

Abstract
Biomaterials derived from protein-based block copolymers are increasingly investigated for potential application in medicine. In particular, recombinant elastin block copolymers provide significant opportunities to modulate material microstructure and can be processed in various forms, including particles, films, gels, and fiber networks. As a consequence, biological and mechanical responses of elastin-based biomaterials are tunable through precise control of block size and amino acid sequence. In this review, the synthesis of a set of elastin-mimetic triblock copolymers and their diverse processing methods for generating material platforms currently applied in medicine will be discussed.
1. Introduction
Recombinant elastin-mimetic protein polymers have recently emerged as a promising class of biomaterial in a number of biomedical applications. The ability to form as a variety of platforms such as gels [1-3], films [4,5], or nanofibers [6,7] demonstrate the versatility of these protein polymers with significant potential applications in medicine, including drug delivery [1,8,9], or as implants and coatings of medical devices [10,11].
Elastin, along with collagen, is a major protein component of extracellular matrix (ECM), which is found in many tissues such as skin, lung, artery, ligament and cartilage. Native elastin is composed of elastomeric domains and crosslinking domains. The elastomeric domains are rich in hydrophobic amino acids, and have repetitive tetra-, penta-, and hexapeptides sequences, including VPGG, VPGVG, and APGVGV, respectively. The crosslinking domains are rich in alanines and lysines, which are involved in the crosslinking reactions. Native elastin is an extremely insoluble matrix protein and forms fiber networks due to interaction between hydrophobic segments. Pentapeptide, Val-Pro-Gly-Val-Gly, is a common repeat unit within the elastomeric domain that is responsible for viscoelastic properties. Genetic engineering has made it possible to create novel recombinant elastin-mimetic protein polymers composed of pentapeptide repeats, [Val-Pro-Xaa-Yaa-Gly]n (where Xaa is Glycine or Alanine and Yaa can be any amino acid besides proline, respectively), that can undergo reversible, temperature-dependent, hydrophobic assembly from aqueous solution in analogy to the phase behavior of native elastin [12-15]. The self-assembling process results in spontaneous phase separation of the protein polymers above a lower critical solution temperature (LCST), which coincides with a conformational rearrangement of the local secondary structure within the pentapeptide motifs. Spectroscopic analysis has demonstrated that the pentameric sequence units undergo a conformational transition from a random coil to β-turns as the temperature approaches the transition temperature (Tt) [13,16]. Substitution of the third amino acid residue of the repeat sequence (Gly to Ala) results in a change of the mechanical response of the material from elastic to plastic. Additionally, amino acid substitution of the fourth position modulates the temperature-dependent phase behavior of the material [17]. These tunable properties lead to the generation of recombinant elastin-mimetic protein polymers that can serve as promising biomaterials for tissue engineering and biomedical applications.
Another strategy to change the properties of elastin-mimetic biomaterials is to design protein multiblock copolymers through genetic engineering of elastin-mimetic polypeptides. The design framework for this new class of elastin-mimetic block copolymer is derived from previous pioneering investigations of pentapeptide repeats [1,2]. We are particularly interested in elastin-mimetic triblock copolymers composed of identical hydrophobic endblocks separated by a central hydrophilic block in which individual blocks show different mechanical, and phase separation properties. In a manner similar to that observed for synthetic block copolymers, the size and chemical properties of individual blocks, especially the midblock, represent important variables in tuning material properties of protein-based block copolymers [18]. In this article, we will review the synthesis and related processing methodologies for elastin-mimetic triblock copolymers and present potential applications of these biomaterials in medicine.
2. Design and synthesis of elastin-mimetic block copolypeptides
2.1. Elastin-mimetic polypeptides
A variety of the pentapeptide repeat derived from elastomeric or plastic sequence [Ile/Val-Pro-(Xaa)-(Yaa)-Gly] exhibits tunable mechanical and thermodynamic properties that rely upon amino acid substitution in a position-specific manner (Table 1). We have recently employed these elastin-mimetic polypeptides for the construction of triblock copolymers, which have robust viscoelastic and mechanical responses [3-5,7,10,19]. The elastomeric polypeptide (E) is derived from combination of the pentapeptide sequences of [Val1-Pro2-Gly3-(Yaa)4-Gly5] format, in which the fourth residue is responsible for the transition temperature (Tt) of the polypeptide. Incorporation of glutamic acid significantly increases values of Tt in aqueous solutions. Compared with E4, transition temperatures of E1-3 in a dilute aqueous protein solution are not observed at temperatures as high as 75°C due to the presence of polar residues. The plastic polypeptide, [(Ile/Val)1-Pro2-Ala3-Val4-Gly5], is derived from the parent (Val1-Pro2-Gly3-Va4l-Gly5) pentapeptide sequence which displays dramatic changes in the macromolecular properties, particularly mechanical behavior and structure, between the two protein polymers. First, significant differences were observed in mechanical response between the γ-irradiation crosslinked hydrogel materials derived from the two pentapeptide repeat sequences, X20-poly(GVGVP) and -poly(AVGVP). Young's modulus of the latter polypeptide is 2.0×108 dynes/cm2, which is approximately two orders of magnitude greater than that of the former polypeptide (1.0×106 dynes/cm2) [20]. In addition, Reguera et al. have demonstrated through DSC and turbidity experiments that poly(VPAVG) displayed a significant hysteresis behavior during heating and cooling process (ΔT = 25 °C) in which restoration to the soluble state of the protein polymer required a high under-cooling (Tt = 6 °C for cooling) [21]. It also has a different enthalpy value (ΔH) for heating (34 J g-1) and cooling (66 – 91 J g-1). In contrast, the elastomeric polypeptide, poly(VPGVG), shows reversible behavior in which the transition temperature and the enthalpy values are almost identical during heating and cooling cycles. The structural difference between the two polypeptides is correlated with the observed difference in reversibility of the phase transition. NMR, ATR FTIR and Raman studies of poly(VPAVG) indicate that the Pro-Ala sequence motif forms a β-turn structure, but the other residues not involved in β-turn formation form a β-sheet structure through the hydrogen bonding interactions that can more stabilize the plastic polypeptide under cooling than the elastomeric polypeptide. Above difference in macromolecular properties could be used for design and synthesis of elastin-mimetic block copolymers comprised of blocks derived from the elastomeric and plastic sequences that have similar properties to synthetic thermoplastic elastomers [2-7,18]. An alteration in mechanical response of elastomeric polypeptides can be achieved by replacing glycine in the third position with alanine having an additional methyl group, which results in mechanical responses of a plastic-like nature. The plastic polypeptide (P) is relatively less diverse and derived from one of the pentapeptide sequences [(Ile/Val)1-Pro2-Ala3-Val4-Gly5] or their combination.
Table 1. Transition temperatures for elastin-mimetic block sequences.
| Polypeptide sequence | Transition temperature (Tt)† | Mechanical property | |
|---|---|---|---|
| E1 | [(VPGVG)2VPGEG(VPGVG)2]30 | >75°C | Elastomeric-like |
| E2 | [(VPGVG)2VPGEG(VPGVG)2]48 | >75°C | |
| E3 | [(VPGAG)2VPGEG(VPGAG)2]28 | >75°C | |
| E4 | [(VPGIG)5]15 | 16°C | |
| P1 | [(IPAVG)2VPAVG(IPAVG)2]16 | 25°C | Plastic-like |
| P2 | [(IPAVG)5]33 | 21°C | |
| P3 | [(IPAVG)5]16 | 26°C | |
Tt is for an aqueous solution at 1 mg/mL. The Tt of P1 is an estimate based on the measurement of Tt from a triblock copolymer system with identical P1 endblocks separated by central E1 block [5].
2.2. Elastin-mimetic triblock copolymers
The rational design strategy for elastin-mimetic amphiphilic block copolymers was derived from prior investigations [12-15,17]. Specifically, one block comprised of [(VPGAG)2(VPGEG)(VPGAG)2] repeats displays elastomeric behavior and is hydrophilic due to the presence of glutamic acid that is chargeable at physiological pH, while the other block containing [(I/V)PAVG] repeats displays thermoplastic behavior and consists of non-polar, hydrophobic residues. Synthetic block copolymers composed of hydrophilic and hydrophobic segments display phase separation behavior due to the solubility differences in aqueous solution [18]. In a similar manner, protein-based block copolymers composed of individual blocks having different mechanical properties and chemical polarity, can undergo phase separation from aqueous solution, which subsequently results in self-assembly of identical blocks. We have recently described the synthesis of a series of elastin-mimetic di- (P-P) and triblock (PEP) copolymers composed of a hydrophilic elastomeric block (E) and hydrophobic endblocks (P) that display plastic-like mechanical responses (Table 2) [3-5,7,10,19].
