

Abstract
Choline acetyltransferase (ChAT; EC 2.3.1.6) catalyzes the reversible synthesis of acetylcholine (ACh) from acetyl CoA and choline at cholinergic synapses. Mutations in genes encoding ChAT affecting motility exist in Caenorhabditis elegans and Drosophila, but no CHAT mutations have been observed in humans to date. Here we report that mutations in CHAT cause a congenital myasthenic syndrome associated with frequently fatal episodes of apnea (CMS-EA). Studies of the neuromuscular junction in this disease show a stimulation-dependent decrease of the amplitude of the miniature endplate potential and no deficiency of the ACh receptor. These findings point to a defect in ACh resynthesis or vesicular filling and to CHAT as one of the candidate genes. Direct sequencing of CHAT reveals 10 recessive mutations in five patients with CMS-EA. One mutation (523insCC) is a frameshifting null mutation. Three mutations (I305T, R420C, and E441K) markedly reduce ChAT expression in COS cells. Kinetic studies of nine bacterially expressed ChAT mutants demonstrate that one mutant (E441K) lacks catalytic activity, and eight mutants (L210P, P211A, I305T, R420C, R482G, S498L, V506L, and R560H) have significantly impaired catalytic efficiencies.
Congenital myasthenic syndromes (CMS) are heterogeneous disorders caused by postsynaptic, synaptic, and presynaptic defects. Recent studies identified CMS caused by mutations in the postsynaptic acetylcholine receptor (AChR) and in synaptic acetylcholinesterase (reviewed in ref. 1), but the molecular basis of presynaptic CMS has not been elucidated to date. One form of presynaptic CMS presents at birth or in the neonatal period with hypotonia, variable eyelid ptosis, severe bulbar weakness causing dysphagia, and respiratory insufficiency with cyanosis and apnea. If the infant survives, the symptoms improve, but the crises recur abruptly with infections, fever, excitement, vomiting, or overexertion. During crises, episodes of apnea can cause sudden death or anoxic brain injury. Between crises, patients may have only mild or no myasthenic symptoms. When weakness is absent, it can be induced readily by exercise (2–10). The crises can be prevented or mitigated by anticholinesterase drugs. In 1960, Greer and Schotland (2) described the clinical features of the disease, and in 1975 Conomy et al. (3) referred to it as “familial infantile myasthenia.” Because all CMS can be familial and because most CMS presents in infancy, the term “familial infantile myasthenia” has become a source of confusion (11). We therefore refer to this disease as CMS with episodic apnea (CMS-EA).
Previous studies of CMS-EA revealed no endplate AChR or acetylcholinesterase deficiency but suggested impaired resynthesis or vesicular packaging of ACh (9, 12). We here show that spontaneous mutations in CHAT, the gene encoding choline acetyltransferase (ChAT; EC 2.3.1.6), cause CMS-EA. In the course of this study, we also determine the genomic structure of 11 previously uncharacterized exons at the 3′ end of CHAT and identify an S transcript in human spinal cord.
Materials and Methods
Clinical Data.
Five patients, now 26, 40, 5, 6, and 4 years old, were studied. Four were male and one was female. All had myasthenic symptoms since birth or early infancy, negative tests for anti-AChR antibodies, and abrupt episodic crises of increased weakness, bulbar paralysis, and apnea precipitated by undue exertion, fever, or excitement. Electromyographic studies in the five patients between crises revealed no decremental response on 2-Hz stimulation of motor nerves, but such a response appeared after a conditioning train of 10-Hz stimuli for 5 min. Patient 2 had three affected siblings, and patient 4 had two; three of the five affected siblings died during febrile episodes, and one died suddenly without apparent cause. Previous studies of intercostal muscle specimens from patients 1 and 2 revealed no endplate AChR deficiency, a normal miniature endplate potential (MEPP) amplitude in rested muscle, and an abnormal decrease of the MEPP amplitude after 5 min stimulation at 10 Hz, suggesting a defect in ACh resynthesis or vesicular packaging (9, 12).
Endplate Studies.
Intercostal muscle specimens were obtained from patients 3 and 4 and a control subject without muscle disease who was undergoing thoracic surgery. In patient 5, CMS-EA was suspected on clinical grounds. All human studies were in accordance with the guidelines of the Institutional Review Board of the Mayo Clinic.
The number of AChRs per endplate was measured with 125I-labeled α-bungarotoxin (13). MEPP and EPP recordings and estimates of the number of transmitter quanta released by nerve impulse were carried out as described (13). MEPP recordings were obtained before and within 5 min after 10-Hz stimulation for 5 min. EPP recordings were obtained from curarized muscle fibers before and within 1 min after 10-Hz stimulation for 5 min.
Sequencing Procedures.
