

Abstract
Encapsulation of macromolecules within lipid vesicles has the potential to drive biological discovery and enable development of novel, cell-like therapeutics and sensors. However, rapid and reliable production of large numbers of unilamellar vesicles loaded with unrestricted and precisely-controlled contents requires new technologies that overcome size, uniformity, and throughput limitations of existing approaches. Here we present a high-throughput microfluidic method for vesicle formation and encapsulation using an inkjet printer at rates up to 200 Hz. We show how multiple high-frequency pulses of the inkjet’s piezoelectric actuator create a microfluidic jet that deforms a bilayer lipid membrane, controlling formation of individual vesicles. Variations in pulse number, pulse voltage, and solution viscosity are used to control the vesicle size. As a first step toward cell-like reconstitution using this method, we encapsulate the cytoskeletal protein actin and use co-encapsulated microspheres to track its polymerization into a densely entangled cytoskeletal network upon vesicle formation.
Introduction
Advances in microfluidic technology and molecular cell biology have raised considerable interest in the study of chemical and biological reactions encapsulated in small volumes.1–5 Use of small reaction volumes has several advantages over conventional macroscopic tubes and wells, including length scales that are comparable to those of cells, minimal use of reagents like purified proteins, the ability to carry out multiple reactions in parallel, and efficient formation and handling of individual reaction volumes using microfluidic devices. Previously, small biological reactors have consisted of oil/water emulsions,6,7 where the reactions either take place in aqueous droplets within a continuous oil phase (single emulsions)8 or the aqueous droplet is surrounded by an oil shell within a continuous aqueous phase (double emulsions) and produced by either continuous flows or microfluidic jets.9,10 Both single and double emulsions can be generated in large numbers with uniform size and controlled contents, and they are both useful for forming condensed particles and shells,7 as well as performing high-throughput biochemical assays such as PCR-based screens and directed evolution studies.11
Emulsions, however, are not appropriate containers for cell-like encapsulated systems because they lack a lipid bilayer membrane. Encapsulation within lipid bilayer membranes is fundamental to all cells, providing the appropriate boundary conditions for cellular processes such as motility and shape change. Therefore, further progress in the development of small-volume reaction systems capable of cell-like interactions with the external environment and biologically-relevant internal spatial organization will require new high-throughput methods for producing unilamellar vesicles.
Several approaches for lipid vesicle encapsulation of biochemical solutions have been demonstrated, the most common of which include swelling,12 electroformation,13 injection of pre-formed vesicles,14 and reverse phase evaporation15 or emulsion.16 However, important limitations such as lack of size control, poor encapsulation of high molecular weight species, and multilamellarity of the vesicle boundary have prevented their widespread adoption for cell-like encapsulation.12 In particular, the reverse emulsion technique has successfully encapsulated purified proteins17,18 as well as cytoplasmic extract,19 but vesicles formed often include solvent in the membranes,17 are formed with low efficiency, and have non-uniform sizes.16 It has recently been shown that double emulsion droplets can gradually be “ripened” to remove the oil shell and form vesicles.20 However biological encapsulants will be exposed to solvents, and vesicles may not be unilamellar due to excess lipid content, complicating attempts to include membrane proteins or perform biochemical reactions after encapsulation.
In a previous report, we showed that it was possible to use a precisely-controlled microfluidic jet propelled by a piezo-actuated syringe to deform a lipid bilayer membrane into a giant unilamellar vesicle capable of incorporating membrane proteins.21 While this system successfully demonstrated the formation of unilamellar vesicles by microfluidic jetting, it was severely limited in the throughput of vesicle formation and did not achieve variation of vesicle size or production of cell-sized vesicles.
Here we report the development of a system capable of simultaneously forming and loading unilamellar vesicles at rates up to 200 Hz using microfluidic inkjet printing. Fluid loaded into a piezoelectric-actuated inkjet is accelerated through a micron-scale nozzle to create a high-speed liquid jet. We take advantage of the precision, capacity for control, and high-frequency displacements inherent to piezoelectric inkjets, a technology originally developed for printing applications. Using this approach we achieve high-throughput vesicle production and control vesicle size over a range of approximately 10 to 400 μm in diameter corresponding to more than four orders of magnitude in volume. Since the vesicles can be imaged immediately after their formation, reaction dynamics and bilayer membrane interactions can be followed from initiation. The ability to rapidly form multiple vesicles of equal size also enables large numbers of experiments to be conducted simultaneously so that statistically significant behavior can be identified. This combination of attributes makes inkjet printing of unilamellar vesicles a simple, robust, and rapid method for generating small reaction volumes with cell-like membrane boundaries.
To demonstrate how inkjet formation of vesicles can be used to create and control biological reactions, we loaded giant unilamellar vesicles with purified monomeric actin and tracer particles and initiated actin polymerization within the vesicles during encapsulation. Immediately after formation of the vesicles, we observed the dramatic increase in particle confinement that accompanied the actin network assembly into an entangled network. Cellular reconstitution studies such as these, along with pharmaceutical challenges that require encapsulation of multiple components, will benefit from inkjet formation of unilamellar vesicles as a platform for discovery.