Table 2. Representative sequences of elastin-mimetic triblock copolymers.
| Triblock polypeptide sequence | [X] | |
|---|---|---|
| C5 | [VPAVG[(IPAVG)4(VPAVG)16IPAVG]- VPGVG[X]VPGVG- |
[(VPGVG)2VPGEG(VPGVG)2]30 |
| B9 | [VPAVG[(IPAVG)4(VPAVG)16IPAVG] | [(VPGVG)2VPGEG(VPGVG)2]48 |
| B10 | [(VPAVG](IPAVG)4] [(IPAVG)5]33-[X]- [(VPAVG)(IPAVG)4][(IPAVG)5]33VPGVG |
IPGAG(VPGAG)VPGEG(VPGAG)2[(VPGA G)2VPGEG (VPGAG)2]20 |
| LysB10 | VPAVGKVPAVG[(VPAVG](IPAVG)4][(IPAVG)5]33(IPAV G)-[X]-[(VPAVG)(IPAVG)4][(IPAVG)5]33IPAVGKAAKA |
IPAVGKAAK(VPGAG)[(VPGAG)2VPGEG (VPGAG)2]28(VPAVG)KAAK(VPGAGG) |
| R4 | VPAVGKVPAVG[(IPAVG)5]16(IPAVG)-[X]- VPGAG[(IPAVG)5]16(IPAVG)VPAVGKAAKA |
IPAVGKAAK(VPGAG)VPGIG[(VPGIG)5]15 (VPGIG)(VPAVG)KAAK(VPGAG) |
2.2.1. Elastin-mimetic triblock copolymers without crosslinking domain
We have synthesized recombinant elastin-mimetic triblock copolymers incorporating identical end blocks of hydrophobic plastic sequence separated by a central hydrophilic elastomeric block, which distinct phase transitions and mechanical responses for each of the block types [3]. These materials have capacity to form physical crosslinks through microphase separation with formation of stable protein network structure through the self-assembly of hydrophobic endblocks above their transition temperature. The synthetic strategy and method of for assembly of a concatamerized gene encoding elastomeric or plastic block have been described by Wright et al. in a prior review [1]. In brief, convergent assembly of triblock copolymers from plasmids containing individual plastic (P) and elastomeric (E) blocks is outlined in Figure 1. The pair of recombinant plasmids pPN and pPC encoding the N-terminal and C-terminal plastin blocks, respectively, was digested with restriction enzymes, SexA I and Xma I to afford two fragments. Ligation of the fragment from pPN and the fragment from pPC afforded the recombinant plasmid pPP, which encodes plastin diblock with a single SexA I restriction site separating into two P blocks. Plasmid pPP was then cleaved by restriction enzyme SexA I that enables the insertion of a variety of central elastomeric blocks. Subsequently a concatemer encoding the elastomeric repeat sequence, E, was inserted into the compatible SexA I site of pPP to afford a plasmid, pPEP, encoding an elastin-mimetic triblock protein polymer.
Figure 1. Scheme for for generating a triblock protein polymer with physical crosslinking sites.
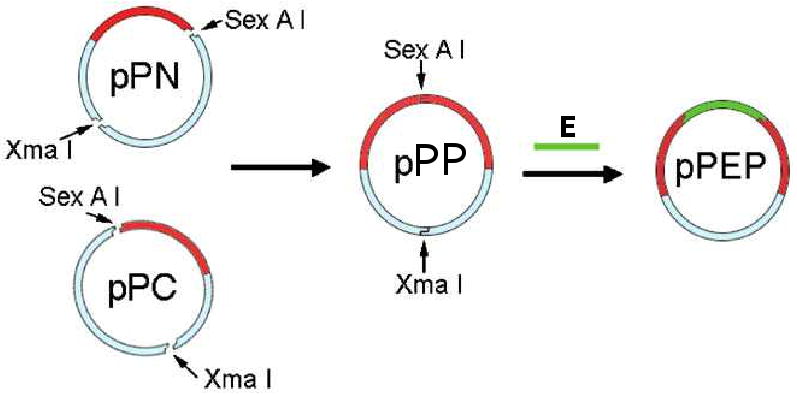
Synthesis of genes encoding amphiphilic elastin-mimetic triblock copolymers by assembly of a gene encoding a central hydrophilic block and identical plastic endblocks. (Reprinted from [3] with permission of American Chemical Society)
2.2.2. Elastin-mimetic triblock copolymers with crosslinking domain
Although elastin-mimetic block copolymers form physical crosslinks by self-assembly of hydrophobic blocks [4,5], physical networks may be deformed when subject to an external stress. Thus, potential limitations may exist in the application of elastin-based materials, solely stabilized by physical crosslinks, in tissue engineering or regenerative medicine. Thus, we have postulated that a chemically crosslinked triblock protein polymer may provide a strategy to maintain morphological integrity and enhance the mechanical response of materials under external loading forces. Tropoelastin occurs in its native form as elastic fiber networks enzymatically crosslinked through lysine residues [22,23]. Likewise, introduction of lysine residues into elastin-mimetic protein polymers provides free reactive amines for crosslinking using a variety of approaches. Characteristically, crosslinking of elastin-based biomaterials has been investigated via use of gamma irradiation [13,24], enzymes [25] and chemical crosslinkers, including isocyanates, glutaraldehyde, hydroxymethylphosphine, and genipin [24,26-30].
We have recently designed and synthesized a new class of recombinant elstin-mimetic triblock copolymers, LysB10 and R4 containing lysine residues between each plastic and elastomeric blocks (H2N-K-P-KAAK-E-KAAK-P-KAAK) (Fig. 2) [19]. LysB10 was designed with two identical hydrophobic endblocks, consisting of 33 repeats of the plastic pentapeptide sequence, [IPAVG], separated by hydrophilic midblock consisting of 28 repeats of the elastomeric pentapeptide sequence, [(VPGVG)2(VPGEG)(VPGVG)2]. In addition, R4 was designed with two identical hydrophobic endblock, consisting of 16 repeats of the plastic pentapeptide sequence, [IPAVG], separated by more hydrophobic midblock, consisting of 15 repeats of the elastomeric pentapeptide sequence, [VPGIG]. While both midblocks would display elastomeric behavior when chemically cross-linked, the transition temperature of midblock of R4 is significantly different from that of LysB10 due to the incorporation of hydrophobic residue, isoleucine, in which R4 is considered as predominantly hydrophobic polymer. We employed two lysine-containing polylinkers that can introduce two free amines at ends of each block. The lysine insert (I) encodes the crosslinking sequence, K-A-A-K, which is inserted between the plastic and elastomeric blocks. The lysine adaptor (A) encodes a single N-terminal lysine residue (H2N-VPAVG-K) and two C-terminal lysine residues (K-A-A-K). Plasmids containing the lysine insert and lysine adaptor are designated as pI and pA, respectively.
Figure 2. Representation of a triblock protein polymer with chemical and physical crosslinking sites.
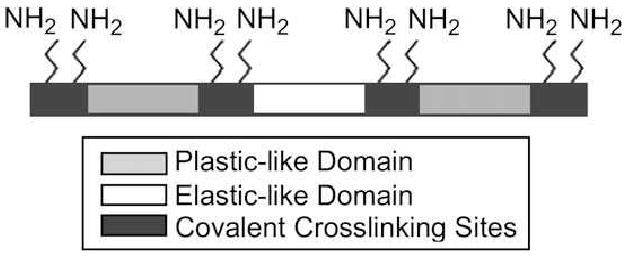
Crosslinkable elastin-mimetic triblock copolymers containing eight free amines placed at the end of each block that are available for chemical crosslinking. (Reprinted from [19] with permission of Elsevier)
Concatamers encoding the plastic block, P, were inserted into respective restriction sites, Bbs I and BsmB I, of pI to afford two plasmids, pIP and pPI. Analogously, elastomeric concatamers, E, were inserted into the BsmB I site of the lysine insert in the pPI to afford a diblock plasmid, pPIE. Two plasmids were constructed from pI, such that the order of the restriction sites, Bbs I and BsmB I, respectively, employed for the insertion of the plastic concatamer, P. The two recombinant plasmids pIP and pPIE were digested with Bbs I/Xma I and BsmB I/Xma I, respectively, to afford two fragments containing plastin blocks to be ligated each other. As the restriction site Xma I digests within the Zeocin coding region of pZErO-1, only clones containing the correctly assembled triblock ensure the selective propagation of ligation products. Ligation of the fragment from pIP and the fragment from pPIE afforded the recombinant plasmid pPIEIP, which encoded triblock copolymer with lysine domains between plastic and elstomeric blocks. The triblock gene, PIEIP, was inserted into a single BsmB I site within the lysine adaptor of plasmid, pA, to afford the final triblock gene product with lysine crosslinking domains, pAPIEIPA (Fig. 3).