PCR-amplified fragments were purified by the QIAquick 8 PCR Purification kit (Qiagen, Chatsworth, PA). Plasmids were purified by the QIAprep Spin Miniprep kit (Qiagen, Chatsworth, CA). PCR products and plasmids were sequenced with an ABI 377 DNA sequencer (Applied Biosystems) by using fluorescently labeled dideoxy terminators.
Analysis of Genomic Structure of CHAT.
The genomic structure of exons R to 7 of the human CHAT gene was reported previously (14–16). To determine the remaining genomic structure, we synthesized 34 cDNA primers based on the published cDNA sequence (17) and amplified overlapping genomic DNA segments by using long-distance PCR.
Mutation Analysis.
DNA was isolated from blood by using a QIAamp DNA Blood kit (Qiagen). By using the genomic sequence determined above, we synthesized 18 pairs of PCR primers to amplify and directly sequence 18 exons and their flanking intronic or untranslated regions. After identifying mutations in CHAT, we screened the patients' relatives for the observed mutations by restriction analysis. We used allele-specific PCR to screen for the identified missense mutations in 400 normal alleles.
cDNA Cloning of Human CHAT.
We used nested reverse transcription (RT)–PCR to amplify the entire coding region of the M transcript of human CHAT (nucleotides 90–2425; GenBank accession no. AF305907) from human spinal cord cDNA (Quick-Clone cDNA, CLONTECH). We used spinal cord cDNA because ChAT is synthesized in anterior horn cells and reaches the nerve terminal by slow axonal transport (18, 19). The attempt to clone the M transcript unexpectedly amplified an S transcript. The RT-PCR products were cloned into a pGEM-T vector (Promega) and were sequenced to confirm absence of PCR artifacts.
Construction of Expression Vectors.
Presence of EcoRI sites in both the untranslated region of exon S and pGEM-T enabled us to clone the EcoRI-digested S transcript into pRBG4, a cytomegalovirus-based expression vector (20). Naturally occurring or artificial CHAT mutations were introduced into CHAT cDNA in pRBG4 by using the QuickChange Site-Directed Mutagenesis kit (Stratagene). The presence of desired mutations and the absence of unwanted mutations was confirmed by sequencing the entire insert.
We next amplified by PCR the coding region for 70-kDa ChAT (nucleotides 578-2470 of S transcript, GenBank accession no. AF305908) from wild-type and mutant CHAT cDNAs in pRBG4 and cloned them into the bacterial-expression vector pCRT7/NT (Invitrogen), which introduces six N-terminal histidine residues. We also cloned the coding region of the wild-type 74-kDa ChAT into pCRT7/NT and replaced the translational start site of 70-kDa ChAT with alanine to suppress the production of 70-kDa ChAT.
Immunoblot Analysis of ChAT Expressed in COS Cells.
COS-7 cells were grown and transfected 1 day after spreading by the DEAE-dextran method (21) with 10 μg of wild-type or mutant CHAT cDNA and 2 μg of pSV-β-galactosidase control vector (Promega) per 10-cm dish. COS cells were extracted 3 days after transfection with 900 μl per dish of Reporter lysis buffer (Promega). Aliquots of each extract (10 μl ) were separated on 10% NuPage Bis-Tris gel (Invitrogen) and transferred electrophoretically to nitrocellulose membrane (Trans-Blot, Bio-Rad). The membrane was immunostained with affinity purified goat anti-human ChAT antibody (Chemicon International) and alkaline phosphatase-conjugated rabbit-anti-goat IgG (Southern Biotechnology Associates). Plate-to-plate variations of transfection efficiency were corrected for by measuring the β-galactosidase activities of transfected cells. The National Institutes of Health's IMAGE 1.61 software was used for densitometric analysis.
Expression and Purification of Recombinant ChAT Expressed in Bacteria.
BL21(DE3)pLysS (Invitrogen) was transformed with pCRT7/NT-CHAT and grown overnight at 37°C in 2 ml LB medium containing 100 μg/ml ampicillin and 34 μg/ml chloramphenicol. The entire overnight culture was transferred to 100 ml LB medium containing 100 μg/ml ampicillin, and incubation was continued at 25°C. When A600 of the culture reached 0.5 (≈7 h), isopropyl-1-thio-β-D-galactopyranoside was added to a final concentration of 0.5 mM, and growth continued overnight at 25°C. The bacterial cell pellet from 50-ml culture medium was suspended in 4 ml lysis buffer containing 50 mM sodium phosphate (pH 7.4), 300 mM NaCl, 10 mM imidazole, 1 mM PMSF, 0.1 mM benzamidine, 1 mM MgSO4, and 11 units per ml DNase I and was disrupted by freezing and thawing three times. A mixture of the supernatant of the lysate and 1 ml Ni-nitrilotriacetic acid (NTA) agarose (Qiagen) was shaken at 1,000 rpm at 4°C for 1 h. Next, Ni-NTA agarose was washed three times with 4 ml of 50 mM sodium phosphate (pH 7.4), 300 mM NaCl, and 20 mM imidazole. ChAT peptide was eluted with 1 ml of 50 mM sodium phosphate (pH 7.4), 300 mM NaCl, and 250 mM imidazole and diluted with 9 ml of 20 mM sodium phosphate (pH 7.4). The diluted eluate was loaded to 0.5-ml Amicon DyeMatrex blue B gels (Millipore), washed with 5 ml of 20 mM sodium phosphate (pH 7.4), and eluted with 1 ml of linear gradient of 0.2–2.0 M NaCl in 20 mM sodium phosphate (pH 7.4). Elution fractions of 0.2–0.8 M NaCl were combined and concentrated/desalted with Centricon YM-30 (Millipore) and resuspended in 300 μl 20 mM sodium phosphate (pH 7.4). Protein concentrations were determined by densitometric assay of Coomassie G-250-stained SDS/PAGE against IgG-free BSA as a standard. We obtained 29–101 μg of ChAT peptide from 50 ml culture medium.