Materials and methods
Reagents
1,2-Diphytanoyl-sn-glycero-3-phosphocholine (DPhPC) lipid was purchased dissolved in chloroform from Avanti Polar Lipids, resuspended in n-decane (Sigma-Aldrich) at a concentration of 25 mg/ml, and stored at −20 °C with a dessicant (Drierite). Sucrose and glucose were prepared at approximately 400 mOsm. Ficoll PM 400 (Amersham Biosciences) solutions was prepared at 7.5–8.75 weight percent, and 300 mM of glucose was added to aid in visualization during jetting. Actin was purified from rabbit muscle acetone powder,22 and purity was confirmed by sodium dodecyl sulfate polyacrylamide gel electrophoresis. Actin concentration was determined by UV absorbance and BCA assays.
Rheometry
Viscosity of solutions was measured at low shear rates (18–50 s−1) using a Gemini rheometer (Malvern Instruments) with a concentric cylinder geometry.
Inkjet device and vesicle formation
Inkjets and drive electronics were from MicroFab Technologies. Inkjets used home built glass orifices installed by MicroFab. Orifices were made from glass capillary stock (0.6 mm inner diameter and 0.75 mm outer diameter) and formed by pulling the capillaries into micropipettes with sharp tips (P97 Flaming/Brown Micropipette Puller, Sutter Instruments). Micropipettes were refined using a Microforge MFG-3 (MicroData Instruments) and sanded to the desired orifice inner diameter, 10 μm. Disposable syringes were used to load inkjets with the jetted solution from the rear and provided constant perfusion of this solution (approximately 10 μL/min) during experiments. Loaded inkjets were inserted into bilayer lipid membrane chambers and positioned with less than 200 μm between the nozzle orifice and bilayer (Supplementary Fig. 1†). Multiple fluid pulses from the print head (12–30) were used to form vesicles. Application of a trapezoidal voltage pulse from the drive triggered each pulse. Identical pulses repeated at 20 kHz had a rise time of 3 μs, amplitude of 25–35 V, duration at this amplitude of 35 μs (to maximize velocity), and fall time of 3 μs. Vesicle formation was recorded using high-speed microscopy (500–30,000 frames per second, Photron 1024 PCI). Osmotically balanced (~400 mOsM) solutions of sucrose in the nozzle and glucose surrounding the bilayer were used to achieve optical contrast and sinking of the vesicles to the chamber bottom for observation.
Planar bilayer lipid membranes
Vesicles were formed from planar lipid bilayer membranes constructed by contacting monolayers in a double-well chamber.21,23,24 Bilayers were formed at the intersection of chamber wells (30–80 μL each), where chambers had the shape of an “infinity” symbol, and the bilayer was formed at the waist of the pattern. However, chamber wells of arbitrary shape are possible, where circular, ellipsoidal, and rectangular shapes have been used. Chambers were made using a laser cutter (Versa Laser) typically from 3 mm acrylic sheets (TAP plastics) and bonded to acrylic cover slips (0.2 mm thick, Astra Products) using acrylic cement (TAP plastics). A hole of approximately 1.5 mm diameter was drilled on one end of the chamber, perpendicular to the bilayer plane, to facilitate insertion and positioning of the inkjet nozzle. Adhesive (Locktite 495) adhered a thin (0.26 mm) sheet of natural rubber (McMaster-Carr) to the external surface of this hole to provide a seal. A needle was used to make a small hole in the rubber sheet for insertion of the inkjet nozzle. To form bilayer lipid membranes, approximately 12 μL of lipid solution (25 mg/mL DPhPC dissolved in n-decane) was pipetted into the chamber. Aqueous droplets were pipetted sequentially into each of the fluid chambers and rapidly came into contact, forming the bilayer lipid membrane at their interface.
Actin network polymerization
To polymerize 9.2 μM actin in the lumen of GUVs (assuming 50% dilution during encapsulation), the inkjet was loaded with 385 mOsm sucrose, 5 mM Tris HCl pH 7.8, 0.2 mM CaCl2, 0.01% NaN3, 2 mM ATP pH 7.5, 18.4 μM actin, and 300x dilution of 2.63% solids-latex Fluoresbrite Polychromatic red 0.5 μm diameter microspheres (Polysciences, Inc.). Microspheres were sonicated for 5 minutes prior to use in order to break up aggregates. The solution was filtered prior to addition of the microspheres, ATP, and actin. The solution in the droplets surrounding the bilayer lipid membrane contained 200 mM KCl, 4 mM MgCl2, and 2 mM EGTA pH 7.5. The vesicles were formed at 22 volts and 18 pulses, and bead motion was monitored between 15 and 40 minutes from the time of encapsulation. For the control experiment used to assess microsphere diffusion in the absence of an actin network, solutions A and B were prepared. Solution A contained 385 mOsm sucrose, 5 mM Tris HCl pH 7.8, 0.2 mM CaCl2, 0.01% NaN3, 333x dilution of sonicated 2.63% solids-latex Fluoresbrite Polychromatic red 0.5 micron microspheres. Solution B contained 200 mM KCl, 4 mM MgCl2, 2 mM EGTA pH 7.5. Solutions A and B were mixed in a 1:1 ratio and added to a homemade chamber (0.1 mm × 5 mm × 20 mm) consisting of a glass coverslip adhered to a glass slide using double-sided tape, then sealed with VALAP (1:1:1 of vaseline, lanoline, and paraffin) before imaging. Bead diffusion was recorded at 100x using a CoolSnap HQ Camera (Photo-metrics). Bead tracking was done using the Track Points application in Metamorph v7.1 (Molecular Devices), and diffusion curves were calculated using MatLab software (MathWorks).
