

Abstract
In Candida albicans, ergosterol biosynthetic genes, including ERG11, which encodes the target of azole antifungal drugs, are regulated by the transcriptional regulator Upc2p. To initially characterize the promoter of the UPC2 gene, 5′ rapid amplification of cDNA ends was used to identify two transcriptional initiation sites upstream of the ATG codon. The regions within the UPC2 promoter required for azole regulation of the UPC2 promoter were then identified using nested deletions fused to a luciferase reporter which were tested for azole inducibility in wild-type (WT) and upc2Δ/upc2Δ strains. Two distinct regions important for azole induction were identified: a Upc2p-dependent region (UDR) between bp −450 and −350 upstream of the ATG codon and a Upc2p-independent region (UIR) between bp −350 and −250 upstream of the ATG codon. Within the UDR, loss or mutation of the sterol response element (SRE), so named because of homology to the Saccharomyces cerevisiae Upc2p binding site, resulted in a decrease in both basal and induced expression in the WT strain but did not affect azole inducibility in the upc2Δ/upc2Δ deletion strain. Gel shift analyses using the DNA binding domain of Upc2p confirmed binding of the protein to two SRE-related sequences within the UPC2 promoter, with strongest binding to the UDR SRE. Detailed gel shift analyses of the UDR SRE shows that Upc2p binds to a bipartite element within the UPC2 promoter, including the previously identified SRE and a new, adjacent element, the short direct repeat (SDR), with partial homology to the SRE.
The pathogenic yeast Candida albicans causes oral, vaginal, and systemic diseases in immunocompromised hosts and vaginal infection in immune-competent hosts. Significant mortality is seen with systemic disease, which occurs most commonly in neutropenic patients, such as those receiving transplant chemotherapy. The most frequently used antifungals for treatment of oral candidiasis are azoles, which act by targeting the ergosterol biosynthesis enzyme lanosterol 14-α-demethylase (Erg11p). Overexpression of ERG11 and other genes in the ergosterol biosynthetic pathway (ERG genes) has been associated with decreased susceptibility to azoles in clinical isolates of C. albicans (19). Upc2p is a global transcriptional activator of the ERG genes in Saccharomyces cerevisiae and in C. albicans and has been shown to upregulate gene expression as a compensatory mechanism in response to inhibition of ergosterol biosynthesis. Deletion of UPC2 in C. albicans leads to a decrease in tolerance to azole drugs, and UPC2 gain-of-function mutations that lead to ERG11 overexpression have been identified in azole-resistant clinical isolates (5, 7). Upc2p is thought to activate gene expression by binding to a DNA sequence known as the sterol response element (SRE) within the promoters of target genes in both S. cerevisiae and C. albicans (11, 15). The SRE contains a 7-bp conserved core sequence that is essential for Upc2p binding and activation in response to azole drugs (11, 12). However, the full binding site for Upc2p in these promoters has never been fully characterized.
Work done with S. cerevisiae has shown that an SRE within the S. cerevisiae UPC2 (ScUPC2) promoter itself is important for anaerobic induction of ScUPC2 expression (2). Ablation of this SRE caused a decrease in the fold induction of a UPC2-lacZ reporter in response to anaerobicity but did not completely eliminate induction. Additionally, there was a modest increase in the basal, or uninduced, level of UPC2-lacZ expression from the reporter in which the SRE was mutated. These data suggest a number of important conclusions. First, the decreased inducibility of the UPC2-lacZ construct lacking the SRE suggests that Upc2p acts at its own promoter to control UPC2 gene expression in an autoregulatory mechanism. Second, the higher basal level of expression when the SRE is lacking may represent relief from repression, suggesting that the autoregulatory mechanism is one in which Upc2p represses its own transcription. Importantly, the observation that mutation of the SRE does not completely eliminate the inducibility of the ScUPC2 promoter indicates that an additional, novel factor may also activate transcription of the ScUPC2 promoter.
Data obtained using a C. albicans upc2Δ/upc2Δ strain have similarly suggested that C. albicans Upc2p (CaUpc2p) regulates its own expression together with another factor (9). That work demonstrated that even in the absence of endogenous CaUpc2p, a UPC2-RLUC reporter was inducible in response to various sterol-depleting conditions, suggesting that another transcription factor activates CaUPC2 expression. The putative SRE in the CaUPC2 promoter is located between bp −437 and −431 upstream of the translational start (Fig. 1). It is presumed that CaUpc2p acts through this binding site, although this has not been tested directly. Recent chromatin immunoprecipitation (ChIP)-on-ChIP experiments using hemagglutinin-tagged CaUpc2p have demonstrated that CaUpc2p indeed binds to its own promoter, although the location of binding within this promoter or its effects on transcriptional activity were not studied (23).
Fig. 1.

UPC2 promoter sequence and transcriptional initiation sites. (A) Diagram of the UPC2 promoter region. Arrows pointing to the right indicate the putative starts of transcription at bp −361 and −148 upstream of the ATG codon, the start of the coding region (gray box at right). The UDR and UIR of the promoter are shown as gray boxes. The SDR, SRE-A, and SRE-B are shown as black boxes. Below the diagram, the lines indicate the region of the promoter included in each construct, as labeled on the right. The X in the 450-m construct represents the mutation of SRE-A. At the right, the level of promoter activity is shown for each construct when it is expressed in the WT or upc2Δ/upc2Δ strain. This diagram is not drawn to scale but reflects the relative positions of the elements in the promoter. (B) The promoter sequence is shown for bp −500 upstream of the ATG initiation codon (bold and italic). Sequence analysis of 5′ RACE products indicated 5′ start of transcription at bp −361 and −148 (bold and underlined) relative to the translational start. Putative consensus TATA boxes are upstream of each site (bold), suggesting that there are two independent transcriptional starts. The putative SRE-A at bp −437 to −431 and the putative SRE-B at bp −183 to −177 are underlined. The sequences within the SRE and SDR that have been identified as important for Upc2p binding are overlined.
In this study, we performed deletion and binding analyses to demonstrate that CaUpc2p binds to SRE and SRE-like sequences within the UPC2 promoter, demonstrating transcriptional autoregulation. This work determines which regions of the CaUPC2 promoter are required for azole inducibility in wild-type (WT) C. albicans as well as a C. albicans upc2Δ/upc2Δ strain. Regions of the promoter required for Upc2p-dependent and Upc2p-independent azole induction have been identified. Most importantly, the work shows that a separate and adjacent Upc2p binding element, termed the short direct repeat (SDR), is also important for activation of the UPC2 promoter. This work contributes to our understanding of how CaUpc2p acts to regulate genes required for the response to antifungal drugs that target ergosterol biosynthesis.
