

Abstract
Some Gram-negative bacteria express a novel enzyme with lysine-ɛ-oxidase (LOD) activity (EC 1.4.3.20). The oxidation of l-Lys generates, among other products, hydrogen peroxide, which confers antimicrobial properties to this kind of enzyme and has been shown to be involved in cell death during biofilm development and differentiation. In addition to LOD, the melanogenic marine bacterium Marinomonas mediterranea, which forms part of the microbiota of the marine plant Posidonia oceanica, expresses two other oxidases of biotechnological interest, a multicopper oxidase, PpoA, with laccase activity and a tyrosinase named PpoB, which is responsible for melanin synthesis. By using both lacZ fusions with the lodAB promoter and quantitative reverse transcription-PCR (qRT-PCR), this study shows that the hybrid sensor histidine kinase PpoS regulates LOD activity at the transcriptional level. Although PpoS also regulates PpoA and PpoB, in this case, the regulatory effect cannot be attributed only to a transcriptional regulation. Further studies indicate that LOD activity is induced at the posttranscriptional level by l-Lys as well as by two structurally similar compounds, l-Arg and meso-2,6-diaminopimelic acid (DAP), neither of which is a substrate of the enzyme. The inducing effect of these compounds is specific for LOD activity since PpoA and PpoB are not affected by them. This study offers, for the first time, insights into the mechanisms regulating the synthesis of the antimicrobial protein lysine-ɛ-oxidase in M. mediterranea, which could be important in the microbial colonization of the seagrass P. oceanica.
l-Amino acid oxidases (LAOs) are enzymes able to catalyze the oxidative deamination of amino acids, generating a keto acid, ammonia, and hydrogen peroxide. The generation of the last compound confers antimicrobial and antitumoral properties to these enzymes (3, 20), which are also of considerable biotechnological interest in processes such as the design of biosensors (1, 43) and biotransformations (41).
LAOs have been described for different organisms throughout the phylogenetic scale from bacteria to animals (19). Regarding microbial enzymes, it has been proposed that fungal LAOs are involved in the utilization of amino acids as nitrogen source (6, 37). The physiological roles of LAOs in bacteria are little known, although different functions have been suggested, bearing in mind the different final products of the reaction. For example, it has been proposed that the alpha-keto acids generated after the deamination of amino acids can act as siderophores in some bacterial groups (10). By analogy with the physiological role of LAOs in eukaryotic microorganisms, it has been proposed that bacterial LAOs might also be involved in nitrogen and/or amino acid metabolism, although definitive evidence is lacking (13). Finally, it was recently described that an LAO is used by the lactic acid bacterium Streptococcus oligofermentans to generate hydrogen peroxide, which is involved in the process of microbial competition with the cariogenic Streptococcus mutans (42).
The melanogenic marine bacterium Marinomonas mediterranea is a component of the microbiota of the marine plant Posidonia oceanica, which is of great importance in Mediterranean Sea ecosystems (12). It synthesizes an antimicrobial protein that was initially named marinocine and was later named LodA (24, 25). LodA is a novel enzyme (EC 1.4.3.20) very specific for l-Lys that catalyzes the reaction l-lysine + O2 + H2O→2-aminoadipate 6-semialdehyde + NH3 + H2O2.
Accordingly, the reaction catalyzed by LodA is an amino acid oxidation of l-Lys in the epsilon position (15), unlike the oxidation in the alpha position catalyzed by the l-lysine-α-oxidase synthesized by Trichoderma viride (20). The gene lodA, encoding the lysine oxidase, forms part of an operon with a second gene, named lodB, which encodes a protein required for the expression of lysine-ɛ-oxidase (LOD) activity (16).
Orthologues of LodA have been detected in a group of proteobacteria (25). For instance, in Pseudoalteromonas tunicata, the orthologue was named AlpP due to its autolytic properties (18). In microorganisms expressing orthologues of LodA, these proteins are involved in the process of biofilm development and differentiation. The hydrogen peroxide generated by LOD activity is responsible for cell death in the center of microcolonies, a process that facilitates the subsequent differentiation and dispersal of cells from the biofilm (27). Despite the importance of LOD activity in biofilm formation and its potential biotechnological interest, very little is known about the mechanisms regulating its expression in bacteria. In P. tunicata, AlpP is regulated by a homologue of ToxR from Vibrio species and CadC from Escherichia coli, named WmpR (white mutant phenotype regulator). The name of this regulator is based on the fact that, in addition to regulating the synthesis of AlpP and other bioactive molecules, a mutation in this gene abolishes pigment synthesis in P. tunicata (11).
In addition to LOD, M. mediterranea expresses two other oxidases, more specifically polyphenol oxidases (PPOs), of biotechnological interest. The first PPO, PpoA, is a multicopper oxidase showing laccase activity, whose physiological role is unknown (35, 39). The second PPO, PpoB, is a tyrosinase, which shows tyrosine hydroxylase (TH) and 3,4-dihydroxyphenylalanine (DOPA) oxidase (DO) activities, which are involved in melanin pigment synthesis from l-tyrosine (23). Regarding the regulation of the oxidase activities in M. mediterranea, it was previously reported that M. mediterranea T103, which is mutated in PpoS, a hybrid sensor histidine kinase, does not show LOD activity, although the mechanisms involved in the process were not described (25). Strain T103 also shows a decrease in PPO activities and a nonpigmented phenotype (26), in an intriguing similarity with the phenotype of the WmpR mutant of P. tunicata (11).
The aim of this study was to gain insight into the mechanisms regulating LOD activity in M. mediterranea compared with PPO regulation. The results could offer clues on the physiological function of the enzymes in the context of the natural environment of this bacterium.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. Marinomonas mediterranea (MMB-1R) and mutant strains derived from it were usually grown in marine broth and marine agar 2216 (Pronadisa). Another complex medium used was MMC (14). In some experiments the chemically defined medium MNG, MNGL, or MNL was used. MNG is a modification of MN (17), in which the 11.8 mM glutamate used as a carbon and nitrogen source is replaced by 30 mM glucose and 3 mM glutamate. MNGL is MNG plus 3 mM l-Lys. Instead of the glutamate found in MN, MNL contains 30 mM glucose as a carbon source and 3 mM l-Lys as a nitrogen source. When indicated, other amino acids or nitrogenated compounds were added to MNG. Marinomonas strains were incubated in liquid medium at 25°C and 130 rpm. Escherichia coli strains were grown in Luria-Bertani (LB) medium at 37°C. When required, the media were supplemented with an appropriate antibiotic.