Results and discussion
Formation of single unilamellar lipid vesicles with multiple pulses of an inkjet
We used a drop-on-demand inkjet to produce a microfluidic jet that deforms a planar bilayer lipid membrane into unilamellar vesicles that separate from the planar membrane (Fig. 1a). The inkjet generates multiple discrete pulses using a cylindrical piezoelectric actuator surrounding a fluid filled nozzle. The actuator contracts and expands radially, producing pressurization and rarefaction waves in the fluid. As demonstrated by Bogy et al., application of appropriate voltage pulses to such devices results in the ejection of fluid from the device due to the constructive interference of traveling pressurization waves (and destructive interference of traveling rarefaction waves) within the nozzle.25 In our work, multiple, closely spaced (20 kHz) voltage pulses applied to the inkjet produced a set of fluid pulses. As they traveled in and entrained the surrounding fluid, these fluid pulses combined to form a growing vortex ring structure26 (Fig. 1b and Movie 1†) that was capable of deforming the bilayer lipid membrane to form vesicles (Fig. 1c and Movie 2†). Examination of selected frames from a high-speed video (22,500 frames per second) recorded during jetting shows the formation and addition of individual fluid pulses into a composite vortex ring (Fig. 1b). Vesicle formation occurs via a process similar to the one we observed for a single pulse jet flow generated by a piezo-actuated syringe.21 Briefly, membrane deformation (t1, Fig. 1c), membrane collapse (t2–t4, Fig. 1c), and vesicle separation (t5–t6, Fig. 1c) stages were observed over about 5 ms, typically. Vesicles formed were highly monodisperse (for example 194.1 μm ± 4.6 μm average diameter and first standard deviation, N = 50 vesicles formed consecutively). Vesicles contain a mixture of the fluid loaded in the inkjet and the fluid surrounding the inkjet, which is entrained as the vortex ring forms and grows. Isolated vortex rings have been shown to contain 25–40% entrained volume.27 We expect a somewhat lower fraction of entrained fluid to be encapsulated inside vesicles because the vortex ring is partially confined by the lipid membrane as it grows.
Fig. 1.

Formation of lipid vesicles with an inkjet. (a) Each vesicle is formed by a series of inkjet pulses that form a single microfluidic jet, where pauses between pulse sets determine the frequency of vesicle formation. (b) Multiple inkjet pulses (15 pulses at 30 V constant amplitude) build to form a single traveling vortex ring as depicted in sequential frames recorded at 22,500 frames per second (222, 311, 356, 444, 578, 711, 889 μs). (c) A vesicle is formed by a series of 17 inkjet pulses at 50 V, as depicted in sequential frames recorded at 5,000 frames per second (0.8, 1.4, 2.2, 3.0, 4.2, 4.8, 9.4 ms). Scale bars are 100 μm.
Control of vesicle formation using inkjet parameters
Use of an inkjet to form vesicles with multiple pulses provides the opportunity to control the vesicle formation process by tuning multiple inkjet parameters. Two inkjet parameters expected to directly influence the resulting vesicle formation process are (i) the pulse amplitude and (ii) the number of pulses used to form each vesicle. We expect these parameters to correlate strongly with the vortex velocity and volume, respectively. We began by investigating the impact of pulse amplitude and number on the initial motion of the vortex ring leading edge, a property of the jet that can be conveniently measured by video microscopy (Fig. 2a, b). In all cases, the travel velocity decayed rapidly due to the effects of fluid entrainment and drag. When the pulse amplitude was increased with fixed pulse number, the primary result was an increase in vortex ring travel velocity (increase in slope of curves in Fig. 2a). In contrast, when the number of pulses increased at constant pulse amplitude, the initial travel velocity (initial slope of curves in Fig. 2b) remained roughly constant while the duration of travel increased. In this way, pulse amplitude appears to control the vortex travel velocity, while pulse number appears to control the duration of travel.
Fig. 2.