Figure 3. Scheme for generating a triblock protein polymer with chemical and physical crosslinking sites.

Synthesis of gene encoding amphiphilic elastin-mimetic triblock copolymers with crosslinking domain via assembly of each gene encoding a central hydrophilic block and identical hydrophobic endblocks.
3. Drug delivery applications
A variety of drug delivery platforms, such as nano/microparticles [31-33], hydrogels [34-36], and films [37-40], have been developed from synthetic block copolymers for the purpose of local and controlled delivery of bioactive molecules. Synthetic block copolymers with distinct block polarity can self-assemble into a range of supramolecular structures that facilitate the loading of hydrophobic drugs into local hydrophobic microenvironments. Likewise, elastin-mimetic triblock copolymers undergo phase separation through self-assembly of hydrophobic plastic blocks to afford the formation of hydrophobic aggregates ranging in dimensions from the nano- to the micron scale. The phase transition can be readily modulated by variation of size, composition and sequence of the polypeptides that comprise the blocks, which enables precise control of molecular self-assembly through subtle changes in temperature, pH, and ionic strength of the aqueous solution. Nature of the aggregates can be varied from discrete nanoparticles to extended networks through appropriate choice of the block copolymer architecture. In addition, diverse platforms, including micellar nanoparticles [41], hydrogels [42] and films [3] that incorporate drugs can be formed through appropriate processing of these materials. Amphiphilic elastin-mimetic triblock copolymers hold significant promise as drug delivery vehicles. However, the use of these materials for direct medical applications still remains unexplored.
3.1. Micelles
Polymeric micelles can be formed through the self-assembly of amphiphilic block copolymers to form a compact core surrounded by hydrophilic shell, into diverse structures including rods, cylinders, spheres and vesicles [43-46]. Micellar nano- and microparticle systems have recently been of significant interest for both drug delivery and bioimaging applications. Most elastin-mimetic micelles reported so far are based on amphiphilic diblock copolymer systems comprised of hydrophilic block and hydrophobic block to form spherical nanoparticle [1,47]. In our own studies, the elastin-mimetic triblock copolymer, B9, was synthesized with a transition temperature of ∼21°C [3,41]. Dynamic light scattering (DLS) measurements of dilute solutions of B9 demonstrated a monodisperse size distribution consistent with a spherical shape with an RH ranging between 90 and 120 nm (Fig. 4). Comparison of RH values of protein micelles formed at low and high temperature revealed the presence of thermally sensitive size trigger, such that micelles decreased in size upon temperature elevation. Similarly, D'Souza et al have reported the micelle formation of dilute C5 triblock copolymer solutions with RH value of 75 nm measured by DLS at 15 °C [48].
Figure 4. Thermally responsive protein micelles regulated through a reversible switch in protein secondary structure.
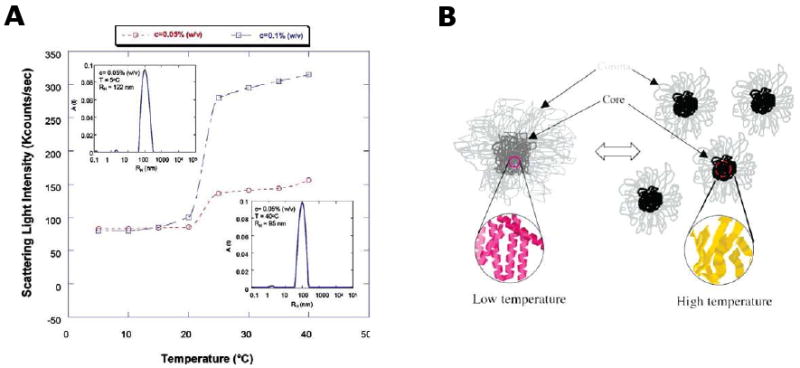
(A) Scattering light intensity as a function of temperature and protein polymer concentration. (B) Schematic representation for nanoparticle formation from elastin-mimetic triblock copolymers in which size and core density of micelles show temperature-dependency Figures adapted from [39]. (Reprinted from [41] with permission of American Chemical Society)
Encapsulation of a hydrophobic molecule within the micelle core was confirmed using a polarity-sensitive fluorescent hydrophobic molecule, pyrene [46]. A significant enhancement of fluorescent intensity of pyrene was obtained when the solution temperature was increased from 15 to 30 °C consistent with the formation of an increasingly hydrophobic core. Given the potential of protein-based block copolymer systems to provide a biocompatible and biodegradable vehicle for drug delivery [1,41,42,47-50], it is possible to envision the use of stimuli responsive micelles as targeted drug delivery systems for pharmacological and cosmetic applications.
3.2. Injectable gels
Stimulus-responsive polymeric materials have been developed for drug delivery, cell encapsulation and tissue engineering in a variety of platforms. Among them, temperature-responsive polymers exhibiting a lower critical solution temperature (LCST) are attractive as injectable materials [51-53]. The most widely used and well-known temperature-responsive polymers are poly(N-isopropylacrylamide) (pNIPAAm)-based block copolymers and the poloxamer (Pluronics®). They can be injected as solutions mixed with drugs or cells below the transition temperature of the polymer with gel formation at body temperature [34-36].
Elastin-mimetic polypeptides that undergo temperature-dependent phase transition facilitate processing as injectable formats with various drugs. Adams et al. have recently examined elastin-like polypeptides (ELP) for sustained release of antibiotics, for use in orthopedic applications [54]. The release of vancomycin was prolonged over 20 days and only 50% was released. Shamji et al. have developed ELPs for perineural injection that provided the possibility for controlled release of immunomodulator therapeutics [55]. Hart et al. have studied the release of model compounds, such as theophylline, vitamin B12 and ovalbumin from a triblock copolymer, C5 [42].
We have demonstrated that elastin-mimetic triblock copolymers undergo reversible gelation [2,5,7]. Above the gel point, the shear storage (G′) and loss modulus (G″) increase while tan δ (G″/G′) decreases, consistent with the formation of a viscoelastic hydrogel. We have revealed that difference in midblock size and pentapeptide sequence critically affect on viscoelastic and mechanical properties of the materials [5,7,19,56]. Elastin-mimetic triblock copolymers, B9 and C5, were designed with identical elastomeric midblock sequences, [(VPGVG)2(VPGEG)(VPGVG)2], and exactly same block length of two plastic endblocks ([(IPAVG)4(VPAVG)16IPAVG]), but the midblock length of B9 (n=48, n: number of monomer unit) is 62 % longer than that of C5 (n=30). Rheological analysis of both gels has shown that B9 exhibited an increase in viscosity compared to C5. Gel forming temperature of B9 triblock copolymer (18 °C) is higher than that of C5 (14.8 °C). The tan δ measured for P-P diblock copolymer, which consists of two identical plastic endblocks divided by short peptide consisting of two repeats of the elastomeric pentapeptide sequence, (VPGVG)2, was nearly four fold greater than that of B9 and ten fold than C5. The rheology data demonstrate that elastin-mimetic triblock copolymer becomes more viscoelastic as midblock size is larger. Likewise, to further enhance the properties of these materials, changes in endblock size resulted in significant changes in thermal and mechanical properties. Elastin-mimetic triblock copolymer, B10, (n=33) was designed with nearly twice larger hydrophobic endblocks than B9 (n=16), but exactly same midblock in size. Gel forming temperature of concentrated solutions for B10 was five degrees lower than that for B9 in good agreement with difference in the transition temperature of the dilute protein solutions due to an increase in the endblock size. The rheological behavior of B10 solutions is different from that of B9. The difference between values of shear storage and loss modulus for B10 was two orders of magnitude less than that observed for B9. The decrease of tan δ is consistent with formation of a viscoelastic gel. In this regard, the mechanical properties of the injectable hydrogel and the release rate of hydrophobic drugs, therapeutic proteins and genes for gene therapy application may be controlled through changes in midblock size and endblock size as well.