Measurement of Enzyme Activity and Treatment of Kinetic Data.
ChAT activity was measured under steady-state conditions by the radioactive assay of Fonnum (22). Initial reaction velocities were determined at 37°C in a 40-μl reaction mixture containing 5–1,200 μM [1-14C]acetyl-CoA (AcCoA; NEN), 0.1–3.5 mM choline, 0.2–10 ng ChAT protein, 50 mM sodium phosphate (pH 7.4), 250 mM NaCl, 1 mM EDTA, and 0.5 mg/ml BSA.
The kinetic mechanism of the ChAT reaction follows a random Theorell-Chance mechanism in which a low but finite amount of ternary complex exists (23). Accordingly, the initial velocity of the reaction is defined by Eq. 1,
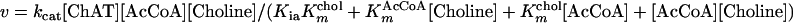 |
1 |
where v is the observed velocity, kcat is the turnover number (Vmax/[ChAT]), Kia is the dissociation constant for the AcCoA–enzyme complex, and KmAcCoA and Kmchol are the Michaelis–Menten constants. The kinetic constants for the equation were determined by weighted nonlinear regression using a SIGMAPLOT 5 computer program (SPSS, Chicago) and were confirmed by using the FIT function of MLAB (Civilized Software, Bethesda).
For the L210P mutant, for which the obtainable concentration of AcCoA is much lower than KmAcCoA, Eq. 1 reduces to Eq. 2:
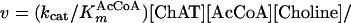 |
2 |
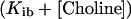 |
where Kib is equivalent to Kia⋅Kmchol/KmAcCoA in Eq. 1. Similarly, for the R560H mutant, for which the obtainable concentration of choline is much lower than Kmchol, Eq. 1 reduces to Eq. 3:
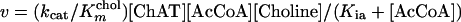 |
3 |
The catalytic efficiencies (kcat/KmAcCoA and kcat/Kmchol) in Eqs. 2 and 3 were calculated by nonlinear regression using SIGMAPLOT 5 and were confirmed by MLAB.
Results
Endplate Studies in Patients 3 and 4.
The number of α-bungarotoxin binding sites per endplate was 6.02 × 106 in patient 3 (age 5 years) and 6.13 × 106 in patient 4 (age 6 years). These values were lower than in 13 adult controls (12.82 ± 2.84 × 106, mean ± SD) but were similar to that in a 6-year-old control (6.9 × 106). In both patients, the MEPP amplitude was slightly lower than normal, and the quantal content of EPP was normal in rested muscle. In both patients, 10-Hz stimulation for 5 min reduced the amplitude of the MEPP by 50% but had no effect on the quantal content of the EPP (Table 1). The stimulation-dependent decrease of the MEPP amplitude was consistent with insufficient resynthesis or vesicular packaging of ACh in response to increased functional demand. Candidate proteins for this type of defect would be the presynaptic high-affinity choline transporter (24, 25), ChAT, the vesicular ACh transporter (vAChT; ref. 26), or the vesicular proton pump (27). Direct sequencing of the VACHT in patients 1–4 revealed no mutations (data not shown). We next searched for mutations in CHAT.
Table 1.
Endplate studies
| Patient 3 | Patient 4 | Control* | |
|---|---|---|---|
| MEPP amplitude before | 0.77 ± 0.04 | 0.88 ± 0.06 | 1.22 ± 0.062 |
| stimulation, mV† | (n = 20) | (n = 17) | (n = 12) |
| MEPP amplitude after | 0.38 ± 0.04 | 0.41 ± 0.04 | 1.09 ± 0.043 |
| stimulation, mV† | (n = 5) | (n = 10) | (n = 13) |
| EPP quantal content | 28.94 ± 2.44 | 28.91 ± 2.9 | 36.5 ± 3.9 |
| before stimulation‡ | (n = 18) | (n = 10) | (n = 7) |
| EPP quantal content | ND | 28 ± 3.1 | 39.3 ± 6.09 |
| after stimulation‡ | (n = 10) | (n = 7) |
Values shown are means ± SEM. T = 30°C; n = number of fibers; ND, not determined.
