

Abstract
Haemophilus ducreyi must adapt to the environment of the human host to establish and maintain infection in the skin. Bacteria generally utilize stress response systems, such as the CpxRA two-component system, to adapt to hostile environments. CpxRA is the only obvious two-component system contained in the H. ducreyi genome and negatively regulates the lspB-lspA2 operon, which encodes proteins that enable the organism to resist phagocytosis. We constructed an unmarked, in-frame H. ducreyi cpxA deletion mutant, 35000HPΔcpxA. In human inoculation experiments, 35000HPΔcpxA formed papules at a rate and size that were significantly less than its parent and was unable to form pustules compared to the parent. CpxA usually has kinase and phosphatase activities for CpxR, and the deletion of CpxA leads to the accumulation of activated CpxR due to the loss of phosphatase activity and the ability of CpxR to accept phosphate groups from other donors. Using a reporter construct, the lspB-lspA2 promoter was downregulated in 35000HPΔcpxA, confirming that CpxR was activated. Deletion of cpxA downregulated DsrA, the major determinant of serum resistance in the organism, causing the mutant to become serum susceptible. Complementation in trans restored parental phenotypes. 35000HPΔcpxA is the first H. ducreyi mutant that is impaired in its ability to form both papules and pustules in humans. Since a major function of CpxRA is to control the flow of protein traffic across the periplasm, uncontrolled activation of this system likely causes dysregulated expression of multiple virulence determinants and cripples the ability of the organism to adapt to the host.
Haemophilus ducreyi causes chancroid, a sexually transmitted genital ulcer disease endemic in Africa, Asia, and the tropics (36). Chancroid is a public health problem because it facilitates the transmission and acquisition of human immunodeficiency virus type 1 (HIV-1) (36).
To determine how H. ducreyi causes infection, we developed a human inoculation model in which strain 35000HP (HP, human passaged) and its derivatives are inoculated into the skin of the arm of healthy adult volunteers via puncture wounds (24). Papules develop within 24 h and either spontaneously resolve or evolve into pustules within 2 to 5 days, simulating natural infection. Within 24 h, polymorphonuclear leukocytes (PMN) and macrophages traffic into the wounds on collagen and fibrin scaffolds, surrounding the organism (7). The phagocytes coalesce into an abscess that eventually erodes the epidermis (7, 8). Throughout experimental infection, H. ducreyi organisms are found in the abscess and associate with PMN and macrophages, both of which fail to ingest the organism (7). Similar relationships between H. ducreyi and host cells are maintained in natural ulcers (9). Thus, H. ducreyi must adapt to the hostile environment of an abscess to establish and maintain infection in the human host.
We recently used selective capture of transcribed sequences (SCOTS) (6) and microarrays of the SCOTS-derived sequences (unpublished) to identify genes important to the survival of H. ducreyi in pustules. We found that several hundred bacterial transcripts were differentially regulated in pustules relative to their expression levels in vitro, suggesting that H. ducreyi senses and responds to the host environment.
Gram-negative bacteria generally utilize multiple stress response systems to adapt to changing environments (32, 34). Of these systems, only genes encoding homologues of the alternative sigma factor RpoE and CpxRA are contained in the genome of 35000HP (GenBank accession no. AE017143). Although some Gram-negative bacteria contain over 30 two-component systems (43), CpxRA is the only obvious intact two-component system recognized in the H. ducreyi genome. Microarrays of the SCOTS-derived transcripts indicated that several H. ducreyi homologues of the Escherichia coli CpxRA regulon were upregulated in pustules (unpublished), suggesting that this system is functioning during human infection.
H. ducreyi expresses the antiphagocytic proteins LspA1 and LspA2, whose expression is required for virulence in human volunteers and whose secretion is mediated by LspB (23, 25). Recently, Labandeira-Rey et al. showed that a cpxR deletion mutant has increased expression of LspB and LspA2 relative to 35000HP and that recombinant CpxR binds to promoter sequences preceding the lspB-lspA2 operon (25). These data suggest that CpxRA has a direct effect on the expression of LspB and LspA2 and is linked to pathogenesis (25).
In E. coli, CpxA is a sensor that spans the cytoplasmic membrane and has autokinase, kinase, and phosphatase activities (32, 34, 41). In the absence of envelope stress, the default status of CpxA is as a phosphatase, and the system is inactive (41). CpxA autophosphorylates in response to membrane stress, and activated CpxA donates its phosphoryl group to CpxR, a response regulator, forming CpxR-P. CpxR-P binds to conserved DNA sequences preceding approximately 100 genes, enhancing or inhibiting their transcription (14). In changing environments, the phosphorylation status of CpxR is tightly modulated by CpxA to control membrane traffic and organism integrity (33). In cpxA deletion mutants, CpxR accepts phosphate groups from small molecules donors, such as acetyl phosphate (Ac-P), and cannot be dephosphorylated, leading to excess CpxR-P and activating the system (41).
In the present study, we constructed an H. ducreyi cpxA deletion mutant, which was unable to initiate infection in humans. Deletion of cpxA led to the downregulation of lspB-lspA2, suggesting that CpxR was activated in this mutant. Deletion of cpxA also resulted in the downregulation of DsrA, an outer membrane protein (OMP) that is the major determinant of serum resistance in this organism (16). The data suggest uncontrolled activation of CpxR downregulates several major virulence determinants of H. ducreyi and cripples the ability of the organism to survive in vivo.
MATERIALS AND METHODS
Bacterial strains, plasmids, oligonucleotides, and culture conditions.
