

Abstract
In shark heart, the Na+–Ca2+ exchanger serves as a major pathway for both Ca2+ influx and efflux, as there is only rudimentary sarcoplasmic reticulum in these hearts. The modulation of the exchanger by a β-adrenergic agonist in whole-cell clamped ventricular myocytes was compared with that of the Na+–Ca2+ exchanger blocker KB-R7943. Application of 5 μM isoproterenol and 10 μM KB-R7943 suppressed both the inward and the outward Na+–Ca2+ exchanger current (INa−Ca). The isoproterenol effect was mimicked by 10 μM forskolin. Isoproterenol and forskolin shifted the reversal potential (Erev) of INa−Ca by approximately −23 mV and −30 mV, respectively. An equivalent suppression of outward INa−Ca by KB-R7943 to that by isoproterenol produced a significantly smaller shift in Erev of about −4 mV. The ratio of inward to outward exchanger currents was also significantly larger in isoproterenol- than in control- and KB-R7943-treated myocytes. Our data suggest that the larger ratio of inward to outward exchanger currents as well as the larger shift in Erev with isoproterenol results from the enhanced efficacy of Ca2+ efflux via the exchanger. The protein kinase A-mediated bimodal regulation of the exchanger in parallel with phosphorylation of the Ca2+ channel and enhancement of its current may have evolved to satisfy the evolutionary needs for accelerated contraction and relaxation in hearts of animals with vestigial sarcoplasmic Ca2+ release stores.
The β-adrenergic agonists are known to potentiate the force of cardiac contraction by enhancing Ca2+ current (ICa) and Ca2+ release from the internal calcium store secondary to cAMP-dependent phosphorylation of the Ca2+ channel (1–3). Relaxant effect of catecholamines (4), on the other hand, appears to be mediated by phosphorylation of phospholamban and troponin C resulting in stimulation of the Ca2+ pump (5, 6) and decreased myofilament Ca2+ sensitivity (7–9). Similar to the mammalian myocardium, the shark heart also exhibits some of the features associated with β-agonist response, namely enhancement of ICa (10) and twitch force, and suppression of maintained contracture tension (11). In light of previous reports showing little ultrastructural and functional evidence for Ca2+ release stores in shark heart (12, 13), the β-adrenergic-induced suppression of KCl-induced contractures (11) may be somewhat surprising. In shark heart the Na+–Ca2+ exchanger appears to serve as a major pathway of both Ca2+ influx and efflux (13). Here we report on a cellular mechanism by which the β-adrenergic agonists suppress the influx and efflux mode of the Na+–Ca2+ exchanger, but enhance the efficacy of Ca2+ efflux (Iinward/Ioutward) via the exchanger. To gain insight into the mechanism by which isoproterenol regulates the Na+–Ca2+ exchanger, we compared the effects of this hormone with that of the exchanger blocker KB-R7943 (14). Our experiments reveal that in shark myocytes this bimodal regulation of the exchanger may decrease the Ca2+ load of the myocytes during experimentally or hormonally induced prolonged membrane depolarizations, resulting in uncoupling of the duration of the action potential from contraction (11).
Materials and Methods
Cell Isolation.
Single ventricular cells were isolated from hearts of dogfish sharks (Squalus acanthias) by using previously described procedures (10). Briefly, male dogfish (2–7 kg) were immobilized by complete spinal pithing. Hearts were removed and mounted in a Langendorff apparatus. The two major coronary vessels and aorta were cannulated and perfused with oxygenated Ca2+-free elasmobranch solution containing (in mM) 270 NaCl, 4 KCl, 3 MgCl2, 0.5 KH2PO4, 0.5 Na2SO4, 350 urea, 10 N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (Hepes), 0.5 glucose, pH 7.2, at 30°C for 10–15 min. The heart was then perfused for 15 min with Ca2+-free elasmobranch solution containing 1 mg/ml collagenase (type A, Boehringer Mannheim) and 0.2 mg/ml protease (type XIV, Sigma), and was washed free of enzyme with 0.2 mM CaCl2-containing elasmobranch solution for 10 min. The ventricle was then cut free of the cannula and gently agitated in 0.2 mM Ca2+-containing solution to disperse the cells. Cell yields varied greatly between 20% and 80% depending on the efficacy of the coronary perfusion.
Experimental Protocol and Data Analysis.