For 164 unstimulated muscle fibers of 15 other controls, the MEPP amplitude was 1.00 ± 0.025 mV.
Corrected to a resting potential of −80 mV and for a fiber diameter of 50 μm.
Recordings before and after 10-Hz stimulation were obtained from the same fibers of curarized muscle at 1 Hz with correction for resting membrane potential of −80 mV, nonlinear summation, and non-Poisson release.
Genomic Structure and Mutation Analysis of CHAT.
Preparatory to sequencing CHAT, we wished to determine its genomic structure. CHAT is in the cholinergic gene locus at 10q11.2 that also comprises VACHT in the first intron of CHAT (26). The genomic structure of CHAT between exons R and 7 was reported previously (14–16); we therefore determined the genomic structure of the remaining 11 exons (Fig. 1). Comparison of CHAT with rat Chat (28) revealed size identity and structural homology of exons 6–17 and structural homology of exons R, 5, and 18.
Figure 1.

Genomic structure and alternative transcripts of human CHAT and 10 identified mutations. CHAT and VACHT are encoded in the cholinergic gene locus (26). Four alternative CHAT transcripts (R, N1, N2, and M) have been reported (15, 17, 41). The S transcript is a moiety identified in the spinal cord. All transcripts encode a 70-kDa ChAT. M transcript encodes an additional 83-kDa ChAT and S transcript codes for an additional 74-kDa ChAT. Vertical shading indicates untranslated regions of VACHT. Diagonal shading show untranslated regions of CHAT. For M and S transcripts, diagonal shading indicates untranslated regions for yielding 83- and 74-kDa ChATs, respectively. Arrows indicate translational start sites. Exons and introns are drawn to indicated scale.
Next, we sequenced 18 exons and their flanking intronic or untranslated regions directly by using patients' blood DNA and identified two heterozygous mutations in each patient (Table 2 and Figs. 1 and 2). Restriction analysis of DNA from relatives revealed that a living affected sibling harbors both mutations, the asymptomatic parents carry a single mutation, and other unaffected relatives carry either no or a single mutation (data not shown). Therefore, each mutation is heteroallelic and recessive. None of the nine missense mutations was detected in 400 normal alleles. All nine missense mutations alter residues conserved in mouse, rat, and pig (29–31; see Fig. 2). Patients 1 and 2 are also heterozygous for 358G/A (A120T), a polymorphism detected in 17/60 in normal alleles.
Table 2.
Identified CHAT mutations
| Patient | Nucleotide change* | Amino acid change* | Effects |
|---|---|---|---|
| 1 | 523insCC | Frameshift | No expression |
| 931C→G | P211A | Very high KmAcCoA | |
| 2 | 1371G→A | E441K | Low expression, no catalytic activity |
| 1516G→T | V506L | High KmAcCoA | |
| 3 | 1444A→G | R482G | High KmAcCoA |
| 1679G→A | R560H | Very high KmAcCoA and Kmchol | |
| 4 | 629T→C | L210P | Very high KmAcCoA and Kmchol |
| 1493C→T | S498L | High KmAcCoA | |
| 5 | 914T→C | I305T | Low expression, high KmAcCoA |
| 1258C→T | R420C | Low expression, very high KmAcCoA and Kmchol |
Nucleotide and codon numbers start from the translational start site of 83-kDa ChAT.
Figure 2.

Alignment of the amino acid sequences of human 83-kDa ChAT and of single isoforms identified in pig, mouse, and rat ChAT. Divergent N-terminal ends of the human 74- and 70-kDa ChATs are shown also. Dashes indicate residues identical to the human 83-kDa ChAT. Gaps are inserted for alignment. The right column indicates codon numbers. Arrowheads indicate the 10 recessive mutations identified in five patients. The arrowhead under 523insCC points to the first residue substituted by the frameshift.
Identification of an S Transcript in Human Spinal Cord.
For expression studies of wild-type and mutant ChATs, we cloned CHAT cDNA by RT-PCR. RT-PCR amplification with primers designed to amplify the M transcript from a human spinal cord cDNA library yielded a transcript (S transcript) with a 70-bp insertion (exon S) between exons M and 5 (Fig. 1). Sequencing of 14 plasmids cloned from four different RT-PCR reactions failed to reveal the M transcript. Expression of the S transcript in COS cells showed two distinct proteins (Fig. 3). Replacement of the initial ATG methionine codons with GCG alanine codons identified two translational start sites of the S transcript, yielding 74- and 70-kDa ChATs (Fig. 3). The kinetic properties of the purified recombinant 74-kDa ChAT expressed in Escherichia coli are identical essentially with those of 70-kDa ChAT (data not shown).
Figure 3.