The bacterial strains and plasmids used in the present study are included in Table 1. Selected oligonucleotide primers are listed in Table 2 . All H. ducreyi strains were grown on chocolate agar plates supplemented with 1% IsoVitaleX and incubated with 5% CO2. For the human inoculation experiments, H. ducreyi samples were grown in a proteose peptone broth-based medium with 5% heat-inactivated fetal calf serum as described previously (23). In other experiments, H. ducreyi was grown in a Columbia broth-based medium with 2.5% heat-inactivated fetal calf serum as described previously (25). Except as indicated, all H. ducreyi cultures were grown at 33°C. When appropriate, the media were supplemented with chloramphenicol, spectinomycin, or kanamycin at 0.3, 200, or 20 μg/ml. E. coli strains were grown in Luria-Bertani broth or plates at 37°C, except for strain DY380, which was grown or maintained at 32°C and induced to express λ recombinase at 42°C.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5α | Strain used for general cloning procedures | Invitrogen |
| DY380 | DH10B derivative containing a defective λ prophage in which the red, bet, and gam genes are controlled by the temperature-sensitive λcI857 repressor | 26 |
| H. ducreyi | ||
| 35000HP | Human passaged variant of strain 35000 | 2 |
| 35000HPΔcpxR | 35000HPcpxR::cat insertion/deletion mutant | 25 |
| 35000HPΔcpxA | 35000HPcpxA unmarked, in-frame deletion mutant | This study |
| FX517 | 35000dsrA::cat insertion mutant | 16 |
| MF35000 | Spontaneous mutant of 35000HP that expresses upregulated OmpP2B | 31 |
| Plasmids | ||
| pRSM2072 | H. ducreyi suicide vector | 11 |
| pLS88 | H. ducreyi shuttle vector | 15 |
| pML141 | pLS88 derivative containing the cpxA ORF flanked by 265 bp upstream and 17 bp of downstream sequences cloned into a SacII site | M. Labandeira-Rey and E. Hansen |
| pRSM2947 | Plasmid containing the origin of replication and kanamycin resistance gene from pLS88 and the FLP recombinase gene from pFT-A | 38 |
| pRSM2975 | Derivative of pRSM2947 containing a point mutation conferring a temperature-sensitive phenotype in H. ducreyi | This study |
| pRB157 | pLS88 derivative containing a ΩAmp cartridge, followed by a BglII site for insertion of promoter sequences and a promoterless GFP cassette derived from pGreenTIR (29) | This study |
| pKF1 | pRB157 derivative containing the lspB promoter region | This study |
| pKF2 | pRB157 derivative containing the dsrA promoter region | This study |
TABLE 2.
Oligonucleotides used in this study
| Primer | 5′ to 3′ sequencea |
|---|---|
| P1 | ATCCCTCGCAAGTTCGGGGGATTTTCTCTTTTTAAAGTATTTAATATATGATTCCGGGGATCCGTCGACC |
| P2 | ATAGACCGCTTTACCTAAAATGTATTAAATTATTCAAGCCAGAGTGGTAGTGTAGGCTGGAGCTGCTTCG |
| P3 | TATCTGCTCGGAGTTGCTTATCT |
| P4 | ACCGTTTCACCATTCTCGTT |
| P5 | AACGTTACCTTCAGCAAGCGGTTC |
| P6 | GCCGTTTGGGATCGTCGAGTGTATA |
| P7 | GCCATTCCTAATTTCGATGCGCGTTC |
| P8 | TTTGAGTGGCTACAGAAAGGCGAC |
| P9 | CCAAAGGTACCAAGCGTGTCTGTATCCCAC |
| P10 | GGGGATATCATTTAAATACAAGGTCAATGCTCGGC |
| P11 | GGAAGATCTCGCTACATCAGTTA |
| P12 | GGAAGATCTATTTGTTAAAGTGCTCAC |
| P13 | GGAAGATCTATTGGAGTGGACCAGGACAGCATT |
| P14 | GGAAGATCTAATCACCTCATTAAGTAAATAAT |
| P15 | CCTTCACCCTCTCCACTGACAG |
| P16 | TGGACTTGGTCTAATGAAGGCGGT |
| P17 | AAACGCCAGGAGCATATGTCACGA |
Boldfaced text represents sequences with homology to the mutagenic cassette. Underlining indicates regions corresponding to restriction enzyme sites as described in text. All H. ducreyi sequences are from GenBank accession no. AE017143.
Construction and complementation of an unmarked, in frame H. ducreyi cpxA deletion mutant.
We developed tools to apply “recombineering” methodology (4) to make unmarked, in frame deletion mutants in H. influenzae (38) and applied this methodology to H. ducreyi. In brief, 70-bp primers were designed to facilitate construction of 35000HPΔcpxA. The forward primer (P1) included 47 bp upstream of cpxA plus the ATG codon. The reverse primer (P2) included 21 bp at the 3′ end of cpxA, including the TAA codon plus 29 bp of the downstream region. The 3′ end of the primers contained 20 bp complementary to regions 5′ and 3′ of a spectinomycin (spec) resistance cassette flanked by flippase (FLP) recognition target (FRT) sites, which had been cloned into pRSM2832 (38). PCR of pRSM2832 with the 70-bp primers yielded a 2-kb amplicon that contained the spec cassette flanked by FRT sites and 50 bp of DNA homologous to regions 5′ of and 3′ of H. ducreyi cpxA.
The cpxRA operon and ∼1.8-kb of DNA 5′ and 3′ of these genes was amplified by PCR using chromosomal DNA as a template, Easy-A polymerase (Stratagene, La Jolla, CA), and primers 3 and 4. The amplicon was cloned into pGEM-T Easy and then transformed into E. coli DY380, which contains λ recombinase (26). Induction of λ recombinase generated plasmids where cpxA was replaced with the mutagenic cassette, except for the cpxA start codon and the terminal 21 bp of the cpxA open reading frame (ORF). A SpeI fragment containing the insertionally inactivated cpxA gene and flanking DNA was ligated into the suicide vector pRSM2072 (11). The construct was confirmed by sequencing and then electroporated into 35000HP. Cointegrates were selected on chocolate agar plates containing spectinomycin and then resolved by passage on plates containing spectinomycin and X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) (11). Allelic exchange was confirmed by colony PCR.
pRSM2947 contains a temperature sensitive replicon derived from pLS88, a kanamycin resistance cassette and FLP recombinase under the control of a tet repressor (38). pRSM2975, a derivative of pRSM2947, whose origin was further altered by a point mutation of G to A at base 2151 of pLS88 (15), so that the plasmid was unable to replicate in H. ducreyi at 35°C, was transformed into the mutant. Transformants were selected at 32°C on chocolate agar containing kanamycin. Transformants were grown in broth containing kanamycin at 32°C to mid-log phase, and anhydrotetracycline (200 ng/ml) was added to induce expression of the FLP recombinase. After 2 h, the cultures were diluted on chocolate agar plates that were incubated at 35°C for 48 h to cure the plasmid. Colonies were replica plated on chocolate agar, as well as chocolate agar supplemented with spectinomycin or kanamycin to identify clones that lost both the temperature-sensitive plasmid and the spec cassette. The plasmid and the spec cassette were lost in 16 of 343 (4.3%) of the clones; 5 clones had a deletion of the expected size by colony PCR. One clone was designated 35000HPΔcpxA. Sequence analysis confirmed that cpxA had been replaced by a short ORF that consisted of the start codon of cpxA, 81 bp encoding a FLP scar peptide, and the last 21 bp of cpxA, including its stop codon.
