

Abstract

Substituted dihydropyrans, easily accessed from a commercially available glycal, undergo acid-catalyzed rearrangement to provide highly functionalized isochroman and dioxabicyclooctane scaffolds.
Diversity oriented synthesis (DOS) has proven to be an exceptional strategy for exploring chemical space.1 Synthetic approaches employing the principles of DOS address variations in three aspects of molecular structure – skeleton, substitution, and stereochemistry.2 Following the tenets of DOS, controlled skeletal rearrangement processes offer rapid access to diverse, stereochemically rich frameworks.3 As a method to access skeletal diversity, we recently reported the rearrangement of stereochemically well-defined dihydropyrans derived from glycals such as 1 to afford highly substituted tetrahydrofurans (Scheme 1).4 In this transformation, dihydropyrans (2) underwent Au(III)-mediated ionization at the anomeric C–O bond to form an allylic carbocation intermediate (3) which was trapped by the C6 hydroxyl generating tetrahydrofurans 4. We sought to exploit this reactivity by incorporating nucleophiles at different positions of precursor pyrans (5).5 We anticipated that this design might allow rapid access to a series of diverse skeletons (e.g. 6 and 7) by changing the nature of the substituents at C1, C4, and C6 in the dihydropyran substrates. Herein, we demonstrate the realization of this concept employing terminating groups at C4 that dictate various reaction pathways involving Friedel-Crafts and cation-olefin cyclizations.
Scheme 1.

Skeletal Diversification Strategies Based on Rearrangement of Substituted Dihydropyrans
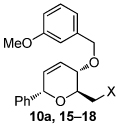
We initially focused our efforts on the development of a general synthesis of the appropriate dihydropyran substrates containing benzyl or allyl ethers at C4. d-Glucal-derived diol 84 was converted to substrates 10a–d and 11a–e in a straightforward manner (Scheme 2a). An aryl ether (cf. Table 2, entry 11) was synthesized via allylic alkylation of 3,4-dimethoxyphenol with the corresponding allylic carbonate using Pd2(dba)3 and (S,S)-DACH phenyl Trost ligand under microwave conditions.6,7 Ferrier reaction of tri-O-acetyl-d-glucal (1) using ((4-(tert-butyl)phenyl)-ethynyl)trimethylsilane was used to access an alkynyl dihydropyran (cf. Table 2, entry 12).5
Scheme 2.

a) General Synthesis of Dihydropyran Substrates and b) Initial Attempt at Pyran Rearrangement
Table 2.
Dihydropyran Rearrangements Resulting from Friedel-Crafts Alkylationa
| entry | substrate | product (% yield)b | entry | substrate | product (% yield)b |
|---|---|---|---|---|---|
 |
 |
9 | 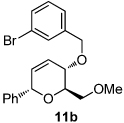 |
 |
|
| 1 | X= OH (10a) | 83; (19:23 = 2.8:1) | 10 | 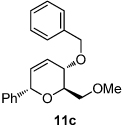 |
 |
| 2 | OTBS (15) | 95; (19:23 = 5.5:1; X = OH) | |||
| 3 | OAc (16) | 98; (20:24 = 2:1) | |||
| 4 | Br (17) | 93; (21) | |||
| 5 | N3 (18) | 80; (22:25 = 4.5:1) | |||
 |
 |
11 | 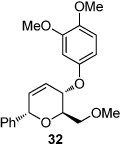 |
 |
|
| 6 | R = OMe (10b) | 26 (97) | 12 | 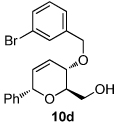 |
 |
| 7 | R = Me (10c) | 27 (81) | |||
| 8 | 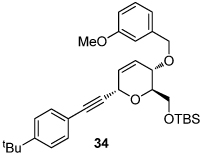 |
 |
Conditions: Sc(OTf)3 (20 mol %), Bu4NPF6 (20 mol %), CH3NO2, 0 °C to rt, 30 min.
Isolated yield and ratio.
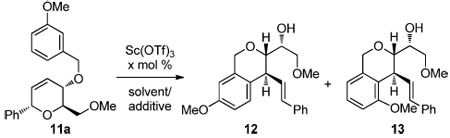
Dihydropyran 11a was selected to investigate the trapping of proposed intermediate 3 (Scheme 2b) using Lewis acid catalysis.8 A preliminary screen revealed that scandium(III) triflate was the optimal Lewis acid for this transformation.9 Indeed, exposure of 11a to 100 mol % Sc(OTf)3 in CH2Cl2 provided isochroman regioisomers 12 and 13 (5.5:1) in 95% combined yield.
