wilson D, Htoo AK, Hendersen T, Liu SF. Chronic intermittent hypoxia activates nuclear factor-kappaB in cardiovascular tissues in vivo. Biochem Biophys Res Commun. 2006;343:591–596. doi: 10.1016/j.bbrc.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 42.Selmi C, Montano N, Furlan R, Keen CL, Gershwin ME. Inflammation and oxidative stress in obstructive sleep apnea syndrome. Exp Biol Med (Maywood) 2007;232:1409–1413. doi: 10.3181/0704-MR-103. [DOI] [PubMed] [Google Scholar]
- 43.Yamauchi M, Tamaki S, Tomoda K, Yoshikawa M, Fukuoka A, Makinodan K, Koyama N, Suzuki T, Kimura H. Evidence for activation of nuclear factor kappaB in obstructive sleep apnea. Sleep Breath. 2006;10:189–193. doi: 10.1007/s11325-006-0074-x. [DOI] [PubMed] [Google Scholar]
- 44.Tasali E, Ip MS. Obstructive sleep apnea and metabolic syndrome: alterations in glucose metabolism and inflammation. Proc Am Thorac Soc. 2008;5:207–217. doi: 10.1513/pats.200708-139MG. [DOI] [PubMed] [Google Scholar]
- 45.Foufelle F, Ferré P. [Regulation of carbohydrate metabolism by insulin: role of transcription factor SREBP-1c in the hepatic transcriptional effects of the hormone] J Soc Biol. 2001;195:243–248. [PubMed] [Google Scholar]
- 46.Ahmed MH, Byrne CD. Modulation of sterol regulatory element binding proteins (SREBPs) as potential treatments for non-alcoholic fatty liver disease (NAFLD) Drug Discov Today. 2007;12:740–747. doi: 10.1016/j.drudis.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 47.Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 48.Ferré P. The biology of peroxisome proliferator-activated receptors: relationship with lipid metabolism and insulin sensitivity. Diabetes. 2004;53 Suppl 1:S43–S50. doi: 10.2337/diabetes.53.2007.s43. [DOI] [PubMed] [Google Scholar]
- 49.Svegliati-Baroni G, Candelaresi C, Saccomanno S, Ferretti G, Bachetti T, Marzioni M, De Minicis S, Nobili L, Salzano R, Omenetti A, et al. A model of insulin resistance and nonalcoholic steatohepatitis in rats: role of peroxisome proliferator-activated receptor-alpha and n-3 polyunsaturated fatty acid treatment on liver injury. Am J Pathol. 2006;169:846–860. doi: 10.2353/ajpath.2006.050953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 51.Berg AH, Combs TP, Scherer PE. ACRP30/adiponectin: an adipokine regulating glucose and lipid metabolism. Trends Endocrinol Metab. 2002;13:84–89. doi: 10.1016/s1043-2760(01)00524-0. [DOI] [PubMed] [Google Scholar]
- 52.Stefan N, Vozarova B, Funahashi T, Matsuzawa Y, Weyer C, Lindsay RS, Youngren JF, Havel PJ, Pratley RE, Bogardus C, et al. Plasma adiponectin concentration is associated with skeletal muscle insulin receptor tyrosine phosphorylation, and low plasma concentration precedes a decrease in whole-body insulin sensitivity in humans. Diabetes. 2002;51:1884–1888. doi: 10.2337/diabetes.51.6.1884. [DOI] [PubMed] [Google Scholar]
- 53.Savvidou S, Hytiroglou P, Orfanou-Koumerkeridou H, Panderis A, Frantzoulis P, Goulis J. Low serum adiponectin levels are predictive of advanced hepatic fibrosis in patients with NAFLD. J Clin Gastroenterol. 2009;43:765–772. doi: 10.1097/MCG.0b013e31819e9048. [DOI] [PubMed] [Google Scholar]
- 54.Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, Furukawa S, Tochino Y, Komuro R, Matsuda M, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007;56:901–911. doi: 10.2337/db06-0911. [DOI] [PubMed] [Google Scholar]