3.3. Films
Drug-eluting films can be used in a variety of medical applications. In such applications, polymeric films containing drugs can be implanted as drug delivery system at specific sites in the body. Controlling nano/microscale structure of the films provides a means to tailor drug release [37]. Examples for drug-eluting films include degradable biomedical devices [57,58], film coatings on medical devices [38,39] and film-based implants [40]. The potential versatility of elastin-mimetic amphiphilic triblock copolymers for drug delivery application can be expanded by processing the protein polymer into drug-loaded film by a casting/solvent evaporation process [3]. Our investigations demonstrate that the generation of large triblock protein polymers offers a unique opportunity to systematically modify material microstructure on both nano- and meso-length scales with tailored mechanical behavior and drug elution rates.
Mechanical behavior of the triblock B9 depends on processing conditions, including solvent, temperature and pH (Figure 5). Conditions that promote phase separation afford materials with greater elastomeric character or plastic character. Trifluoroethanol (TFE) solvates both blocks, which promotes the interpenetration of blocks while water acts as a selective solvent for the hydrophilic midblock, which results in phase separation of the hydrophobic endblocks. Additionally, mechanical properties of elastin-mimetic triblock copolymers are dependent on the length of midblock. The observed difference in previous rheological behavior of B9 and C5 hydrogels extends to uniaxial stress-strain behavior measured from B9 and C5 films. For mechanical response only minor difference in Young's modulus (0.03-0.05 MPa) and ultimate tensile strength (0.78-0.96 MPa) was observed while strain at failure was strikingly increased for B9 films with larger midblock size than C5.
Figure 5. Uniaxial mechanical responses of elastin-mimetic protein polymer films.
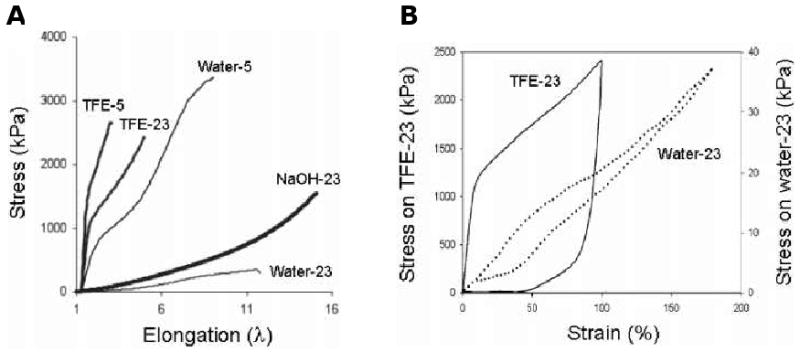
(A) Stress-strain curve for B9 films cast from water, TFE or NaOH at 5 and 23°C and rehydrated in PBS. (B) Hysteresis curves for B9 cast from water and TFE at 23°C. (Reprinted from [3] with permission of American Chemical Society)
The mesoscopic morphology of hydrated B9 films in PBS cast from water and TFE was imaged by cryo-HRSEM (Fig. 6). Interestingly, water cast films were characterized by an open-cell microstructure with half-micron size pores, whereas TFE cast film featured fibrous morphology. In TFE cast films, phase separation occurred prior to the film formation and coalescence of particles led to the fiber-based morphology.
Figure 6. Microstructure of elastin-mimetic triblock protein polymer hydrogels.
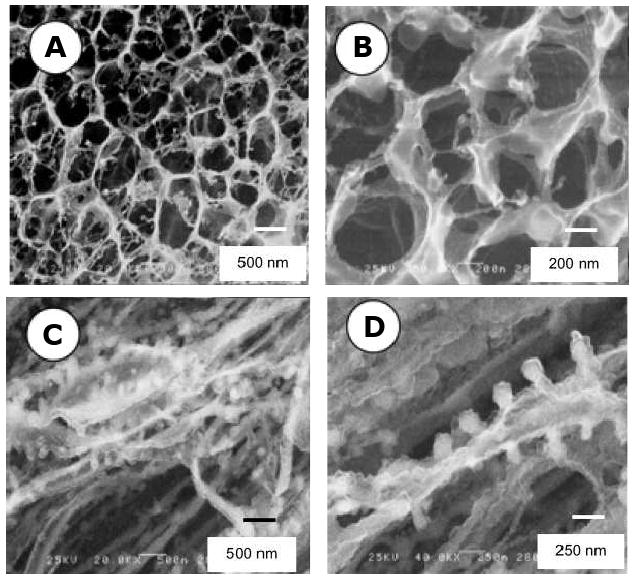
Cryo-HRSEM micrographs of B9 films initially cast from water (A,B) or TFE (C,D) and rehydrated in PBS at room temperature. (Reprinted from [3] with permission of American Chemical Society)
We have measured the release rate of a lipophilic hydrophobic drug from microphase separated films of amphiphilic elastin-mimetic triblock copolymer, B9 (Fig. 7). Sphingosine-1-phosphate (S1P) used as a model drug is a potent anti-inflammatory agent composed of a long hydrocarbon chain and a charged head group and activates cell surface G-protein coupled receptors (S1P receptors). The release rate of S1P from water-cast films was approximately 30 fold higher than that from TFE-cast films due to differences in process induced changes in microscale structure demonstrated by solid state NMR spectroscopy.
Figure 7. Elution rates of an amphiphilic compound from protein copolymer films.
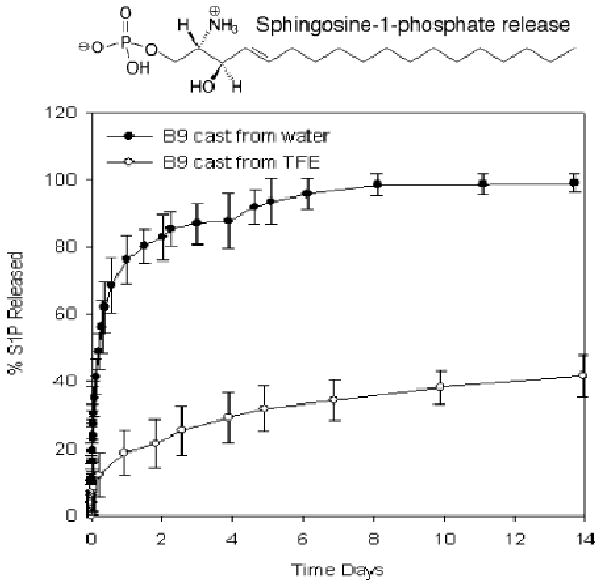
Drug release rates are dependent upon film processing conditions that influence protein microstructure and microphase separation of protein blocks. The in vitro release profile is presented for S1P from hydrated B9 films cast from water or TFE at room temperature. (Reprinted from [3] with permission of American Chemical Society)
4. Implant applications
Elastin-based biomaterials have been processed as films [10,37-40], fibers [7,59], and hydrogels [34-36,56] for a variety of implant applications.
4.1. Films
Cardiovascular disease remains one of the leading causes of death in the United States. Two major synthetic polymeric materials clinically used for vascular grafts are expanded polytetrafluoroethylene (ePTFE) and Dacron. Although both synthetic grafts have performed successfully for diameters larger than 6 mm, these devices remain limited for small-diameter (4 mm) vascular applications due to thrombus formation and the development of anastomotic intimal hyperplasia. Thus, the need for generating small-diameter vascular graft has become increasingly important for vascular implant. To overcome these problems, synthetic grafts have been coated with biocompatible materials, such as albumin, heparin, and prostacyclin analogues that may inhibit platelet adhesion and aggregation [60-63]. As one of the major structural components in the vascular wall, native elastin has elastic properties and little platelet adhesion. Motivated by that observation, elastin-based materials have been used as an alternative surface coating on synthetic vascular grafts to improve long-term patency and function [11,64]. Ito et al have coated Dacron vascular grafts with elastin with inhibition of smooth muscle cell migration that contributes to intimal hyperplasia [65]. Woodhouse et al. reported coating a recombinant human elastin polypeptide on the surface of catheters and demonstrated reduced platelet deposition and activation in vitro and delayed catheter occlusion in vivo [11]. Our group examined the blood contacting properties of an elastin-mimetic triblock copolymer, B9, when coated as a thin film on the surface of a 4 mm diameter ePTFE vascular graft [10]. The results of comparative studies among un-coated control, impregnated and coated grafts demonstrate the fabrication of B9-coated ePTFE vascular grafts. Impregnation and coating of B9 on ePTFE grafts have been monitored by Coomassie dye staining, water contact angles, and high resolution SEM (HRSEM) (Fig. 8). For example, B9-coated sample was well stained with the blue dye and remained stably after a constant flow of PBS buffer for a day, whereas the control was not stained. HRSEM images after impregnation and coating suggest disappearance of fibril structure of un-coated ePTFE and development of an open-cell microstructure after multi-layer coating. Vascular grafts with or without a luminal coating of B9 were studied in baboons using an ex vivo femoral arteriovenous shunt model in which surfaces were exposed to blood flow at 100 mL/min (Fig. 9A). Deposition of 111In-labeled platelets was measured for a period of 1 hour [66]. In vivo studies demonstrated minimal thrombogenicity when B9 is coated on the lumen of a small diameter vascular graft in comparison with un-coated ePTFE grafts (Fig. 9B). A remarkable decrease in fibrinogen adsorbed on the B9-coated grafts (0.03±0.02 mg/cm2) was observed (p<0.05) when compared to un-coated control grafts (1.44±0.75 mg/cm2). As a result, this elastin-mimetic triblock copolymer, B9, exhibited a strong potential for use as a thromboresistant coating for small diameter vascular grafts. In addition, the results of our study indicate that elastin-mimetic triblock copolymers in Table 2 would be a valuable vascular biomaterial for implantable grafts. Successful implantable vascular graft material also requires cell adhesion onto vascular grafts. Cell adhesion can be obtained by incorporating cell-binding sequences into the elastin-mimetic sequences. Heilshorn et al. have demonstrated incorporation of the cell-binding domains into elastin-like polypeptide (aECM protein) enabled the adhesion of human vein endothelial cells (HUVEC) and cells were attached under stresses similar to the in vivo system [67]. Nagapudi et al. have also confirmed that amphiphilic drugs could be incorporated into B9 as an additional mechanism to control biological responses [3]. We anticipate that elastin-mimetic triblock copolymers hold significant potential in a range of implant applications, including synthetic vascular graft coatings and drug-eluting films coated on medical devices, especially drug-eluting stent coating [39].