Dissociated myocytes were placed in a chamber on the stage of an inverted microscope and superfused with 2 mM Ca2+-containing elasmobranch solution. Whole-cell currents were measured with a 3- to 5-MΩ pipette attached to the input of a patch clamp amplifier (model 8900, Dagan Instruments, Minneapolis). The pipette solution contained (in mM) 200 KCl, 60 NaCl, 300 urea, 10 Hepes, 5 MgATP, 10 tetraethylammonium chloride, 0.2 EGTA, pH 7.2 with KOH. To record Na+–Ca2+ exchanger current (INa-Ca), the external solutions were supplemented with 10 μM nifedipine to block Ca2+ channels, 0.1 mM BaCl2 to block inwardly rectifying K+ channels, and 5 mM CsCl to block inwardly rectifying and delayed rectifying K+ channels. β-Adrenergic agonist and a the Na+–Ca2+ exchanger blocker 2-{2-[4-(4-nitrobenzyloxy)phenyl]ethyl}isothiourea methanesulfonate (KB-R7943) were added to this solution in appropriate concentrations. NiCl2 was also used in external solution to block INa-Ca. Alternate switching between various external solutions was accomplished rapidly (<50 ms) by using an electronically controlled multibarrel puffing system (15). The time course of hypothetical changes in intracellular Ca2+ concentration ([Ca2+]i) associated with the activity of the exchanger was constructed by digital integration of the traces of INa-Ca envelopes by the equation
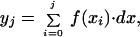 |
1 |
where f(xi) is the Ni2+-sensitive INa-Ca envelope current (pA), and yj (fC) and dx (ms) are digitized quantity of charge and 0.3, respectively. The digitized value provides an estimate of the total charge transferred in a 3 Na+ for 1 Ca2+ exchanger stoichiometry. To measure total rise in cellular Ca2+, cell volume was calculated from the measured cell membrane capacitance of shark ventricular myocytes by assuming 1 for surface-to-volume ratio. Experiments were carried out at room temperature. The data were collected, stored, and analyzed on a personal computer using pCLAMP 5.5.1 (Axon Instruments, Foster City, CA) and origin (Microcal Software, Northampton, MA) software. Data are represented as the mean ± SEM and n is number of cells.
Results
Characterization of Na+–Ca2+ Exchange Current in Shark Ventricular Myocytes.
Fig. 1 A–C illustrates the procedure used to isolate the inward and outward components of current carried by the Na+–Ca2+ exchanger in a single voltage-clamped shark ventricular myocyte in which K+ and Ca2+ current were blocked (see legend). As the duration of clamp pulse was prolonged, the outward current activated at +60 mV slowly decayed, and the tail currents accompanying the repolarization of membrane to −80 mV became enhanced (Fig. 1A). Exposure of myocytes to 5 mM Ni2+ blocked both the slowly decaying outward current and the accompanying inward tail current (Fig. 1B). Subtraction of current envelopes obtained in the presence and absence of Ni2+ revealed a slowly decaying outward current on depolarization and expanding inward tail current on repolarization (Fig. 1C), consistent with previous measurements of INa-Ca (13, 16, 17). The pulse protocol of Fig. 1D employs a procedure that activates INa-Ca at +60 mV, then measures its voltage dependence with a repolarizing ramp pulse (17). The application of 5 mM Ni2+ blocked the outward current induced by depolarization to +60 mV by 56.7% ± 2.9% (n = 12; Table 1). The density of the outward exchanger current at 50 ms into the depolarizing pulse to +60 mV was 5.63 ± 0.8 pA/pF (n = 14), after subtraction of the Ni2+-resistant current. The voltage dependence of INa-Ca was measured by subtraction of currents induced by the ramp protocol in the presence and absence of Ni2+. It should be noted that, because the kinetics of the exchanger current may not be at their steady-state values during the ramp pulse, an accurate estimation of the voltage dependence of INa-Ca may be compromised. In addition, because the inward exchanger currents during the envelope pulse protocol decay by about 60% over 30 ms, it is likely that the 100-ms ramp protocol used might somewhat underestimate the inward exchanger current. Despite these constraints, the reversal potential could be reliably measured with this procedure. The Ni2+-blockable ramp current showed a reversal potential (Erev) of −29.1 ± 5.7 mV (n = 9), providing for a calculated Ca2+ equilibrium potential (ECa) at +60 mV, using the equation
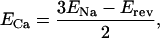 |
2 |
where ENa is 38.6 mV (extracellular Na+ concentration, [Na+]o, = 270 mM and [Na+]i = 60 mM). The calculated ECa for Erev of −29.1 mV was about 72.5 mV. Under these conditions the effective [Ca2+]i calculated from the Nernst equation was 7.1 μM at 2 mM [Ca2+]o at the end of a 50 ms depolarizing pulse. Significant myocyte shortening was noted at these [Ca2+]i values. There was considerable variability (−20 to −50 mV) in the reversal potential of INa-Ca after a 50-ms depolarizing pulse to +60 mV, resulting most likely from the variability in free cytosolic Ca2+ concentrations in different cells.
Figure 1.