TABLE 1.
Strains, plasmids, transposons, and primers used in this study
| Strain, plasmid, or primer | Description and/or relevant genotype or primera | Reference or source |
|---|---|---|
| Strains | ||
| M. mediterranea | ||
| MMB-1R | Wild type; spontaneously Rifr | 39 |
| T103 | MMB-1R ppoS::Tn10 Kmr | 26 |
| MMB1LAC0 | MMB-1R Ω mini-Tn10 Gmr (trp′-′lacZ) | 26 |
| T103LAC0 | T103 Ω mini-Tn10 Gmr (trp′-′lacZ) | 26 |
| MMB1LACL1 | MMB-1R Ω mini-Tn10 Gmr φ[PlodAB(252 bp)-lacZ] | This study |
| MMB1LACL2 | MMB-1R, Ω mini-Tn10 Gmr φ[PlodAB(115 bp)-lacZ] | This study |
| T103LACL1 | T103 Ω mini-Tn10 Gmr φ[PlodAB(252 bp)-lacZ] | This study |
| LD (lodAB mutant) | MMB-1R ΔlodAB | P. Lucas-Elío et al., unpublished results |
| LDAB | LD Ω mini-Tn10 Gmr (lodAB) | 16 |
| LDAHa/B-1 | LDB Ω mini-Tn10 Kmr (lodA-Ha-Ha-His6) | 16 |
| E. coli S17-1(λpir) | Kmr::Tn7 Tpr SmrrecA ths hsdRM+; λpir phage lysogen RP4::Mu::Km Tn7 | 8 |
| Plasmids | ||
| pBFU8 | pBSL 182 mini-Tn10 Gmr φ(PppoA-lacZ); delivery vector | 26 |
| pBPlod1 | pBSL182 mini-Tn10 Gmr φ[PlodAB(252 bp)-lacZ]; delivery vector | This study |
| pBPlod2 | pBSL182 mini-Tn10 Gmr φ[PlodAB(115 bp)-lacZ]; delivery vector | This study |
| Primers | ||
| MAREV3 | 5′-GCCCATAAAGAAGGTTGCC-3′ | |
| AAP | 5′-GGCCACGCGTCGACTAGTACGGGIIGGGIIGGGIIG-3′ | Invitrogen |
| MAREV4 | 5′-CACCCAATACAATAAGGCG-3′ | |
| AUAP | 5′-GGCCACGCGTCGACTAGTAC-3′ | Invitrogen |
| MAREVsal | 5′-CACTTGTCCCTGTCGACG-3′ | |
| MARDIReco3 | 5′-GGTTGAATTCTGTTATTGAGACGGTC-3′ | |
| MARDIReco4 | 5′-CGTTGGGGAATTCAGAGAAACAGCCTC-3′ | |
| MAREV3bam | 5′-TGTTGATAACTAGGATCCATAAACCGTT-3′ | |
| GmREV2 | 5′-GGACCAGTTGCGTGAGCG-3′ | |
| LacZREV | 5′-GGTTTTCCCAGTCACGACG-3′ |
For the sequence of primer AAP, “I” refers to deoxyinosine. Engineered restriction sites are underlined.
Identification of the transcriptional start site for lodAB by 5′ rapid amplification of cDNA ends (5′-RACE).
To obtain total RNA samples from M. mediterranea strains, they were cultured in MN, and 1 ml was harvested early in the stationary phase (optical density at 600 nm [OD600] of ∼0.95). The culture was immediately stabilized with RNAprotect bacterial reagent (Qiagen), according to the manufacturer's instructions, by brief vortexing, followed by incubation for 5 min at room temperature and centrifugation for 10 min at 5,000 × g. The pellet was resuspended in 0.5 ml of lysozyme (1 mg/ml) and incubated for 5 min at room temperature prior to RNA extraction using the RNeasy Midi kit (Qiagen). In order to remove DNA contamination from the samples, the preparations were treated for 2 h at 37°C with 2 units of RNase-free DNase (Fermentas), followed by incubation at the same temperature for 90 min with 2.4 units of proteinase K (Fermentas). DNA was partitioned into the organic phase by three consecutive acid phenol-chloroform extractions followed by ether extraction. RNA was finally recovered by ethanol precipitation. RNA quantification was performed spectrophotometrically at 260 nm, and RNA purity was checked by using the ratio of A260/A280 (34). The RNA was stored at −80°C before analysis.
Approximately 2.8 μg of total RNA was reverse transcribed using SuperScript II RNase H reverse transcriptase (Invitrogen, CA) according to the manufacturer's protocol. Briefly, the RNA solution containing 0.5 μM primer MAREV4 and 0.5 mM deoxynucleoside triphosphate (dNTP) mix was incubated at 65°C for 5 min and then quickly chilled on ice. The reverse transcription was performed in the supplier's buffer with 10 mM dithiothreitol (DTT), 1 μl of RNase Out reagent (Invitrogen), and 200 U of SuperScript II reverse transcriptase at 42°C for 50 min followed by heat inactivation at 70°C for 15 min. After this reaction, 2 units of RNase H was added, and the samples were incubated for 20 min at 37°C.
Next, about half of the cDNA obtained from the previous reaction was tailed with dCTP and 30 units of terminal deoxynucleotidyl transferase (Fermentas), according to the instructions of the manufacturer. The PCR amplification of the dC-tailed cDNA was performed as described for the 5′-RACE system for rapid amplification of cDNA ends (Invitrogen Life Technologies) using primers MAREV3 and AAP (abridged anchor primer) in the first round and MAREVsal and AUAP (abridged universal amplification primer) for the second round of PCRs (Table 1). The transcription initiation site for the lodAB operon was determined by direct sequencing, using primer MAREVsal, of the amplification product by the Centro de Apoyo a la Investigación y Desarrollo (CAID), Research and Development Support Center, University of Murcia.
Single-copy transcriptional fusion construction.
