

Abstract
Neuronal proteins are transported to either the axon or dendrites through the action of kinesin motors; however understanding of how cytoskeletal elements steer these cargo-motor complexes to one compartment or the other has remained elusive. Three recent developments, the discovery of an actin-based filter within the axon initial segment, the identification of the pivotal role played by myosin motors in dendritic targeting, and the determination of the properties of a kinesin motor that cause it to prefer axonal to dendritic microtubules, have now provided a structural framework for understanding polarized targeting in neurons.
One hundred years agoRamon Y Cajal speculated that “an axipetal polarization of the protoplasma” could explain why electrical signals flow within neurons from dendrites to axons(1–2). Although the language is now antiquated, the concept that inherently polarized structural elements determine the direction of information flow in neurons is well accepted. Such structural polarization is exemplified by the synapse, in which synaptic vesicles line up on the axonal (presynaptic) side and neurotransmitter receptors are concentrated on the dendritic (postsynaptic) side. The postsynaptic component of the synapse acquires its unique protein distribution as a result of the one way flow of newly synthesized proteins from the cell body to dendrites(3). As with the polarized flow of chemical signals, it is logical to assume that the polarized trafficking of newly synthesized proteins is determined by intrinsically polarized structures, in this case structures within the neuronal cytoskeleton. However, the nature of these structures has been unknown. Recently, three papers have been published that identify cytoskeletal elements essential for neuronal polarityand provide a framework for understanding how these structures might mediate polarized targeting of neuronal proteins(4–6).
Decades of work, some leading to the Nobel prize, has shown that encoded within the primary amino acid sequences of proteins are the signals that determine their ultimate subcellular localization; for instance there are well-defined protein sequences that cause localization to the secretory pathway or to the nucleus(7–9). Similar signals determine the localization of proteins to the apical and basolateral membrane domains of polarized epithelial cells (10). Inspired by these discoveries, neurobiologists have identified signals that are necessary and sufficient for targeting of proteins to axons or dendrites(11–13). However, because the proteins with which these signals interact have proven difficult to identify, the mechanisms by which they mediate targeting have remained obscure(14). Another avenue for understanding polarized targeting involves studying the transport process itself. For instance, kinesin molecules serve as the motors that transport vesicles containing transmembrane proteins to the axon and dendrites(15). Furthermore, certain kinesin motors interact specifically with dendritic proteins and are confined to the dendrites, suggesting that dendritic targeting might occur simply as a result of the preference of individual kinesin motors for dendritically-projecting microtubules(16–18). However, the motor domains of dendritically localized kinesins do not appear to have an intrinsic preference for dendritically-versusaxonally-projecting microtubules, suggesting that targeting of proteins to dendrites depends on additional cues (19).
Three groups have recently taken a fresh approach to this question by looking for differences in the cytoskeletal structure of the axon and dendrites that could cause proteins to be excluded from one of these two compartments (4–6). Such a structural asymmetry was identified in the course of a simple, yet ingenious experiment performed by Song et al. (6), in which different sized dextran molecules were injected into the cell body and their subsequent localization was monitored. Strikingly, high molecular weight dextran did not enter the distal axon but localized predominantly in the somatodendritic region, suggesting that a molecular sieve existed at the axon initial segment. Further experiments showed that this filter sorted vesicles that contained various transmembrane proteins, and were associated with various kinesin motors, based on both of these components. How might such a filter work? Both Song et al. and Lewis et al. (5) found that an intact actin network was necessary for polarized targeting in neurons, suggesting that actin might form the filter. Based on this hypothesis, Lewis et al. looked at the contribution of myosin motors to polarized targeting and found that interaction with Myosin Va is both necessary and sufficient to mediate targeting of transmembrane proteins to dendrites. Finally, close observation of the localization of dendritic proteins showed their presence in the proximal, but not the distal, axon. Together these observations suggest that within the axon initial segment there is a structure consisting of polarized actin filaments that point out of the axon, preventing vesicles carrying dendritic proteins and bearing activated, + end-directed myosins from entering the axon (Fig. 1). This model is consistent with observations made a decade ago that a structure within the axon initial segment acts as a diffusion barrier that prevents surface proteins from moving between polarized neuronal compartments (20).
Fig. 1. A model for dendritic targeting.