Control of vesicle formation through variation of the amplitude and number of inkjet pulses used to form each vesicle. (a) Vortex ring displacement as a function of time for fixed pulse number (15 pulses) and a range of pulse amplitudes: 10 V (red), 15 V (green), 20 V (violet), 25 V (blue), 30 V (magenta). (b) Vortex ring displacement as a function of time for fixed pulse amplitude (30 V) and a range of pulse numbers: 1 (red), 3 (green), 5 (violet), 9 (blue), 13 (magenta), 15 pulses (gold). For all ring displacements, the standard deviation between displacement values was less than 2% for N = 3. (c) Vesicle diameter as a function of pulse number for fixed pulse amplitude (35 V). Solid line is a power law curve fit with power 1/3, leading coefficient 75.3 μm, and R2 of 0.917. (d) Minimum number of pulses to create a vesicle as a function of pulse amplitude in volts. (e) Diameter of vesicle formed using minimum number of pulses, as recorded in part d, as a function of pulse amplitude in volts.
We next examined the impact of pulse amplitude and number on the vesicle diameter. We held the pulse amplitude at 35 volts and varied the pulse number at fixed separation between the bilayer and nozzle (Fig. 2c). Vesicles were first formed using 16 pulses. We found that the vesicle diameter increased monotonically with increasing number of pulses from approximately 187 μm (16 pulses) to 210 μm (21 pulses). The relationship between pulse number and vesicle diameter was fit reasonably well (R2 = 0.917, N = 18) by a power law relation with exponent 1/3, as would be expected if the vesicle volume varied proportionally with pulse number. However, the ideal fit (R2 = 0.965) was achieved with a somewhat larger exponent (0.429 ± 0.04) indicating that vesicle volume increased with pulse number faster than could be accounted for by a linear relationship. This additional growth likely arose from fluid entrainment, which had more time to evolve as the pulse number increased, increasing the duration of the composite pulse. We examined the combined impact of variations in pulse number and amplitude on vesicle diameter, identifying the minimum number of pulses needed to create a vesicle, 7–39, over a range of pulse amplitudes, 25–45 volts (Fig. 2d). We found that the minimum number of pulses decreased with increasing pulse amplitude, and the vesicle diameter decreased with increasing pulse amplitude (Fig. 2e) from approximately 210 to 140 μm. Here as pulse velocity increased with increasing amplitude, the amount of membrane deformation achieved by each pulse increased such that fewer pulses were needed to form a vesicle, and the resulting vesicle was smaller.
Formation of cell-sized vesicles through variation of fluid viscosity
Some experiments using small encapsulated volumes, particularly those aimed at reconstituting specific cellular processes, would benefit from vesicle dimensions on the scale of typical single cells. Vesicle diameters in the range of 1–50 μm would better emulate most eukaryotic and some bacterial cells, extending the range of encapsulation applications. Since vesicle volume is defined during the encapsulation process by collapse of the deformed membrane around the jetted fluid, we hypothesized that differences in viscosity on either side of the bilayer membrane would alter the collapse dynamics and resulting vesicle size.
First, we found that jetting of a more viscous solution (8.75 w/v% ficoll 400 and 300 mM sucrose dissolved in water ~5 cp) while maintaining an inviscid solution on both sides of the bilayer (300 mM glucose dissolved in water ~1 cp) led to a dramatic increase in vesicle diameter (380 μm diameter, data not shown).
Next, we investigated whether jetting a less viscous fluid into a more viscous fluid would lead to the formation of a smaller vesicle. The fluid in the jet (300 mM sucrose solution) and on the jet side of the bilayer (300 mM glucose solution) had a viscosity of approximately 1 cp, while the viscosity of the fluid on the opposing side of the bilayer had approximately 3.9 cp viscosity (7.5 w/v% ficoll 400 and 300 mM glucose solution) at low shear rates as measured by rheometry (Fig. 3a). We observed that vesicles of significantly smaller size (approximately 8–110 μm) were formed in this configuration (Fig. 3b and Movie 3†). Notably, vesicles were formed during retraction of the membrane rather than during expansion, and the process of formation was characterized by an asymmetric retraction, not observed in the absence of a viscosity differential. Specifically, lateral retraction of the membrane sides occurred more rapidly than axial retraction of the membrane, such that the aspect ratio (axial extent to lateral extent) of the membrane protrusion increased during retraction until a vesicle was formed.
Fig. 3.