Plasmids pBPlod1 and pBPlod2, containing a transcriptional fusion between a fragment of the PlodAB promoter of 252 bp (from positions −225 to +27) and 115 bp (from position −88 to +27), respectively, and the lacZ gene were created by using pBFU8, a plasmid containing a transcriptional fusion between PppoA and the lacZ gene inserted into a mini-Tn10 transposon (26), as a backbone. The PCR products carrying the fragments comprising the promoter region were generated by the amplification of M. mediterranea genomic DNA with primers MARDIReco3 and MAREV3bam to create pBPlod1 and with primers MARDIReco4 and MAREV3bam to create pBPlod2 (Table 1). These products were digested with EcoRI and BamHI, cutting the restriction sites introduced into the primers, and cloned between the corresponding sites of pBFU8, thus replacing the PppoA promoter region in this vector and generating a transcriptional fusion between PlodAB and the lacZ gene. Plasmid pBGAL, containing a promoterless lacZ gene, was used as a control (26). The transposons were transferred from E. coli S17-1 (λpir) to M. mediterranea MMB-1R and T103 by transposon mutagenesis as previously described (39). Several mutant strains, Rifr, Gmr, and Amps strains, with a single transposon insertion were selected, and their growths and β-galactosidase activities were compared to select a representative strain.
Cell extract and extracellular fraction preparation.
Cultures of M. mediterranea strains were harvested at different times by centrifugation at 13,000 × g for 5 min, and the supernatant obtained was considered the extracellular fraction. To obtain cell extracts, the pellet was resuspended in the same volume of 0.1 M phosphate buffer (pH 7), disrupted by sonication as previously described (38), and centrifuged at 13,000 × g for 5 min, discarding the remaining cell debris.
Lysine oxidase activity.
To estimate the LOD activity of LodA in the supernatant of the cultures, a fluorimetric assay with Amplex Red reagent (Molecular Probes) was used (15). The assay mixture (100 μl) contained 2 mM l-Lys in a solution containing 0.05 M phosphate buffer (pH 7.4), 0.05 mM Amplex Red, 0.1 U/ml of horseradish peroxidase (HRP), and 10 μl of the supernatant of the culture. Reactions were carried out for 15 min in 96-well enzyme-linked immunosorbent assay (ELISA) plates. Amplex Red oxidation was monitored with a Fluostar Optima microplate reader (BMG Labtech) using an excitation filter of 550 nm and an emission filter of 590 nm. Background fluorescence due to the slow spontaneous oxidation of the fluorochrome in the absence of l-Lys was subtracted. For each sample, all measurements were made in duplicate. Specific activities were normalized according to the optical density of the culture and expressed as relative fluorescent units (RFU)·min−1·OD600−1·ml−1.
Polyphenol oxidase activity.
PPO activities were determined in 96-well plates at a final volume of 250 μl. Total cell extracts were prepared as previously described (38). l-Tyrosine and l-DOPA oxidase activities (TH and DO, respectively) were determined by monitoring the oxidation of 2 mM l-tyrosine or l-DOPA at 475 nm in 0.1 M phosphate buffer (pH 5.0). For TH activity, 25 μM l-DOPA was added to the assay mixture to eliminate the lag period. When required, these activities were also assayed in the presence of 0.02% SDS (THSDS and DOSDS, respectively) (38). Dimethoxyphenol oxidase (DMPO) activity, generated by the laccase, was determined by monitoring the oxidation of 2 mM dimethoxyphenol (DMP) at 468 nm in 0.1 M phosphate buffer (pH 5.0). The reactions were carried out for 15 min. In all cases, 1 unit was defined as the amount of enzyme that catalyzes the transformation of 1 micromole of substrate per minute at 37°C. Specific activities were normalized according to the milligrams of protein present in each sample, which were measured by using the QuantiPro bicinchoninic assay kit (Sigma).
β-Galactosidase activity.
The permeabilization of cells and β-galactosidase measurements were performed according to the protocol described previously by Miller (28). In addition, an approach that allowed the continuous measurement of the A420 in microwell plates was also used. For this method, cultures were centrifuged at 13,000 × g for 2 min and resuspended in the same volume of buffer Z (28). Permeabilization of the cells was achieved by treatment with 4% chloroform and 0.002% SDS. Next, 60 μl of permeabilized cells was mixed with 140 μl of buffer Z and 50 μl of 4 mg/ml o-nitrophenyl-β-d-galactopyranoside (ONPG). Depending on the activity of the samples, variable amounts of permeabilized cells and buffer Z were added. The increase in the A420 was recorded with a Thermo Multiskan spectrum spectrophotometer measuring 60 cycles of 30 s. The increase in absorbance registered was corrected to compensate for the differences in absorbances of the o-nitrophenol in buffer Z before and after the addition of Na2CO3 according to the Miller method. The values obtained by using both methods were similar.
Polyacrylamide gel electrophoresis.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was carried out under denaturing conditions by using a method described previously by Laemmli (21), with 8% acrylamide in the separating gel and 3% acrylamide in the stacking gel. The running buffer contained 0.3% Tris, 1.44% glycine, and 0.1% SDS (pH 8.3). The samples were mixed in a 2:1 (vol/vol) ratio with loading buffer (3 M 2-mercaptoethanol, 0.18 M Tris-HCl [pH 6.8], 15% glycerol, 0.075% bromophenol blue, and 9% SDS) and heated at 90°C for 10 min. Gels were stained by using a silver stain kit (Bio-Rad Laboratories) and fixed, keeping the gels in 5% (vol/vol) acetic acid for 15 min.
Western blot and densitometric analysis.
Cellular extracts of strain LDAHa/B-1, which expresses LodA (with a hemagglutinin [HA] epitope and polyhistidine fused to its C terminus) (16), were separated by SDS-PAGE and transferred onto an Immobilon-P transfer membrane (Millipore) using a Trans-Blot SD semidry transfer system (Bio-Rad) at a constant current of 22 V. After the transfer, the membrane was blocked with PBST buffer (0.5% milk powder in 1 mM KH2PO4, 10 mM Na2HPO4, 137 mM NaCl, 2.7 mM KCl, and 0.1% [vol/vol] Tween 20 [pH 7.4]) for 1 h at room temperature. Next, the membrane was incubated with the primary antibody (HA-probe, 1:200) for 120 min at room temperature and washed three times for 10 min each with PBST buffer. Subsequently, the membrane was incubated with an HRP-conjugated secondary antibody (goat anti-mouse IgG-HRP, 1:2,000) for 60 min at room temperature. The membrane was then washed and developed by chemiluminescence using an ECL kit (GE Healthcare) and exposure to Amersham ECL Hyperfilms. Densitometric analysis of the films was performed by using ImageJ software.