Encouraged by this result, we set out to decrease catalyst loading (Table 1). Reducing the amount of Sc(OTf)3 to 25 mol % in CH2Cl2 resulted in a longer reaction time and low yields of 12/13 (entry 1). Substituting CH3NO2 for CH2Cl2, the former a cation stabilizing and Lewis acid activating solvent,10 enabled use of 20 mol % catalyst while maintaining the reaction rate (entry 2). A desire to further improve the catalytic efficiency of the reaction led us to salts having non-coordinating anions have been shown to activate Lewis acid catalysts.11 Conducting the rearrangement in the presence of Bu4NPF6 increased the rate of the reaction. As a control, conducting the reaction in the absence of Sc(OTf)3 resulted in recovery of 11a (entry 6).
Table 1.
Optimization of Catalytic Reaction Conditionsa
 | |||
|---|---|---|---|
| entry | x | solvent/additive (equiv) | % yield (12:13)b |
| 1 | 25 | CH2Cl2 / none | 72; 5:1c |
| 2 | 20 | MeNO2 / none | 94; 3.7:1 |
| 3 | 10 | MeNO2 / Bu4NPF6 (2) | 97; 3.3:1d |
| 4 | 2 | MeNO2 / Bu4NPF6 (2) | 85; 2.8:1 |
| 5 | 20 | MeNO2 / Bu4NPF6 (0.2) | 93; 3.6:1d |
| 6 | 0 | MeNO2 / Bu4NPF6 (2) | -e |
| 7 | 0 | MeNO2 / TfOH (0.2) | 87; 2.5:1d |
| 8 | 20 | MeNO2 / Bu4NPF6 (0.2)/ DTBMP (1) |
-e |
| 9 | 20 | MeNO2 / Bu4NPF6 (0.2)/ 3 Å MS |
-e |
All reactions conducted at 0 °C to rt for 2 h unless otherwise noted.
Isolated yield and ratios.
rt, 17 h.
time = 30 min.
no reaction. DTBMP = 2,6-di-tert-butyl-4-methylpyridine.
Triflic acid (TfOH) has been shown to be an active catalyst in reactions employing metal triflates.12 Accordingly, conducting the reaction with TfOH (20 mol %) provided a slightly lower yield and ratio of 12 and 13 in comparison to Sc(OTf)3 (entry 7). Furthermore, inclusion of 2,6-di-tert-butyl-4-methylpyridine (DTBMP) as an acid scavenger completely inhibited the reaction (entry 8). Addition of 3Å molecular sieves to the reaction to eliminate adventitious water also resulted in the recovery of 11a (entry 9). Taken together, our results strongly evaluate Bu4NPF6 as additive (entries 3–5). Organic suggest involvement of TfOH as a viable catalyst for the rearrangement.
A possible mechanism for the dihydropyran rearrangement is illustrated in Scheme 3. We propose intial Sc(OTf)3− or TfOH-promoted ionization of the anomeric C–O bond to afford a highly stabilized allyl cation similar to our previously reported Au(III) catalyzed process.4 Subsequent Friedel-Crafts alkylation13 likely proceeds through a chair-like transition state (14) in which both the hydroxymethyl ether and styryl substituents are oriented equatorially. This reactive conformer would lead to the observed trans stereochemistry of the newly formed pyran ring of products 12 and 13.
Scheme 3.

Proposed Mechanism of the Dihydropyran Rearrangement
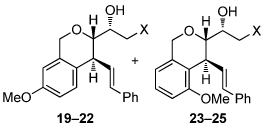
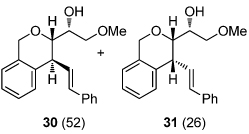
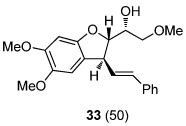
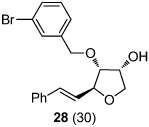
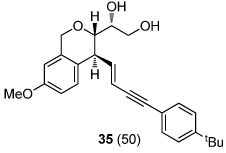
We next explored the substrate scope of the Friedel-Crafts reaction employing 20 mol % Sc(OTf)3 and 20 mol % Bu4NPF6 in CH3NO2 as standard conditions (Table 2). Functional group compatibility at C6 was first examined. Notably, in the case of substrates containing a competing nucleophile at C6, Friedel-Crafts alkylation was observed as the preferred pathway (80–95% yield, entries 1,2, and 5). Dihydropyrans containing either acetate or bromide functionality at C6 also rearranged efficiently (entries 3–4). Within the C4 benzyl ether series, electron-rich derivatives produced the corresponding isochromans effectively (entries 6 and 7). On the other hand, 3-bromobenzyl ether substrate 10d afforded the ring contraction product 28 in low yield (entry 8). Epimerization at C1 of the corresponding methyl ether derivative 11b (entry 9) provided support for our proposed mechanism. The neutral benzyl ether substrate 11c regained Friedel-Crafts alkylation reactivity producing a 2:1 mixture of trans:cis substituted isochromans in 78% yield (entry 10). Although moderately successful, rearrangement of C4 aryl ether 32 provided the distinct dihydrobenzofuran scaffold 33 (entry 11). Finally, the C1-alkynyl dihydropyran substrate 34 (entry 12) rearranged in good yield to afford isochroman enyne 35.