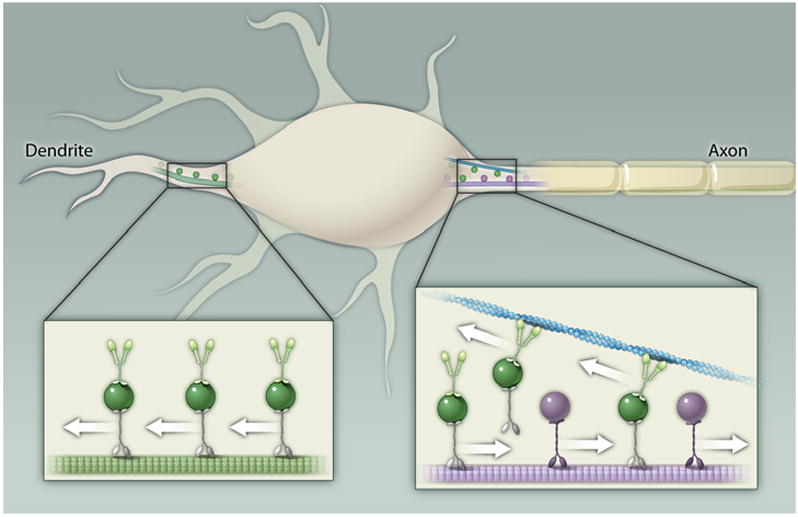
Vesicles containing dendritic proteins (dark green) associate with both kinesin motors and Myosin Va (light green) and move along axonally projecting microtubules (purple) until Myosin Va interacts with actin filaments (blue). The orientation of these filaments causes Myosin Va and the vesicle to which it is attached to move toward the cell body. Vesicles containing axonal proteins (dark purple) do not associate with Myosin Va and, thus, are allowed to move to the distal axon unimpeded. In the dendrite, the absence of a vesicle filter allows vesicles carrying dendritic proteins to move distally. Purple denotes detyrosinated microtubules, which are found in the axon, and light green denotes tyrosinated microtubules, which are found in the dendrites. Kif5 is indicated in purple, other kinesins in gray.
When does the axon filter form? According to Song et al., in dissociated neuronal cultures it forms between 3 and 5 days in vitro, at least one day subsequent to the initial elongation of the process that becomes the axon, the quintessential event in neuronal polarization (21). Thus, the vesicle filter appears to form after the neuron has become polarized, which raises the question of how polarized proteins are sorted prior to its establishment. An answer to this question may be found in data presented by Konishi and Setou (4). This paper describes how a particular region of the kinesin Kif5 causes this motor protein to bind with a relatively low affinity to microtubules modified by tyrosination, a process that occurs more commonly in dendrites (22). Tyrosination occurs subsequent to detyrosination, a ubiquitous process in which the final (C-terminal) tyrosine of α-tubulin is removed through the action of tubulin carboxypeptidase, an enzyme that is regulated by phosphorylation (23–24). Because tubulin carboxypeptidase specifically acts on intact microtubules, most microtubules in regions of high stability such as the axon tend to be detyrosinated (25–26). Conversely, the α-tubulin C-terminal tyrosine is added back to the microtubule through tubulin tyrosine ligase (TTL), which acts preferentially on α-β dimers, which predominate in areas of reduced microtubule stability such as the dendrites. TTL itself contains binding sites for such kinases as protein kinase A (PKA), protein kinase C (PKC), casein kinase II (CKII) and tyrosine kinase and thus is likely also regulated by its phosphorylation state (27).
Tailless Kif5 is targeted to the axon immediately after axonal specification and is thought to carry proteins, such as c-Jun activated-terminal kinase, that propagate the process of neuronal polarization (28–29). Thus, early in the process of neuronal specification, before formation of the axon vesicle filter, detyrosination of microtubules in the axon could cause Kif5 to bind preferentially to axonal microtubules. Subsequent to formation of the axon vesicle filter, it appears that microtubule-based mechanisms continue to play a role in polarized targeting. Song et al. found that the ease with which a vesicle carrying a particular protein passes through the axon filter depends on which kinesin isoform is propelling it in addition to the identity of the protein itself. Moreover, Konishi and Setou found that blocking Kif5 function by expressing a dominant negative variant of it caused neurons to have more than one axon, indicating that it is necessary for maintenance of polarity. Defining the relative contribution of microtubule-based versus actin-based polarity mechanisms--as well as signal transduction pathways that could connect the two--will be important areas for future investigation.
Beyond addressing fundamental questions in neuroscience, the discoveries reported in these three papers are likely to find practical applications as they drive the development of new tools to target proteins to specific neuronal compartments. An example of this is illustrated in the work of Lewis et al., who demonstrated that by fusing a short myosin binding motif to Channelrhodopsin-2 (ChR2), a light-activated ion channel, they could cause the expressed channel to localize specifically to dendrites of pyramidal cells inmouse cortex (5, 30). Dendritic localization, in this case, improves the utility of this Channelrhodopsin for tracing neuronal circuits, and should be of wide interest to systems electrophysiologists (31). It can be expected that in the future, as more and different molecules are identified that can be used to regulate neuronal activities, strategies to control the subcellular targeting of these proteins will become increasingly important. The basic mechanisms identified in these papers, and those to come, lay the foundation for a new level of control over neuronal protein function.
Acknowledgments
I thank Su Hyun Kwon for creating the figure and Emily Liman and Tommy Lewis for help editing the manuscript. This work was supported by NIH grants (NS-41963 and MH-71439).