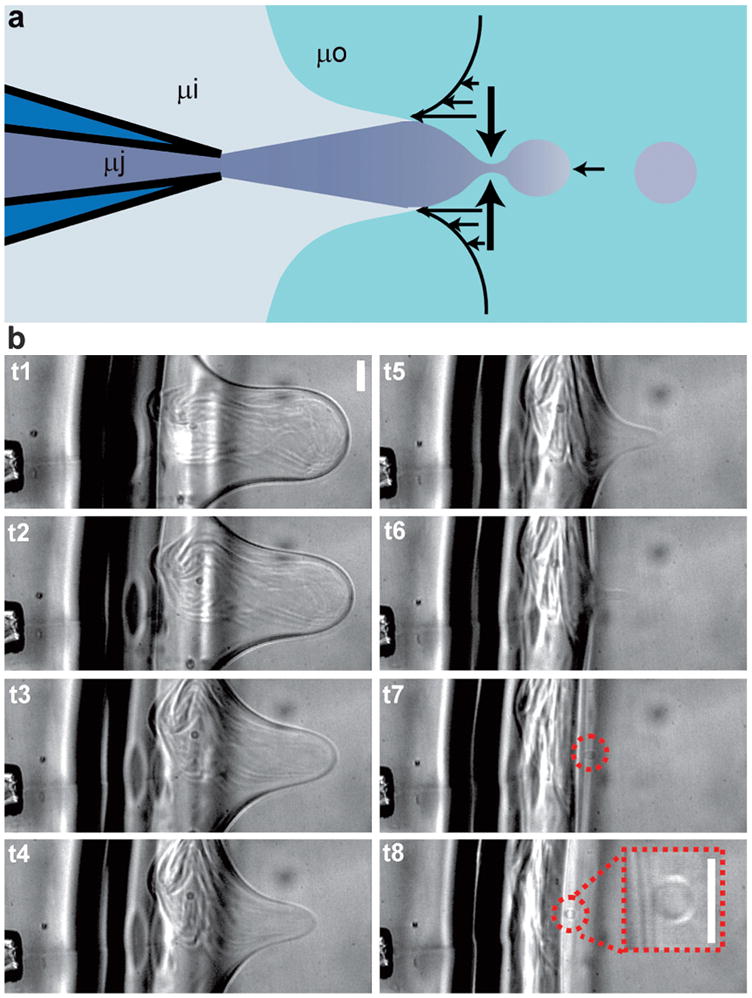
Formation of cell-sized vesicles using a viscosity differential across the bilayer lipid membrane. (a) By placing a solution of elevated viscosity (approximately 3.9 cp) on the side of the bilayer opposite the inkjet, it is possible to manipulate the shear rates on each side of the bilayer during membrane collapse such that small, cell-sized vesicles are formed (8–110 μm diameter). The viscosities of the jetted solution, surrounding solution, and opposite solution are labeled as μj, μi, and μo, respectively. (b) Formation of small cell-sized vesicles as depicted by sequential frames recorded at 7500 frames per second (0.53, 2.00, 2.80, 3.87, 4.53, 5.20, 5.73, 6.27 ms) using inkjet pulses at 36 volts amplitude. (b inset) Enlargement of cell-sized vesicle from a separate formation at higher (20x) magnification. Scale bars are 50 μm.
Vesicle formation is successful when the lateral retraction of membrane sides comes to completion before the axial retraction of the membrane, allowing a portion of the membrane area to separate. The observered decrease in vesicle size with increase in viscosity differential is expected considering that each membrane retraction process, axial and lateral, depends on viscosity. Axial retraction involves dragging the vesicle bolus through the surrounding fluid and is therefore slowed down by increasing the viscosity of the fluid on the side of the bilayer opposite the jet. Lateral retraction requires expulsion of the encapsulated fluid through the bolus neck and is thus sped up by decreasing the viscosity of the fluid on the jet side of the bilayer (in and around the jet). Therefore, when a relatively higher viscosity solution is used in the jet, the rate of axial retraction increases relative to the rate of lateral retraction such that longer, more powerful ejections are required to form vesicles, and the resulting vesicles are larger. In contrast, use of a lower viscosity in the jet than on the opposing side of the membrane decreases the rate of axial retraction in comparison to lateral retraction, resulting in an asymmetric contraction of the membrane that enables formation of smaller vesicles. Cell-sized vesicles formed using this viscosity differential were monodisperse with a typical variation in diameter of approximately 10% within populations made using the same jetting parameters (relative viscosity, voltage, pulse number, and nozzle-bilayer separation). It is likely that these parameters can be varied in order to maintain a specific vesicle size while accommodating variations in the composition and viscosity of encapsulated solution. However, interdependencies between these parameters have not been characterized quantitatively.
High-throughput inkjet formation of vesicles
Use of an inkjet enables rapid production of large numbers of vesicles, facilitating statistical studies of system behavior and emergent properties, similar to the study of a population of cells. Analogous to the continuous production of ink drops during document printing, high-throughput formation of vesicles takes advantage of the high bandwidth of piezoelectric actuators. Each vesicle is formed by a set of closely spaced (20 kHz) pulses and the temporal spacing of pulse sets determines the rate of vesicle formation. We investigated vesicle formation at rates from 0.5 Hz to 200 Hz (Fig. 4), seeking to understand the limitations on formation rate.
Fig. 4.