To complement 35000HPΔcpxA in trans, we electroporated the mutant with pML141, which was kindly provided by Maria Labandeira-Rey and Eric Hansen (University of Texas, Southwestern). pML141 contains the cpxA ORF flanked by 265 bp upstream and 17 bp of downstream sequences cloned into the SacII site of pLS88. As controls, we also transformed 35000HP and 35000HPΔcpxA with pLS88.
Human inoculation experiments.
Dedicated stocks of 35000HP and 35000HPΔcpxA were prepared according to FDA guidelines (BB-IND 13064). Five healthy adult volunteers (four females and one male; one Hispanic and four black; mean age ± the standard deviation [SD] 33 ± 14 years) over 21 years of age participated in the study. Subjects gave informed consent for participation and for HIV serology, in accordance with the human experimentation guidelines of the U.S. Department of Health and Human Services and the Institutional Review Board of Indiana University-Purdue University of Indianapolis. The experimental challenge protocol, preparation and inoculation of the bacteria, calculation of the estimated delivered dose (EDD), clinical observations, surface cultures, definitions of clinical endpoints, biopsies, and antibiotic treatment of the volunteers were carried out exactly as described previously (24). To account for the correlation among sites within the same individual, comparison of papule and pustule formation rates for the two strains were performed using a logistic regression model with generalized estimating equations (GEE), as described previously (37). The GEE sandwich estimate for the standard errors was used to calculate the 95% confidence intervals (95% CI) for these rates except when the rate was zero and the estimate did not exist. For this case, we calculated the exact binomial confidence interval based on the number of subjects rather than sites, as described previously (37).
To confirm that bacteria isolated from the inocula, surface cultures, or biopsies had the intended phenotype, colonies were replica plated and grown on nitrocellulose filters. The filters were probed with amplicons corresponding to dnaE or to the deleted sequences of cpxA. The dnaE probe was made using primers P5 and P6 and the cpxA probe was made by using primers P7 and P8. The probes were labeled with digoxigenin using a DIG DNA labeling kit (Roche Applied Sciences, Penzberg, Germany) and detected with the DIG Easy Hyb protocol (Roche Applied Sciences) according to the manufacturer's instructions.
CpxR reporter constructs.
pRB157 is a pLS88 derivative that contains a BglII site for insertion of promoter sequences preceding a promoterless green fluorescent protein (GFP) cassette. pRB157 was constructed by amplifying the origin of replication and streptomycin resistance gene from pLS88 by PCR using the primers P9 and P10. These primers contain KpnI, EcoRV, and SwaI sites. The resulting 3.2-kb amplicon was then digested with KpnI and ligated to a KpnI/SmaI fragment from pGreenTIR (29) containing a promoterless gfp cartridge. To stop possible plasmid promoter-based transcription into the promoterless gfp, an ΩAmp cartridge from pKT254Ω-Ap (17) was inserted into the SwaI site upstream of the gfp ORF. Then, a BglII linker, consisting of dimers of the oligonucleotide 5′-CAGATCTG-3′ was ligated into the EcoRV site to yield pRB157, which contains the promoterless gfp construct in which DNA fragments with BglII-compatible ends could be inserted in front of the gfp ORF. The lspB-lspA2 (301 bp) and the dsrA (178 bp) promoter regions were amplified by PCR and cloned into the BglII site to form pKF1 and pKF2, using the primer pairs P11-P12 and P13-P14, respectively. The orientation of the promoters with respect to the gfp cassette was confirmed by PCR by using each promoter-specific forward primer and a reverse primer (P15) that hybridized to the gfp sequences downstream of the BglII site. Each of the reporter constructs was electroporated into 35000HP, 35000HPΔcpxA, and 35000HPΔcpxR. Transformants of each strain harboring a specific reporter were grown in broth overnight, diluted into fresh media, and grown to mid log phase. Cells were harvested at various time points during exponential growth. Whole-cell lysates were analyzed by Western blots probed with an anti-GFP polyclonal serum (42) and with the peptidoglycan-associated lipoprotein (PAL)-specific monoclonal antibody, 3B9 (18). PAL is constitutively expressed by H. ducreyi and served as a control for protein loading. For each strain, the level of expression of GFP normalized to PAL was determined by densitometry using Adobe Photoshop CS4 (Adobe Systems, Inc., San Jose, CA).
RNA isolation and real-time PCR.
Bacterial RNA was prepared from mid-log phase organisms by using the TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. The RNA was treated twice with DNase I (Ambion, Austin, TX) for 1 h at 37°C and then purified by using the RNeasy system (Qiagen, Valencia, CA). The integrity of the samples was confirmed with an Agilent Bioanalyser (Agilent Technologies, Palo Alto, CA). After optimizing primers so that their efficiencies were >95%, we examined the level of transcript expression in RNA isolated from 35000HP and 35000HPΔcpxA. Reactions utilizing bacterial RNA, the dsrA primers P16 and P17 and dnaE primers P5 and P6, and the one-step QuantiTect SYBR green reverse transcription-PCR (RT-PCR) kit (Qiagen) were performed in triplicate as described previously (19). The levels of expression were determined by using an ABI Prism 7000 sequence detector (Applied Biosystems, Carlsbad, CA). The data were expressed as the fold change of dsrA relative to the level of dnaE using the following equation: ratio = (EdsrA)ΔCTdsrA (35000HP − 35000HPΔcpxA)/(EdnaE)ΔCTdnaE (35000HP − 35000HPΔcpxA), where E is the amplification efficiency (equal to 10−1/slope) and ΔCT is the change in cycle threshold (30).
Phenotypic comparisons.
OMPs and lipooligosaccharides (LOS) and were prepared from 35000HP and 35000HPΔcpxA and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis as previously described (18). Western blots of whole-cell lysates of 35000HP(pLS88), 35000HPΔcpxA(pML141), and 35000HPΔcpxA(pLS88) and FX517, a dsrA insertion/deletion mutant, were probed with an anti-DsrA polyclonal serum (16), kindly provided by Christopher Elkins (University of North Carolina, Chapel Hill), or an anti-OmpP2B polyclonal serum (31), kindly provided by Anthony Campagnari (State University of New York at Buffalo). As a control for protein loading, each blot was probed with 3B9 (18).
Bactericidal assays.
Bactericidal assays were performed exactly as described previously (1, 5), except a single healthy donor was the source of normal human serum (NHS). In the first set of experiments, we compared the survival in 50% NHS of plate-grown 35000HP, 35000HPΔcpxA, and FX517, which is serum susceptible (16). In the second set of experiments, we compared the survival of 35000HP(pLS88), 35000HPΔcpxA(pLS88), and 35000HPΔcpxA(pML141). The data were reported as the percent survival in active NHS compared to that in heat-inactivated serum [(geometric mean CFU in active NHS/geometric mean CFU in heat-inactivated NHS) × 100]. Each experiment was repeated five times, and the arithmetic mean and SD of the percent survival were calculated. Comparison of the strains was performed by using paired Student t tests. With the Bonferroni adjustment for multiple comparisons, a P value of <0.017 was considered significant for these assays.