Figure 8. An ePTFE vascular graft coated with an elastin-mimetic protein polymer.

(A) Macroscopic photographs of unstained (left) and Coomassie-stained (right) graft samples: plain ePTFE, after B9 impregnation, after multilayer B9 deposition, and after exposure to PBS for 24 at 37°C at a defined flow rate. (B) SEM images of ePTFE vascular grafts processed by critical point drying. (Reprinted from [10] with permission of Elsevier)
Figure 9. Examination of the blood compatibility of small diameter (4 mm) ePTFE vascular grafts coated with an elastin-mimetic protein polymer.
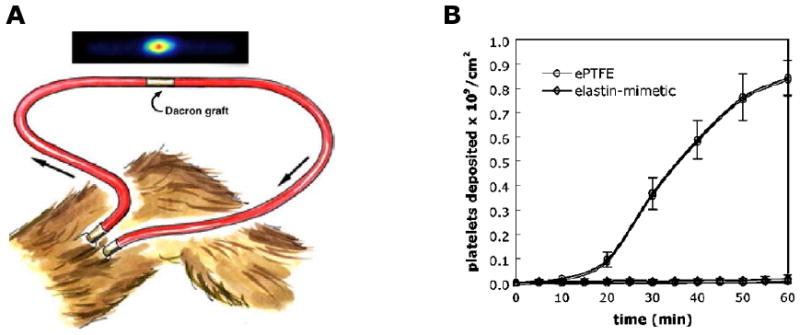
(A) Schematic illustration of an ex vivo femoral arteriovenous shunt used to assess platelet deposition in a baboon model [63]. (B) Platelet deposition normalized by surface area over a 60-min time period (n=6). (Reprinted from [10] with permission of Elsevier)
4.2. Fibers
As biomaterials to fabricate artificial organs and engineered tissues, elastin provides a great opportunity to effectively tailor targeted mechanical and biological behavior to obtain a desired clinical response [68]. Its versatility as a scaffold material includes its ability to be processed into fiber networks. Electrospinning is often used for preparing protein fibers with diameters in the hundreds of nanometers [69]. The assembly of recombinant elastin fiber networks provides an important new design strategy for generating tubular constructs for a clinically durable small diameter arterial substitute. Huang et al. first demonstrated that electrospining could be utilized to fabricate recombinant elastin-mimetic polypeptides derived from pentameric repeat sequence [(VPGVG)4VPGKG] into nonwoven fabrics consisting of fiber networks that mimic native elastin scaffolds [70]. Similarly, electrospinning has been recently employed to fabricate 2-D and 3-D fiber networks from elastin-mimetic triblock copolymer with fiber size dependent on processing conditions (Fig. 10). Tubular constructs have been produced by fiber deposition on a collecting mandrel with controlled rotational and translational speeds. Of interest, thermal annealing at 60 °C has been observed to changes the mechanical response of electrospun elastin protein polymer networks with a two fold increase in Young's modulus (0.366±0.05 MPa) and 30 % in tensile strength comparable to that within an arterial wall (Y ∼0.3 MPa) [59,71].
Figure 10. Fabrication of a small diameter vascular graft from electrospun elastin-mimetic protein polymers.

SEM micrographs of fibers spun from a 10-wt% solution of B9 in TFE at a magnification of (A) 1000× and (B) 10,000×. Fiber networks produced from an electrospun recombinant elastin-mimetic triblock copolymer. (Reprinted from [7] with permission of Elsevier) (C) Cryo-HRSEM micrograph of fiber networks electrospun from a 12-wt% solution of B9 in TFE. (D) A tubular conduit was fabricated from electrospun protein fibers using a rotating mandrel as the collecting apparatus. (Reprinted from [59] with permission of Koninklijke Brill)
4.3. Hydrogels
Hydrogels are complex 3-D network of polymer that are water-insoluble, but can highly absorb water. For clinical applications, hydrogels can be used as implants in surgical sutures, artificial organs, membranes, drug delivery systems and contact lenses. Voldrich et al have reported successful use of poly(glycol monomethacrylate) hydrogels as implants in human with nasal plastic surgery [72]. Other examples include poly(methyl methacrylate) (PMMA) for ocular implants, poly(lactide) and poly(glycolide) for surgical sutures and silicone as breast implants. As clinical implants, hydrogels should have properties such as, most importantly, biocompatibility, mechanical behavior and biodegradability of a polymer scaffold. Similarly, the rate and extent of protein polymer biodegradation is essential in assessing the suitability of elastin-mimetic protein polymers for implant applications. However, relatively few studies have characterized in vivo response to elastin or elastin-mimetic biomaterials. Mithieux et al. demonstrated that after a 13-week period chemically crosslinked recombinant human elastin subcutaneous implants were surrounded by a fibrous capsule with a minimal to moderate inflammatory response [71]. Rincon et al. have recently prepared microparticles derived from pentameric sequence poly(VPAVG) and evaluated biocompatibility after subcutaneous and intravitreal injections. A significant inflammatory response was not observed over a 1 month period [73]. Most recently, Mithieux et al. have shown that recombinant human tropoelastin formed an irreversible hydrogel at pH 10 through association of spherical particles [74]. After subcutaneous injection, 2 weeks after implantation the hydrogel was surrounded by a fibrous capsule with a mild inflammatory response.
Our lab has synthesized several different types of elastin-mimetic triblock copolymers, B9, LysB10, and R4, in which the latter two copolymers have chemically crosslinkable sites. In designing those triblock copolymers, the midblock structure of R4 is significantly different from two other triblocks. Despite the presence of an elastomeric midblock, the R4 midblock is remarkably more hydrophobic than LysB10. As apparent by a considerably higher tan δ and complex viscosity, rheological analysis demonstrated that R4 gels were more viscoelastic than LysB10 gels. This difference indicates that amino acid sequence of midblock is also an important factor to determine the viscoelastic properties of the triblock copolymers. Using these protein polymers, we generated hydrogels as in situ forming gels for injection and as cylindrically cast gels for implants. In vivo responses for periods exceeding one year were studied in a murine model. Injectable hydrogels can be locally introduced using a syringe and gel at body temperature. They can serve as filler materials in defect sites as well as serve to promote wound healing through the incorporation of bioactive molecules, such as drugs, growth factors, and cells through mixing [75]. Moreover, in-situ gel forming and injectable triblock copolymers can serve as an embolic agent for a minimally invasive, non-surgical procedure. The triblock hydrogels may be able to apply to a blood vessel or vessel wall such as a wall rupture or aneurysms in a soluble form that subsequently can form physically or chemically crosslinked hydrogels. Therefore, the triblock hydrogel displaces or prevents further blood flow in a region. Injectable B9 hydrogels were implanted into either the subcutaneous space or the peritoneal cavity in a mouse model to evaluate early in vivo responses (Fig. 11). After injection, samples in subcutaneous and peritoneal locations were optically transparent. All implants were surrounded by a thin fibrous capsule without cellular infiltration. Macrophages were present along the periphery consistent with a mild inflammatory response. FACS analysis of peritoneal lavage fluid demonstrated similar number and type of inflammatory cells when compared to those mice without an implant.