Isolation of Na+–Ca2+ exchanger current (INa-Ca) in shark ventricular myocytes. (A) Superimposed membrane currents obtained in external solution containing Ca2+- and K+-channel inhibitors (10 μM nifedipine, 0.1 mM Ba2+, and 5 mM Cs+) in response to voltage-clamp protocol shown at the top. (B) Membrane currents obtained under the same experimental conditions but after external 5 mM Ni2+. (C) Ni2+-sensitive component of the current (INa-Ca) obtained after subtraction of current records shown in B from those in A. (D) Original current traces obtained in the absence and presence of 5 mM Ni2+ in response to ramp voltage-clamp protocol shown above the original current traces. (E) Current density–voltage relation for Ni2+-sensitive INa-Ca constructed from ramp-current records shown in D.
Table 1.
Effect of various agents on outward INa-Ca density and Na+–Ca2+ exchanger reversal potential (Erev) in shark ventricular myocytes
| Agent | Iout, pA/pF | 5 mM Ni2+
|
10 μM
KB-R7943
|
INa-Ca (Iout − Iout(Ni)), pA/pF | ΔINa-Ca, inhibition, % | ΔErev, mV | ||
|---|---|---|---|---|---|---|---|---|
| Iout(Ni), pA/pF | Inhibition, % | Iout(KB-R7943), pA/pF | Inhibition, % | |||||
| Control | 8.86 ± 0.76 (12) | 3.84 ± 1.9 (12) | 56.7 ± 2.9 (12) | 3.38 ± 1.6 (4) | 62.5 ± 6.4 (4) | 5.42 ± 0.42 (12) | ||
| Isoproterenol (5 μM) | 6.94 ± 1.47* (3) | 2.88 ± 0.63 (3) | 57.7 ± 3.5 (3) | 4.06 ± 1.2† (3) | 24.0 ± 7.2 (3) | −22.9 ± 1.8 (3) | ||
| Forskolin (10 μM) | 5.65 ± 0.76* (2) | 2.61 ± 0.22 (2) | 54.1 ± 1.5 (2) | 3.04 ± 0.54† (2) | 44.4 ± 0.95 (2) | −29.6 ± 3.7 (2) | ||
Iout was measured in each cell at 50 ms after the onset of depolarization to +60 mV during ramp protocol (Fig. 1D) and was normalized to the cell capacitance to yield current density (pA/pF). ΔINa-Ca represents a difference between INa-Ca in control and INa-Ca in the presence of isoproterenol or forskolin. ΔErev is the amount of change in Erev of the INa-Ca in the presence of isoproterenol or forskolin compared with that in control. Data are represented as the mean ± SEM (number of cells). *, Significantly different from control Iout at P < 0.05;
, significantly different from control INa-Ca at P < 0.05.
Modulation of Exchanger Activity by β-Adrenergic Signaling Pathway.
Application of 5 μM isoproterenol suppressed the magnitude of Ni2+-sensitive outward INa-Ca) induced by a 50-ms depolarizing pulse from −80 to +60 mV by 24.0% ± 7.2% (n = 3) (Fig. 2 A and B; see Table 1 for details). In the absence or presence of isoproterenol, 5 mM Ni2+ rapidly and reversibly suppressed the outward current. The slow kinetics of the isoproterenol-induced effect on INa-Ca is consistent with the activation of a second messenger-signaling pathway. To examine whether this effect was mediated by the binding of the hormone to the β-adrenoreceptor and the activation of the adenylate-cyclase dependent signaling pathway, adenylate cyclase was directly activated by forskolin. Forskolin mimicked the isoproterenol-suppressive effect on INa-Ca (Fig. 2 C and D), such that the outward INa-Ca generated at +60 mV was suppressed by 44.4% ± 0.95% (n = 2) (see Table 1 for details). Although the absolute outward current after Ni2+ exposure was smaller in the presence of isoproterenol or forskolin than in control myocytes (Fig. 2 A–D), the fraction of current inhibited by Ni2+ in control or isoproterenol-treated myocytes was similar (≈55%, Table 1). Thus, the Ni2+ effect does not appear to be modified by the phosphorylation state of the exchanger protein.
Figure 2.

The effects of isoproterenol (ISO), forskolin (FSK), and KB-R7943 on INa-Ca in shark ventricular myocytes. (A) Ramp voltage-clamp protocol and corresponding original currents obtained in control external solution (trace 1), after application of 5 mM Ni2+ (trace 2), after addition of 5 μM ISO (trace 3), and after treatment of 5 mM Ni2+ in the presence of ISO (trace 4). (B) Time course of changes of the steady-state outward current density at +60 mV in response to voltage-clamp pulses shown in A. (C) Ramp voltage-clamp protocol and corresponding currents obtained in control (trace 1), after 5 mM Ni2+ (trace 2), after addition of 10 μM FSK (trace 3), and after treatment with 5 mM Ni2+ in FSK (trace 4). (D) Time course of changes of the steady-state outward current density at +60 mV shown in C. The effect of KB-R7943 on INa-Ca in shark ventricular myocytes. (E) Ramp voltage-clamp protocol and corresponding currents obtained in control (trace 1), in 5 mM Ni2+ (trace 2), after addition of 10 μM KB-R7943 (trace 3), and after 5 mM Ni2+ in KB-R7943 (trace 4). (F) Time course of changes of the steady-state outward current density at +60 mV shown in E. Pulses were delivered at a frequency of 0.1 Hz. Numbers along the experimental points in B, D, and F mark traces shown in A, C, and E, respectively. Iout was measured at 50 ms after the onset of depolarization to +60 mV and normalized to the cell capacitance to yield current density (pA/pF).