- 55.Chen B, Lam KS, Wang Y, Wu D, Lam MC, Shen J, Wong L, Hoo RL, Zhang J, Xu A. Hypoxia dysregulates the production of adiponectin and plasminogen activator inhibitor-1 independent of reactive oxygen species in adipocytes. Biochem Biophys Res Commun. 2006;341:549–556. doi: 10.1016/j.bbrc.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 56.Fasshauer M, Kralisch S, Klier M, Lossner U, Bluher M, Klein J, Paschke R. Adiponectin gene expression and secretion is inhibited by interleukin-6 in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2003;301:1045–1050. doi: 10.1016/s0006-291x(03)00090-1. [DOI] [PubMed] [Google Scholar]
- 57.Snyder EM, Carr RD, Deacon CF, Johnson BD. Overnight hypoxic exposure and glucagon-like peptide-1 and leptin levels in humans. Appl Physiol Nutr Metab. 2008;33:929–935. doi: 10.1139/H08-079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grosfeld A, Andre J, Hauguel-De Mouzon S, Berra E, Pouyssegur J, Guerre-Millo M. Hypoxia-inducible factor 1 transactivates the human leptin gene promoter. J Biol Chem. 2002;277:42953–42957. doi: 10.1074/jbc.M206775200. [DOI] [PubMed] [Google Scholar]
- 59.Sanner BM, Kollhosser P, Buechner N, Zidek W, Tepel M. Influence of treatment on leptin levels in patients with obstructive sleep apnoea. Eur Respir J. 2004;23:601–604. doi: 10.1183/09031936.04.00067804. [DOI] [PubMed] [Google Scholar]
- 60.Kapsimalis F, Varouchakis G, Manousaki A, Daskas S, Nikita D, Kryger M, Gourgoulianis K. Association of sleep apnea severity and obesity with insulin resistance, C-reactive protein, and leptin levels in male patients with obstructive sleep apnea. Lung. 2008;186:209–217. doi: 10.1007/s00408-008-9082-x. [DOI] [PubMed] [Google Scholar]
- 61.Ulukavak Ciftci T, Kokturk O, Bukan N, Bilgihan A. Leptin and ghrelin levels in patients with obstructive sleep apnea syndrome. Respiration. 2005;72:395–401. doi: 10.1159/000086254. [DOI] [PubMed] [Google Scholar]
- 62.Yasumasu T, Takahara K, Nakashima Y. Hypoxia inhibits leptin production by cultured rat adipocytes. Obes Res. 2002;10:128. doi: 10.1038/oby.2002.20. [DOI] [PubMed] [Google Scholar]
- 63.Schmoller A, Voss M, Gehring H, Rudolf S, Schweiger U, Schultes B, Oltmanns KM. Short Hypoxia Does not Affect Plasma Leptin in Healthy Men under Euglycemic Clamp Conditions. Int J Endocrinol. 2009;2009:270698. doi: 10.1155/2009/270698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chaiban JT, Bitar FF, Azar ST. Effect of chronic hypoxia on leptin, insulin, adiponectin, and ghrelin. Metabolism. 2008;57:1019–1022. doi: 10.1016/j.metabol.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 65.Keijer J, van Schothorst EM. Adipose tissue failure and mitochondria as a possible target for improvement by bioactive food components. Curr Opin Lipidol. 2008;19:4–10. doi: 10.1097/MOL.0b013e3282f39f95. [DOI] [PubMed] [Google Scholar]
- 66.Bruce KD, Cagampang FR, Argenton M, Zhang J, Ethirajan PL, Burdge GC, Bateman AC, Clough GF, Poston L, Hanson MA, et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology. 2009;50:1796–1808. doi: 10.1002/hep.23205. [DOI] [PubMed] [Google Scholar]
- 67.Nakatani Y, Kaneto H, Kawamori D, Yoshiuchi K, Hatazaki M, Matsuoka TA, Ozawa K, Ogawa S, Hori M, Yamasaki Y, et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J Biol Chem. 2005;280:847–851. doi: 10.1074/jbc.M411860200. [DOI] [PubMed] [Google Scholar]