MATERIALS AND METHODS
Strains and growth conditions.
C. albicans strain BWP17 (ura3::λ434/ura3::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG). (20) and its derivative, D-6 (upc2::URA3/upc2::ARG4) (15), were transformed with UPC2-RLUC expression constructs containing the nourseothricin resistance marker SAT1. For FCR1 analysis, a C. albicans strain containing insertion mutations within both alleles of the FCR1 open reading frame was obtained from an insertion mutant library from the laboratory of A. Mitchell. C. albicans laboratory strain SC5314 (6), the parental strain of BWP17, D-6, and the FCR1 mutant, was used for RNA extraction for analysis of UPC2 transcription initiation sites. Growth of strains for all experiments was determined in CSM (0.75 g CSM [Bio 101 Inc., Vista, CA], 5.0 g ammonium sulfate, 1.7 g yeast nitrogen base without amino acids or ammonium sulfate, and 20 g dextrose per liter) or YEPD (10 g yeast extract, 20 g peptone, and 20 g dextrose per liter) as indicated.
Creation of constructs containing UPC2 promoter deletions for transcriptional fusions with the Renilla reniformis luciferase reporter.
Construction of nested deletions of the UPC2 promoter that drives RLUC expression was carried out as described for the UPC2-750-RLUC construct, created in the vector pCRW3 (9, 17). Briefly, oligonucleotides were designed (see Table S1 in the supplemental material) and used in PCR to amplify a specific region of the UPC2 promoter from the upstream ATG codon to a designated point from the full-length clone pUPC2-RLUC (9). The promoter fragments were then cloned upstream of the luciferase gene in the vector pCRW3-SAT1 (Fig. 1A) (9). To create the 450-bp UPC2-RLUC reporter construct containing the mutated SRE, a QuikChange II site-directed mutagenesis kit (Stratagene, La Jolla, CA) was used with oligonucleotides UPC2SRE-F and UPC2SRE-R (see Table S1).
Construction of C. albicans strains expressing UPC2-luciferase reporters.
C. albicans parental strains (see Table S2 in the supplemental material) were transformed using the lithium acetate heat shock method described previously (9) with modifications. Briefly, 500 μl of YEPD overnight cultures was diluted into 50 ml of fresh YEPD and grown for 5 h at 30°C with shaking at 180 rpm. Cells were prepared by pelleting, one wash with sterile water, and resuspension in 0.1 M lithium acetate in Tris-EDTA (TE), pH 7.5 (Li-TE). NsiI-linearized plasmid DNA (5 to 10 μg) and 10 μg carrier DNA (sheared herring sperm DNA; Invitrogen) were used for each transformation. Cells were incubated overnight with DNA in 40% polyethylene glycol 3350 in Li-TE, heat shocked for 30 min at 42°C, and washed once in 1 M sorbitol. This was followed by 4 h of growth at 30°C with shaking at 180 rpm in YEPD in the absence of nourseothricin selection, after which cells were plated on YEPD plates containing 200 μg/ml nourseothricin as described previously (13).
PCR and Southern blot screening of C. albicans transformants.
To confirm proper integration of the constructs, genomic DNA was prepared from the C. albicans transformants grown overnight in YEPD-NAT (nourseothricin, 200 μg/ml), using glass bead lysis as described previously (8). Transformants were initially screened for positive integration of NsiI-digested pUPC2-RLUC at the ADE2 locus by using the oligonucleotides ADE2 and RLUC (see Table S1 in the supplemental material). PCR-positive transformants were then confirmed by Southern blotting to contain the pUPC2-RLUC construct in a single copy. Briefly, 10 μg of genomic DNA was digested with KpnI overnight, run on a 0.7% agarose gel, and blotted overnight to a nitrocellulose membrane. The blot was probed with a 32P-end-labeled RLUC oligonucleotide probe (see Table S1). Transformants containing pUPC2-RLUC in a single copy at the ADE2 locus were used for the luciferase assay.
Luciferase assay of UPC2-RLUC activity.
Luciferase assays were performed as described previously (17) with modifications (16). Azole induction was assessed by growing strains expressing UPC2-RLUC constructs in the presence and absence of 100 μg/ml fluconazole (FLC; stock concentration of 3.0 mg/ml; Pfizer, New York, NY). The data shown are representative of three independent experiments, and in all cases, identical patterns of induction were seen between experiments. The values reported are ratios of luciferase activity in the presence of FLC relative to those in the absence of FLC, and thus, data were not compared statistically between experiments. Intra-assay variability was tested and in all cases was shown to be minimal; between 3 independent replicates in the same assay, variability was always lower than 10% and most often was below 5%.
Quantitative real-time reverse transcriptase PCR (QRT-PCR) to determine UPC2 expression levels.
To assay UPC2 transcript levels, overnight cultures of C. albicans BWP17 and fcr1/fcr1 strains were used to inoculate 25 ml of CSM to an optical density at 600 nm of 0.1 and cultures were grown for 6 h in the presence and absence of 100 μg/ml FLC. RNA was extracted as described previously (3). RNA quantity and integrity were checked using agarose gel electrophoresis. To perform QRT-PCR analysis, cDNA was synthesized from 200 ng of total RNA. First, RNA was DNase treated with RQ1 DNase (Promega Corp., Madison, WI) according to the manufacturer's recommendation. The DNase-treated RNA was then used as the template in cDNA synthesis reactions by using a Quantiscript reverse transcription kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. Real-time PCR was performed using the iTaq (Bio-Rad, Hercules, CA) SYBR green system on an ABI 7000 (Applied Biosystems) by following the manufacturer's recommended protocol.
5′ RACE analysis of UPC2 transcriptional initiation sites.
Transcriptional initiation sites within the UPC2 promoter were mapped using the 5′ RACE system for rapid amplification of cDNA ends (Invitrogen, Carlsbad, CA). RNA was extracted as described for QRT-PCR analysis. For cDNA synthesis, 5 μg of total RNA was reverse transcribed using the UPC2-specific oligonucleotide UPC2 RACE cDNA (see Table S1 in the supplemental material). The 5′ RACE PCR was performed using the UPC2-specific primer UPC2 RACE PCR (see Table S1) and the cDNA end anchoring primer provided by the manufacturer (Invitrogen). Resulting products were analyzed by agarose gel electrophoresis and sequencing.
Electrophoretic mobility shift assays (EMSA).