Fig. 3 shows that both isoproterenol and forskolin significantly suppressed the inward and outward INa-Ca at most potentials tested, and shifted its reversal potential toward more negative potentials (Fig. 3 A and B). The changes in Erev (ΔErev, Table 1) in the presence of isoproterenol or forskolin were −22.9 ± 1.8 mV (n = 3) and −29.6 ± 3.7 (n = 2), respectively. These findings suggest that stimulation of the β-adrenergic/adenylate cyclase pathway results in substantial reduction (≈2.5-fold with isoproterenol and ≈3.1-fold with forskolin) in [Ca2+]i in shark ventricular myocytes. The apparent enhancement of INa-Ca in the small range of potentials spanning its reversal potential (Fig. 3 A and B) occurs, in part, from the reduction of [Ca2+]i, as the Ca2+ influx on the exchanger is suppressed at more positive potentials. Consistent with this idea, in myocytes where [Ca2+]i was buffered no apparent enhancement of INa-Ca was observed (17).
Figure 3.

The effects of isoproterenol (ISO), forskolin (FSK), and KB-R7943 on voltage dependence of INa-Ca. (A) Voltage dependence of INa-Ca in control and ISO-containing solutions. (B) Voltage dependence of INa-Ca in control and FSK-containing solutions. (C) Voltage dependence of INa-Ca in control and KB-R7943-containing solutions. Current–voltage relations for control INa-Ca and INa-Ca in the presence of ISO, FSK, or KB-R7943 were constructed from records during the ramp pulse shown at the top of Fig. 2A after subtraction of Ni2+-resistant current.
Effect of KB-R7943 on Na+–Ca2+ Exchanger.
Figs. 2 and 3 also compare the effect of isoproterenol on INa-Ca with that of KB-R7943, a cardiac Na+–Ca2+ exchanger blocker (14). The application of 10 μM KB-R7943 produced gradual suppression of INa-Ca within 3 min of the application of the drug (Fig. 2F). The maximal suppressive effect of KB-R7943 was equivalent to that of 5 mM Ni2+ (Fig. 2E, traces 2 and 4; Table 1). To compare the KB-R7943 and isoproterenol effects on the Erev of INa-Ca, we selected values that produced equivalent (≈20%) inhibitory levels with the two drugs (compare point 3 of Fig. 2F with point 3 of Fig. 2B). The KB-R7943 suppressive effect was accompanied by only a −3.8 ± 0.6 mV (n = 4) shift of INa-Ca reversal potential compared with −22.9 ± 1.8 mV (n = 3) for isoproterenol (Table 1). Thus, although the level of suppression of outward exchanger current was equivalent with the two drugs, the decrease in the [Ca2+]i, as calculated from the magnitude of the shift in the reversal potential, was markedly larger with isoproterenol. This finding implies that isoproterenol may differentially regulate the exchanger as compared with KB-R7943.
Bimodal Regulation of Na+–Ca2+ Exchanger by Isoproterenol.
To quantitate the efficacy of the β-adrenergic regulatory effect on the Ca2+ influx or efflux mode of the exchanger, we used the “envelope” pulse protocol, by which both the inward and outward component of Ca2+ transport on the exchanger could be independently assessed. Fig. 4 illustrates the effect of isoproterenol on the Ni2+-sensitive envelope of the exchanger-activated currents. In the presence of isoproterenol both the inward and outward current components were reduced (Fig. 4B). Clearly, the decreased Ca2+ influx via the Na+–Ca2+ exchanger into the myocyte would by itself reduce the Ca2+ efflux on the exchanger during the repolarization period. Nevertheless, the efficacy of Ca2+ efflux on the exchanger in the presence of isoproterenol appeared to be enhanced when corrected for the reduced Ca2+ influx. This was reflected in the larger ratio of the inward to outward (Iinward/Ioutward) INa-Ca in the isoproterenol-treated myocytes (Fig. 4C). We also measured the magnitude of Ni2+-sensitive outward current of each episode at 25 ms after the onset of depolarization. The amplitude of the peak Ni2+-sensitive inward current was measured at 3 ms after the start of repolarization because the fast capacitive transients may contaminate the ionic current in the early part of the tail currents. In quantifying the Ca2+ influx and efflux modes of the exchanger, we found that as the duration of the depolarizing pulse increased gradually the ratio of Iinward/Ioutward in control and isoproterenol-treated myocytes increased, albeit with different slopes (Fig. 4C). It is likely that the higher [Ca2+]i during the longer depolarizing pulses facilitates the Ca2+ efflux mode of the exchanger, through activation of its Ca2+ regulatory site (18, 19). In isoproterenol-treated myocytes, in fact, the slope [(Iinward/Ioutward)/s] of the relation (reflecting the efficacy of Ca2+ efflux via the exchanger, Fig. 4C) was larger (11.0 ± 1.2, n = 3) than that (7.22 ± 1.0, n = 7) found in control myocytes. Quantification of the total Ni2+-sensitive charge (Q) transported by the exchanger in the inward and outward direction similarly showed that isoproterenol increased the fraction of inward to outward charge (Qinward/Qoutward) by ≈2-fold, Fig. 4D. This finding therefore suggests more efficient Ca2+ extrusion by the exchanger in the presence of isoproterenol.