Immunoblot identifying translational start sites. (Left) Wild type. A Met-to-Ala substitution at the putative translational start site of 74-kDa ChAT prevents expression of the 74-kDa ChAT (Center). A similar Met-to-Ala substitution of 70-kDa ChAT prevents expression of the 70-kDa ChAT (Right).
Expression Studies in COS Cells.
To study the effects of the mutations at the protein level, we performed immunoblot analysis of wild-type and mutant ChAT proteins expressed in COS cells. Densitometric analysis of immunoblots showed no expression of the 523insCC mutant, and 30, 49, and 26% expression of wild-type for E441K, I305T, and R420C mutants, respectively (Fig. 4).
Figure 4.

Immunoblot demonstrating expression of wild-type and mutant ChATs in COS cells. Expression levels are normalized for cotransfected β-galactosidase activity and compared with that of wild-type. Bars indicate means and standard errors of three to four transfections. * indicates significant difference from wild type (P < 0.01). The 74- to 70-kDa ChAT ratios for the mutants were not significantly different from that of wild type.
Kinetic Analysis of ChAT Mutants Expressed in E. coli.
To determine whether the mutations affect the kinetic properties of ChAT, we expressed recombinant wild-type and mutant ChATs in E. coli by using truncated S transcripts that yield only 70-kDa ChAT. SDS/PAGE of purified recombinant ChATs revealed a single band of 74 kDa comprising the 4-kDa hexahistidine tag and 70-kDa ChAT (Fig. 5). The 523insCC mutant did not express in E. coli. The E441K mutant showed no catalytic activity over a range of substrate concentrations. The eight remaining mutants all showed abnormal activation with AcCoA or choline or both, a catalytic efficiency for AcCoA that varied from 1–49% of wild type, and an overall catalytic efficiency that ranged 0.2–64% of wild type (Table 3 and Fig. 6).
Figure 5.

SDS/PAGE of purified wild-type and mutant recombinant ChATs expressed in E. coli stained with Coomassie G-250.
Table 3.
Kinetic parameters of wild-type and mutant ChAT enzymes
| kcat, s−1 | KmAcCoA, μM | Kmchol, mM | kcat/KmAcCoA* | kcat/(KmAcCoA⋅Kmchol)* | |
|---|---|---|---|---|---|
| wild type | 99.6 ± 3.5 | 26.8 ± 1.9 | 0.478 ± 0.036 | 1.00 | 1.00 |
| L210P | 35.0 ± 2.2† | n.d.‡ | 2.693 ± 0.280† | 0.09 | 0.02† |
| P211A | 418.8 ± 15.9 | 287.9 ± 28.6 | 0.514 ± 0.046 | 0.39 | 0.36 |
| I305T | 60.4 ± 3.4 | 70.6 ± 6.5 | 0.590 ± 0.064 | 0.23 | 0.19 |
| R420C | 176.8 ± 42.9 | 461.8 ± 134.4 | 1.018 ± 0.389 | 0.10 | 0.05 |
| R482G | 99.0 ± 6.9 | 91.7 ± 10.7 | 0.500 ± 0.067 | 0.29 | 0.28 |
| S498L | 88.6 ± 1.5 | 94.6 ± 7.2 | 0.418 ± 0.018 | 0.25 | 0.29 |
| V506L | 125.8 ± 7.4 | 68.4 ± 7.7 | 0.370 ± 0.053 | 0.49 | 0.64 |
| R560H | 34.1 ± 1.3§ | 870.8 ± 62.4§ | n.d.¶ | 0.011§ | 0.002§ |
Values indicate estimate ± standard error of estimate.
Catalytic efficiency (kcat/KmAcCoA) and the overall catalytic efficiency [kcat/(KmAcCoA⋅Kmchol)] are normalized with respect to wild type. In wild type, kcat/KmAcCoA = 3.72 × 106 s−1⋅M−1 and kcat/(KmAcCoA⋅Kmchol) = 7.77 × 109 s−1⋅M−2.
Apparent values calculated at 116 μM AcCoA.
Not determined because KmAcCoA exceeds practical concentration range of AcCoA. Catalytic efficiency is calculated from Eq. 2 (see Materials and Methods).
Apparent values are calculated at 3.5 mM choline.
Not determined because Kmchol exceeds practical concentration range of choline. Catalytic efficiency is calculated from Eq. 3 (see Materials and Methods).
Figure 6.

Individually scaled kinetic landscapes of wild-type and mutant ChATs. Dots represent observed values. Meshes show curves fitted to Eq. 1, except for panels B and I in which the curves are fitted to Eqs. 2 and 3, respectively (see Materials and Methods).
Discussion
In this study, we identify and functionally characterize loss-of-function mutations in CHAT as the cause of a highly fatal presynaptic myasthenic syndrome in humans. That CHAT became a candidate gene for CMS-EA was owing to in vitro-electrophysiologic studies that pointed to a defect in ACh resynthesis or vesicular filling. Defects in the presynaptic high-affinity choline transporter (24, 25), the vesicular ACh transporter (26), or the vesicular proton pump (27) may have similar phenotypic consequences, but no mutations in humans of these proteins have been detected to date.