Analysis of gene expression by quantitative reverse transcription (RT)-PCR (qRT-PCR).
mRNA Levels from several genes, lodA, ppoA, and ppoB, were determined for the M. mediterranea wild-type strain and the T103 mutant. Both strains were cultured in MNG and MNGL until the stationary phase was reached (OD600 of ∼0.9). Two independent cultures were set for each condition and treated separately to provide the statistics of the results. RNA was isolated as described above for the 5′-RACE technique. First-strand cDNA was synthesized from 500 ng of RNA using random primers and the QuantiTect reverse transcription kit (Qiagen) according to the manufacturer's instructions. This step was performed with a CT-320 thermal cycler (Techne). Control reactions without reverse transcriptase were added to confirm the absence of contaminating DNA in the RNA samples. None of these controls gave a fluorescent signal with real-time PCR, confirming that the DNase treatment had removed all genomic DNA.
The PCRs were run on a 7500 real-time PCR system (Applied Biosystems). The PCR of each sample was set in duplicate for each of two independent cultures. These reaction mixtures contained the primers at 0.05 μM, SYBR green PCR master mix (Applied Biosystems) (half the total volume), and different dilutions of the cDNA (see below). The pairs of primers used for the amplification of each gene (Table 2) were designed by using Primer Express software v. 2.0 (Applied Biosystems). The PCR amplification protocol started with an initial denaturation step at 95°C for 10 min, followed by 40 cycles of denaturing at 95°C for 15 s and annealing/elongation at 60°C for 1 min. Immediately after the final PCR cycle, a melting-curve analysis was made to determine the specificity of the reaction by observing the melting temperature of the product. The cycle threshold (CT) for each PCR was determined by use of the 7500 system SDS v 1.4 software (Applied Biosystems), which automatically set the threshold signal at the log phase of the amplification curve. Several dilutions of each cDNA sample were assayed for every gene of interest in order to obtain a linear regression between the CT values (ranging from 20 to 30 cycles) and the log10 of the cDNA concentration. The amplification efficiency was calculated for each target gene from the slope of that linear regression according to the formula E = 10(−1/slope).
TABLE 2.
Genes amplified and primers used for qRT-PCR
| Gene (GenBank accession no.) | Primer | Sequence | Length of PCR product (bp) |
|---|---|---|---|
| lodAB (AY968053) | LODADIR1 | 5′-TGGAGTTTACAGAACCCCGTGA-3′ | 101 |
| LODAREV1 | 5′-TCACGTTCAGGTGTAAGCCCA-3′ | ||
| ppoA (AF184209) | PPOADIR1 | 5′-TAGTTGGCCCAACCTTGAAGC-3′ | 105 |
| PPOAREV1 | 5′-CAGAGCACAAAAAGTTTGGCG-3′ | ||
| ppoB (AY052787) | PPOBDIR1 | 5′-AATAATCCGGCTCTCAACCCC-3′ | 108 |
| PPOBREV1 | 5′-TTCCTTCATCCATAGCCCTGG-3′ | ||
| 16S rRNA (NR_024908) | 16SRNADIR1 | 5′-CTCCATGAAGTCGGAATCGCT-3′ | 103 |
| 16SRNAREV1 | 5′-GCAATCAACTCCCATGGTGTG-3′ |
The housekeeping 16S rRNA gene was used as the control to verify that there were equal amounts of target cDNA in all samples. In order to validate the expression stability of 16S rRNA as a reference gene for RT-PCR, the regression curve representing the CT values against the dilution of the cDNA sample was calculated for all the different media and M. mediterranea strains used. In all cases, cDNA samples had to be diluted to 10−3 in order to obtain CT values with a maximum range of 22 to 25, a small variation taking into account the number of steps involved in the process. Thus, the CT values can be considered similar regardless of the medium or strain used, indicating that 16S RNA was expressed at an identical level in all the samples and thus could be used as an appropriate reference gene. The relative expression (RE) of the target genes lodAB, ppoA, and ppoB compared to that of the reference 16S rRNA gene was calculated by using the E values and the crossing-point deviation (ΔCT) for each gene, medium, and strain by using the following equation (32):
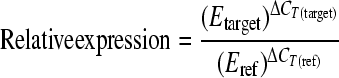 |
where ΔCT is the difference between the cycle threshold for the reference or target gene under the assay conditions (MNGL medium) compared with that under control conditions (MNG medium).
RESULTS
Identification of the transcriptional start site of lodAB and construction of a transcriptional fusion between PlodAB and lacZ.
To study the transcriptional regulation of the lodAB operon coding for LOD activity (16), its transcriptional start was first determined by 5′-RACE. It was identified at a cytosine located 54 bp upstream from the start codon of lodA. The putative −10 region is TTAACT, and the −35 region is TGATAC (see Fig. S1 in the supplemental material). The three oxidase activities so far studied for M. mediterranea, LOD, laccase (encoded by ppoA), and tyrosinase (encoded by ppoB), show some common features, such as growth phase regulation (16, 26). Another common feature is that strain T103, which is mutated in the hybrid sensor histidine kinase PpoS, shows low levels of LOD activity and of the laccase and the tyrosinase (25, 26). Accordingly, in order to detect possible common regulatory elements, the promoters of the different M. mediterranea genes encoding oxidase activities were compared (Fig. S1). A substantial degree of similarity was seen between the promoters of ppoA and ppoB and between them and the sigma 70 housekeeping promoters in the −35 (TTGXXX) and −10 (TATXXT) hexamers. On the contrary, there was little sequence similarity between the lodAB promoter and the other promoters.
Next, a transcriptional fusion between the promoter of the lodAB operon and the lacZ gene was constructed. Two versions of PlodAB differing in length, as indicated in Fig. S1 in the supplemental material, were amplified by PCR, cloned upstream of a promoterless lacZ gene, and later mobilized to M. mediterranea. No differences in transcriptional activity were observed between the two versions of the promoter in the media assayed, MNG and MNGL (data not shown). Given these results, in the following experiments, only strain MMB1LACL1, which contained the longer version of the promoter from nucleotides −225 to +27, was used.