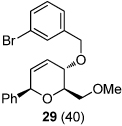
To investigate alternative reaction pathways accessible via the allylic cation, other π-terminating substituents at C4 were examined (Scheme 4). By replacing the benzyl group at C4 with an allyl group, we hoped to observe a sequential process where an initial cation-olefin cyclization14 would provide a tertiary carbocation that could undergo further transformations. Under the optimized conditions, α-styrenyl ether 11d afforded the dioxabicyclo[2.2.2]-octane 38 in 63% yield. Presumably, this reaction proceeds through trapping of the stabilized tertiary carbocation, arising from a cation-olefin cyclization, by the newly formed secondary metal-alkoxide (37). Interestingly, rearrangement of the related 2-methallyl ether substrate 11e resulted in formation of an unexpected product which was characterized as the dioxabicyclo[3.2.1]-octane 40. The structure of 40 was confirmed by X-ray analysis of a crystalline 2,4-dinitrophenylhydrazone derivative.6 In this case, the tertiary carbocation 39 apparently undergoes a 1,2-hydride shift (migration of the appropriately aligned H1 provides the major diastereomer observed) resulting in formation of an oxocarbenium ion, which is trapped by the metal alkoxide leading to 40.
Scheme 4.

Rearrangements of substituted allyl ethers and mechanistic rationale.
In summary, we have demonstrated divergent rearrangements of glycal-derived dihydropyrans to afford a series of structurally distinct frameworks. Isochroman skeletons were obtained by Friedel-Crafts trapping of allylic cations generated from the acid-catalyzed opening of a dihydropyran. Dioxabicyclo[2.2.2]- and dioxabicyclo[3.2.1]octanes have been accessed in a process involving nucleophilic attack on the cation generated from olefin cyclizations. Expansion of the rearrangement chemistry to cascade processes, as well as library synthesis applications, is currently underway and will be reported in future publications.
Supplementary Material
Acknowledgment
We gratefully acknowledge the NIGMS CMLD initiative (P50-GM067041) for financial support and the National Science Foundation for the purchase of the Waters high resolution mass spectrometer (CHE-0443618) used in this work. We also thank Dr. Paul Ralifo (Boston University) for NMR assistance and Dr. Jeff Bacon (Boston University) for X-ray crystallographic analysis.
Footnotes
Supporting Information Available Experimental procedures and characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Select reviews: Liao Y, Hu V, Wu J, Zhu Q, Donovan M, Fathi R, Yang Z. Curr. Med. Chem. 2003;10:2285. doi: 10.2174/0929867033456738. Reayi A, Arya P. Curr. Opin. Chem Biol. 2005;9:240. doi: 10.1016/j.cbpa.2005.04.007. Thomas GL, Wyatt EE, Spring DR. Curr. Opin. Drug Discovery Dev. 2006;9:700. Grabowski K, Baringhaus K-H, Schneider G. Nat. Prod. Rep. 2008;25:892. doi: 10.1039/b715668p. Cordier C, Morton D, Murrison S, Nelson A, O'Leary-Steele C. Nat. Prod. Rep. 2008;25:719. doi: 10.1039/b706296f. Nielsen TE, Schreiber SL. Angew. Chem. Intl. Ed. 2008;47:48. doi: 10.1002/anie.200703073.