Previous studies identified recessive loss-of-function mutations of ChAT in invertebrates. Homozygous cha-1 mutants of C. elegans have marked ChAT deficiency, small size, uncoordinated behavior, and resistance to cholinesterase inhibitors. In mutants retaining 10% of wild-type enzyme activity, the residual enzyme has an altered Km for both choline and AcCoA (32). Drosophila Cha mutants are temperature sensitive, becoming paralyzed at a restrictive temperature of 32°C (33). No spontaneous ChAT mutations of vertebrates have been identified, but studies of Chat knockout mice were reported recently (34). Homozygous null mice lacking ChAT are stillborn and have underdeveloped muscles; heterozygous mice have 40–50% ChAT activity and seem normal. Similarly, in our series, heterozygous parents are unaffected even when they carry a nonfunctional allele such as 523insCC or E411K. Patient 2, however, who is a compound heterozygote for 523insCC and V506L, is symptomatic, although the kinetics of the V506L mutant are the least abnormal among the catalytically active mutants (see Fig. 6H and Table 3). This observation implies that even a small decrease of ChAT activity below 50% can impair neuromuscular transmission when the functional demand for ACh resynthesis is increased.
Effects of the CHAT Mutations.
The 523insCC mutation is frameshifting and does not express in COS cells (see Fig. 4) or E. coli. The E441K protein expresses at 29% of wild type in COS cells, but the purified mutant is devoid of catalytic activity. Interestingly, the residue corresponding to the adjacent H442 in Drosophila is essential for affinity for AcCoA (35). Of the eight enzymatically active mutants, seven predominantly affect affinity for AcCoA and five have a minor or no effect on affinity for choline (see Table 3).
R420C and I305T, both in patient five, resulted in the most severe phenotype in our series. Protein expression of these mutants is 26 and 49% of wild type in COS cells (see Fig. 4). The KmAcCoA of the R420C mutant is 17-fold higher than wild type but carries a high standard error owing to incomplete saturation of the enzyme with up to 0.16 mM AcCoA (Table 2 and Fig. 6E). Interestingly, an artificial arginine-to-alanine mutation of a corresponding residue in rat (see Fig. 2) does not affect kinetics of the reverse ChAT reaction significantly (36). The KmAcCoA for I305T is 2.6-fold higher than that of wild type. The severe phenotypic consequences in patient five probably owe to combined effects of reduced expression and kinetic abnormalities of both mutants.
The L210P mutant exhibits no saturation over a practical range of AcCoA concentrations, indicating an extremely high KmAcCoA (Table 3 and Fig. 6B). An adjacent mutation, P211A, also results in a very high KmAcCoA (Table 3 and Fig. 6C). These findings implicate L210 and P211 as residues that participate in governing affinity for AcCoA.
The R560H mutation markedly reduces affinity for AcCoA and shows no saturation with 3.5 mM choline (Table 3 and Fig. 6I). A residue corresponding to human R560 in rat (see Fig. 2) interacts with 3′ phosphate of CoA (36). R560 is also close to the VDN residues at 567–569; the corresponding residues in rat participate in determining affinity for choline (37).
For the three remaining mutants (R482G, S498L, and V506L), KmAcCoA is 3.4-, 3.5-, and 2.6-fold higher than that of wild type, respectively, and the catalytic efficiency for AcCoA (kcat/KmAcCoA) is diminished to 29, 25, and 49% of wild type (Table 3 and Fig. 6 F–H).
CHAT S transcript.
The S transcript isolated from human spinal cord has two functional translational start sites that encode 74- and 70-kDa ChATs (Figs. 1 and 3). The 74-kDa ChAT differs from 70-kDa ChAT by 36 extra N-terminal residues that have no known functional significance. That the boundaries of exon S have high consensus values for splicing (38) and that the two translational start sites are used in COS cells consistently (Figs. 1 and 3) argue that the S transcript is not an RT-PCR artifact or immature mRNA. However, the level or site of expression of the 74-kDa ChAT in humans is not known. The S transcript also harbors the translational start site of 83-kDa ChAT (see Fig. 1). However, the 83-kDa start site within the S transcript is of doubtful significance, because after 112 codons it leads to a stop codon within exon S.
Possible Reasons for Selective Vulnerability of the Neuromuscular Synapse.