PpoS is involved in the transcriptional regulation of LOD activity.
Several experiments were performed to study the effect of PpoS on the transcriptional regulation of lodAB in M. mediterranea. First, the levels of LOD activity were compared in the wild-type and T103 (PpoS−) strains grown in MNGL medium. For the wild-type strain, LOD activity was maximal in this medium, while strain T103 showed no detectable level of LOD activity under the same conditions (data not shown). Next, the transcriptional fusion of PlodAB with lacZ was mobilized into strain T103, generating strain T103LACL1. In this strain the transcription from PlodAB in MNGL medium was clearly repressed, suggesting that PpoS is required for the transcriptional expression of lodAB (Fig. 1 A). In order to confirm this result, qRT-PCR was used to compare mRNA levels of the M. mediterranea genes encoding oxidase activities known to be downregulated in the strain mutated in PpoS. It was observed that the mutation in PpoS affected the genes studied in different ways. While ppoB, which encodes the tyrosinase, was not affected and ppoA, encoding the laccase, was only partially affected, the transcriptional activity was completely repressed in the case of lodA (Fig. 1B), in agreement with the results obtained with the PlodAB-lacZ fusions.
FIG. 1.

Comparative analysis of lodAB transcriptional levels between the wild-type strain and the ppoS mutant strain. (A) Expression of a PlodAB::lacZ transcriptional fusion. β-Galactosidase activities of strains MMB1LACL1 (squares) and T103LACL1 (PpoS−) (triangles) are represented by continuous lines. Growth is indicated by discontinuous lines. The β-galactosidase activity of control strains MMB1LACL0 and T103LACL0 has been subtracted from each strain. (B) Relative expression, determined by qRT-PCR, of the lodA, ppoA, and ppoB genes of mutant strain T103 (PpoS−) during the early stationary phase of growth in MNGL compared to mRNA levels of the wild-type strain under the same conditions.
LOD activity is induced by l-Lys, l-Arg, and meso-2,6-diaminopimelic acid (DAP).
The effect of different compounds on LOD activity was studied by adding these compounds to MNG, a chemically defined medium with low basal LOD activity. In a first series of experiments, the effect of l-Lys, which is the substrate of the enzyme (15), was assayed. LOD activity in the supernatants of M. mediterranea cultures was determined at the stationary phase of growth, when the protein is secreted into the extracellular medium (16). The amino acid had a clear inducing effect on the activity, the strength of which was concentration dependent (Fig. 2 A). The induction of LOD activity by l-Lys was not the result of the increased availability of nutrients since the addition of similar amounts of l-glutamic acid, which can be used as a sole nitrogen and carbon source, did not have the same effect (Fig. 2A).
FIG. 2.

Effect of l-Lys on LOD activity in M. mediterranea. (A) Extracellular LOD activity of strain MMB1-R at 30 h of growth. Gray scale bars are MNG medium with increasing l-Lys concentrations of 0 mM, 0.1 mM, 0.5 mM, 1. 25 mM, and 3 mM (increasingly dark). The striped bars correspond to a culture in MNG plus 3 mM glutamate. (B) LodA cellular levels detected by Western blotting with anti-HA antibody in cell extracts of M. mediterranea LDAHa/B-1 in MNG and MNGL media in the late exponential phase of growth (10 h). The same amount of protein (0.336 μg) was loaded into each well. The graph represents a density scan of the 95-kDa bands shown at the top for each condition.
The effect of other amino acids and different nitrogenated compounds on the induction of LOD activity in MNG medium was also studied. It was observed that the activity was strongly induced by l-Arg and DAP at levels comparable to those for l-Lys (Table 3). Interestingly, l-Arg and DAP are not substrates of LOD in spite of their structural similarity to l-Lys (16).
TABLE 3.
Effects of different nitrogenated compounds on the induction of LOD activity in M. mediterraneaa
| Nitrogenated compound | LOD activity (%)b |
|---|---|
| l-Lysine | 100 (MNGL medium) |
| l-Arginine | 99.16 |
| l-Isoleucine | 40.27 |
| l-Serine | 27.45 |
| l-Glycine | 17.81 |
| l-Leucine | 10.98 |
| l-Valine | 9.64 |
| l-Glutamine | 8.04 |
| l-Histidine | 4.56 |
| Glutamic acid | 4.34 (MNG medium) |
| l-Alanine | 3.64 |
| l-Asparagine | 1.56 |
| l-Threonine | 0.47 |
| l-Methionine | 0.37 |
| l-Proline | 0.19 |
| l-Phenylalanine | 0.05 |
| Aspartic acid | ND |
| l-Cysteinec | ND |
| l-Tryptophan | ND |
| l-Tyrosine | ND |
| Diaminopimelic acid | 61.38 |
| 5-Aminovaleric acid | 25.71 |
| Lysine-methylester | 14.98 |
| l-Lysine | 10.07 |
| 6-Aminocaproic Acid | 8.62 |
| Putrescine | 7.42 |
| 2-Aminoadipic acid | 5.4 |
| 5-Hydroxy-l-lysine | 4.85 |
| LSKL tetrapeptide | 4.52 |
| Cadaverine | 2.94 |
| Pipecolic acid | 2.45 |
The indicated compounds were added to MNG medium at a final concentration of 3 mM, and the cultures were incubated for 30 h.
The LOD (RFU·min−1·OD600−1·ml−1) is expressed as a percentage with respect to the activity detected in medium with l-Lys. ND, not detected.
No growth was observed in this medium.
The effect of the addition of l-Lys, l-Arg, and DAP on M. mediterranea laccase and tyrosinase activities was also investigated (Fig. 3). It was observed that these activities were not induced by these compounds, indicating that their effect is specific for LOD and does not affect the other two oxidases studied.
FIG. 3.