Figure 4.

The effect of isoproterenol (ISO) on the ratio of the inward to outward INa-Ca. Superimposed traces of INa-Ca in control (A) and in the presence of 5 μM ISO-containing solutions (B) obtained by subtraction of 5 mM Ni2+-resistant component of current. Voltage-clamp pulses from −80 to +60 mV were applied by increasing duration from 25 to 150 ms in 25-ms increments. (C) The relationship between the pulse duration and the ratio (mean ± SEM, n = 7 in control and n = 3 in isoproterenol; different cells from the different animals) of the maximum current amplitude of inward tail current (Iinward) to that of outward current (Ioutward) in control (CON) and in the presence of ISO. (D) The relationship between the pulse duration and the ratio (mean ± SEM, n = 3; different cells from the different animals) of the charge (Q) carried during the tail current to that during the pulse in CON and in the presence of ISO. The amplitude of the peak inward or outward current and the charge were measured at 3 ms after the start of depolarization or repolarization because a fast capacitive current could contaminate the ionic current in the early part of repolarization pulse.
Fig. 5 illustrates the effect of isoproterenol on the time course of rise in [Ca2+]i, constructed from digital integration of INa-Ca traces of Fig. 4 A and B, using a 3 Na+ to 1 Ca2+ exchanger stoichiometry. As the duration of depolarizing pulse became longer both the Ca2+ influx and efflux via the exchanger appeared to increase (Fig. 5A). In the presence of isoproterenol the peak [Ca2+]i was much smaller than that found in control solution (Fig. 5 A and B), consistent with suppression of Ca2+ influx via the exchanger. However, the decay of intracellular Ca2+ transients during repolarization appeared to be faster in isoproterenol-treated myocytes (see normalized traces, Fig. 5C), consistent with idea that isoproterenol also enhances the efficacy of Ca2+ extrusion via the exchanger.
Figure 5.

The effect of isoproterenol (ISO) on intracellular calcium concentration in shark ventricular myocyte. (A) Time courses of calculated rise in total intracellular calcium concentration ([Ca2+]i) constructed from digital integration of the traces of INa-Ca envelopes shown in Fig. 4 A and B to provide and estimate of the total charge transferred, using a 3 Na+ to 1 Ca2+ exchanger stoichiometry. (B) Superimposed digitized calcium changes constructed from the currents during the 150-ms depolarizing pulse in the presence and absence of ISO. (C) Normalized [Ca2+]i constructed from the records shown in B for control (CON) and ISO-exposed myocytes.
Comparing the effect of isoproterenol and KB-R7943 on the envelope of the exchanger currents and their ratios showed that 10 μM KB-R7943 was as effective as Ni2+ in suppressing both the inward and outward INa-Ca (Fig. 6), such that the ratio of Iinward to Ioutward in KB-R7943-treated myocytes was the same as that of control cells (data not shown). These findings suggest that the larger ratio of inward to outward exchanger currents observed in isoproterenol-treated myocytes results from a unique regulatory mechanism of the Ca2+ efflux mode of the exchanger.
Figure 6.

The effect of KB-R7943 on INa-Ca. (A) Superimposed envelope currents obtained in control external solution in response to voltage-clamp protocol shown at the top. (B and C) Membrane currents obtained under the same experimental conditions but after application of external 5 mM Ni2+ and 10 μM KB-R7943, respectively.