That none of our CMS-EA patients had symptoms referable to the central or autonomic nervous system is of special interest, because it implies a larger safety margin for central and autonomic synapses than for neuromuscular synapses. Reasons for selective vulnerability of the neuromuscular synapse may be owing to differences in presynaptic levels of ChAT, choline, or AcCoA, rates of choline uptake, or rates of ACh release under conditions of increased neuronal impulse flow. Presently, there is no evidence that tissue-specific isoforms explain the selective vulnerability of neuromuscular transmission. Although there are five alternative CHAT transcripts (Fig. 1) with at least three different promoters in human (39), and CHAT expression may be regulated by different control mechanisms in brain, spinal cord, and autonomic ganglia, the observed mutations are in the shared coding region of the recognized ChAT isoforms. Moreover, the 83- and 70-kDa ChATs synthesize ACh at similar rates (40), and the kinetic properties of the 74- and 70-kDa ChATs are identical (see Results). Therefore, tissue specific expression of ChAT isoforms cannot explain the tissue-specific vulnerability, but tissue-specific vulnerability could be affected by promoters of differing strength producing different levels of ChAT expression in different tissues.
Acknowledgments
This work was supported by National Institutes of Health Grant NS6277 (to A.G.E.) and a Muscular Dystrophy Association grant (to A.G.E.).
Abbreviations
- CMS
congenital myasthenic syndrome
- ACh
acetylcholine
- AChR
ACh receptor
- CMS-EA
CMS associated with episodic apnea
- ChAT
choline acetyltransferase
- EPP
endplate potential
- MEPP
miniature EPP
- RT
reverse transcription
- AcCoA
acetyl CoA
Footnotes
References
- 1.Engel A G, Ohno K, Sine S M. In: Myasthenia Gravis and Myasthenic Disorders. Engel AG, editor. New York: Oxford Univ. Press; 1999. pp. 251–297. [Google Scholar]
- 2.Greer M, Schotland M. Pediatrics. 1960;26:101–108. [PubMed] [Google Scholar]
- 3.Conomy J P, Levisohn M, Fanaroff A. J Pediatr (Berlin) 1975;87:428–429. doi: 10.1016/s0022-3476(75)80651-2. [DOI] [PubMed] [Google Scholar]
- 4.Fenichel G M. Arch Neurol (Chicago) 1978;35:97–103. doi: 10.1001/archneur.1978.00500260035009. [DOI] [PubMed] [Google Scholar]
- 5.Robertson W C, Chun R W M, Kornguth S E. Arch Neurol (Chicago) 1980;37:117–119. doi: 10.1001/archneur.1980.00500510075018. [DOI] [PubMed] [Google Scholar]
- 6.Smit L M, Barth P G. Dev Med Child Neurol. 1980;22:371–374. doi: 10.1111/j.1469-8749.1980.tb03718.x. [DOI] [PubMed] [Google Scholar]
- 7.Baptist E C, Landes R V, Sturman J K. South Med J. 1985;78:201–202. doi: 10.1097/00007611-198502000-00022. [DOI] [PubMed] [Google Scholar]
- 8.Gieron M A, Korthals J K. Arch Neurol (Chicago) 1985;42:143–144. doi: 10.1001/archneur.1985.04060020053015. [DOI] [PubMed] [Google Scholar]
- 9.Mora M, Lambert E H, Engel A G. Neurology. 1987;37:206–214. doi: 10.1212/wnl.37.2.206. [DOI] [PubMed] [Google Scholar]
- 10.Zammarchi E, Donati M A, Masi S, Sarti A, Castelli S. Childs Nerv Syst. 1994;10:347–349. doi: 10.1007/BF00335178. [DOI] [PubMed] [Google Scholar]
- 11.Deymeer F, Serdaroglu P, Özdemir C. Neuromuscul Disord. 1999;9:129–130. doi: 10.1016/s0960-8966(99)00004-8. [DOI] [PubMed] [Google Scholar]
- 12.Engel A G, Lambert E H. Electroencephalogr Clin Neurophysiol Suppl. 1987;39:91–102. [PubMed] [Google Scholar]
- 13.Engel A G, Nagel A, Walls T J, Harper C M, Waisburg H A. Muscle Nerve. 1993;16:1284–1292. doi: 10.1002/mus.880161204. [DOI] [PubMed] [Google Scholar]
- 14.Hahm S H, Chen L, Patel C, Erickson J, Bonner T I, Weihe E, Schafer M K, Eiden L E. J Mol Neurosci. 1997;9:223–236. doi: 10.1007/BF02800504. [DOI] [PubMed] [Google Scholar]
- 15.Chireux M A, Le Van Thai A, Weber M J. J Neurosci Res. 1995;40:427–438. doi: 10.1002/jnr.490400402. [DOI] [PubMed] [Google Scholar]