PPO activities of M. mediterranea MMB-1R incubated for 30 h in MNG (open bars) and MNG supplemented with 3 mM l-Lys (MNGL) (solid bars), 3 mM l-Arg (shaded bars), or 3 mM DAP (striped bars). TH, tyrosine hydroxylase activity; DO, DOPA oxidase activity; the subscript SDS indicates that those activities have been determined in the presence of SDS. DMPO, dimethoxyphenol oxidase activity. THSDS activity is due mainly to the tyrosinase (PpoB), while DMPO activity is characteristic of the laccase (PpoA). TH, DO, and DOSDS activities are a result of both PPOs in M. mediterranea.
l-Lys, l-Arg, and DAP can be used as nitrogen sources regardless of LOD activity.
Recent results from our group have shown that LOD activity, prior to being secreted into the supernatants, is detected in M. mediterranea cells (16). Since this activity releases ammonium and our results here have shown that it is induced by different amino acids, the possible involvement of LOD activity in amino acid metabolism was explored. In a series of experiments, the capacity of M. mediterranea MMB-1R (wild-type strain) and LD (a mutant strain with a complete deletion of the lodAB operon) to use l-Lys, l-Arg, or DAP as nutrients was assayed. It was observed that none of them can be used as a single carbon or nitrogen source (data not shown). In contrast, all three compounds can be used as a nitrogen source when glucose was provided as a carbon source. It is important that under these conditions, no differences in growth were found between strains MMB-1R and LD (see Fig. S2 in the supplemental material). These results indicate that in M. mediterranea strains with the lodAB operon deleted, alternative pathways must be involved when l-Lys and similar compounds are used as nitrogen sources.
l-Lys induces an increase in LodA cellular levels.
The higher levels of LOD activity in the supernatant of MNGL cultures than in MNG could be due to an increase in enzyme synthesis or to higher secretion levels. Since LodA enzymatic activity is not easily detected in cell extracts, in order to study cell LodA levels regardless of its enzymatic activity, a strain named LDAHa/B-1, containing LodA fused to a hemagglutinin epitope (16), was used. Western blot analysis was used to determine cellular levels of LodA at 10 h of growth, when the maximal concentration of LodA is observed, prior to its secretion (16). The results obtained indicate that cell extracts from cultures in MNGL showed larger amounts of LodA than did cells from MNG medium (Fig. 2B). This result suggests that the induction of LOD activity by l-Lys is the result of an increase in enzyme synthesis.
The effect of inducing compounds on LOD activity is exerted at the posttranscriptional level.
The results obtained with strains carrying a PlodAB::lacZ transcriptional fusion revealed that lodAB transcription increased during the exponential growth phase, reaching a maximum at the beginning of the stationary phase and decreasing thereafter, whether or not l-Lys was present in the medium (Fig. 4 A). A similar result was obtained with l-Arg and DAP, when it was observed that the β-galactosidase activities at the beginning of the stationary phase in media containing these compounds were 117% ± 5% and 106% ± 40%, respectively, of the activity observed with MNG.
FIG. 4.

Effect of l-Lys on the transcriptional regulation of lodAB in M. mediterranea. (A) β-Galactosidase activity of M. mediterranea MMB1LACL1, with a PlodAB::lacZ transcriptional fusion, in MNG medium (open symbols) and MNGL (closed symbols). β-Galactosidase activity is represented as continuous lines, and growth was measured as the OD600 (discontinuous lines). The β-galactosidase activity of control strain MMB1LACL0 in each medium has been subtracted. (B) Relative expression levels, determined by qRT-PCR, of mRNA from the lodA, ppoA, and ppoB genes of M. mediterranea during the early stationary phase of growth in MNGL medium referred to MNG medium.
In order to study the transcriptional regulation of lodAB by l-Lys using a complementary approach, the relative levels of lodAB mRNA were determined by qRT-PCR experiments in which the mRNA levels of ppoA and ppoB were also determined. These measurements indicate that the expression levels of all target genes, lodA, ppoA, and ppoB, were similar in MNGL and MNG cultures in the early stationary phase of growth (Fig. 4B). In the case of ppoA and ppoB, the results agree with the lack of induction of the activities encoded by these genes after the addition of l-Lys (Fig. 3). Regarding lodAB, this result and the determination of transcriptional activity observed using the lacZ fusion described above (Fig. 4A) indicate that the addition of l-Lys does not have a significant effect on lodAB transcription. Thus, the increase of LOD synthesis and activity observed when l-Lys is present in the medium (Fig. 2) is due to posttranscriptional regulatory mechanisms that do not affect mRNA levels.
DISCUSSION
A group of Gram-negative bacteria, including, among others, Marinomonas mediterranea and Pseudoalteromonas tunicata, express an antimicrobial protein with LOD activity. This protein has been implicated in biofilm development and differentiation. LOD generates hydrogen peroxide, which is responsible for cell death in the center of microcolonies, a process which facilitates the subsequent dispersal of cells from the biofilm (27). In order to understand the mechanisms involved in the process of biofilm differentiation, greater insight into the regulation of LOD activity is necessary. For Pseudoalteromonas tunicata, it was described that mutants in the transcriptional regulator WmpR showed lower levels of a group of fouling inhibitors, including an antimicrobial protein with LOD activity, AlpP (11). Further characterization of the WmpR mutant strain revealed differences from the wild-type strain in the expression of proteins and transcripts involved in a variety of cellular processes (40).
WmpR is a ToxR-like transcriptional regulator, the first one of which was characterized for Vibrio cholerae (29). ToxR is a transmembrane protein that senses environmental changes and activates the expression of a group of genes collectively named the ToxR regulon (9). In some cases, ToxR binds directly to the promoter of the regulated genes, but in other cases, ToxR regulation is mediated by ToxT (9). In the case of AlpP regulation by WmpR, the precise mechanism of regulation has not been characterized. In this study it has been shown that M. mediterranea LOD activity is regulated by a different regulatory mechanism in which PpoS participates. PpoS is a hybrid sensor kinase containing additional modules fused to the sensor kinase, such as a response regulator plus a phosphotransferase domain (26). PpoS shows a strong similarity to some kinases that are highly conserved in the Gammaproteobacteria, which have received different names depending on the microorganism studied (GacS, BarA, etc.) (22). These types of proteins participate in phosphorelay systems, which are a more complex version of two-component regulatory systems (2). Regulation by these proteins can be exerted at the transcriptional level as well as posttranscriptionally, with the mediation, in many cases, of small RNAs (22).