Discussion
Our results describe a mechanism by which β-adrenergic agonists enhance the efficacy of Ca2+ efflux even when Ca2+ influx through the Na+–Ca2+ exchanger is reduced. The bimodal regulation of the exchanger in shark ventricular myocytes provides a possible mechanism for the intriguing findings that in intact ventricular strips, β-adrenergic agonists, while potentiating the phasic component of contraction, suppress the maintained component of tension (11). It has been previously suggested (13, 17) that activation of Ca2+ channel produces the initial rapid influx of Ca2+, whereas the later phase of contraction is supported by the slower influx of Ca2+ via the Na+–Ca2+ exchanger. In this scheme the simultaneous adrenergic-induced enhancement of ICa and the suppression of Ca2+ influx on the exchanger may provide for the observed enhancement of the rate of rise of twitch early in the action potential (20) and for the dissociation of contraction from the duration of depolarization later on in the action potential (4, 11). In addition, the adrenergic enhancement of the efficacy of Ca2+ efflux via the Na+–Ca2+ exchanger, described here, may contribute significantly to the decay of intracellular Ca2+ and acceleration of relaxation. Such differential and temporal modulation of Ca2+ influx and efflux may have both developmental and evolutionary significance for hormonal regulation of cellular Ca2+ homeostasis. This mechanism may have particularly evolved in hearts, where sarcoplasmic reticulum (SR) is poorly developed and physiological Ca2+ release pools are absent (13). In this respect, in the amphibian and neonatal mammalian hearts in which SR is also poorly developed, the β-adrenergic agonists appeared to similarly enhance the twitch and suppress KCl-induced contractures (21, 22), consistent with the enhancement of ICa and suppression of Ca2+ influx on the exchanger (17, 23). In frog ventricular myocytes, the ratio of the inward to outward exchanger current did not increase, nor did the slope of the relationship between the ratio and the duration of the depolarization change (17). In guinea-pig ventricular myocytes incubated at 37°C, β-adrenergic agonists were shown to stimulate both the Ca2+ influx and efflux mode of the exchanger via the protein kinase A (PKA)-mediated signaling pathway (24). We have been unable to reproduce these data in rat myocytes at room temperature. Nevertheless, the enhancement of exchanger current in this report was not accompanied by a shift in Erev, as Ca2+ was highly buffered by the dialyzing solution (24).
In the present result we show that β-adrenergic signaling pathway induces a shift of Erev toward more negative potential (Fig. 2 A and B). We have proposed that isoproterenol- and forskolin-induced decrease in [Ca2+]i may be responsible for the negative shift in the Erev, because in highly Ca2+-buffered cells (9 mM [EGTA]i and 6.17 mM CaCl2; free [Ca2+]i = 200 nM) the negative shift in Erev was prevented (17), even though isoproterenol continued to suppress the outward Na+–Ca2+ exchanger current (17). Because Erev may also change in response to the alteration of exchanger stoichiometry (25) we considered whether a change in stoichiometry could be responsible for such a shift. If the stoichiometry for Na+ to Ca2+ increases from 3:1 to 4:1 because of the reduction of [Ca2+]i, the reversal potential would shift toward positive potentials, as predicted theoretically and reported experimentally (25). Therefore, the negative shift in the Erev in the presence of isoproterenol is unlikely to be related to the change in the exchanger stoichiometry.
We also compared the regulation of exchanger by KB-R7943 with that of isoproterenol. KB-R7943 has been reported to block the outward Na+–Ca2+ exchanger current more potently than the inward current under unidirectional ionic conditions, possibly by binding to the Ca2+-binding conformation of the exchanger (26). Under the bidirectional ionic conditions, KB-R7943 has been reported to inhibit the inward and outward exchanger currents with equal potency (27). Fig. 6 shows that 10 μM KB-R7943 blocked both the inward and outward exchanger current as effectively as did Ni2+. However, when the suppressions of outward exchanger current by isoproterenol and KB-R7943 were equivalent, the shift in the Erev was smaller with KB-R7943 compared with isoproterenol, suggesting less reduction in [Ca2+]i with this drug. In addition, the ratio of the inward to outward exchanger current, and the slope of the relation (reflecting the efficacy of Ca2+ efflux via the exchanger, Fig. 4C) was larger in isoproterenol than in control or KB-R7943-treated cells. The data support the idea that suppression of influx mode of the exchanger is not a prerequisite for the enhancement of Ca2+ efflux mode of the exchanger, suggesting that isoproterenol may independently enhance the efficacy of the Ca2+ efflux mode of the exchanger.
The mechanism for the enhancement of the efficacy of Ca2+ efflux by isoproterenol, even at lower [Ca2+]i, is not as yet clear. One possibility to consider is that PKA-induced phosphorylation may increase the affinity of the exchanger for its ionic substrates. An ATP-dependent enhancement of Na+–Ca2+ exchanger by an increase in the affinity of the exchanger for its substrates and the regulatory site has been already reported in the squid giant axon and cardiac myocytes (28–30). The effect of ATP in the squid giant axon requires internal Ca2+, hydrolyzable ATP, and the phosphorylation process, suggesting involvement of a Ca2+-dependent protein kinase (31, 32). The outward exchanger current in CHO cells expressing Na+–Ca2+ exchanger was also stimulated by hyperphosphorylation process, but the mechanism does not appeared to be mediated by protein kinases (33). Although phosphorylation has been implicated previously in adult mammalian exchanger (34) and a possible PKA-dependent phosphorylation site has been identified on the cloned protein (35), experiments on giant excised patches from cardiac myocytes suggest that the ATP effect is mediated by phosphatidylinositol 4,5-bisphosphate (PIP2) generated from phosphatidylinositol (PI) (36). In frog heart, the existence of a nine-amino acid exon, containing a putative nucleotide-binding domain (23) not present in the mammalian heart, appears to be responsible for the cAMP-dependent suppression of the exchanger (17, 23). Whether a similar structural component is present in the shark heart isoform of the exchanger remains to be determined. The functional data, presented here, suggest that the shark exchanger may also contain additional regulatory sites that serve as molecular determinants responsible for the unusual bimodal regulation of the exchanger.