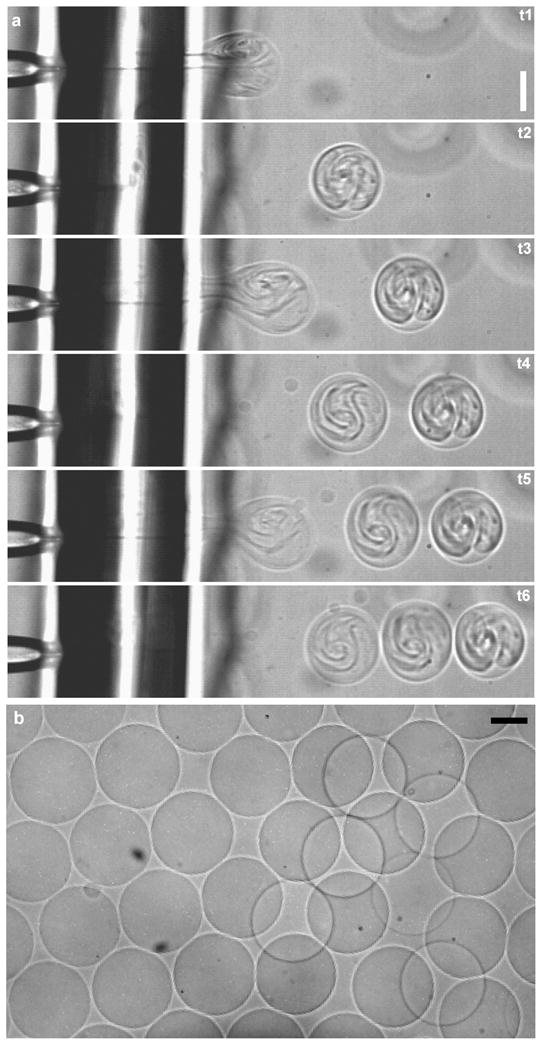
Formation of multiple vesicles by inkjet printing. (a) Three vesicles are formed at a rate of 200 Hz, as depicted in sequential frames recorded at 5,400 frames per second (1.11, 3.33, 6.67, 9.82, 11.67, 13.70 ms) using 16 inkjet pulses per vesicle at 33 V amplitude. (b) A population of vesicles formed by high throughput inkjet printing. Scale bars are 100 μm.
At low formation frequencies, we found that continuous vesicle formation was possible. At a rate of 0.5 Hz, the inkjet printing method was able to consistently form hundreds of vesicles (Movie 4†). Cell-sized vesicles formed using a viscosity differential across the bilayer were also produced at this rate (data not shown). Upon increasing the rate of vesicle formation above 1 Hz, we found that some of the supporting oil matrix was included in the vesicle boundaries after 10–15 solvent-free vesicles were formed (dark contrast appearing at vesicle edges in Movie 5† after 10 vesicles are formed). It is interesting to note that the vesicles formed before oil was observed have a total surface area of the same order as that of the bilayer lipid membrane (~1 mm2). Based on this observation it appears likely that the inclusion of oil in the vesicle boundaries after 10–15 vesicles were formed indicates that the rate of bilayer membrane regrowth from the oil phase was not keeping up with the rate of membrane removal at formation rates above 1 Hz.
At higher frequencies, multiple solvent-free vesicles could still be formed, though oil fouled the vesicles after only a few (less than 15) were made. When small batches of vesicles are formed, the rate of membrane regrowth no longer limits the vesicle production rate. Instead, the formation rate should be limited by the time required for a single vesicle to form and the lipid membrane to relax back to its original position, which is about 5 ms. In agreement with this idea, we were able to form several solvent-free vesicles at rates up to 200 Hz (Fig. 4a and Movie 6†). After several vesicles are formed at this rate, a pause in vesicle formation of 2–4 seconds allows the bilayer to regrow. In this way, multiple batches of several solvent-free vesicles each were formed with an inter-batch spacing of 2–4 seconds and an inter-vesicle spacing of as little as 5 ms (200 Hz). This maximum rate of vesicle formation may be particularly useful if the rate of membrane regrowth can be increased such that it is no longer limiting, and it therefore likely represents an upper limit for vesicle production rates.
Real-time assembly and interrogation of actin networks inside vesicles
Encapsulation of biological molecules with defined composition and concentration is a significant technical challenge that has limited progress on reconstituting cellular processes in vitro. To demonstrate the utility of vesicle formation and loading by inkjet printing for reconstitution experiments, we loaded and polymerized the cytoskeletal protein actin in the lumen of a GUV (Fig. 5). A bilayer lipid membrane was set up with actin polymerization buffer on each side. Purified actin and latex beads (used to probe solution rheology) were loaded into the inkjet nozzle in monomeric form, and a vesicle containing a mixture of actin monomers (~0.39 mg/mL monomeric actin), beads, and polymerization buffer was formed (Fig. 5a). Entrainment of actin polymerization buffer during vesicle formation initiated actin polymerization only in the lumen of the GUV, avoiding clogging the inkjet nozzle and temporally defining the start of the reaction such that actin polymerization could be observed immediately following encapsulation.
Fig. 5.

Controlled assembly and interrogation of actin networks in lipid vesicles. (a) A mixture of actin monomers and 500 nm fluorescent beads are co-encapsulated in vesicles by microfluidic encapsulation with an inkjet. A polymerization buffer is entrained and mixed with the beads and monomers during encapsulation such that network assembly begins only after vesicles are formed. Diffusion of fluorescent beads is a probe for network viscosity. (b) Free diffusion of individual fluorescent beads (trails marked in red) over 5 s. Scale bar is 10 μm. (c) Diffusion of individual fluorescent beads confined by actin network polymerization (trails marked in red over the same period). Scale bar is 10 μm. (d) Plot of mean squared bead displacement with (blue) and without (red) actin network confinement as a function of time. (e) Zoomed plot of mean squared bead displacement after actin-network assembly. The traces comes from three vesicles and represents the average from 30 beads (10 beads within each vesicle).