Figure 11. In vivo responses to elastin-mimetic protein polymer implants.

Short-term host implant responses after (A) subcutaneous and (B) peritoneal injection of B9 in a mouse model. Subcutaneous implants were examined 3 weeks and peritoneal implants 1 week after implantation. (Reprinted from [56] with permission of Elsevier)
Chemically crosslinkable triblock copolymer gels have also been produced as cylindrical implants [19,56]. Over a 3-week period, implants retained their original structure and demonstrated the presence of a mild inflammatory response with macrophages at the periphery of the gels.
Evaluation of integrity for a physically crosslinked B9 hydrogel implanted subcutaneously mice demonstrated a decrease in gel dimensions over a 1 year period through MR image analysis (Fig. 12). During the first 2-3 months after implantation, cross-section area and gel volume decreased by 30 to 40% with little change thereafter. The equilibrium water content of the B9 hydrogel is approximately 82 % which is high water-content. At initial implantation, water desorption from the hydrogel may cause the change in gel volume after implantation. After a certain time point, water desorption equilibrium occurs and then the volume change is not observed. At one year samples remained intact with limited inflammatory response, fibrous capsule formation, and cellular infiltration into gels. Cryo-HRSEM of 1-year implants at the time of removal revealed that the microstructure of the gel displayed an open-cell morphology similar to that observed before implantation.
Figure 12. Serial MR imaging of subcutaneous protein polymer implants over 1 year.
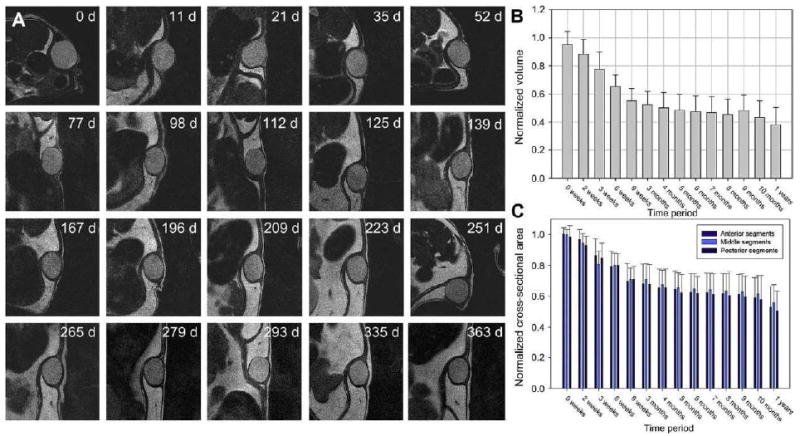
(A) Representative serial images of B9 implants over a 1-year period. (B) Normalized implant volumes and (C) implant cross-section areas were determined over a 1-year implant period (n=7, mean ± SD). (Reprinted from [56] with permission of Elsevier)
5. Conclusions
Elastin-mimetic block copolymers are a promising new class of biomaterials for the drug delivery and implant applications. These materials exhibit high level of biocompatibility and a tunable range of biomechanical properties as a result of the precise control of pentapeptide sequence, block sequence, and processing conditions. A variety of opportunities exists in the processing of these protein polymers into nanoparticles, hydrogels, films or fibers for a number of implant applications in orthopedics, as well as in plastics, cardiovascular, and general surgery. These applications can be further extended by incorporating bioactive drugs, proteins, and cells, within or onto the protein polymer network.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wright ER, Conticello VP. Self-assembly of block copolymers derived from elastin-mimetic polypeptide sequences. Adv Drug Deliv Rev. 2002;54(8):1057–73. doi: 10.1016/s0169-409x(02)00059-5. [DOI] [PubMed] [Google Scholar]
- 2.Wright ER, McMillan RA, Cooper A, Apkarian RP, Conticello VP. Thermoplastic elastomer hydrogels via self-assembly of an elastin-mimetic triblock polypeptide. Advanced Functional Materials. 2002;12(2):149–154. [Google Scholar]
- 3.Nagapudi K, Brinkman WT, Leisen J, Thomas BS, Wright ER, Haller C, Wu XY, Apkarian RP, Conticello VP, Chaikof EL. Protein-based thermoplastic elastomers. Macromolecules. 2005;38(2):345–354. [Google Scholar]
- 4.Wu X, Sallach R, Haller CA, Caves JA, Nagapudi K, Conticello VP, Levenston ME, Chaikof EL. Alterations in physical cross-linking modulate mechanical properties of two-phase protein polymer networks. Biomacromolecules. 2005;6(6):3037–44. doi: 10.1021/bm0503468. [DOI] [PubMed] [Google Scholar]
- 5.Wu X, Sallach RE, Caves JM, Conticello VP, Chaikof EL. Deformation responses of a physically cross-linked high molecular weight elastin-like protein polymer. Biomacromolecules. 2008;9(7):1787–94. doi: 10.1021/bm800012x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagapudi K, Brinkman WT, Leisen JE, Huang L, McMillan RA, Apkarian RP, Conticello VP, Chaikof EL. Photomediated solid-state cross-linking of an elastin-mimetic recombinant protein polymer. Macromolecules. 2002;35(5):1730–1737. [Google Scholar]
- 7.Nagapudi K, Brinkman WT, Thomas BS, Park JO, Srinivasarao M, Wright E, Conticello VP, Chaikof EL. Viscoelastic and mechanical behavior of recombinant protein elastomers. Biomaterials. 2005;26(23):4695–4706. doi: 10.1016/j.biomaterials.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 8.Herrero-Vanrell R, Rincon AC, Alonso M, Reboto V, Molina-Martinez IT, Rodriguez-Cabello JC. Self-assembled particles of an elastin-like polymer as vehicles for controlled drug release. J Control Release. 2005;102(1):113–22. doi: 10.1016/j.jconrel.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 9.Dreher MR, Raucher D, Balu N, Michael Colvin O, Ludeman SM, Chilkoti A. Evaluation of an elastin-like polypeptide-doxorubicin conjugate for cancer therapy. J Control Release. 2003;91(1-2):31–43. doi: 10.1016/s0168-3659(03)00216-5. [DOI] [PubMed] [Google Scholar]
- 10.Jordan SW, Haller CA, Sallach RE, Apkarian RP, Hanson SR, Chaikof EL. The effect of a recombinant elastin-mimetic coating of an ePTFE prosthesis on acute thrombogenicity in a baboon arteriovenous shunt. Biomaterials. 2007;28(6):1191–7. doi: 10.1016/j.biomaterials.2006.09.048. [DOI] [PubMed] [Google Scholar]
- 11.Woodhouse KA, Klement P, Chen V, Gorbet MB, Keeley FW, Stahl R, Fromstein JD, Bellingham CM. Investigation of recombinant human elastin polypeptides as non-thrombogenic coatings. Biomaterials. 2004;25(19):4543–53. doi: 10.1016/j.biomaterials.2003.11.043. [DOI] [PubMed] [Google Scholar]
- 12.van Hest JC, Tirrell DA. Protein-based materials, toward a new level of structural control. Chem Commun (Camb) 2001;19:1897–904. doi: 10.1039/b105185g. [DOI] [PubMed] [Google Scholar]
- 13.Urry DW, Shaw RG, Prasad KU. Polypentapeptide of elastin: temperature dependence of ellipticity and correlation with elastomeric force. Biochem Biophys Res Commun. 1985;130(1):50–7. doi: 10.1016/0006-291x(85)90380-8. [DOI] [PubMed] [Google Scholar]
- 14.Luan CH, Parker TM, Gowda DC, Urry DW. Hydrophobicity of amino acid residues: differential scanning calorimetry and synthesis of the aromatic analogues of the polypentapeptide of elastin. Biopolymers. 1992;32(9):1251–61. doi: 10.1002/bip.360320914. [DOI] [PubMed] [Google Scholar]
- 15.Urry DW, Hugel T, Seitz M, Gaub HE, Sheiba L, Dea J, Xu J, Parker T. Elastin: a representative ideal protein elastomer. Philos Trans R Soc Lond B Biol Sci. 2002;357(1418):169–84. doi: 10.1098/rstb.2001.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamaoka T, Tamura T, Seto Y, Tada T, Kunugi S, Tirrell DA. Mechanism for the phase transition of a genetically engineered elastin model peptide (VPGIG)40 in aqueous solution. Biomacromolecules. 2003;4(6):1680–5. doi: 10.1021/bm034120l. [DOI] [PubMed] [Google Scholar]