RESULTS
An H. ducreyi cpxA deletion mutant is impaired in its ability to infect human volunteers.
In H. ducreyi 35000HP, cpxR and cpxA are located in an operon whose gene order is mazG, cpxR, cpxA, and HD1471 (25). We constructed an unmarked, in-frame cpxA deletion mutant, 35000HPΔcpxA, using recombineering methodology (4, 38). Sequence analysis showed that the cpxA gene was replaced by a short ORF that encoded a 34-amino-acid peptide and that cpxR and HD1471 were in frame. As determined by quantitative RT-PCR, the expression level of HD1471 was unchanged in strain 35000HPΔcpxA compared to strain 35000HP. The growth rates of 35000HPΔcpxA and 35000HP in broth were identical. Gram stains and colonial morphology suggested there were no differences in chaining or clumping of the mutant and the parent. The LOS profiles of the strains were identical (data not shown).
To test whether 35000HPΔcpxA was virulent in humans, we inoculated two groups of volunteers. The first group of three subjects was inoculated with an EDD of 55 CFU of 35000HP at three sites on one arm and with 38, 75, and 150 CFU of 35000HPΔcpxA at three sites on the other arm. The second group of two subjects was inoculated with 42 CFU of 35000HP at three sites on one arm and with 65, 130, and 261 CFU of 35000HPΔcpxA at three sites on the other arm. Overall, papules formed at 93.3% (95% CI, 80.7 to 99.9%) of parent sites and 60% (95% CI, 35.2 to 84.8%) of mutant sites (P = 0.039) (Table 3). After 24 h of infection, the mean area of the papules at mutant-inoculated sites was 2.2 ± 2.0 mm2, while the mean area of the papules at parent-inoculated sites was 15.1 ± 10.1 mm2 (P < 0.0001). Pustules formed at 53.5% (95% CI, 28.1 to 78.6%) of sites inoculated with the parent and 0% (95% CI, 0 to 45.0%) of sites inoculated with the mutant (P = 0.0003). Thus, deletion of cpxA severely impaired the ability of H. ducreyi to initiate infection and survive in vivo.
TABLE 3.
Response to inoculation with live H. ducreyi
| Volunteera (gender) | Observation period (days) | Strainb | Dose, (CFU)c | No. of initial papules | Final outcome of papule |
|
|---|---|---|---|---|---|---|
| No. resolved | No. of pustules | |||||
| 361 (F) | 6 | P | 55 | 3 | 3 | |
| M | 38-150 | 3 | 3 | |||
| 362 (M) | 5 | P | 55 | 3 | 3 | |
| M | 38-150 | 1 | 3 | |||
| 363 (F) | 7 | P | 55 | 2 | 1 | 2 |
| M | 38-150 | 2 | 3 | |||
| 364 (F) | 5 | P | 42 | 3 | 2 | 1 |
| M | 65-261 | 0 | 3 | |||
| 366 (F) | 6 | P | 42 | 3 | 1 | 2 |
| M | 65-261 | 3 | 3 | |||
Volunteers 361, 362, and 363 were inoculated in the first group; volunteers 364 and 366 were inoculated in the second group. M, male; F, female.
P, parent strain 35000HP; M, mutant strain 35000HPΔcpxA.
38-150, one dose each of 38, 75, and 150 CFU; 65-261, one dose each of 65, 130, and 261 CFU.
Surface cultures obtained at follow-up visits grew H. ducreyi from 27% of the parent-inoculated and 0% of the mutant-inoculated sites. All colonies recovered from the parent sites (n = 102) and colonies from the parent (n = 70) and mutant (n = 67) inocula were tested for the presence of cpxA and dnaE sequences by colony hybridization. The dnaE probe hybridized to all of the colonies, whereas the cpxA probe hybridized only to the colonies obtained from the parent-inoculated sites or the parent inocula. Thus, there was no evidence of cross-contamination between mutant-inoculated and parent-inoculated sites.
CpxR is activated in 35000HPΔcpxA.
In the absence of CpxA, CpxR accepts phosphate groups from alternative donors such as Ac-P and cannot be dephosphorylated (41). Relative to the wild type, CpxR-P accumulates in cpxA deletion mutants and activates the Cpx regulon when organisms are grown in noninducing conditions, such as logarithmic growth (41).
CpxR downregulates the lspB-lspA2 operon in H. ducreyi (25). To test whether CpxR was activated in 35000HPΔcpxA, the lspB promoter region was cloned upstream of a reporter gene encoding GFP to form pKF1. We transformed pKF1 into 35000HP, 35000HPΔcpxA and 35000HPΔcpxR and grew the strains to mid log phase. As determined by Western blotting, the level of GFP expression was downregulated in 35000HPΔcpxA relative to 35000HP and 35000HPΔcpxR (Fig. 1), suggesting that CpxR was activated in 35000HPΔcpxA.
FIG. 1.
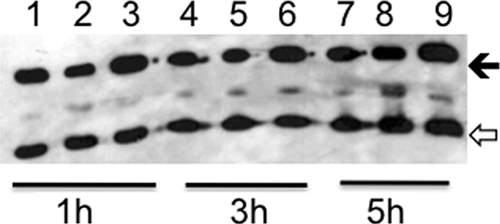
Regulation of lspB promoter activity in the cpxA and cpxR deletion mutants during exponential growth. Whole-cell lysates of 35000HP(pKF1) (lanes 1, 4, and 7), 35000HPΔcpxA(pKF1) (lanes 2, 5, and 8), and 35000HPΔcpxR(pKF1) (lanes 3, 6, and 9) were probed with an anti-GFP polyclonal serum and with the PAL-specific monoclonal antibody, 3B9. Cells were obtained after 1 h (lanes 1 to 3), 3 h (lanes 4 to 6), and 5 h (lanes 7 to 9) of growth. The solid arrow indicates GFP, and the open arrow indicates PAL. Normalized to PAL, the relative levels of expression of GFP for the cpxA mutant were 55, 76, and 75% that of the parent, respectively, after 1, 3, and 5 h of growth. The relative levels of expression of GFP for the cpxR mutant were 103, 123, and 98% that of the parent, respectively, after 1, 3, and 5 h of growth. The blot is representative of two independent experiments.