To determine whether Upc2p binds to sequences within the UPC2 promoter, the genomic sequence encoding the Upc2p DNA binding domain (DBD) (amino acids 1 to 152) was amplified by PCR using primers UPC2DBD 5′ and UPC2DBD 3′ (see Table S1 in the supplemental material) and cloned into the vector pET-28B, which adds a His tag to the N terminus of the protein. The construct was then expressed in Escherichia coli and the recombinant DBD protein (rDBD) purified using His columns (Ni-NTA Fast Start kit, catalog no. 30600; Qiagen, Valencia, CA). The purified and tagged rDBD was then used to perform EMSA. Two 65-bp Cy5-labeled complementary oligonucleotides spanning UPC2 SRE-A or the SRE upstream of ERG11 (see Table S1) were used to detect electrophoretic mobility shifts in the presence of the rDBD. The single-stranded oligonucleotides were annealed by heating to 100°C and then cooling to room temperature at a concentration of 10 μM in 1× TE to generate double-stranded probes. Competition experiments were performed with unlabeled 65-bp double-stranded DNA (dsDNA) oligonucleotides listed in Table S1 that were annealed at a concentration of 180 μM in 1× TE. Binding reactions were performed for 30 min at room temperature under the following reaction conditions: 4 mM Tris, 4 mM MgCl2, 10 μM ZnSO4, 5% glycerol, 40 nM NaCl, 0.5 μg of herring sperm DNA, 0.25 μg Upc2p-rDBD, and 10 pmol of labeled probe in a final volume of 20 μl. For competition experiments, a 25× concentration of unlabeled dsDNA was added to the reaction. Reactions were run at 20 to 25 mA on a 6% polyacrylamide gel that was prerun in 1× Tris-borate-EDTA for 30 min.
RESULTS
UPC2 transcription is initiated at two distinct sites in response to azole drug treatment.
The transcriptional initiation sites (specifically, the mRNA start sites) in the UPC2 promoter were mapped using 5′ RACE. Transcription initiation was analyzed in SC5314, the parental strain of both the wild type, BWP17, and the upc2Δ/upc2Δ strain, D-6. 5′ RACE analysis amplified two bands of approximately 450 bp and 250 bp (Fig. 2, lane RACE). Sequencing of these two fragments showed the 5′ termini of the two bands to be at bp −361 and −148 upstream of the ATG start of translation (Fig. 1B). The intensity of the two bands suggests that the −148 initiation site is the predominant initiation site for the UPC2 gene. Both of these transcriptional start sites contain putative consensus TATA boxes within a reasonable distance upstream, and no ATG codons are present downstream of the −361 initiation site until the presumed initiation codon (Fig. 1B), suggesting that RNAs that initiate at both sites act to initiate translation of Upc2p.
Fig. 2.

5′ RACE analysis reveals two 5′ termini for the UPC2 transcript. RNA from SC5314 was prepared and used as the template in 5′ RACE analysis. Resulting PCR products were analyzed using gel electrophoresis on a 1.5% agarose gel. Bands were extracted from the gel, and 5′ termini were determined by DNA sequencing. Lanes are from two sections of the same gel. Bands are labeled for the position within the gene (bp −361 and −148) and not the size of the gel fragment.
Upc2p-dependent region (UDR) required for azole inducibility located between bp 450 and 350 upstream of the translational start.
Previous studies have suggested that UPC2 transcription in S. cerevisiae is autoregulated (1, 4). Work from this laboratory has shown that in addition to autoregulation by Upc2p, an additional transcription factor controls UPC2 expression in C. albicans (9). To identify the regions of the UPC2 promoter that are required for azole inducibility, nested deletions of 100 bp were made from bp 750 to 250 upstream of the UPC2 translation initiation codon (Fig. 1A). These promoter fragments were cloned upstream of the Renilla reniformis luciferase reporter (RLUC) in the plasmid pCRW3. The reporter constructs were introduced into BWP17 and assayed for luciferase activity after 48 h of exposure to 100 μg/ml FLC. The 48-h time point was chosen, as induction becomes more prominent at this time point relative to earlier time points, presumably due to depletion of intracellular sterol stores prior to drug exposure (9). The nested deletions within the UPC2 promoter were also introduced into the upc2Δ/upc2Δ strain, D-6, to differentiate Upc2p-dependent and Upc2p-independent promoter regions.
The 100-bp deletions were tested for RLUC activity in response to 48 h of FLC exposure in both the wild-type and upc2Δ/upc2Δ strains. Deletions between bp 750 and 450 upstream of the start did not significantly alter UPC2-RLUC inducibility in response to 100 μg/ml FLC in either strain, showing 18-fold induction in response to azoles for all fragment sizes in this region (data not shown). Loss of the 100-bp region between bp 450 and 350, however, led to a dramatic decrease in inducibility (85% reduction) in the wild-type strain, BWP17, after 48 h of FLC exposure (Fig. 3).
Fig. 3.
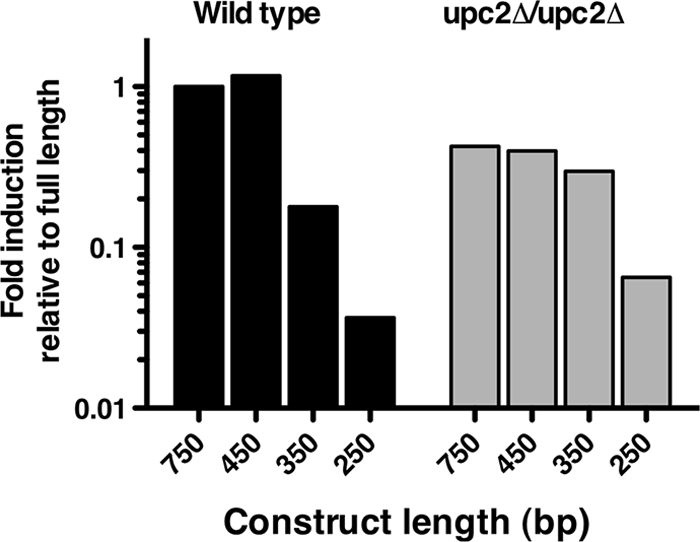
Azole inducibility of 100-bp nested deletions of the UPC2-RLUC reporter in the WT and the UPC2 homozygous deletion strain. Deletion constructs of 100 bp are listed on the x axis, with the length of the construct representing the distance upstream of the ATG codon. Luciferase assays were performed after 48 h of growth in the presence and absence of 100 μg/ml FLC. Data are presented as the proportion of azole induction of RLUC activity of each deletion construct relative to the activity in the full-length promoter construct. Wild-type BWP17 is shown in black and the upc2Δ/upc2Δ strain in gray.