- 17.Urry DW, Luan CH, Parker TM, Gowda DC, Prasad KU, Reid MC, Safavy A. Temperature of Polypeptide Inverse Temperature Transition Depends on Mean Residue Hydrophobicity. Journal of the American Chemical Society. 1991;113(11):4346–4348. [Google Scholar]
- 18.Spontak RJ, Patel NP. Thermoplastic elastomers: fundamentals and applications. Current Opinion in Colloid & Interface Science. 2000;5(5-6):334–341. [Google Scholar]
- 19.Sallach RE, Cui W, Wen J, Martinez A, Conticello VP, Chaikof EL. Elastin-mimetic protein polymers capable of physical and chemical crosslinking. Biomaterials. 2009;30(3):409–22. doi: 10.1016/j.biomaterials.2008.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Urry DW. Protein-based materials with a profound range of properties and applications: The elastin ΔTt hydrophobic paradigm. Birkhauser; Boston: 1997. pp. 133–177. [Google Scholar]
- 21.Reguera J, Lagaron JM, Alonso M, Reboto V, Calvo B, Rodriguez-Cabello JC. Thermal behavior and kinetic analysis of the chain unfolding and refolding and of the concomitant nonpolar solvation and desolvation of two elastin-like polymers. Macromolecules. 2003;36(22):8470–8476. [Google Scholar]
- 22.Urry DW, Parker TM. Mechanics of elastin: molecular mechanism of biological elasticity and its relationship to contraction. J Muscle Res Cell Motil. 2002;23(5-6):543–59. [PubMed] [Google Scholar]
- 23.Vrhovski B, Weiss AS. Biochemistry of tropoelastin. Eur J Biochem. 1998;258(1):1–18. doi: 10.1046/j.1432-1327.1998.2580001.x. [DOI] [PubMed] [Google Scholar]
- 24.Lee J, Macosko CW, Urry DW. Elastomeric polypentapeptides cross-linked into matrixes and fibers. Biomacromolecules. 2001;2(1):170–9. doi: 10.1021/bm0000900. [DOI] [PubMed] [Google Scholar]
- 25.Kagan HM, Tseng L, Trackman PC, Okamoto K, Rapaka RS, Urry DW. Repeat polypeptide models of elastin as substrates for lysyl oxidase. J Biol Chem. 1980;255(8):3656–9. [PubMed] [Google Scholar]
- 26.Bellingham CM, Lillie MA, Gosline JM, Wright GM, Starcher BC, Bailey AJ, Woodhouse KA, Keeley FW. Recombinant human elastin polypeptides self-assemble into biomaterials with elastin-like properties. Biopolymers. 2003;70(4):445–55. doi: 10.1002/bip.10512. [DOI] [PubMed] [Google Scholar]
- 27.Vieth S, Bellingham CM, Keeley FW, Hodge SM, Rousseau D. Microstructural and tensile properties of elastin-based polypeptides crosslinked with genipin and pyrroloquinoline quinone. Biopolymers. 2007;85(3):199–206. doi: 10.1002/bip.20619. [DOI] [PubMed] [Google Scholar]
- 28.Nowatzki PJ, Tirrell DA. Physical properties of artificial extracellular matrix protein films prepared by isocyanate crosslinking. Biomaterials. 2004;25(7-8):1261–7. doi: 10.1016/s0142-9612(03)00635-5. [DOI] [PubMed] [Google Scholar]
- 29.Trabbic-Carlson K, Setton LA, Chilkoti A. Swelling and mechanical behaviors of chemically cross-linked hydrogels of elastin-like polypeptides. Biomacromolecules. 2003;4(3):572–80. doi: 10.1021/bm025671z. [DOI] [PubMed] [Google Scholar]
- 30.Lim DW, Nettles DL, Setton LA, Chilkoti A. In situ cross-linking of elastin-like polypeptide block copolymers for tissue repair. Biomacromolecules. 2008;9(1):222–30. doi: 10.1021/bm7007982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiellini F, Piras AM, Errico C, Chiellini E. Micro/nanostructured polymeric systems for biomedical and pharmaceutical applications. Nanomed. 2008;3(3):367–93. doi: 10.2217/17435889.3.3.367. [DOI] [PubMed] [Google Scholar]
- 32.Kwon GS, Okano T. Soluble self-assembled block copolymers for drug delivery. Pharm Res. 1999;16(5):597–600. doi: 10.1023/a:1011991617857. [DOI] [PubMed] [Google Scholar]
- 33.Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. Biodegradable long-circulating polymeric nanospheres. Science. 1994;263(5153):1600–3. doi: 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- 34.Temenoff JS, Mikos AG. Injectable biodegradable materials for orthopedic tissue engineering. Biomaterials. 2000;21(23):2405–12. doi: 10.1016/s0142-9612(00)00108-3. [DOI] [PubMed] [Google Scholar]
- 35.Hatefi A, Amsden B. Biodegradable injectable in situ forming drug delivery systems. J Control Release. 2002;80(1-3):9–28. doi: 10.1016/s0168-3659(02)00008-1. [DOI] [PubMed] [Google Scholar]
- 36.Yu L, Ding J. Injectable hydrogels as unique biomedical materials. Chem Soc Rev. 2008;37(8):1473–81. doi: 10.1039/b713009k. [DOI] [PubMed] [Google Scholar]
- 37.Zilberman M, Shifrovitch Y, Aviv M, Hershkovitz M. Structured drug-eluting bioresorbable films: microstructure and release profile. J Biomater Appl. 2009;23(5):385–406. doi: 10.1177/0885328207088261. [DOI] [PubMed] [Google Scholar]
- 38.Langer R. New methods of drug delivery. Science. 1990;249(4976):1527–33. doi: 10.1126/science.2218494. [DOI] [PubMed] [Google Scholar]
- 39.Acharya G, Park K. Mechanisms of controlled drug release from drug-eluting stents. Adv Drug Deliv Rev. 2006;58(3):387–401. doi: 10.1016/j.addr.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 40.Kawatsu S, Oda K, Saiki Y, Tabata Y, Tabayashi K. External application of rapamycin-eluting film at anastomotic sites inhibits neointimal hyperplasia in a canine model. Ann Thorac Surg. 2007;84(2):560–7. doi: 10.1016/j.athoracsur.2007.02.034. discussion 567. [DOI] [PubMed] [Google Scholar]
- 41.Sallach RE, Wei M, Biswas N, Conticello VP, Lecommandoux S, Dluhy RA, Chaikof EL. Micelle density regulated by a reversible switch of protein secondary structure. J Am Chem Soc. 2006;128(36):12014–9. doi: 10.1021/ja0638509. [DOI] [PubMed] [Google Scholar]
- 42.Hart DS, Gehrke SH. Thermally associating polypeptides designed for drug delivery produced by genetically engineered cells. J Pharm Sci. 2007;96(3):484–516. doi: 10.1002/jps.20755. [DOI] [PubMed] [Google Scholar]
- 43.Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv Drug Deliv Rev. 2001;47(1):113–31. doi: 10.1016/s0169-409x(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 44.Gaucher G, Dufresne MH, Sant VP, Kang N, Maysinger D, Leroux JC. Block copolymer micelles: preparation, characterization and application in drug delivery. J Control Release. 2005;109(1-3):169–88. doi: 10.1016/j.jconrel.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 45.Mahmud A, Xiong XB, Aliabadi HM, Lavasanifar A. Polymeric micelles for drug targeting. J Drug Target. 2007;15(9):553–84. doi: 10.1080/10611860701538586. [DOI] [PubMed] [Google Scholar]
- 46.Tang Y, Liu SY, Armes SP, Billingham NC. Solubilization and controlled release of a hydrophobic drug using novel micelle-forming ABC triblock copolymers. Biomacromolecules. 2003;4(6):1636–45. doi: 10.1021/bm030026t. [DOI] [PubMed] [Google Scholar]
- 47.Dreher MR, Simnick AJ, Fischer K, Smith RJ, Patel A, Schmidt M, Chilkoti A. Temperature triggered self-assembly of polypeptides into multivalent spherical micelles. J Am Chem Soc. 2008;130(2):687–94. doi: 10.1021/ja0764862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.D'Souza AJM, Hart DS, Middaugh CR, Gehrke SH. Characterization of the changes in secondary structure and architecture of elastin-mimetic triblock polypeptides during thermal gelation. Macromolecules. 2006;39(20):7084–7091. [Google Scholar]