References and Notes
- 1.Ramon S, Cajal Y. Histologie du systeme nerveux de l’homme et les verteberes. Vol. 1 Imprenta N. Moya; Madrid: pp. 1909–1911. [Google Scholar]
- 2.Changeux JP P. F. Institut Pasteur. Ann N Y Acad Sci. 2001 Apr;929:147. doi: 10.1111/j.1749-6632.2001.tb05713.x. [DOI] [PubMed] [Google Scholar]
- 3.Burack MA, Silverman MA, Banker G. Neuron. 2000;26:465. doi: 10.1016/s0896-6273(00)81178-2. [DOI] [PubMed] [Google Scholar]
- 4.Konishi Y, Setou M. Nat Neurosci. 2009 May;12(5):559. doi: 10.1038/nn.2314. [DOI] [PubMed] [Google Scholar]
- 5.Lewis TL, Jr, Mao T, Svoboda K, Arnold DB. Nat Neurosci. 2009 May;12(5):568. doi: 10.1038/nn.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song AH, et al. Cell. 2009 Mar 20;136(6):1148. doi: 10.1016/j.cell.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 7.Blobel G, Dobberstein B. J Cell Biol. 1975 Dec;67(3):852. doi: 10.1083/jcb.67.3.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blobel G, Dobberstein B. J Cell Biol. 1975 Dec;67(3):835. doi: 10.1083/jcb.67.3.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalderon D, Roberts BL, Richardson WD, Smith AE. Cell. 1984 Dec;39(3 Pt 2):499. doi: 10.1016/0092-8674(84)90457-4. [DOI] [PubMed] [Google Scholar]
- 10.Mostov KE, Verges M, Altschuler Y. Curr Opin Cell Biol. 2000 Aug;12:483. doi: 10.1016/s0955-0674(00)00120-4. [DOI] [PubMed] [Google Scholar]
- 11.Gu C, Jan YN, Jan LY. Science. 2003 Aug 1;301:646. doi: 10.1126/science.1086998. [DOI] [PubMed] [Google Scholar]
- 12.Rivera JF, Ahmad S, Quick MW, Liman ER, Arnold DB. Nat Neurosci. 2003;6:243. doi: 10.1038/nn1020. [DOI] [PubMed] [Google Scholar]
- 13.Kanaani J, et al. J Cell Biol. 2002 Sep 30;158(7):1229. doi: 10.1083/jcb.200205053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arnold DB. Pflugers Arch. 2007 Mar;453:763. doi: 10.1007/s00424-006-0155-5. [DOI] [PubMed] [Google Scholar]
- 15.Hirokawa N. Science. 1998;279:519. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- 16.Setou M, Nakagawa T, Seog DH, Hirokawa N. Science. 2000;288:1796. doi: 10.1126/science.288.5472.1796. [DOI] [PubMed] [Google Scholar]
- 17.Saito N, et al. Neuron. 1997;18:425. doi: 10.1016/s0896-6273(00)81243-x. [DOI] [PubMed] [Google Scholar]
- 18.Hanlon DW, Yang Z, Goldstein LS. Neuron. 1997;18:439. doi: 10.1016/s0896-6273(00)81244-1. [DOI] [PubMed] [Google Scholar]
- 19.Nakata T, Hirokawa N. J Cell Biol. 2003 Sep 15;162:1045. doi: 10.1083/jcb.200302175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Winckler B, Forscher P, Mellman I. Nature. 1999 Feb 25;397:698. doi: 10.1038/17806. [DOI] [PubMed] [Google Scholar]
- 21.Dotti CG, Sullivan CA, Banker GA. J Neurosci. 1988 Apr;8(4):1454. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Witte H, Neukirchen D, Bradke F. J Cell Biol. 2008 Feb 11;180(3):619. doi: 10.1083/jcb.200707042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ersfeld K, et al. J Cell Biol. 1993 Feb;120(3):725. doi: 10.1083/jcb.120.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Contâin MA, et al. Eur J Biochem. 2003 Dec;270(24):4921. doi: 10.1046/j.1432-1033.2003.03893.x. [DOI] [PubMed] [Google Scholar]
- 25.Webster DR, Wehland J, Weber K, Borisy GG. J Cell Biol. 1990 Jul;111(1):113. doi: 10.1083/jcb.111.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khawaja S, Gundersen GG, Bulinski JC. J Cell Biol. 1988 Jan;106(1):141. doi: 10.1083/jcb.106.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Idriss HT. Cell Motil Cytoskeleton. 2000 May;46(1):1. doi: 10.1002/(SICI)1097-0169(200005)46:1<1::AID-CM1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 28.Oliva AA, Jr, et al. J Neurosci. 2006 Sep 13;26(37):9462. doi: 10.1523/JNEUROSCI.2625-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jacobson C, et al. Neuron. 2006 Mar 16;49(6):797. doi: 10.1016/j.neuron.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 30.Nagel G, et al. Proc Natl Acad Sci U S A. 2003 Nov 25;100:13940. doi: 10.1073/pnas.1936192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petreanu L, Huber D, Sobczyk A, Svoboda K. Nat Neurosci. 2007 May;10:663. doi: 10.1038/nn1891. [DOI] [PubMed] [Google Scholar]