In the upc2Δ/upc2Δ strain, the 450-bp fragment shows maximal FLC inducibility of 6.2-fold at 48 h (data not shown). These data are consistent with the overall lower fold induction of UPC2-RLUC in the upc2Δ/upc2Δ strain than in the wild-type strain reported previously (9). The 350-bp UPC2 promoter fragment expressed in the upc2Δ/upc2Δ strain is reduced only 25% after 48 h of FLC exposure, retaining much of the inducibility of the longer, 450-bp construct. The loss of azole induction in the wild-type strain and not the upc2Δ/upc2Δ strain suggests a role for Upc2p-dependent azole inducibility in the promoter region between bp 450 and 350. Because this region appears important only in the presence of Upc2p, we have termed this region the Upc2p-dependent region (UDR). Importantly, the putative SRE within the UPC2 promoter is located within the UDR at bp −437 to −431 upstream of the translational start of UPC2 (Fig. 1).
Upc2p-independent azole-inducible region located between bp 350 and 250 upstream of the translational start.
Because the 350-bp promoter fragment still retained some inducibility in both the wild-type and upc2Δ/upc2Δ strains, further promoter deletion was performed to determine elements required for inducibility downstream of bp 350. Luciferase assays at 48 h revealed that FLC induction was greatly reduced in the 250-bp UPC2 promoter fragment in the wild-type strain (97% reduction) (Fig. 3). Similarly, in the upc2Δ/upc2Δ strain, the 250-bp UPC2-RLUC reporter was not induced by FLC (85% reduction) (Fig. 3). Thus, loss of the promoter sequence between bp 350 and 250 resulted in loss of inducibility independent of whether or not the strain expressed Upc2p (wild type compared to the upc2Δ/upc2Δ strain). Because this 100-bp region is required for the inducibility seen in both the wild-type and UPC2 deletion strains, we have termed it the Upc2p-independent region (UIR).
Deletions within the UDR alter UPC2-RLUC inducibility in the wild-type strain but not in the UPC2 homozygous deletion strain.
Given that the putative SRE within the UPC2 promoter is located within the 100-bp UDR, 25-bp deletions within this region were created to determine whether the promoter containing the SRE was required for azole inducibility (Fig. 1A). Luciferase assays with the deletions in the wild-type strain showed that truncation of the promoter sequence downstream of bp 450 caused a decrease in azole induction in comparison to the 450-bp construct (Fig. 4). It is important to note that all constructs 425 bp or shorter lack the putative SRE at bp −431 to −437, and the loss of induction with the 425-bp construct is consistent with a role for the SRE as an azole-inducible element. All of the truncations between bp 450 and 350 in the upc2Δ/upc2Δ strain were induced to similar levels, indicating that loss of the promoter sequence in this region does not affect induction in the absence of Upc2p (Fig. 4). These data are consistent with the 100-bp deletion analyses showing that azole inducibility within the UDR is altered only in the wild-type strain, in which Upc2p is expressed.
Fig. 4.

Azole inducibility of the Upc2p-dependent region (UDR) in the WT strain and the UPC2 homozygous deletion mutant. Deletion constructs with lengths from 350 to 450 bp upstream of the start of transcription are listed on the x axis. Luciferase assays were performed after 48 h of growth in the presence and absence of 100 μg/ml FLC. Data are presented as the proportion of azole induction of RLUC activity of each deletion construct relative to the activity in the full-length promoter construct. Wild-type BWP17 is shown in black and the upc2Δ/upc2Δ strain in gray.
Mutation of the putative SRE within the UPC2 promoter lowers UPC2-RLUC inducibility.
Previous work has suggested that Upc2p binds to the promoters of target genes through the SRE binding site (11). Mutation of the conserved core sequence within the SRE lowers Upc2p-dependent azole induction of the ERG11 promoter (12). In order to definitively address the role of the putative SRE within the UDR, the SRE within the 450-bp UPC2-RLUC reporter was mutated. The 7-bp core Upc2p binding site sequence 5′-TCGTATA-3′ was altered using site-directed mutagenesis oligonucleotides that replaced guanine with adenine, adenine with cytosine, thymidine with guanine, and cytosine with thymidine (see Table S1 in the supplemental material). The effect of this mutation on UPC2-RLUC azole inducibility was tested in both the wild-type and upc2Δ/upc2Δ strains. When expressed in the wild-type strain, the UPC2-RLUC fusion bearing the mutated SRE showed a 75% reduction in azole inducibility compared with the 450-bp construct with the wild-type SRE (Fig. 5). These data are consistent with the role of the SRE as a cis-acting element required for azole inducibility within the UPC2 promoter. In the upc2Δ/upc2Δ strain, the mutated SRE had a minor effect on azole inducibility (25% reduction), which was already low (Fig. 5).
Fig. 5.
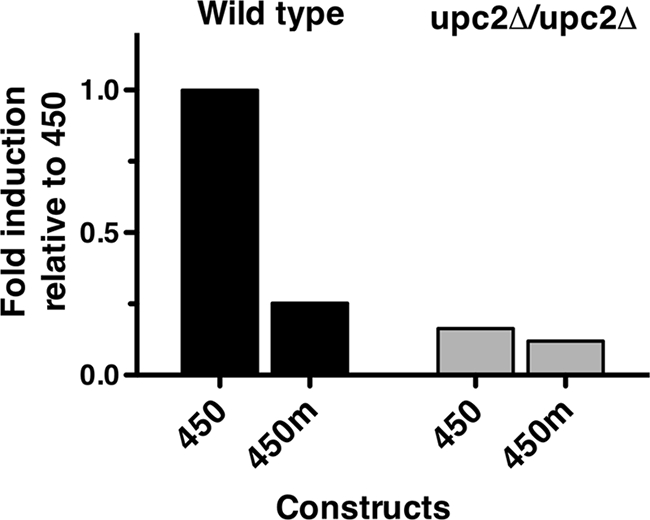
Mutation of the putative SRE lowers the inducibility of UPC2-RLUC in the wild-type strain. The 450-bp luciferase construct (450) and the 450-m construct mutated within the SRE (450m) were assayed in the WT and upc2Δ/upc2Δ strains. Luciferase assays were performed after 48 h of growth in the presence and absence of 100 μg/ml FLC. Data are presented as the proportion of azole induction of RLUC activity of each deletion construct relative to the activity in the WT 450-bp promoter construct. The construct is listed on the x axis. Wild-type BWP17 is shown in black and the upc2Δ/upc2Δ strain in gray.
UPC2 expression is altered in an FCR1 insertion mutant irrespective of FLC exposure.