At long times after vesicle formation (>20 minutes afterward), we observed that all of the encapsulated beads displayed restricted diffusion (Fig. 5c), consistent with the expectation that this concentration of actin monomers should form an entangled network with a mesh size of 0.48 μm, comparable to the bead diameter of 0.49 μm.28 To confirm that this observation was the direct result of actin polymerization, the sample was illuminated with an arc lamp, causing actin to depolymerize due to damage from reactive oxygen species generated as a result of photo-bleaching of the microspheres.29 After 500 ms of exposure, all of the beads were observed to begin diffusing freely again (data not shown). Furthermore, free diffusion of latex beads was observed in the absence of actin monomers (Fig. 5b) and clearly differed from the restricted motion of the encapsulated beads (Fig. 5d,e). Specifically, the slope of the mean squared displacement versus time curve for freely diffusing beads (Fig. 5d, red) is more than 3 orders of magnitude larger than the slope of the curve for vesicle encapsulated beads restricted by entangled actin filaments (Fig. 5d,e blue).
This experiment demonstrates the ability of microfluidic vesicle formation to perform controlled encapsulation of active protein solutions and initiate biomolecular reactions inside lipid vesicles. Further, controlled encapsulation and polymerization of actin within a unilamellar lipid vesicle is an important step toward reconstitution of the cytoskeleton and its membrane interactions. Reconstitution of cytoskeletal structures on lipid vesicles has emerged as a powerful approach for investigating the interplay between force generating polymer networks and deformable lipid membrane substrates.30,31 While cortical actin networks have recently been assembled in the lumen of GUVs,18 reconstitution of complex membrane bound cytoskeletal structures has been limited by the challenge of encapsulating known ratios of actin, actin binding proteins and membrane bound crosslinkers and nucleation factors. Direct control of vesicle size and content by microfluidic encapsulation with an inkjet offers a new platform for quantitatively investigating how specific proteins influence the architecture of membrane bound cytoskeletal and related questions in the emerging area of cellular reconstitution.
Conclusions
New experimental techniques are needed to enable the assembly of lipid vesicle encapsulated systems with increasing sophistication and similarity to living systems. In this paper, we demonstrate a high-throughput microfluidic encapsulation method using an inkjet for unilamellar vesicle formation and loading, which enables the unrestricted encapsulation of biological macromolecules and synthetic particles. We show that uniformly sized vesicles can be produced at rates up to 200 Hz, enabling studies on large populations of confined bioreactors. Further, through manipulation of inkjet parameters and fluid viscosity, we show that vesicles size can be controlled over a wide range (fewer than 10 μm to greater than 400 μm in diameter), which encompasses the size of many cells, a capability that will enable in vitro reconstitution of cellular structures and processes.
As a demonstration of in vitro reconstitution within inkjet-formed vesicles, we polymerized actin monomers upon encapsulation with 500 nm latex beads in the lumen of a unilamellar vesicle. This study exploits an inherent feature of our technique, reproducible mixing between the droplet volume and the jetted volume, to deliver the polymerization initiators during vesicle formation and to provide a passive readout of the mechanical properties of the actin network by bead diffusion. Further, this approach can be generalized to initiate many other reactions at the time of vesicle formation.
By facilitating further development of cell-like encapsulated systems, inkjet-based vesicle production will provide new opportunities for fundamental understanding of biological processes and advancement of primitive cell studies.5,19 Additionally, the capacity of vesicle encapsulated systems to incorporate components and processes borrowed from biological cells will promote the design of increasingly sophisticated cell-like assays11 and drug delivery tools.32
Acknowledgments
The authors are grateful to Malvern Instruments for use of the Gemini rheometer and C.S. Dutcher of the Muller lab for assistance with viscosity measurements. D.L.R. acknowledges fellowship support from the Natural Sciences and Engineering Research Council of Canada. This work was supported by the Cell Propulsion Lab, a National Institutes of Health Nano-medicine Development Center at UC Berkeley and UCSF (to D.A.F.). Sandia is a multiprogram laboratory operated by Sandia Corporation, a Lockheed Martin Company, for the United States Department of Energy’s National Nuclear Security Administration under contract DE-AC04-94AL85000.
Footnotes
Electronic supplementary information (ESI) available: Six movies and a schematic representation of the inkjet vesicle encapsulation system.