- 2.(a) Schreiber SL. Science. 2000;287:1964. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]; (b) Schreiber SL. Chem. Eng. News. 2003;302:613. [Google Scholar]; (c) Tan DS. Chem. Biol. 2007;2:483. [Google Scholar]; (d) Spandl RJ, Bender A, Spring DR. Org. Biomol. Chem. 2008;6:1149. doi: 10.1039/b719372f. [DOI] [PubMed] [Google Scholar]; (e) Shaw JT. Nat. Prod. Rep. 2009;26:11. doi: 10.1039/b814468k. [DOI] [PubMed] [Google Scholar]
- 3.(a) Schreiber SL, Burke MD. Angew. Chem. Intl. Ed. 2004;43:46. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]; (b) Kesavan S, Panek JS, Porco JA., Jr Org. Lett. 2007;9:5203. doi: 10.1021/ol7023778. [DOI] [PubMed] [Google Scholar]; (c) Su S, Porco JA., Jr Org. Lett. 2007;9:4983. doi: 10.1021/ol702176h. [DOI] [PubMed] [Google Scholar]; (d) Garcia-Cuadrado D, Barluenga S, Winssinger N. Chem. Commun. 2008:4619. doi: 10.1039/b807869f. [DOI] [PubMed] [Google Scholar]; (e) Aguilar-Moncayo M, Ortiz-Mellet C, Garcia-Fernandez JM, Garcia-Moreno MI. J. Org. Chem. 2009;74:3595. doi: 10.1021/jo900231b. [DOI] [PubMed] [Google Scholar]
- 4.Yeager AR, Min GK, Porco JA, Jr, Schaus SE. Org. Lett. 2006;8:5065. doi: 10.1021/ol0618252. [DOI] [PubMed] [Google Scholar]
- 5.For selected applications of glycals in synthesis see:Heck reaction Bedjeguelal K, Bolitt V, Sinou D. Synlett. 1999;6:762. Pauson-Khand reaction Kubota H, Lim J, Depew KM, Schreiber SL. Chem. Biol. 2002;9:265. doi: 10.1016/s1074-5521(02)00099-6. Radical reactions Fraser-Reid B, López JC. Curr. Org. Chem. 2009;13:532.
- 6.Trost BM, Toste FD. J. Am. Chem. Soc. 1998;120:815. [Google Scholar]
- 7.See Supporting Information for complete experimental details.
- 8.For select syntheses of isochromans via intramolecular Friedel-Crafts-type reactions see: Martin OR. Carbohydr. Res. 1987;171:211. doi: 10.1016/s0008-6215(00)90888-7. Ahmer M, Bloch R. Synth. Commun. 1992;22:1417. Fearnly SP, Tidwell MW. Org. Lett. 2002;4:3797. doi: 10.1021/ol026494h. Chandrasekhar S, Khatun S, Rajesh G, Reddy R. Tetrahedron Lett. 2009;50:6693.
- 9.For applications of lanthanide triflates to Friedel-Crafts reactions, see: Walker M. U.S. Patent. 2002 March 26;6:362–375. Yoon MY, Kim JH, Choi DS, Shin US, Lee JY, Song CE. Adv. Synth. Catal. 2007;349:1725. Dzuda A, Marks TJ. J Org. Chem. 2008;73:4004. doi: 10.1021/jo800158k. Rueping M, Nachtsheim BJ. Beilstein J. Org. Chem. 2010;6(No. 6) doi: 10.3762/bjoc.6.6.
- 10.(a) Hayashi E, Takahashi Y, Itoh H, Yoneda N. Bull. Chem. Soc. Jpn. 1993;66:3520. [Google Scholar]; (b) Hachiya I, Moriwaki M, Kobayashi S. Tetrahedron Lett. 1995;36:409. [Google Scholar]
- 11.(a) Malona JA, Colbourne JM, Frontier AJ. Org. Lett. 2006;8:5664. doi: 10.1021/ol062403v. [DOI] [PubMed] [Google Scholar]; (b) Kim JH, Lee JW, Shin US, Lee JY, Song CE. Chem. Commun. 2007;44:4683. doi: 10.1039/b712060e. [DOI] [PubMed] [Google Scholar]
- 12.(a) Fillion E, Fishlock D. Org. Lett. 2003;5:4653. doi: 10.1021/ol035817m. [DOI] [PubMed] [Google Scholar]; (b) Dumeunier R, Markó IE. Tetrahedron Lett. 2004:825. [Google Scholar]; (c) Rosenfeld DC, Shekhar S, Takemiya A, Utsunomiya M, Hartwig JF. Org. Lett. 2006;8:4179. doi: 10.1021/ol061174+. [DOI] [PubMed] [Google Scholar]
- 13.For select examples of Friedel-Crafts alkylation of allylic cations see: Ohwada T, Yamagata N, Shudo K. J. Am. Chem. Soc. 1991;113:1364. Mitchell WL, Kocovsky P. J. Org. Chem. 1999;64:2751. doi: 10.1021/jo982178y. Malkov AV, Spoor P, Vinader V, Kocovsky P. J. Org. Chem. 1999;64:5308. doi: 10.1021/jo990372u. Huang J-W, Shi M. Tetrahedron Lett. 2003;44:9343. Li J-H, Liu W-J, Yin D-L. Synth. Commun. 2004;34:3161. Bandini M, Eichholzer A, Kotrusz P, Umani-Ronchi A. Adv. Synth. Catal. 2008;350:531. Hu B, Xing S, Wang Z. Org. Lett. 2008;10:5481. doi: 10.1021/ol802301e.
- 14.For a review of related polyolefin cyclizations, see: Yoder RA, Johnston JN. Chem. Rev. 2005;105:4730. doi: 10.1021/cr040623l.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.