We have also considered other possible mechanisms that, if activated by β-adrenergic pathway, could produce an apparent enhancement of Ca2+ efflux. In mammalian hearts, the PKA-dependent phosphorylation of phospholamban and the subsequent stimulation of the Ca2+ pump (5, 6), in addition to decreased myofilaments Ca2+ sensitivity (7–9), are thought to mediate the relaxant properties of β-adrenergic pathway. Supposing that the acceleration of Ca2+-ATPase in the sarcoplasmic reticulum or plasma membrane reduces [Ca2+]i during relaxation, the level of Ca2+ efflux via the Na+–Ca2+ exchanger might be underestimated because of the decreased [Ca2+]i by such mechanisms. On the other hand, a decrease in myofilament sensitivity to Ca2+ by the PKA pathway might increase [Ca2+]i, thereby accelerating Ca2+ efflux via the exchanger.
The possible bimodal regulation of the exchanger by PKA-mediated phosphorylation in parallel with enhancement of Ca2+ current may allow for the temporal shift in delivery of contractile Ca2+ from a slow (exchanger-mediated) to a faster (Ca2+ channel-mediated) pathway. β-Adrenergic-induced suppression of the Ca2+ influx and enhancement of the extrusion mode of the exchanger, in shark heart lacking significant intracellular Ca2+ release pools, may have evolved to meet the evolutionary requirement for faster contraction and accelerated relaxation in animals subjected to fight, fright, and flight. Whether such a PKA-dependent-regulatory mechanism can be made to operate by genetic manipulation of the mammalian Na+–Ca2+ exchanger when the exchanger is overexpressed (37, 38) in heart failure remains to be tested.
Acknowledgments
We thank Dr. L. Cleemann for helpful discussion. We also thank Dr. J. R. Ahn for providing the program for the rapid digital integration and Ana Dorfman for technical assistance. This work was supported by Grant HL-16152 from the National Institutes of Health.
Abbreviations
- [Ca2+]i
intracellular Ca2+ concentration
- PKA
protein kinase A
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.041327398.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.041327398
References
- 1.Reuter H. Nature (London) 1983;301:569–574. doi: 10.1038/301569a0. [DOI] [PubMed] [Google Scholar]
- 2.Kameyama M, Hofmann F, Trautwein W. Pflügers Arch. 1985;405:285–293. doi: 10.1007/BF00582573. [DOI] [PubMed] [Google Scholar]
- 3.Spurgeon H A, Stern M D, Baartz G, Raffaeli S, Hansford R G, Talo A, Lakatta E G, Capogrossi M C. Am J Physiol. 1990;258:H574–H586. doi: 10.1152/ajpheart.1990.258.2.H574. [DOI] [PubMed] [Google Scholar]
- 4.Kavaler F, Morad M. Circ Res. 1966;18:492–501. doi: 10.1161/01.res.18.5.492. [DOI] [PubMed] [Google Scholar]
- 5.Lindemann J P, Jones L R, Hathaway D R, Henry B G, Watanabe A M. J Biol Chem. 1983;258:464–471. [PubMed] [Google Scholar]
- 6.Wegener A D, Simmerman H K B, Lindemann J P, Jones L R. J Biol Chem. 1989;264:11468–11474. [PubMed] [Google Scholar]
- 7.McClellan G B, Winegrad S. J Gen Physiol. 1978;72:737–764. doi: 10.1085/jgp.72.6.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Endoh M, Blinks J R. Circ Res. 1988;62:247–265. doi: 10.1161/01.res.62.2.247. [DOI] [PubMed] [Google Scholar]
- 9.McIvor M E, Orchard C H, Lakatta E G. J Gen Physiol. 1988;92:509–529. doi: 10.1085/jgp.92.4.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maylie J, Morad M. Am J Physiol. 1995;269:H1695–H1703. doi: 10.1152/ajpheart.1995.269.5.H1695. [DOI] [PubMed] [Google Scholar]
- 11.Maylie J, Morad M. MDIBL Bull. 1979;19:87. [Google Scholar]
- 12.Maylie J, Nunzi M G, Morad M. MDIBL Bull. 1979;19:84–87. [Google Scholar]