PpoS was named after its initial description as a sensor protein regulating polyphenol oxidase (PPO) activities and melanin synthesis in M. mediterranea (26). Interestingly, the P. tunicata regulator of LOD activity, WmpR, also regulates pigment synthesis, although the pigments synthesized by P. tunicata are not melanins (11). The possible relationship between pigment synthesis and antimicrobial expression has also been described for other marine bacterial groups, such as the Roseobacter clade of bacteria, for which several studies have shown that the production of antimicrobial compounds is induced under growth conditions in which brown pigments are synthesized (5, 33). In actinomycetes, such a synthesis of pigments is considered an indication of a rich secondary metabolism (30).
M. mediterranea PpoS regulates different cellular processes such as LOD and PPO activities. However, the data obtained in this study clearly indicate that those processes are regulated at different levels. First, the effect of the PpoS mutation on LOD activity is stronger than that on PPO activities. In mutant strain T103 (PpoS−), LOD activity was undetectable, and the transcriptional activity of lodAB, determined using both lacZ transcriptional fusions and qRT-PCR, was negligible. These results indicate that PpoS regulates lodAB transcriptionally, most probably through the action of a still-unidentified response regulator. In contrast, the effect of the mutation of PpoS on ppoA and ppoB expression is less strong. Second, the differences in the promoter sequences of lodAB compared with those of ppoA and ppoB also indicate that they must be recognized by different transcriptional regulatory elements. Overall, these results suggest that PPO and LOD activities are independent processes that share some common regulatory mechanisms but might respond to different environmental conditions.
A second important difference between PPOs and LOD regulation is that, unlike PPOs, LOD is induced by l-Lys as well as by other structurally similar compounds, such as l-Arg and DAP. No differences in growth between the wild-type strain and mutant strain LD, with a deletion of the lodAB operon, were observed for media containing l-Lys, l-Arg, or DAP as single nitrogen sources. This result is not surprising in the case of l-Arg and DAP, since they are not substrates of LOD, but clearly indicates that the nitrogen metabolism routes for those compounds do not involve LOD activity. In the case of l-Lys, the data obtained indicate that LOD activity is not required for nitrogen metabolism under the conditions assayed, although the possibility that it plays a role in the metabolism of l-Lys under certain conditions cannot be completely ruled out. In conclusion, although l-amino acid oxidase (LAO) activities have been related to nitrogen metabolism in different microbial groups such as fungi (7, 37) and marine phytoplankton (31), the lysine oxidase synthesized by M. mediterranea does not seem to participate in that process.
The effect of l-Lys, l-Arg, and DAP on LOD activity is posttranscriptional, since no differences in mRNA levels between media with and those without those compounds were found by using qRT-PCR or β-galactosidase activity for a strain containing a lacZ fusion with PlodAB. Bacterial riboswitches can be used to regulate, at both transcriptional and posttranscriptional levels, genes involved in l-Lys metabolism and transport through the recognition of the so-called L box (36). Moreover, some analogues to l-Lys can be recognized by the system (4). Interestingly, sequence analysis of the lodAB operon did not reveal any sequence similarity with the above-mentioned L box, and thus, the molecular mechanism for the posttranscriptional regulation in the presence of l-Lys does not seem to involve a riboswitch and remains to be determined.
The induction of LOD activity by l-Lys, l-Arg, and DAP could indicate that these compounds act as a signal of a particular physiological situation. The natural inducer may well be l-Lys, since it is the substrate of the enzyme, while the others would act simply because of the striking structural similarity of all these compounds containing a side chain with at least three carbons and an amino group. M. mediterranea is a component of the microbiota associated with the marine plant Posidonia oceanica (12). One possibility is that the presence of l-Lys reflects an environment in close proximity to the plant that would release the amino acid. However, l-Lys is secreted by M. mediterranea and P. tunicata biofilms (27). Accordingly, a possible model is that cells in the biofilm secrete l-Lys, which induces the synthesis of lysine oxidase. When cell growth is limited, the enzyme is released, and cell death takes place in the center of the microcolonies, facilitating biofilm differentiation. According to this model, LOD activity is not related with nitrogen metabolism. Its most relevant activity would be antimicrobial, and in this capacity, it would act on the population of producing cells and perhaps on some competing microbiota of Posidonia oceanica.
Supplementary Material
Acknowledgments
This work has been supported by grant BIO2007-64082 from the Ministerio de Ciencia e Innovación, España. L.R.M.-Q. is a recipient of a fellowship of the Universidad Centroccidental Lisandro Alvarado (Venezuela).