- 49.Wang X, Yucel T, Lu Q, Hu X, Kaplan DL. Silk nanospheres and microspheres from silk/pva blend films for drug delivery. Biomaterials. 2009 doi: 10.1016/j.biomaterials.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin J, Zhu J, Chen T, Lin S, Cai C, Zhang L, Zhuang Y, Wang XS. Drug releasing behavior of hybrid micelles containing polypeptide triblock copolymer. Biomaterials. 2009;30(1):108–17. doi: 10.1016/j.biomaterials.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 51.Yoshino K, Kadowaki A, Takagishi T, Kono K. Temperature sensitization of liposomes by use of N-isopropylacrylamide copolymers with varying transition endotherms. Bioconjug Chem. 2004;15(5):1102–9. doi: 10.1021/bc034205j. [DOI] [PubMed] [Google Scholar]
- 52.Liu L, Sheardown H. Glucose permeable poly (dimethyl siloxane) poly (N-isopropyl acrylamide) interpenetrating networks as ophthalmic biomaterials. Biomaterials. 2005;26(3):233–44. doi: 10.1016/j.biomaterials.2004.02.025. [DOI] [PubMed] [Google Scholar]
- 53.Zhang XZ, Chu CC. Preparation of thermosensitive PNIPAAm hydrogels with superfast response. Chem Commun (Camb) 2004;3:350–1. doi: 10.1039/b313073h. [DOI] [PubMed] [Google Scholar]
- 54.Adams SB, Jr, Shamji MF, Nettles DL, Hwang P, Setton LA. Sustained release of antibiotics from injectable and thermally responsive polypeptide depots. J Biomed Mater Res B Appl Biomater. 2009;90(1):67–74. doi: 10.1002/jbm.b.31254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shamji MF, Whitlatch L, Friedman AH, Richardson WJ, Chilkoti A, Setton LA. An injectable and in situ-gelling biopolymer for sustained drug release following perineural administration. Spine (Phila Pa 1976) 2008;33(7):748–54. doi: 10.1097/BRS.0b013e3181695773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sallach RE, Cui W, Balderrama F, Martinez AW, Wen J, Haller CA, Taylor JV, Wright ER, Long RC, Jr, Chaikof EL. Long-term biostability of self-assembling protein polymers in the absence of covalent crosslinking. Biomaterials. 31(4):779–91. doi: 10.1016/j.biomaterials.2009.09.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zilberman M, Eberhart RC. Drug-eluting bioresorbable stents for various applications. Annu Rev Biomed Eng. 2006;8:153–80. doi: 10.1146/annurev.bioeng.8.013106.151418. [DOI] [PubMed] [Google Scholar]
- 58.Lao LL, Venkatraman SS. Paclitaxel release from single and double-layered poly(DL-lactide-co-glycolide)/poly(L-lactide) film for biodegradable coronary stent application. J Biomed Mater Res A. 2008;87(1):1–7. doi: 10.1002/jbm.a.31706. [DOI] [PubMed] [Google Scholar]
- 59.Sallach RE, Leisen J, Caves JM, Fotovich E, Apkarian RP, Conticello VP, Chaikof EL. A permanent change in protein mechanical responses can be produced by thermally-induced microdomain mixing. J Biomater Sci Polym Ed. 2009;20(11):1629–44. doi: 10.1163/156856208X386228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Joseph G, Sharma CP. Prostacyclin immobilized albuminated surfaces. J Biomed Mater Res. 1987;21(7):937–45. doi: 10.1002/jbm.820210709. [DOI] [PubMed] [Google Scholar]
- 61.van der Giessen WJ, van Beusekom HM, Eijgelshoven MH, Morel MA, Serruys PW. Heparin-coating of coronary stents. Semin Interv Cardiol. 1998;3(3-4):173–6. [PubMed] [Google Scholar]
- 62.Buller CE, Dzavik V, Carere RG, Mancini GB, Barbeau G, Lazzam C, Anderson TJ, Knudtson ML, Marquis JF, Suzuki T, Cohen EA, Fox RS, Teo KK. Primary stenting versus balloon angioplasty in occluded coronary arteries: the Total Occlusion Study of Canada (TOSCA) Circulation. 1999;100(3):236–42. doi: 10.1161/01.cir.100.3.236. [DOI] [PubMed] [Google Scholar]
- 63.al-Khaffaf H, Charlesworth D. Albumin-coated vascular prostheses: A five-year follow-up. J Vasc Surg. 1996;23(4):686–90. doi: 10.1016/s0741-5214(96)80050-4. [DOI] [PubMed] [Google Scholar]
- 64.Dutoya S, Verna A, Lefebvre F, Rabaud M. Elastin-derived protein coating onto poly(ethylene terephthalate). Technical, microstructural and biological studies. Biomaterials. 2000;21(15):1521–9. doi: 10.1016/s0142-9612(99)00274-4. [DOI] [PubMed] [Google Scholar]
- 65.Ito S, Ishimaru S, Wilson SE. Application of coacervated alpha-elastin to arterial prostheses for inhibition of anastomotic intimal hyperplasia. ASAIO J. 1998;44(5):M501–5. doi: 10.1097/00002480-199809000-00036. [DOI] [PubMed] [Google Scholar]
- 66.Hanson SR, Kotze HF, Savage B, Harker LA. Platelet interactions with Dacron vascular grafts. A model of acute thrombosis in baboons. Arteriosclerosis. 1985;5(6):595–603. doi: 10.1161/01.atv.5.6.595. [DOI] [PubMed] [Google Scholar]
- 67.Heilshorn SC, DiZio KA, Welsh ER, Tirrell DA. Endothelial cell adhesion to the fibronectin CS5 domain in artificial extracellular matrix proteins. Biomaterials. 2003;24(23):4245–4252. doi: 10.1016/s0142-9612(03)00294-1. [DOI] [PubMed] [Google Scholar]
- 68.Chaikof EL, Matthew H, Kohn J, Mikos AG, Prestwich GD, Yip CM. Biomaterials and scaffolds in reparative medicine. Ann N Y Acad Sci. 2002;961:96–105. doi: 10.1111/j.1749-6632.2002.tb03057.x. [DOI] [PubMed] [Google Scholar]
- 69.Boland ED, Matthews JA, Pawlowski KJ, Simpson DG, Wnek GE, Bowlin GL. Electrospinning collagen and elastin: preliminary vascular tissue engineering. Front Biosci. 2004;9:1422–32. doi: 10.2741/1313. [DOI] [PubMed] [Google Scholar]
- 70.Huang L, McMillan RA, Apkarian RP, Pourdeyhimi B, Conticello VP, Chaikof EL. Generation of synthetic elastin-mimetic small diameter fibers and fiber networks. Macromolecules. 2000;33:2989–2997. [Google Scholar]
- 71.Mithieux SM, Rasko JE, Weiss AS. Synthetic elastin hydrogels derived from massive elastic assemblies of self-organized human protein monomers. Biomaterials. 2004;25(20):4921–7. doi: 10.1016/j.biomaterials.2004.01.055. [DOI] [PubMed] [Google Scholar]
- 72.Voldrich Z, Tomanek Z, Kopecek J. Long-Term Experience with Poly(Glycol Monomethacrylate) Gel in Plastic Operations of Nose. Journal of Biomedical Materials Research. 1975;9(6):675–685. doi: 10.1002/jbm.820090612. [DOI] [PubMed] [Google Scholar]
- 73.Rincon AC, Molina-Martinez IT, de Las Heras B, Alonso M, Bailez C, Rodriguez-Cabello JC, Herrero-Vanrell R. Biocompatibility of elastin-like polymer poly(VPAVG) microparticles: in vitro and in vivo studies. J Biomed Mater Res A. 2006;78(2):343–51. doi: 10.1002/jbm.a.30702. [DOI] [PubMed] [Google Scholar]
- 74.Mithieux SM, Tu Y, Korkmaz E, Braet F, Weiss AS. In situ polymerization of tropoelastin in the absence of chemical cross-linking. Biomaterials. 2009;30(4):431–5. doi: 10.1016/j.biomaterials.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 75.Kretlow JD, Klouda L, Mikos AG. Injectable matrices and scaffolds for drug delivery in tissue engineering. Advanced Drug Delivery Reviews. 2007;59(4-5):263–273. doi: 10.1016/j.addr.2007.03.013. [DOI] [PubMed] [Google Scholar]