References
- 1.Sessa G, Weissmann G. J Biol Chem. 1970;245:3295–301. [PubMed] [Google Scholar]
- 2.Fygenson DK, Marko JF, LA Phys Rev Lett. 1997;79:4497–4500. [Google Scholar]
- 3.Tawfik DS, Griffiths AD. Nat Biotechnol. 1998;16:652–6. doi: 10.1038/nbt0798-652. [DOI] [PubMed] [Google Scholar]
- 4.Miyata H, Nishiyama S, Akashi K, Kinosita K., Jr Proc Natl Acad Sci U S A. 1999;96:2048–53. doi: 10.1073/pnas.96.5.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mansy SS, Schrum JP, Krishnamurthy M, Tobe S, Treco DA, Szostak JW. Nature. 2008;454:122–5. doi: 10.1038/nature07018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gunther A, Jensen KF. Lab Chip. 2006;6:1487–503. doi: 10.1039/b609851g. [DOI] [PubMed] [Google Scholar]
- 7.Teh SY, Lin R, Hung LH, Lee AP. Lab Chip. 2008;8:198–220. doi: 10.1039/b715524g. [DOI] [PubMed] [Google Scholar]
- 8.Hosokawa K, Fujii T, Endo I. Anal Chem. 1999;71:4781–4785. [Google Scholar]
- 9.Utada AS, Lorenceau E, Link DR, Kaplan PD, Stone HA, Weitz DA. Science. 2005;308:537–41. doi: 10.1126/science.1109164. [DOI] [PubMed] [Google Scholar]
- 10.Funakoshi K, Suzuki H, Takeuchi S. J Am Chem Soc. 2007;129:12608–9. doi: 10.1021/ja074029f. [DOI] [PubMed] [Google Scholar]
- 11.Kelly BT, Baret JC, Taly V, Griffiths AD. Chem Commun (Camb) 2007:1773–88. doi: 10.1039/b616252e. [DOI] [PubMed] [Google Scholar]
- 12.Walde P. Curr Opin Colloid Interface Sci. 1996;1:638–644. [Google Scholar]
- 13.Angelova M, Dimitrov D. Faraday Discuss. 1986;81:303–311. [Google Scholar]
- 14.Karlsson M, Nolkrantz K, Davidson MJ, Stromberg A, Ryttsen F, Akerman B, Orwar O. Anal Chem. 2000;72:5857–62. doi: 10.1021/ac0003246. [DOI] [PubMed] [Google Scholar]
- 15.Szoka F, Jr, Papahadjopoulos D. Proc Natl Acad Sci U S A. 1978;75:4194–8. doi: 10.1073/pnas.75.9.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pautot S, Frisken BJ, Weitz DA. Langmuir. 2003;19:2870–2879. doi: 10.1073/pnas.1931005100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takiguchi K, Yamada A, Negishi M, Tanaka-Takiguchi Y, Yoshikawa K. Langmuir. 2008;24:11323–11326. doi: 10.1021/la802031n. [DOI] [PubMed] [Google Scholar]
- 18.Pontani LL, van der Gucht J, Salbreux G, Heuvingh J, Joanny JF, Sykes C. Biophys J. 2009;96:192–8. doi: 10.1016/j.bpj.2008.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noireaux V, Libchaber A. Proc Natl Acad Sci U S A. 2004;101:17669–74. doi: 10.1073/pnas.0408236101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shum HC, Lee D, Yoon I, Kodger T, Weitz DA. Langmuir. 2008;24:7651–7653. doi: 10.1021/la801833a. [DOI] [PubMed] [Google Scholar]
- 21.Stachowiak JC, Richmond DL, Li TH, Liu AP, Parekh SH, Fletcher DA. Proc Natl Acad Sci. 2008;105:4697–4702. doi: 10.1073/pnas.0710875105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spudich JA, Watt S. J Biol Chem. 1971;246:4866–71. [PubMed] [Google Scholar]
- 23.Funakoshi K, Suzuki H, Takeuchi S. Anal Chem. 2006;78:8169–74. doi: 10.1021/ac0613479. [DOI] [PubMed] [Google Scholar]
- 24.Holden MA, Needham D, Bayley H. J Am Chem Soc. 2007;129:8650–8655. doi: 10.1021/ja072292a. [DOI] [PubMed] [Google Scholar]
- 25.Bogy DB, Talke FE. IBM J Res Dev. 1984;28:314–321. [Google Scholar]
- 26.Maxworthy T. J Fluid Mech. 1972;51:15–32. [Google Scholar]
- 27.Dabiri J, Gharib M. Journal of fluid mechanics. 2004;511:311–331. [Google Scholar]
- 28.Schmidt CF, Baermann M, Isenberg G, Sackmann E. Macromolecules. 1989;22:3638–3649. [Google Scholar]
- 29.Jacobson K, Rajfur Z, Vitriol E, Hahn K. Trends Cell Biol. 2008;18:443–450. doi: 10.1016/j.tcb.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu AP, Richmond DL, Maibaum L, Pronk S, Geissler PL, Fletcher DA. Nature Physics. 2008;4:789–793. doi: 10.1038/nphys1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Osawa M, Anderson DE, Erickson HP. Science. 2008;320:792–794. doi: 10.1126/science.1154520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.LaVan DA, McGuire T, Langer R. Nat Biotechnol. 2003;21:1184–91. doi: 10.1038/nbt876. [DOI] [PubMed] [Google Scholar]