- 68.Ozawa K, Miyazaki M, Matsuhisa M, Takano K, Nakatani Y, Hatazaki M, Tamatani T, Yamagata K, Miyagawa J, Kitao Y, et al. The endoplasmic reticulum chaperone improves insulin resistance in type 2 diabetes. Diabetes. 2005;54:657–663. doi: 10.2337/diabetes.54.3.657. [DOI] [PubMed] [Google Scholar]
- 69.Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, Koromilas A, Wouters BG. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol Cell Biol. 2002;22:7405–7416. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gentile CL, Pagliassotti MJ. The role of fatty acids in the development and progression of nonalcoholic fatty liver disease. J Nutr Biochem. 2008;19:567–576. doi: 10.1016/j.jnutbio.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ahmed MH, Byrne CD. Current treatment of non-alcoholic fatty liver disease. Diabetes Obes Metab. 2009;11:188–195. doi: 10.1111/j.1463-1326.2008.00926.x. [DOI] [PubMed] [Google Scholar]
- 72.Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, Fruchart JC, James WP, Loria CM, Smith SC Jr. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120:1640–1645. doi: 10.1161/CIRCULATIONAHA.109.192644. [DOI] [PubMed] [Google Scholar]
- 73.Vgontzas AN, Bixler EO, Chrousos GP. Sleep apnea is a manifestation of the metabolic syndrome. Sleep Med Rev. 2005;9:211–224. doi: 10.1016/j.smrv.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 74.Coughlin SR, Mawdsley L, Mugarza JA, Calverley PM, Wilding JP. Obstructive sleep apnoea is independently associated with an increased prevalence of metabolic syndrome. Eur Heart J. 2004;25:735–741. doi: 10.1016/j.ehj.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 75.Tkacova R, Dorkova Z, Molcanyiova A, Radikova Z, Klimes I, Tkac I. Cardiovascular risk and insulin resistance in patients with obstructive sleep apnea. Med Sci Monit. 2008;14:CR438–CR444. [PubMed] [Google Scholar]
- 76.Sasanabe R, Banno K, Otake K, Hasegawa R, Usui K, Morita M, Shiomi T. Metabolic syndrome in Japanese patients with obstructive sleep apnea syndrome. Hypertens Res. 2006;29:315–322. doi: 10.1291/hypres.29.315. [DOI] [PubMed] [Google Scholar]
- 77.Lam JC, Lam B, Lam CL, Fong D, Wang JK, Tse HF, Lam KS, Ip MS. Obstructive sleep apnea and the metabolic syndrome in community-based Chinese adults in Hong Kong. Respir Med. 2006;100:980–987. doi: 10.1016/j.rmed.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 78.Shiina K, Tomiyama H, Takata Y, Usui Y, Asano K, Hirayama Y, Nakamura T, Yamashina A. Concurrent presence of metabolic syndrome in obstructive sleep apnea syndrome exacerbates the cardiovascular risk: a sleep clinic cohort study. Hypertens Res. 2006;29:433–441. doi: 10.1291/hypres.29.433. [DOI] [PubMed] [Google Scholar]
- 79.Antonopoulos S, Mikros S, Mylonopoulou M, Kokkoris S, Giannoulis G. Rosuvastatin as a novel treatment of non-alcoholic fatty liver disease in hyperlipidemic patients. Atherosclerosis. 2006;184:233–234. doi: 10.1016/j.atherosclerosis.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 80.Targher G, Bertolini L, Poli F, Rodella S, Scala L, Tessari R, Zenari L, Falezza G. Nonalcoholic fatty liver disease and risk of future cardiovascular events among type 2 diabetic patients. Diabetes. 2005;54:3541–3546. doi: 10.2337/diabetes.54.12.3541. [DOI] [PubMed] [Google Scholar]