Data resulting from ChIP-on-ChIP (chromatin immunoprecipitation followed by microarray analysis) experiments using the CaFcr1p transcription factor identified the UPC2 promoter as a target of CaFcr1p (22). Additionally, the inducible expression of CaFcr1p increased CaUPC2 transcription, suggesting that CaFcr1p may directly regulate CaUPC2 transcription (22). To test this, a C. albicans mutant containing an insertion within both alleles of FCR1 (from an insertion mutant library obtained from the lab of A. Mitchell) as well as the parent strain BWP17 was grown in the presence and absence of 100 μg/ml FLC for 6 h, and RNA was extracted for QRT-PCR analysis. Comparison of UPC2 transcript levels by QRT-PCR shows that both strains induce UPC2 mRNA expression 2.0-fold in response to FLC. When the QRT-PCR results for UPC2 expression are compared between the two strains, however, there is a clear difference in UPC2 transcript levels between BWP17 and the FCR1 mutant. UPC2 expression levels are 12.0-fold higher in BWP17 than in the FCR1 mutant in the absence of drug and 16.2-fold higher in BWP17 than in the FCR1 mutant in the presence of FLC (Fig. 6). These data are consistent with ChIP-on-ChIP experiments showing that Fcr1p binds to the UPC2 promoter and positively regulates UPC2 expression (22).
Fig. 6.

CaUPC2 expression is lower in an fcr1 mutant than in the WT irrespective of FLC exposure. QRT-PCR was performed on RNA from either the WT (BWP17) or the Δfcr1/Δfcr1 insertion mutant strain grown for 6 h in the absence (black bars) and presence (gray bars) of 100 μg/ml FLC. Data are expressed as threshold cycle (ΔΔCT) values, normalized to that of an ACT1 control [2(CT UPC2 − CT ACT1)]. RNA was prepared at 6 h, as higher-quality RNA is obtained at this time point than at 48 h, which is used for luciferase experiments.
Upc2p binding is dependent on two elements within the UPC2 promoter, the SRE and an element containing a short direct repeat (SDR).
To validate whether UPC2 transcriptional autoregulation is due to the direct transcriptional effect of Upc2p binding to its own promoter, gel shift analysis was performed. Two putative SREs can be identified in the UPC2 promoter (Fig. 1). The first, SRE-A, is a perfect SRE (5′-TATACGA-3′ [reverse complement]) and is located between bp −437 and −431 upstream of the ATG codon. The second, SRE-B, is not a perfect SRE (5′-TCGTTAA-3′ [mismatches underlined]) and is located at positions −183 to −177 upstream of the ATG codon, very close to the downstream initiation site, between the putative TATA box and the initiation site (Fig. 1). Both SRE-A and SRE-B were tested for binding to the recombinant Upc2p DNA binding domain (rDBD). The purified rDBD from E. coli was used in electrophoretic mobility shift assays (EMSA) with a 65-mer Cy5-labeled oligonucleotide containing the SRE from the ERG11 promoter, which Upc2p is known to regulate. Strong and specific binding of the rDBD to this element was observed (Fig. 7A, lane 2), consistent with previous work that measured binding to the ERG2 promoter (11). Competition experiments were performed using unlabeled oligonucleotides containing SRE-A and SRE-B from the UPC2 promoter. The competition results show that SRE-A was highly proficient at competing with the ERG11 SRE probe (Fig. 7A, lane 3), consistent with its match to the consensus sequence. Competition was also achieved with SRE-B but was much weaker than that seen for SRE-A (Fig. 7A, lane 5). Similar competition results were observed for SRE-A and SRE-B when they were used in competition studies with a Cy5-labeled oligonucleotide containing SRE-A from the UPC2 promoter (Fig. 7A, lanes 7 to 12). All subsequent experiments were performed using labeled UPC2 SRE-A and unlabeled cold competitors.
Fig. 7.

CaUpc2p binds to the SREs within the UPC2 promoter. (A) An electrophoretic mobility shift assay (EMSA) was performed using the purified Upc2p DNA binding domain (rDBD) and Cy5-labeled oligonucleotides containing the SRE from ERG11 (left) or UPC2 (right). Unlabeled competitors include SRE-A and SRE-B as well as mutated versions of SRE-A and SRE-B. Unlabeled competitors were used at 25-fold molar excess. (B) The percent binding or gel shift of the labeled SRE-A probe (y axis) was determined in the presence of increasing amounts of cold competitor of SRE-A (circles) or SRE-B (squares) (x axis).
Competition experiments were performed with various concentrations of unlabeled SRE-A and SRE-B to determine the 50% inhibitory concentrations (IC50s) for each SRE, which is a measure of the relative affinity of each SRE for the rDBD. The IC50 data are plotted in Fig. 7B and show that the rDBD binds with greater affinity to SRE-A than to SRE-B. The IC50s for SRE-A and SRE-B were 10.7 pmol per reaction and 78.6 pmol per reaction, respectively.
Because SRE-A was shown to be important for Upc2p-dependent transcriptional regulation (Fig. 5), further gel shift analysis focused on SRE-A. Competition experiments in which the SRE-A was mutated (Fig. 7A, lanes 4 and 10) show that mutation of the SRE-A core within the oligonucleotide did not significantly abrogate the oligonucleotide's competition for binding to the SRE from ERG11 (lane 4) or for binding to SRE-A from UPC2 (lane 10). This indicates that there is an additional element(s) within the 65-bp UPC2 SRE-A oligonucleotide that competes for binding to the rDBD. Mutation of SRE-B abolishes competition against the ERG11 SRE (lane 6) and UPC2 SRE-A (lane 12), suggesting that SRE-B is the only element in that oligonucleotide important for rDBD binding.
To identify the region of the SRE-A oligonucleotide that is important for rDBD binding, regions of the oligonucleotide upstream or downstream of SRE-A were mutated, alone or in combination with mutated SRE-A, as diagramed in Fig. 8. The altered oligonucleotides were then tested for competition against the SRE-A-labeled probe for binding to the rDBD. Mutation of either the upstream or downstream region of the oligonucleotide in combination with an intact SRE-A oligonucleotide did not reduce competition (Fig. 8, rows 2 and 4), suggesting that SRE-A alone is sufficient for binding to the rDBD. Mutation of the region downstream of SRE-A in combination with mutation of SRE-A did not alter competition, suggesting that base pairs downstream of SRE-A do not bind to the rDBD (Fig. 8, row 4). However, mutation of the upstream region in combination with mutation of SRE-A completely abolished competition (Fig. 8, row 1), suggesting that SRE-A and another upstream element account for all binding properties. This upstream region was then divided into two segments. Both elements could be mutated with wild-type SRE-A without losing competition (Fig. 8, rows 5 and 6), similar to mutation of SRE-A by itself (Fig. 8, row 7). However, when either of these two regions was mutated in combination with SRE-A, the combined mutations abolished competition (Fig. 8, rows 8 and 9). These two upstream regions were then further divided into a total of four regions. Mutation of shorter upstream segments in combination with mutated SRE-A revealed that only the 10 base pairs directly upstream of SRE-A in addition to the intact SRE-A are required for full competition (Fig. 8, rows 10 to 13).