DsrA expression is repressed and OmpP2B expression is enhanced in 35000HPΔcpxA.
A major function of CpxRA is to control protein traffic across the cytoplasmic membrane. Several OMPs, including DsrA, HgbA, NcaA, and PAL, are required for virulence in humans (24). To discern whether activation of CpxR affected the expression of OMPs, we compared the OMP profiles of 35000HPΔcpxA and 35000HP. 35000HPΔcpxA did not express a 28 kDa OMP, which is the apparent molecular mass of DsrA, a major determinant of serum resistance in the organism (16) (Fig. 2A). 35000HPΔcpxA overexpressed a 43-kDa OMP, which is the apparent molecular mass OmpP2B, a porin expressed by H. ducreyi, which is dispensable for virulence in humans (24, 31) (Fig. 2A). Western blot analysis confirmed that DsrA expression was downregulated and OmpP2B expression was upregulated in 35000HPΔcpxA (Fig. 2B and C), suggesting that CpxR regulates the expression of these OMPs.
FIG. 2.
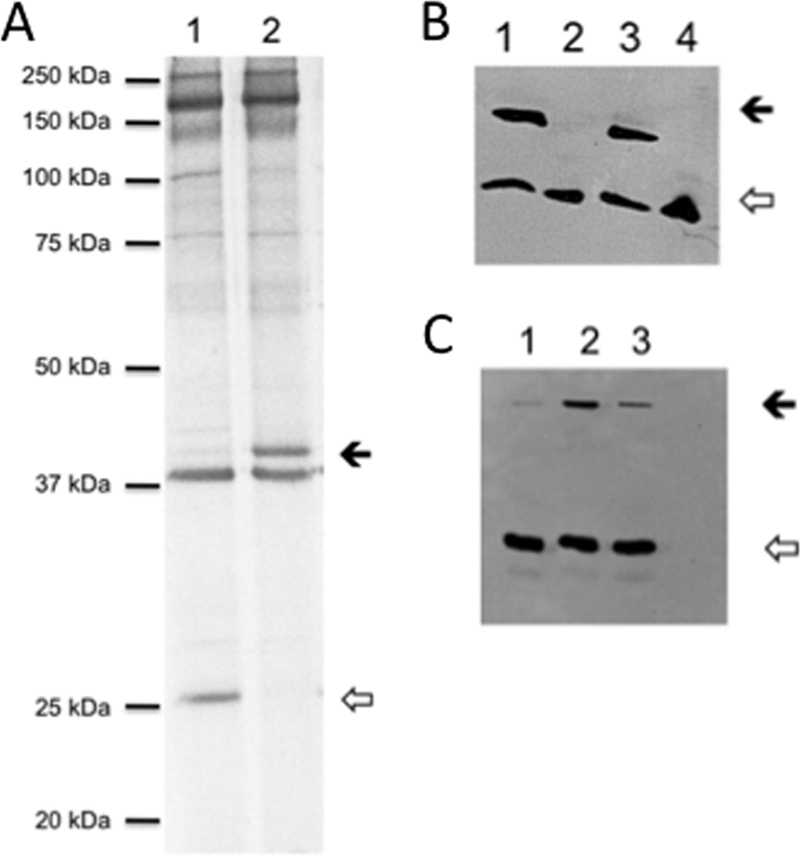
Phenotypes of the cpxA deletion mutant. (A) OMP profiles of 35000HP (lane 1) and 35000HPΔcpxA (lane 2). The closed arrow indicates the 43-kDa band, and the open arrow indicates the 28-kDa band. (B) Whole-cell lysates of 35000HP(pLS88) (lane 1), 35000HPΔcpxA(pLS88) (lane 2), 35000HPΔcpxA(pML1411) (lane 3), and the dsrA mutant FX517 (lane 4) probed with an anti-DsrA polyclonal serum and with the PAL-specific monoclonal antibody, 3B9. The closed arrow indicates DsrA, and the open arrow indicates PAL. In some experiments, a faint DsrA band was detectable in 35000HPΔcpxA(pLS88). (C) Whole-cell lysates probed with an anti-OmpP2B polyclonal serum and with 3B9. Lanes 1 to 3 correspond to the first 3 lanes in panel B. The closed arrow indicates OmpP2B, and the open arrow indicates PAL.
The OMP profile of 35000HPΔcpxA was very similar to that of a spontaneous mutant (MF35000) of 35000HP (31), which exhibits upregulated expression of OmpP2B. However, MF35000 expressed parental levels of DsrA in Western blot (data not shown). Sequence analysis of MF35000 showed that its cpxA and cpxR alleles, ompP2B promoter region, and dsrA promoter region were identical to that of 35000HP (data not shown). Thus, the increased expression of OmpP2B in MF35000 is not likely related to the CpxRA system.
dsrA transcription is regulated by activated CpxR.
Since the expression of DsrA was decreased in strain 35000HPΔcpxA, we determined the nucleotide sequence of dsrA and its promoter region in 35000HPΔcpxA. The sequence was identical to that of 35000HP, indicating that there were no mutations in the dsrA gene or 5′ flanking region. There was a putative CpxR-P recognition sequence (GTAAATAATTGTCAA) at the −29 to −15 position preceding the dsrA ORF. As determined by quantitative RT-PCR, the mean ± the SD level of expression of dsrA transcripts in 35000HPΔcpxA was (13.1 ± 0.6)-fold downregulated relative to that of 35000HP, after normalization to the housekeeping gene, dnaE. To confirm that activated CpxR controlled the expression of dsrA, we cloned the dsrA promoter region (178 bp) upstream of a GFP reporter gene to form pKF2 and transformed 35000HP, 35000HPΔcpxA, and 35000HPΔcpxR with pKF2. GFP expression was downregulated in the 35000HPΔcpxA background relative to 35000HP and 35000HPΔcpxR during logarithmic growth (Fig. 3), confirming that the transcription of dsrA is regulated by CpxR.
FIG. 3.