- 81.Targher G, Bertolini L, Padovani R, Rodella S, Zoppini G, Zenari L, Cigolini M, Falezza G, Arcaro G. Relations between carotid artery wall thickness and liver histology in subjects with nonalcoholic fatty liver disease. Diabetes Care. 2006;29:1325–1330. doi: 10.2337/dc06-0135. [DOI] [PubMed] [Google Scholar]
- 82.Targher G, Bertolini L, Padovani R, Poli F, Scala L, Tessari R, Zenari L, Falezza G. Increased prevalence of cardiovascular disease in Type 2 diabetic patients with non-alcoholic fatty liver disease. Diabet Med. 2006;23:403–409. doi: 10.1111/j.1464-5491.2006.01817.x. [DOI] [PubMed] [Google Scholar]
- 83.Targher G, Bertolini L, Padovani R, Poli F, Scala L, Zenari L, Zoppini G, Falezza G. Non-alcoholic fatty liver disease is associated with carotid artery wall thickness in diet-controlled type 2 diabetic patients. J Endocrinol Invest. 2006;29:55–60. doi: 10.1007/BF03349177. [DOI] [PubMed] [Google Scholar]
- 84.Targher G, Bertolini L, Rodella S, Tessari R, Zenari L, Lippi G, Arcaro G. Nonalcoholic fatty liver disease is independently associated with an increased incidence of cardiovascular events in type 2 diabetic patients. Diabetes Care. 2007;30:2119–2121. doi: 10.2337/dc07-0349. [DOI] [PubMed] [Google Scholar]
- 85.Targher G, Bertolini L, Rodella S, Zoppini G, Zenari L, Falezza G. Associations between liver histology and cortisol secretion in subjects with nonalcoholic fatty liver disease. Clin Endocrinol (Oxf) 2006;64:337–341. doi: 10.1111/j.1365-2265.2006.02466.x. [DOI] [PubMed] [Google Scholar]
- 86.Targher G, Zoppini G, Lippi G, Guidi GC, Muggeo M. Effect of serum gamma-glutamyltransferase and obesity on the risk of dyslipidemia and poor glycemic control in type 2 diabetic patients: cross-sectional findings from the Verona Diabetes Study. Clin Chem. 2007;53:1867–1869; author reply 1869-1870. doi: 10.1373/clinchem.2007.092601. [DOI] [PubMed] [Google Scholar]
- 87.Targher G, Bertolini L, Padovani R, Rodella S, Tessari R, Zenari L, Day C, Arcaro G. Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients. Diabetes Care. 2007;30:1212–1218. doi: 10.2337/dc06-2247. [DOI] [PubMed] [Google Scholar]
- 88.Lévy P, Pépin JL, Arnaud C, Baguet JP, Dematteis M, Mach F. Obstructive sleep apnea and atherosclerosis. Prog Cardiovasc Dis. 2009;51:400–410. doi: 10.1016/j.pcad.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 89.Tomásová H, Lisý V, Trojan S, Stastný F. Effect of short-term or intermittent hypobaric hypoxia on plasma lipids in young rats. Physiol Bohemoslov. 1987;36:361–364. [PubMed] [Google Scholar]
- 90.Li J, Thorne LN, Punjabi NM, Sun CK, Schwartz AR, Smith PL, Marino RL, Rodriguez A, Hubbard WC, O’Donnell CP, et al. Intermittent hypoxia induces hyperlipidemia in lean mice. Circ Res. 2005;97:698–706. doi: 10.1161/01.RES.0000183879.60089.a9. [DOI] [PubMed] [Google Scholar]
- 91.Li J, Nanayakkara A, Jun J, Savransky V, Polotsky VY. Effect of deficiency in SREBP cleavage-activating protein on lipid metabolism during intermittent hypoxia. Physiol Genomics. 2007;31:273–280. doi: 10.1152/physiolgenomics.00082.2007. [DOI] [PubMed] [Google Scholar]