Fig. 8.

CaUpc2p requires an intact SRE and a second element (SDR) for binding. An EMSA was run with the labeled UPC2 SRE-A probe in the presence of cold competitor as shown in the figure. Black bars represent the area that was mutated in the cold competitor, as described in Materials and Methods. Arrows show that the oligonucleotide sequence continues in that direction, as shown in Table S1 in the supplemental material. Competition was determined compared to the gel shift in the absence of competitor. Oligos, oligonucleotides.
To define SRE-A and this upstream region in greater detail, single-base-pair mutations were prepared within SRE-A. These single-base-pair scanning mutations within the SRE were prepared in combination with mutation of the entire upstream element (Fig. 9). The results show that the nucleotides important for SRE-A binding are 5′-(T)ATACGA-3′. Six of the seven bases of the canonical SRE (5′-TATACGA-3′ [reverse complement of 5′-TCGTATA-3′]) are significant, while the 5′ T in SRE-A (0.3′ A in the SRE) appears to be less important by EMSA, despite the conservation of this position in multiple C. albicans genes containing SRE sequences.
Fig. 9.

CaUpc2p binds to SRE-A and an upstream SDR element. An EMSA was performed on the labeled wild-type UPC2 SRE-A in the presence of cold competitor with specific single-base-pair mutations. For the SDR mutations (left), the entire SRE was mutated (underline under SRE sequence). For the SRE mutations (right), the major section of the SDR was mutated (underline under SDR sequence). The percent binding for each residue represents the percent binding of the cold competitor measured when that residue was mutated in the competitor oligonucleotide. Boxed bases indicated proposed base pairs important for Upc2p binding.
Conversely, the upstream element was also analyzed for single-base-pair mutations in combination with a fully mutated SRE-A oligonucleotide (Fig. 9). The bases important in this region are 5′-G(G)AACGA-3′ (the second G appears to be less important). The sequence is similar but not identical to the SRE-A sequence. Both SRE-A and the upstream element contain a short direct repeat sequence, 5′-ACGA-3′ (Fig. 9). The upstream binding region containing 5′-ACGA-3′ was named the short direct repeat (SDR), as the 4-bp 5′-ACGA-3′ sequence is a direct repeat of the same element within the SRE.
DISCUSSION
In this study, promoter deletion analysis has been used to identify cis-acting regions of the UPC2 promoter that are required for induction of expression following exposure to azole drugs. This is the first work that links regulation of the response to azole drugs and Upc2p binding to promoter elements in target genes. In response to antifungal drugs, the prevailing hypothesis is that UPC2 expression is transcriptionally self-regulated by Upc2p binding to the SRE binding site within the UPC2 promoter. Mutation of the SRE in both wild-type and upc2Δ/upc2Δ mutant strains shows that the SRE mutation alters inducibility in a Upc2p-dependent manner. Full-length promoter studies show that in addition to self-regulation, a Upc2p-independent mechanism also contributes to UPC2 azole induction (2, 9). Nested deletions of the UPC2 promoter identify regions of the promoter required for Upc2p-independent inducibility. An FCR1 insertion mutant was used to test the effect of inactivation of FCR1 on UPC2 induction in response to azole drugs. Finally, EMSA analysis indicates that the DNA binding region of CaUpc2p binds directly to sequences containing the SRE and other elements within the UPC2 promoter.
Promoter truncations of 100 bp identified two distinct regions of the UPC2 promoter that are required for azole inducibility. The first is located between bp −450 and −425 upstream of the UPC2 translational start. Deletion of this region results in a significant decrease in azole inducibility only in the WT strain, in which endogenous Upc2p is expressed. The loss of this region lowers the inducibility of the UPC2 promoter in the WT strain to levels similar to those of the full-length promoter expressed in the upc2Δ/upc2Δ mutant. This is consistent with a role for Upc2p activity in controlling induction of the promoter in this 25-bp region, which contains an SRE at bp −431 to −437 upstream of the translational start (Fig. 1). The importance of this region to inducibility and the presence of the SRE at bp −431 to −437 are strongly suggestive that this promoter region is dependent on Upc2p activity, and thus, this region has been termed the UDR.
To define the elements within the UDR that are required for azole inducibility, deletions within the UDR as well as mutation of the putative SRE were created (Fig. 4 and 5). Deletions within the UDR did not lower azole induction in the upc2Δ/upc2Δ deletion mutant, as was seen for the wild-type strain. If Upc2p is required for induction within this region, it is not surprising that induction is not lowered in the upc2Δ/upc2Δ deletion mutant.
Mutation of the SRE provides further evidence that Upc2p is required for azole inducibility in the UDR. The dramatic decrease in induction that results from mutation of the SRE in the WT strain demonstrates that the SRE is an important cis-acting element within the UDR of the UPC2 promoter. Comparison of the effects of the SRE mutant in the WT and upc2Δ/upc2Δ backgrounds provides further support for Upc2p acting through the SRE to induce UPC2 transcription. Mutation of the SRE significantly reduces azole inducibility in the WT strain to levels comparable to those of the nonmutated promoter in the upc2Δ/upc2Δ mutant. When tested in the upc2Δ/upc2Δ deletion strain, mutation of the SRE had very little effect on inducibility. These data indicate that either mutation of the Upc2p binding site or the absence of endogenous Upc2p decreases the inducibility of the UPC2 promoter in response to azole drug exposure.
Analysis of the Upc2p-independent region (UIR) indicates that the Upc2p-independent component of UPC2-RLUC activity is localized to a 50-bp region between bp 300 and 250 before the translational start. The results from the 100-bp truncations initially identified the region between bp 350 and 250 as being required for azole induction in both the WT and upc2Δ/upc2Δ strains (Fig. 3). Additional deletions within the UIR further localized the Upc2p-independent activity to the 50-bp UIR (data not shown). Deletion of this region and the complete upstream region abolished azole induction in both the WT strain and upc2Δ/upc2Δ deletion strain, indicating that elements between bp 300 and 250 are Upc2p independent. This finding is important, as it indicates that a novel, azole-responsive transcription factor is capable of regulating UPC2 transcriptional activation. Creation of linker-scanning mutations within this 50-bp UIR is under way to identify potential binding sites for this novel transcription factor.
A recent report indicates that the transcription factor CaNdt80p plays a role in the regulation of ERG genes and UPC2 transcription, suggesting that Ndt80p may be responsible for transcriptional regulation through the UIR (14). Interestingly, the UPC2 promoter contains a putative Ndt80p binding site (5′-ACACAAA-3′) located within the UDR at bp −421 to −429 upstream of the translational start, suggesting some level of regulation by Ndt80p. Because the Ndt80p binding site is not located within the UIR, there is likely another unidentified transcription factor that regulates azole induction through the UIR.