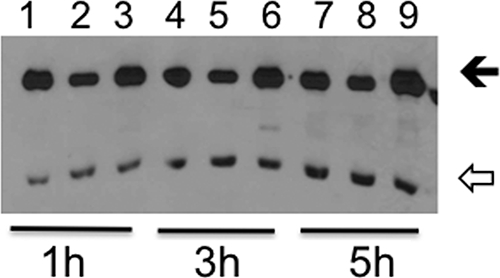
Regulation of dsrA promoter activity in the cpxA and cpxR deletion mutants during exponential growth. Whole-cell lysates of 35000HP(pKF2) (lanes 1, 4, and 7), 35000HPΔcpxA(pKF2) (lane 2, 5, and 8), and 35000HPΔcpxR(pKF2) (lanes 3, 6, and 9) were probed with an anti-GFP polyclonal serum and with the PAL-specific monoclonal antibody, 3B9. Cells were obtained after 1 h (lanes 1 to 3), 3 h (lanes 4 to 6), and 5 h (lanes 7 to 9) of growth. The solid arrow indicates GFP, and the open arrow indicates PAL. Normalized to PAL, the relative levels of expression of GFP for the cpxA mutant were 51, 63, and 82% that of the parent, respectively, after 1, 3, and 5 h of growth. The relative levels of expression of GFP for the cpxR mutant were 107, 126, and 154% that of the parent, respectively, after 1, 3, and 5 h of growth. The blot is representative of two independent experiments.
35000HPΔcpxA is serum sensitive.
We compared the survival of 35000HP, 35000HPΔcpxA, and the DsrA-deficient mutant FX517 in 50% NHS, exactly as described previously (5). The mean percent survivals ± the SD were 83% ± 22% for 35000HP, 8% ± 9% for 35000HPΔcpxA, and 2% ± 4% for FX517 (Fig. 4A). Thus, the downregulation of DsrA in 35000HPΔcpxA correlated with increased serum susceptibility of the mutant.
FIG. 4.

Bactericidal assays. (A) Percent survival of 35000HP, 35000HPΔcpxA, and FX517 in 50% NHS, calculated as follows: (geometric mean CFU in NHS/geometric mean CFU in heat-inactivated NHS) × 100. The P values were as follows: 35000HP versus 35000HPΔcpxA, P = 0.001; 35000HP versus FX517, P = 0.002; and 35000HPΔcpxA versus FX517, P = 0.32. (B) Percent survival of 35000HP(pLS88), 35000HPΔcpxA(pLS88), and 35000HPΔcpxA (pML141) in 50% NHS, calculated as in panel A. The P values were as follows: parent versus mutant, P = 0.0003; parent versus complemented mutant, P = 0.0003; and mutant versus complemented mutant, P = 0.0023. For both panels A and B, values are mean plus the SD from five independent experiments.
Complementation of 35000HPΔcpxA restores parental expression levels of DsrA and OmpP2B and partially restores the serum resistance phenotype.
The cpxA ORF was cloned into the shuttle vector pLS88 to from pML141. Complementation of 35000HPΔcpxA with pML141 in trans restored the expression of DsrA (Fig. 2B). Similarly, complementation restored OmpP2B expression to parental levels (Fig. 2C). We also compared the survival of 35000HP(pLS88), 35000HPΔcpxA(pLS88), and 35000HPΔcpxA(pML141) in 50% NHS. The mean percent survivals ± the SD were 94.8% ± 9.0% for the parent, 2.9% ± 1.7% for the cpxA mutant, and 16.2% ± 3.0% for the complemented mutant (Fig. 4B). Thus, complementation of 35000HPΔcpxA partially restored serum resistance to the mutant.
DISCUSSION
CpxRA is the only obvious intact two-component system contained in the genome of H. ducreyi. When tested in the human infection model, 35000HPΔcpxA was highly impaired in its ability to cause disease. Deletion of the cpxA gene led to downregulation of a lspB reporter gene, strongly suggesting that CpxR was activated in 35000HPΔcpxA. In addition to the downregulation of lspB, dsrA was profoundly downregulated in 35000HPΔcpxA, and the loss of DsrA expression correlated with increased serum susceptibility of the cpxA mutant.
Of 24 H. ducreyi mutants previously evaluated in the human challenge model, 7 were classified as fully attenuated, meaning they were unable to form pustules at doses up to 10-fold that of the parent (24; unpublished results). All of the attenuated mutants, including the dsrA mutant FX517, formed papules at the same rate as the parent (10, 24). 35000HPΔcpxA is the only mutant identified to date that is impaired in its ability to form both papules and pustules. Thus, the attenuation of 35000HPΔcpxA is likely not solely attributable to the downregulation of DsrA.
Hansen and coworkers have shown that the OMP LspB is required for secretion of the antiphagocytic proteins LspA1 and LspA2 across the outer membrane (40). An lspB mutant is as impaired as an lspA1 lspA2 double mutant in its ability to resist phagocytosis by human granulocytic and murine monocyte-macrophage cell lines (39). Although we did not evaluate 35000HPΔcpxA for resistance to phagocytosis, it is highly likely that 35000HPΔcpxA is impaired in this regard.
In E. coli, the conserved sequence for CpxR binding is GTAAA(N)5GTAAA (14). Using a weighted algorithm based on E. coli consensus sequences to search the genome of 35000HP (27), there were 192 putative CpxR targets in H. ducreyi (Y. Liu and S. M. Spinola, unpublished data). In addition to dsrA and lspB, several promoter regions of genes that encode proven virulence determinants in H. ducreyi, including hgbA and sapA, contained putative CpxR recognition sequences. Thus, the cpxA mutant is likely attenuated due to dysregulated expression of multiple gene products.
Labandeira-Rey et al. recently showed that dsrA and ompP2B transcripts are downregulated and upregulated, respectively, when H. ducreyi is grown in medium lacking serum versus medium containing serum (25). H. ducreyi has a higher growth rate in serum replete medium than in serum deplete medium, suggesting that the organism is relatively stressed in the latter condition, which could activate CpxRA (25). Our observation that dsrA was downregulated and ompP2B was upregulated in the cpxA mutant is consistent with these observations.
We inactivated the cpxA gene by use of a “recombineering” strategy, which is especially suitable for making unmarked, in-frame deletion mutants in genes contained in operons. Prior to testing a mutant in an operon in human volunteers, we are required to show that allelic exchange had occurred in the mutant and that the downstream genes are normally transcribed. We are precluded by several regulatory bodies that provide oversight of the human challenge experiments from testing trans-complemented mutants in humans. However, complementation of 35000HPΔcpxA in trans restored parental levels of expression of DsrA and OmpP2B and partially restored serum resistance to the organism, suggesting that the reduced virulence of the mutant is likely due to deletion of cpxA and uncontrolled activation of CpxR.