- 16.Touissaint J L, Geoffroy V, Schmitt M, Werner A, Garnier J M, Simoni P, Kempf J. Genomics. 1992;12:412–416. doi: 10.1016/0888-7543(92)90395-9. [DOI] [PubMed] [Google Scholar]
- 17.Oda Y, Nakanishi I, Deguchi T. Brain Res Mol Brain Res. 1992;16:287–294. doi: 10.1016/0169-328x(92)90237-6. [DOI] [PubMed] [Google Scholar]
- 18.Nixon R A. Curr Opin Cell Biol. 1998;10:87–92. doi: 10.1016/s0955-0674(98)80090-2. [DOI] [PubMed] [Google Scholar]
- 19.Ray K, Perez S E, Yang Z, Xu J, Ritchings B W, Steller H, Goldstein L S B. J Cell Biol. 1999;147:507–517. doi: 10.1083/jcb.147.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohno K, Hutchinson D O, Milone M, Brengman J M, Bouzat C, Sine S M, Engel A G. Proc Natl Acad Sci USA. 1995;92:758–762. doi: 10.1073/pnas.92.3.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Selden R F, Burke-Howie K, Rowe M E, Goodman H M, Moore D D. Mol Cell Biol. 1986;6:3173–3179. doi: 10.1128/mcb.6.9.3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fonnum F. J Neurochem. 1975;24:407–409. doi: 10.1111/j.1471-4159.1975.tb11895.x. [DOI] [PubMed] [Google Scholar]
- 23.Hersh L B, Peet M. J Biol Chem. 1977;252:4796–4802. [PubMed] [Google Scholar]
- 24.Okuda T, Haga T, Kanai Y, Endou H, Ishihara T, Katsura I. Nat Neurosci. 2000;3:120–125. doi: 10.1038/72059. [DOI] [PubMed] [Google Scholar]
- 25.Apparsundaram S, Ferguson S M, George AL, Jr, Blakely R D. Biochem Biophys Res Commun. 2000;276:862–867. doi: 10.1006/bbrc.2000.3561. [DOI] [PubMed] [Google Scholar]
- 26.Erickson J D, Varoqui H, Eiden L E, Schafer M K, Modi W, Diebler M, Weihe E, Rand J, Bonner T I, Usdin T B. J Biol Chem. 1994;269:21929–21932. [PubMed] [Google Scholar]
- 27.Reimer R J, Fon A E, Edwards R H. Curr Opin Neurobiol. 1998;8:405–412. doi: 10.1016/s0959-4388(98)80068-8. [DOI] [PubMed] [Google Scholar]
- 28.Hahn M, Hahn S L, Stone D M, Joh T H. Proc Natl Acad Sci USA. 1992;89:4387–4391. doi: 10.1073/pnas.89.10.4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishii K, Oda Y, Ichikawa T, Deguchi T. Brain Res Mol Brain Res. 1990;7:151–159. doi: 10.1016/0169-328x(90)90092-r. [DOI] [PubMed] [Google Scholar]
- 30.Brice A, Raynaud B, Ansieau S, Coppola T, Weber M J, Mallet J. J Neurosci Res. 1990;23:266–273. doi: 10.1002/jnr.490230304. [DOI] [PubMed] [Google Scholar]
- 31.Berrard S, Brice A, Lottspeich F, Braun A, Barde Y A, Mallet J. Proc Natl Acad Sci USA. 1987;84:9280–9284. doi: 10.1073/pnas.84.24.9280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rand J B, Russell R L. Genetics. 1984;106:227–248. doi: 10.1093/genetics/106.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salvaterra P M, McCaman R E. J Neurosci. 1985;5:903–910. doi: 10.1523/JNEUROSCI.05-04-00903.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brandon E P, Lin W, D'Amour K A, Pizzo D P, Thode S, Dominguez B, Thal L J, Lee K-F, Gage F H. Soc Neurosci Abstr. 2000;26:1089. doi: 10.1523/JNEUROSCI.23-02-00539.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carbini L A, Hersh L B. J Neurochem. 1993;61:247–253. doi: 10.1111/j.1471-4159.1993.tb03561.x. [DOI] [PubMed] [Google Scholar]
- 36.Wu D, Hersh L B. J Biol Chem. 1995;270:29111–29116. doi: 10.1074/jbc.270.49.29111. [DOI] [PubMed] [Google Scholar]
- 37.Cronin C N. J Biol Chem. 1998;273:24465–24469. doi: 10.1074/jbc.273.38.24465. [DOI] [PubMed] [Google Scholar]
- 38.Ohno K, Brengman J M, Felice K J, Cornblath D R, Engel A G. Am J Hum Genet. 1999;65:635–644. doi: 10.1086/302551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eiden L E. J Neurochem. 1998;70:2227–2240. doi: 10.1046/j.1471-4159.1998.70062227.x. [DOI] [PubMed] [Google Scholar]
- 40.Resendes M C, Dobransky T, Ferguson S S, Rylett R J. J Biol Chem. 1999;274:19417–19421. doi: 10.1074/jbc.274.27.19417. [DOI] [PubMed] [Google Scholar]
- 41.Misawa H, Matsuura J, Oda Y, Takahashi R, Deguchi T. Brain Res Mol Brain Res. 1997;44:323–333. doi: 10.1016/s0169-328x(96)00231-8. [DOI] [PubMed] [Google Scholar]