Data presented previously (22) have implicated a transcription factor known as Fcr1p in the regulation of UPC2 activity. CaFCR1 was originally identified due to its ability to complement an S. cerevisiae Δpdr1/Δpdr3 mutant, but its precise molecular function in C. albicans remains unknown (18, 21). The use of an fcr1 insertion mutant, in which both alleles of FCR1 were inactivated, addressed the contribution of Fcr1p to UPC2 azole induction (Fig. 6). QRT-PCR results showing that UPC2 is induced approximately 2.0-fold in both the WT strain and the fcr1 mutant in response to azole drugs demonstrates that UPC2 transcriptional activity is not completely abolished in the fcr1 mutant. When the levels of UPC2 transcription were compared between the fcr1 mutant and the WT strain, however, a clear difference between the two was observed. The decrease in the level of UPC2 transcription in the FCR1 mutant strain compared to that in the WT strain suggests that UPC2 transcription is influenced by Fcr1p. This difference occurs both in the absence and in the presence of FLC, indicating that while UPC2 transcription is azole inducible in the fcr1 mutant, the overall level of UPC2 messages is significantly reduced due to inactivation of the FCR1 locus. The decrease in UPC2 transcription in the fcr1 mutant may be either an indirect consequence of FCR1 inactivation or a direct effect of Fcr1p on UPC2 transcription. ChIP-on-ChIP results confirming that Fcr1p binds directly to the UPC2 promoter support a role for Fcr1p in direct activation of UPC2 transcription (22). This study also shows that FCR1 has a positive transcriptional effect on UPC2 message levels. These results are consistent with a model in which Fcr1p regulates UPC2 expression irrespective of azole exposure. Further work will be performed to identify the region of the UPC2 promoter that is recognized by Fcr1p, either within the UIR or within another region of the promoter.
The transcriptional initiation sites of the UPC2 promoter were mapped using 5′ RACE. Two distinct mRNA start sites were identified, one at bp −148 and one at bp −361 upstream of the ATG translational initiation codon. These 5′ mRNA termini may represent transcription initiation sites or may be the result of posttranscriptional mRNA processing. Both sites are likely to be true initiation sites, as they contain a putative TATA box in close upstream proximity and no ATG codons occur between these initiation sites and the ATG start site of Upc2p. These data may indicate that UPC2 transcription in response to FLC exposure is also regulated at the level of transcription initiation or possibly posttranscriptionally. Loss of a transcriptional initiation site at bp −361 likely affects UPC2-RLUC activity in the promoter analysis. The loss of this site may account for some of the loss in inducibility between the 450-bp and 350-bp promoter regions and may complicate the 100-bp truncation analyses. When the 100-bp deletion results are compared to those obtained with the finer deletions within the UDR, however, it is clear that the important region of inducibility lies between bp −450 and −425 rather than downstream of the putative initiation site at bp −361.
The EMSA studies clearly show that the DNA binding domain of CaUpc2p binds directly to the SREs contained within the UPC2 promoter. Thus, Upc2p directly regulates its own expression via transcriptional control at the UPC2 promoter. The observation that SRE-A binds with higher affinity to Upc2p is consistent with luciferase data showing that loss of SRE-A results in reduced azole induction.
The related SRE, SRE-B, also binds to the rDBD (Fig. 7A), but there are no additional elements surrounding SRE-B that bind to the rDBD (Fig. 7A), and the 250-bp promoter construct that contains SRE-B alone is not azole inducible (Fig. 3). These data suggest that SRE-B is not sufficient to contribute to azole induction (or that SRE-B might require the presence of SRE-A upstream for function).
An additional element, the SDR, was found upstream and in close proximity to SRE-A. This element includes a direct repeat of the short 5′-ACGA-3′ sequence, suggesting that Upc2p binds as a dimer to this region, consistent with many other zinc cluster transcription factors.
Competition studies with single-base-pair scanning mutations have identified several positions that are essential for complete binding of Upc2p. The base pairs within SRE-A that are required are 5′-(T)ATACGA-3′. This indicates that most of the previously described SRE core sequence is required for binding. Another element that was essential is a 7-bp SDR element sequence, 5′-G(G)AACGA-3′, upstream of the SRE. Interestingly, both of these elements contain the short direct repeat sequence [5′-ACGA-3′. The presence of repeat sequences in promoters of genes that are regulated by zinc cluster transcription factors is common, and an imperfect repeat of the SRE in the ERG11 promoter has been defined in C. albicans, Candida glabrata, and Candida krusei (10). These data suggest that Upc2p utilizes bipartite binding sites that can contain either inverted repeats or direct repeats, and further work with SREs from other ERG genes will determine whether this feature is common to all genes that are regulated by Upc2p. This feature implies that either one molecule of Upc2p binds coordinately to both binding sites or, more likely, two molecules of Upc2p bind independently to each element defined in this analysis. Interestingly, the imperfect inverted repeat (INV) in the ERG11 promoter is spaced 13 bp upstream of the SRE and the sequence of the 13 bp is not important to function (12). In the UPC2 promoter, the SDR and SRE-A are separated by 3 bp that are also unimportant for binding (Fig. 9). It may be that DNA topological constraints influence binding, as the spacing between the INV and SRE in the ERG11 promoter differs from the spacing between the SDR and SRE-A in the UPC2 promoter by 10 bp or one turn of the DNA double helix. Future work will be required to elucidate the mechanism of Upc2p binding to these elements and to other elements in Upc2p-regulated genes and will identify whether Upc2p binds to promoters as a dimer.
The data presented in this study support a model for UPC2 transcriptional regulation that has important implications for understanding how C. albicans interacts with antifungals. In previous studies, it has been assumed that UPC2 transcription is controlled solely by an autoregulatory mechanism. The data generated from SRE mutation and use of the upc2Δ/upc2Δ deletion strain support a role for Upc2p in positive autoregulation of UPC2 transcription. In addition to self-regulation, it is clear from these results that additional azole induction elements are present in the promoter, and these elements are both Upc2p dependent (UDR) and Upc2p independent (UIR). This has implications for our understanding of how C. albicans controls the sterol biosynthetic pathway as a part of the azole drug response. The data presented here suggest that an additional transcription factor that has yet to be identified controls sterol biosynthesis through regulation of UPC2 expression. Because expression levels of sterol biosynthesis genes have been shown to affect azole drug resistance, it is clear that transcriptional control of UPC2 affects resistance. The findings presented here implicate both Upc2p and additional factors yet to be described as important contributors to the antifungal drug response and possible therapeutic targets to reduce resistance that arises from overexpression of ergosterol biosynthesis genes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Joachim Morschhauser (University of Wurzburg, Wurzburg, Germany) for providing plasmid pA83 containing the SAT1 marker, David Soll (University of Iowa, Iowa City, IA) for providing the Renilla reniformis luciferase-containing plasmid pCRW3, and Aaron Mitchell (Columbia University, New York, NY) for providing strain BWP17 and the transcription factor mutant library. We thank members of the White laboratory for their valuable comments and support.