- 13.Näbauer M, Morad M. J Physiol. 1992;457:627–637. doi: 10.1113/jphysiol.1992.sp019398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iwamoto T, Watano T, Shigekawa M. J Biol Chem. 1996;271:22391–22397. doi: 10.1074/jbc.271.37.22391. [DOI] [PubMed] [Google Scholar]
- 15.Cleemann L, Morad M. J Physiol. 1991;432:283–312. doi: 10.1113/jphysiol.1991.sp018385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beuckelmann D J, Wier W G. J Physiol. 1989;414:499–520. doi: 10.1113/jphysiol.1989.sp017700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fan J, Shuba Y M, Morad M. Proc Natl Acad Sci USA. 1996;93:5527–5532. doi: 10.1073/pnas.93.11.5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levitsky D O, Nicoll D A, Philipson K D. J Biol Chem. 1994;269:22847–22852. [PubMed] [Google Scholar]
- 19.Matsuoka S, Nicoll D A, Hryshko L V, Levitsky D O, Weiss J N, Philipson K D. J Gen Physiol. 1995;105:403–420. doi: 10.1085/jgp.105.3.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morad M, Weiss J, Cleemann L. Eur J Cardiol. 1978;7:53–62. [PubMed] [Google Scholar]
- 21.Morad M. Science. 1969;166:505–506. doi: 10.1126/science.166.3904.505. [DOI] [PubMed] [Google Scholar]
- 22.Fabiato A, Fabiato F. Ann NY Acad Sci. 1978;307:491–522. doi: 10.1111/j.1749-6632.1978.tb41979.x. [DOI] [PubMed] [Google Scholar]
- 23.Shuba Y M, Iwata T, Naidenov V G, Oz M, Sandberg K, Kraev A, Carafoli E, Morad M. J Biol Chem. 1998;273:18819–18825. doi: 10.1074/jbc.273.30.18819. [DOI] [PubMed] [Google Scholar]
- 24.Perchenet L, Hinde A K, Patel K C R, Hancox J C, Levi A J. Pflügers Arch. 2000;439:822–828. doi: 10.1007/s004249900218. [DOI] [PubMed] [Google Scholar]
- 25.Fujioka Y, Komeda M, Matsuoka S. J Physiol. 2000;523:339–351. doi: 10.1111/j.1469-7793.2000.t01-2-00339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watano T, Kimura J. Br J Pharmacol. 1996;119:555–563. doi: 10.1111/j.1476-5381.1996.tb15708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kimura J, Watano T, Kawahara M, Sakai E, Yatabe J. Br J Pharmacol. 1999;128:969–974. doi: 10.1038/sj.bjp.0702869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Philipson K D, Nicoll D A. Int Rev Cytol. 1993;137C:199–227. [PubMed] [Google Scholar]
- 29.Reeves J P, Condrescu M, Chernaya G, Gardner J P. J Exp Biol. 1994;196:375–388. doi: 10.1242/jeb.196.1.375. [DOI] [PubMed] [Google Scholar]
- 30.Haworth R A, Goknur A B. Circ Res. 1992;71:210–217. doi: 10.1161/01.res.71.1.210. [DOI] [PubMed] [Google Scholar]
- 31.DiPolo R, Beaugé L. Biochim Biophys Acta. 1986;897:347–353. doi: 10.1016/0005-2736(87)90432-9. [DOI] [PubMed] [Google Scholar]
- 32.DiPolo R, Beaugé L. J Physiol. 1993;462:71–86. doi: 10.1113/jphysiol.1993.sp019544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Condrescu M, Hantash B M, Fang Y, Reeves J P. J Biol Chem. 1999;274:33279–33286. doi: 10.1074/jbc.274.47.33279. [DOI] [PubMed] [Google Scholar]
- 34.Caroni P, Carafoli E. Eur J Biochem. 1983;132:451–460. doi: 10.1111/j.1432-1033.1983.tb07383.x. [DOI] [PubMed] [Google Scholar]
- 35.Nicoll D, Bongoni S, Philipson K D. Science. 1990;250:562–565. doi: 10.1126/science.1700476. [DOI] [PubMed] [Google Scholar]
- 36.Hilgemann D W, Ball R. Science. 1996;273:956–959. doi: 10.1126/science.273.5277.956. [DOI] [PubMed] [Google Scholar]
- 37.Hatem S N, Sham J S K, Morad M. Circ Res. 1994;74:253–261. doi: 10.1161/01.res.74.2.253. [DOI] [PubMed] [Google Scholar]
- 38.Flesch M, Schwinger R H G, Schiffer F, Frank K, Sudkamp M, Kuhn-Regnier F, Arnold G, Bohm M. Circulation. 1996;94:992–1002. doi: 10.1161/01.cir.94.5.992. [DOI] [PubMed] [Google Scholar]