- 92.Savransky V, Nanayakkara A, Li J, Bevans S, Smith PL, Rodriguez A, Polotsky VY. Chronic intermittent hypoxia induces atherosclerosis. Am J Respir Crit Care Med. 2007;175:1290–1297. doi: 10.1164/rccm.200612-1771OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Savransky V, Jun J, Li J, Nanayakkara A, Fonti S, Moser AB, Steele KE, Schweitzer MA, Patil SP, Bhanot S, et al. Dyslipidemia and atherosclerosis induced by chronic intermittent hypoxia are attenuated by deficiency of stearoyl coenzyme A desaturase. Circ Res. 2008;103:1173–1180. doi: 10.1161/CIRCRESAHA.108.178533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li J, Bosch-Marce M, Nanayakkara A, Savransky V, Fried SK, Semenza GL, Polotsky VY. Altered metabolic responses to intermittent hypoxia in mice with partial deficiency of hypoxia-inducible factor-1alpha. Physiol Genomics. 2006;25:450–457. doi: 10.1152/physiolgenomics.00293.2005. [DOI] [PubMed] [Google Scholar]
- 95.Steiropoulos P, Tsara V, Nena E, Fitili C, Kataropoulou M, Froudarakis M, Christaki P, Bouros D. Effect of continuous positive airway pressure treatment on serum cardiovascular risk factors in patients with obstructive sleep apnea-hypopnea syndrome. Chest. 2007;132:843–851. doi: 10.1378/chest.07-0074. [DOI] [PubMed] [Google Scholar]
- 96.Robinson GV, Pepperell JC, Segal HC, Davies RJ, Stradling JR. Circulating cardiovascular risk factors in obstructive sleep apnoea: data from randomised controlled trials. Thorax. 2004;59:777–782. doi: 10.1136/thx.2003.018739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gozal D, O’Brien LM. Snoring and obstructive sleep apnoea in children: why should we treat? Paediatr Respir Rev. 2004;5 Suppl A:S371–S376. doi: 10.1016/s1526-0542(04)90066-8. [DOI] [PubMed] [Google Scholar]
- 98.Bradley TD, Floras JS. Obstructive sleep apnoea and its cardiovascular consequences. Lancet. 2009;373:82–93. doi: 10.1016/S0140-6736(08)61622-0. [DOI] [PubMed] [Google Scholar]
- 99.Fletcher EC, DeBehnke RD, Lovoi MS, Gorin AB. Undiagnosed sleep apnea in patients with essential hypertension. Ann Intern Med. 1985;103:190–195. doi: 10.7326/0003-4819-103-2-190. [DOI] [PubMed] [Google Scholar]
- 100.Logan AG, Perlikowski SM, Mente A, Tisler A, Tkacova R, Niroumand M, Leung RS, Bradley TD. High prevalence of unrecognized sleep apnoea in drug-resistant hypertension. J Hypertens. 2001;19:2271–2277. doi: 10.1097/00004872-200112000-00022. [DOI] [PubMed] [Google Scholar]
- 101.Börgel J, Sanner BM, Keskin F, Bittlinsky A, Bartels NK, Büchner N, Huesing A, Rump LC, Mügge A. Obstructive sleep apnea and blood pressure. Interaction between the blood pressure-lowering effects of positive airway pressure therapy and antihypertensive drugs. Am J Hypertens. 2004;17:1081–1087. doi: 10.1016/j.amjhyper.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 102.Peker Y, Kraiczi H, Hedner J, Löth S, Johansson A, Bende M. An independent association between obstructive sleep apnoea and coronary artery disease. Eur Respir J. 1999;14:179–184. doi: 10.1034/j.1399-3003.1999.14a30.x. [DOI] [PubMed] [Google Scholar]
- 103.Mooe T, Franklin KA, Holmström K, Rabben T, Wiklund U. Sleep-disordered breathing and coronary artery disease: long-term prognosis. Am J Respir Crit Care Med. 2001;164:1910–1913. doi: 10.1164/ajrccm.164.10.2101072. [DOI] [PubMed] [Google Scholar]
- 104.Good DC, Henkle JQ, Gelber D, Welsh J, Verhulst S. Sleep-disordered breathing and poor functional outcome after stroke. Stroke. 1996;27:252–259. doi: 10.1161/01.str.27.2.252. [DOI] [PubMed] [Google Scholar]
- 105.Kaneko Y, Hajek VE, Zivanovic V, Raboud J, Bradley TD. Relationship of sleep apnea to functional capacity and length of hospitalization following stroke. Sleep. 2003;26:293–297. doi: 10.1093/sleep/26.3.293. [DOI] [PubMed] [Google Scholar]