This research was funded by NIH NIDCR grants R01 DE11367, R01 DE14161, and R01 DE017078.
Footnotes
Supplemental material for this article may be found at http://ec.asm.org/.
Published ahead of print on 23 July 2010.
REFERENCES
- 1.Abramova N., Sertil O., Mehta S., Lowry C. V. 2001. Reciprocal regulation of anaerobic and aerobic cell wall mannoprotein gene expression in Saccharomyces cerevisiae. J. Bacteriol. 183:2881–2887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abramova N. E., Cohen B. D., Sertil O., Kapoor R., Davies K. J., Lowry C. V. 2001. Regulatory mechanisms controlling expression of the DAN/TIR mannoprotein genes during anaerobic remodeling of the cell wall in Saccharomyces cerevisiae. Genetics 157:1169–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ausubel F. M., Brent R., Kingston R. E., Moore D. D., Seidman J. G., Smith J. A., Struhl K. 1995. Current protocols in molecular biology, vol. 1 John Wiley & Sons, Inc., New York, NY [Google Scholar]
- 4.Davies B. S., Wang H. S., Rine J. 2005. Dual activators of the sterol biosynthetic pathway of Saccharomyces cerevisiae: similar activation/regulatory domains but different response mechanisms. Mol. Cell. Biol. 25:7375–7385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dunkel N., Liu T. T., Barker K. S., Homayouni R., Morschhauser J., Rogers P. D. 2008. A gain-of-function mutation in the transcription factor Upc2p causes upregulation of ergosterol biosynthesis genes and increased fluconazole resistance in a clinical Candida albicans isolate. Eukaryot. Cell 7:1180–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gillum A. M., Tsay E. Y., Kirsch D. R. 1984. Isolation of the Candida albicans gene for orotidine-5′-phosphate decarboxylase by complementation of S. cerevisiae ura3 and E. coli pyrF mutations. Mol. Gen. Genet. 198:179–182 [DOI] [PubMed] [Google Scholar]
- 7.Heilmann C. J., Schneider S., Barker K. S., Rogers P. D., Morschhauser J. 2010. An A643T mutation in the transcription factor Upc2p causes constitutive ERG11 upregulation and increased fluconazole resistance in Candida albicans. Antimicrob. Agents Chemother. 54:353–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoffman C. S., Winston F. 1987. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene 57:267–272 [DOI] [PubMed] [Google Scholar]
- 9.Hoot S. J., Oliver B. G., White T. C. 2008. Candida albicans UPC2 is transcriptionally induced in response to antifungal drugs and anaerobicity through Upc2p-dependent and -independent mechanisms. Microbiology 154:2748–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamping E., Ranchod A., Nakamura K., Tyndall J. D., Niimi K., Holmes A. R., Niimi M., Cannon R. D. 2009. Abc1p is a multidrug efflux transporter that tips the balance in favor of innate azole resistance in Candida krusei. Antimicrob. Agents Chemother. 53:354–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MacPherson S., Akache B., Weber S., De Deken X., Raymond M., Turcotte B. 2005. Candida albicans zinc cluster protein Upc2p confers resistance to antifungal drugs and is an activator of ergosterol biosynthetic genes. Antimicrob. Agents Chemother. 49:1745–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliver B. G., Song J. L., Choiniere J. H., White T. C. 2007. cis-acting elements within the Candida albicans ERG11 promoter mediate the azole response through transcription factor Upc2p. Eukaryot. Cell 6:2231–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reuss O., Vik A., Kolter R., Morschhauser J. 2004. The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene 341:119–127 [DOI] [PubMed] [Google Scholar]
- 14.Sellam A., Tebbji F., Nantel A. 2009. Role of Ndt80p in sterol metabolism regulation and azole resistance in Candida albicans. Eukaryot. Cell 8:1174–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silver P. M., Oliver B. G., White T. C. 2004. Role of Candida albicans transcription factor Upc2p in drug resistance and sterol metabolism. Eukaryot. Cell 3:1391–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song J. L., Harry J. B., Eastman R. T., Oliver B. G., White T. C. 2004. The Candida albicans lanosterol 14-alpha-demethylase (ERG11) gene promoter is maximally induced after prolonged growth with antifungal drugs. Antimicrob. Agents Chemother. 48:1136–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Srikantha T., Klapach A., Lorenz W. W., Tsai L. K., Laughlin L. A., Gorman J. A., Soll D. R. 1996. The sea pansy Renilla reniformis luciferase serves as a sensitive bioluminescent reporter for differential gene expression in Candida albicans. J. Bacteriol. 178:121–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Talibi D., Raymond M. 1999. Isolation of a putative Candida albicans transcriptional regulator involved in pleiotropic drug resistance by functional complementation of a pdr1 pdr3 mutation in Saccharomyces cerevisiae. J. Bacteriol. 181:231–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.White T. C. 1997. The presence of an R467K amino acid substitution and loss of allelic variation correlate with an azole-resistant lanosterol 14alpha demethylase in Candida albicans. Antimicrob. Agents Chemother. 41:1488–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson R. B., Davis D., Mitchell A. P. 1999. Rapid hypothesis testing with Candida albicans through gene disruption with short homology regions. J. Bacteriol. 181:1868–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Znaidi S., De Deken X., Weber S., Rigby T., Nantel A., Raymond M. 2007. The zinc cluster transcription factor Tac1p regulates PDR16 expression in Candida albicans. Mol. Microbiol. 66:440–452 [DOI] [PubMed] [Google Scholar]
- 22.Znaidi S., Weber S., Zin-Al-Abdin O., Bomme P., Saidane S., Drouin S., Lemieux S., De Deken X., Robert F., Raymond M. 2008. Abstr. 9th ASM Conf. Candida Candidiasis, abstr. C255, Jersey City, NJ, 23 to 28 March 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Znaidi S., Weber S., Zin Al-Abdin O., Bomme P., Saidane S., Drouin S., Lemieux S., De Deken X., Robert F., Raymond M. 2008. Genomewide location analysis of Candida albicans Upc2p, a regulator of sterol metabolism and azole drug resistance. Eukaryot. Cell 7:836–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.