CpxRA functions in adhesion, controls regulators of virulence, and acts to counter starvation in several organisms (13, 28, 32). However, CpxR is not required for Salmonella enterica serovar Typhimurium and Y. enterocolitica to infect mice (21, 22) or for Vibrio cholerae to colonize the murine intestine (35). Activation of CpxR in cpxA deletion mutants or cpxA* mutants, which have constitutively active kinase activity, downregulates type III secretion translocators and effectors in E. coli and Yersinia pseudotuberculosis (12, 28), and both cpxA* and cpxA deletion mutants of serovar Typhimurium are highly attenuated in mice (22). These data have led to a theme in the literature that activation of CpxR reduces bacterial pathogenicity by downregulation of virulence determinants (12, 28, 32). Since a major function of CpxRA is to reduce protein traffic to the periplasm in order to relieve cytoplasmic membrane stress, it is not surprising that virulence factors that traverse the periplasm are downregulated in cpxA* and cpxA deletion mutants, as we observed for 35000HPΔcpxA. In contrast, deletion of cpxR causes downregulation of components of the type IV secretion system in Legionella pneumophila and downregulation of lrhA, a positive regulator of virulence determinants in Xenorhabdus nematophila (3, 20). CpxR is required for the virulence of the latter organism in its insect host (20). Taken together, the data suggest that the CpxRA system has different contributions to pathogenesis in different organisms.
In summary, the H. ducreyi cpxA mutant was fully attenuated in its ability to cause disease in human volunteers. The mutant could have been attenuated because its CpxRA system was unable to sense and respond to the host, or because it formed too much CpxR-P and downregulated DsrA, LspB, and LspA2 and dysregulated the expression of multiple other virulence determinants that traverse the cytoplasmic membrane. Future studies will address defining the genes controlled by this regulon and evaluating a cpxR mutant in humans.
Acknowledgments
This study was supported by grants AI31494 and AI27863 to S.M.S. from the National Institutes of Allergy and Infectious Diseases (NIAID). The human challenge trials were supported by grant U19 AI31494 from NIAID and the Indiana Clinical and Translational Sciences Institute and the Indiana Clinical Research Center (UL RR052761).
We have no relevant financial relationships to disclose.
We thank Maria Labandeira-Rey, Eric Hansen, Christopher Elkins, and Anthony Campagnari for sharing strains, plasmids, and antibodies used in this work; Margaret Bauer and Barbara Van Der Pol for their thoughtful criticism of the manuscript; Shelia Ellinger for preparing the regulatory documents for the human trials; and the volunteers who participated in the trial.
Editor: S. R. Blanke
Footnotes
Published ahead of print on 6 July 2010.
REFERENCES
- 1.Abdullah, M., I. Nepluev, G. Afonina, S. Ram, P. Rice, W. Cade, and C. Elkins. 2005. Killing of dsrA mutants of Haemophilus ducreyi by normal human serum occurs via the classical complement pathway and is initiated by immunoglobulin M binding. Infect. Immun. 73:3431-3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al-Tawfiq, J. A., A. C. Thornton, B. P. Katz, K. R. Fortney, K. D. Todd, A. F. Hood, and S. M. Spinola. 1998. Standardization of the experimental model of Haemophilus ducreyi infection in human subjects. J. Infect. Dis. 178:1684-1687. [DOI] [PubMed] [Google Scholar]
- 3.Altman, E., and G. Segal. 2008. The response regulator CpxR directly regulates expression of several Legionella pneumophila icm/dot components as well as new translocated substrates. J. Bacteriol. 190:1985-1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baba, T., T. Ara, M. Hasegawa, Y. Takai, Y. Okumura, M. Baba, K. A. Datsenko, M. Tomita, B. L. Wanner, and H. Mori. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006-2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banks, K. E., K. R. Fortney, B. Baker, S. D. Billings, B. P. Katz, R. S. Munson, Jr., and S. M. Spinola. 2008. The enterobacterial common antigen-like gene cluster of Haemophilus ducreyi contributes to virulence in humans. J. Infect. Dis. 197:1531-1536. [DOI] [PubMed] [Google Scholar]
- 6.Bauer, M. E., K. R. Fortney, A. Harrison, D. M. Janowicz, R. S. Munson, Jr., and S. M. Spinola. 2008. Identification of Haemophilus ducreyi genes expressed during human infection. Microbiology 154:1152-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bauer, M. E., M. P. Goheen, C. A. Townsend, and S. M. Spinola. 2001. Haemophilus ducreyi associates with phagocytes, collagen, and fibrin and remains extracellular throughout infection of human volunteers. Infect. Immun. 69:2549-2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bauer, M. E., and S. M. Spinola. 2000. Localization of Haemophilus ducreyi at the pustular stage of disease in the human model of infection. Infect. Immun. 68:2309-2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bauer, M. E., C. A. Townsend, A. R. Ronald, and S. M. Spinola. 2006. Localization of Haemophilus ducreyi in naturally acquired chancroidal ulcers. Microbe Infect. 8:2465-2468. [DOI] [PubMed] [Google Scholar]
- 10.Bong, C. T. H., R. E. Throm, K. R. Fortney, B. P. Katz, A. F. Hood, C. Elkins, and S. M. Spinola. 2001. A DsrA-deficient mutant of Haemophilus ducreyi is impaired in its ability to infect human volunteers. Infect. Immun. 69:1488-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bozue, J. A., L. Tarantino, and R. S. Munson, Jr. 1998. Facile construction of mutations in Haemophilus ducreyi using lacZ as a counter-selectable marker. FEMS Microbiol. Lett. 164:269-273. [DOI] [PubMed] [Google Scholar]
- 12.Carlsson, K. E., J. Liu, P. J. Edqvist, and M. S. Francis. 2007. Extracytoplasmic-stress-responsive pathways modulate type III secretion in Yersinia pseudotuberculosis. Infect. Immun. 75:3913-3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carlsson, K. E., J. Liu, P. J. Edqvist, and M. S. Francis. 2007. Influence of the Cpx extracytoplasmic-stress-responsive pathway on Yersinia sp.-eukaryotic cell contact. Infect. Immun. 75:4386-4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Wulf, P., A. M. McGuire, X. Liu, and E. C. C. Lin. 2002. Genome-wide profiling of promoter recognition by the two-component response regulator CpxR-P in Escherichia coli. J. Biol. Chem. 277:26652-26661. [DOI] [PubMed] [Google Scholar]
- 15.Dixon, L. G., W. L. Albritton, and P. J. Willson. 1994. An analysis of the complete nucleotide sequence of the Haemophilus ducreyi broad-host-range plasmid pLS88. Plasmid 32:228-232. [DOI] [PubMed] [Google Scholar]
- 16.Elkins, C., K. J. Morrow, and B. Olsen. 2000. Serum resistance in Haemophilus ducreyi requires outer membrane protein DsrA. Infect. Immun. 68:1608-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fellay, R., J. Frey, and H. Krisch. 1987. Interposon mutagenesis of soil and water bacteria: a family of DNA fragments designed for in vitro insertional mutagenesis of Gram-negative bacteria. Gene 52:147-154. [DOI] [PubMed] [Google Scholar]
- 18.Fortney, K. R., R. S. Young, M. E. Bauer, B. P. Katz, A. F. Hood, R. S. Munson, Jr., and S. M. Spinola. 2000. Expression of peptidoglycan-associated lipoprotein is required for virulence in the human model of Haemophilus ducreyi infection. Infect. Immun. 68:6441-6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harrison, A., W. C. Ray, B. D. Baker, D. W. Armbruster, L. O. Bakaletz, and R. S. Munson, Jr. 2007. The OxyR regulon in nontypeable Haemophilus influenzae. J. Bacteriol. 189:1004-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herbert Tran, E. E., and H. Goodrich-Blair. 2009. CpxRA contributes to Xenorhabdus nematophila virulence through regulation of lrhA and modulation of insect immunity. Appl. Environ. Microbiol. 75:3998-4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heusipp, G., K. M. Nelson, M. A. Schmidt, and V. L. Miller. 2004. Regulation of htrA expression in Yersinia enterocolitica. FEMS Microbiol. Lett. 231:227-235. [DOI] [PubMed] [Google Scholar]
- 22.Humphreys, S., G. Rowley, A. Stevenson, M. F. Anjum, M. J. Woodward, S. Gilbert, J. Kormanec, and M. Roberts. 2004. Role of the two-component regulator CpxAR in the virulence of Salmonella enterica serotype typhimurium. Infect. Immun. 72:4654-4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Janowicz, D. M., K. R. Fortney, B. P. Katz, J. L. Latimer, K. Deng, E. J. Hansen, and S. M. Spinola. 2004. Expression of the LspA1 and LspA2 proteins by Haemophilus ducreyi is required for virulence in human volunteers. Infect. Immun. 72:4528-4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janowicz, D. M., S. Ofner, B. P. Katz, and S. M. Spinola. 2009. Experimental infection of human volunteers with Haemophilus ducreyi: 15 years of clinical data and experience. J. Infect. Dis. 199:1671-1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Labandeira-Rey, M., J. R. Mock, and E. J. Hansen. 2009. Regulation of expression of the Haemophilus ducreyi LspB and LspA2 proteins by CpxR. Infect. Immun. 77:3402-3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee, E. C., D. Yu, J. Martinez de Velasco, L. Tessarollo, D. A. Swing, D. L. Court, N. A. Jenkins, and N. G. Copeland. 2001. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73:56-65. [DOI] [PubMed] [Google Scholar]
- 27.Liu, Y., M. W. Taylor, and H. J. Edenberg. 2006. Model-based identification of cis-acting elements from microarray data. Genomics 88:452-461. [DOI] [PubMed] [Google Scholar]
- 28.MacRitchie, D. M., J. D. Ward, A. Z. Nevesinjac, and T. L. Raivio. 2008. Activation of the Cpx envelope stress response downregulates expression of several locus of enterocyte effacement-encoded genes in enteropathogenic Escherichia coli. Infect. Immun. 76:1465-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller, W. G., and S. E. Lindow. 1997. An improved GFP cloning cassette designed for prokaryotic transcriptional fusions. Gene 191:149-153. [DOI] [PubMed] [Google Scholar]
- 30.Pfaffl, M. W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prather, D. T., M. Bains, R. E. W. Handcock, M. J. Filiatrault, and A. A. Campagnari. 2004. Differential expression of porins OmpP2A and OmpP2B of Haemophilus ducreyi. Infect. Immun. 72:6271-6278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raivio, T. L. 2005. Envelope stress responses and Gram-negative bacterial pathogenesis. Mol. Microbiol. 56:1119-1128. [DOI] [PubMed] [Google Scholar]
- 33.Ronnebaumer, K., G. Sander, B. Shutinoski, M. A. Schmidt, and G. Heusipp. 2009. Controlled activation of the Cpx system is essential for growth of Yersinia enterocolitica. FEMS Microbiol. Lett. 296:274-281. [DOI] [PubMed] [Google Scholar]
- 34.Rowley, G., M. Spector, J. Kormanec, and M. Roberts. 2006. Pushing the envelope: extracytoplasmic stress responses in bacterial pathogens. Nat. Rev. Microbiology 4:383-394. [DOI] [PubMed] [Google Scholar]
- 35.Slamti, L., and M. K. Waldor. 2009. Genetic analysis of activation of the Vibrio cholerae Cpx pathway. J. Bacteriol. 191:5044-5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spinola, S. M. 2008. Chancroid and Haemophilus ducreyi, p. 689-699. In K. K. Holmes, P. F. Sparling, W. E. Stamm, P. Piot, J. N. Wasserheit, L. Corey, M. S. Cohen, and D. H. Watts (ed.), Sexually transmitted diseases, 4th ed. McGraw-Hill Book Co., New York, NY.
- 37.Spinola, S. M., K. R. Fortney, B. P. Katz, J. L. Latimer, J. R. Mock, M. Vakevainen, and E. J. Hansen. 2003. Haemophilus ducreyi requires an intact flp gene cluster for virulence in humans. Infect. Immun. 71:7178-7182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tracy, E., F. Ye, B. D. Baker, and R. S. Munson, Jr. 2008. Construction of non-polar mutants in Haemophilus influenzae using FLP recombinase technology. BMC Mol. Biol. 9:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vakevainen, M., S. Greenberg, and E. J. Hansen. 2003. Inhibition of phagocytosis by Haemophilus ducreyi requires expression of the LspA1 and LspA2 proteins. Infect. Immun. 71:5994-6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ward, C. K., J. R. Mock, and E. J. Hansen. 2004. The LspB protein is involved in the secretion of the LspA1 and LspA2 proteins by Haemophilus ducreyi. Infect. Immun. 72:1874-1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wolfe, A. J., N. Parikh, B. P. Lima, and B. Zemaitaitis. 2008. Signal integration by the two-component signal transduction response regulator CpxR. J. Bacteriol. 190:2314-2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao, H., R. B. Thompson, V. Lockatell, D. E. Johnson, and H. L. Mobley. 1998. Use of green fluorescent protein to assess urease gene expression by uropathogenic Proteus mirabilis during experimental ascending urinary tract infection. Infect. Immun. 66:330-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou, L., X. H. Lei, B. R. Bochner, and B. L. Wanner. 2003. Phenotype microarray analysis of Escherichia coli K-12 mutants with deletions of all two-component systems. J. Bacteriol. 185:4956-4972. [DOI] [PMC free article] [PubMed] [Google Scholar]