Footnotes
Published ahead of print on 23 July 2010.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Akyilmaz, E., A. Erdogan, R. Ozturk, and I. Yasa. 2007. Sensitive determination of L-lysine with a new amperometric microbial biosensor based on Saccharomyces cerevisiae yeast cells. Biosens. Bioelectron. 22:1055-1060. [DOI] [PubMed] [Google Scholar]
- 2.Appleby, J. L., J. S. Parkinson, and R. B. Bourret. 1996. Signal transduction via the multi-step phosphorelay: not necessarily a road less traveled. Cell 86:845-848. [DOI] [PubMed] [Google Scholar]
- 3.Barsby, T. 2006. Drug discovery and sea hares: bigger is better. Trends Biotechnol. 24:1-3. [DOI] [PubMed] [Google Scholar]
- 4.Blount, K. F., J. X. Wang, J. Lim, N. Sudarsan, and R. R. Breaker. 2007. Antibacterial lysine analogs that target lysine riboswitches. Nat. Chem. Biol. 3:44-49. [DOI] [PubMed] [Google Scholar]
- 5.Bruhn, J. B., K. F. Nielsen, M. Hjelm, M. Hansen, J. Bresciani, S. Schulz, and L. Gram. 2005. Ecology, inhibitory activity, and morphogenesis of a marine antagonistic bacterium belonging to the Roseobacter clade. Appl. Environ. Microbiol. 71:7263-7270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis, M. A., M. C. Askin, and M. J. Hynes. 2005. Amino acid catabolism by an areA-regulated gene encoding an l-amino acid oxidase with broad substrate specificity in Aspergillus nidulans. Appl. Environ. Microbiol. 71:3551-3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeBusk, R. M., and S. Ogilvie. 1984. Participation of an extracellular deaminase in amino acid utilization by Neurospora crassa. J. Bacteriol. 159:583-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Lorenzo, V., and K. N. Timmis. 1994. Analysis and construction of stable phenotypes in Gram-negative bacteria with Tn5- and Tn10-derived minitransposons. Methods Enzymol. 235:386-405. [DOI] [PubMed] [Google Scholar]
- 9.DiRita, V. J., C. Parsot, G. Jander, and J. J. Mekalanos. 1991. Regulatory cascade controls virulence in Vibrio cholerae. Proc. Natl. Acad. Sci. U. S. A. 88:5403-5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drechsel, H., A. Thieken, R. Reissbrodt, G. Jung, and G. Winkelmann. 1993. Alpha-keto acids are novel siderophores in the genera Proteus, Providencia, and Morganella and are produced by amino acid deaminases. J. Bacteriol. 175:2727-2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Egan, S., S. James, and S. Kjelleberg. 2002. Identification and characterization of a putative transcriptional regulator controlling the expression of fouling inhibitors in Pseudoalteromonas tunicata. Appl. Environ. Microbiol. 68:372-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Espinosa, E., E. Marco-Noales, D. Gomez, P. Lucas-Elio, M. Ordax, N. Garcias-Bonet, C. M. Duarte, and A. Sanchez-Amat. 2010. Taxonomic study of Marinomonas strains isolated from the sea grass Posidonia oceanica, including description of Marinomonas balearica sp. nov. and Marinomonas pollencensis sp. nov. Int. J. Syst. Evol. Microbiol. 60:93-98. [DOI] [PubMed] [Google Scholar]
- 13.Faust, A., B. Geueke, K. Niefind, W. Hummel, and D. Schomburg. 2006. Crystallization and preliminary X-ray analysis of a bacterial L-amino-acid oxidase from Rhodococcus opacus. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 62:279-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernández, E., A. Sanchez-Amat, and F. Solano. 1999. Location and catalytic characteristics of a multipotent bacterial polyphenol oxidase. Pigment Cell Res. 12:331-339. [DOI] [PubMed] [Google Scholar]
- 15.Gómez, D., P. Lucas-Elío, A. Sanchez-Amat, and F. Solano. 2006. A novel type of lysine oxidase: L-lysine-epsilon-oxidase. Biochim. Biophys. Acta 1764:1577-1585. [DOI] [PubMed] [Google Scholar]
- 16.Gómez, D., P. Lucas-Elío, F. Solano, and A. Sanchez-Amat. 2010. Both genes in the Marinomonas mediterranea lodAB operon are required for the expression of the antimicrobial protein lysine oxidase. Mol. Microbiol. 75:462-473. [DOI] [PubMed] [Google Scholar]
- 17.Hernandez-Romero, D., P. Lucas-Elio, D. Lopez-Serrano, F. Solano, and A. Sanchez-Amat. 2003. Marinomonas mediterranea is a lysogenic bacterium that synthesizes R-bodies. Microbiology 149:2679-2686. [DOI] [PubMed] [Google Scholar]
- 18.James, S. G., C. Holmstrom, and S. Kjelleberg. 1996. Purification and characterization of a novel antibacterial protein from the marine bacterium D2. Appl. Environ. Microbiol. 62:2783-2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson, P. M., C. E. Kicklighter, M. Schmidt, M. Kamio, H. Yang, D. Elkin, W. C. Michel, P. C. Tai, and C. D. Derby. 2006. Packaging of chemicals in the defensive secretory glands of the sea hare Aplysia californica. J. Exp. Biol. 209:78-88. [DOI] [PubMed] [Google Scholar]
- 20.Kusakabe, H., K. Kodama, A. Kuninaka, H. Yoshino, H. Misono, and K. Soda. 1980. A new antitumor enzyme, L-lysine alpha-oxidase from Trichoderma viride. Purification and enzymological properties. J. Biol. Chem. 255:976-981. [PubMed] [Google Scholar]
- 21.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 22.Lapouge, K., M. Schubert, F. H. Allain, and D. Haas. 2008. Gac/Rsm signal transduction pathway of gamma-proteobacteria: from RNA recognition to regulation of social behaviour. Mol. Microbiol. 67:241-253. [DOI] [PubMed] [Google Scholar]
- 23.López-Serrano, D., F. Solano, and A. Sanchez-Amat. 2004. Identification of an operon involved in tyrosinase activity and melanin synthesis in Marinomonas mediterranea. Gene 342:179-187. [DOI] [PubMed] [Google Scholar]
- 24.Lucas-Elío, P., P. Hernández, A. Sanchez-Amat, and F. Solano. 2005. Purification and partial characterization of marinocine, a new broad-spectrum antibacterial protein produced by Marinomonas mediterranea. Biochim. Biophys. Acta 1721:193-203. [DOI] [PubMed] [Google Scholar]
- 25.Lucas-Elío, P., D. Gómez, F. Solano, and A. Sanchez-Amat. 2006. The antimicrobial activity of marinocine, synthesized by Marinomonas mediterranea, is due to hydrogen peroxide generated by its lysine oxidase activity. J. Bacteriol. 188:2493-2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lucas-Elío, P., F. Solano, and A. Sanchez-Amat. 2002. Regulation of polyphenol oxidase activities and melanin synthesis in Marinomonas mediterranea: identification of ppoS, a gene encoding a sensor histidine kinase. Microbiology 148:2457-2466. [DOI] [PubMed] [Google Scholar]
- 27.Mai-Prochnow, A., P. Lucas-Elio, S. Egan, T. Thomas, J. S. Webb, A. Sanchez-Amat, and S. Kjelleberg. 2008. Hydrogen peroxide linked to lysine oxidase activity facilitates biofilm differentiation and dispersal in several gram-negative bacteria. J. Bacteriol. 190:5493-5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller, J. H. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 29.Miller, V. L., R. K. Taylor, and J. J. Mekalanos. 1987. Cholera toxin transcriptional activator ToxR is a transmembrane DNA binding protein. Cell 48:271-279. [DOI] [PubMed] [Google Scholar]
- 30.Omura, S., H. Ikeda, J. Ishikawa, A. Hanamoto, C. Takahashi, M. Shinose, Y. Takahashi, H. Horikawa, H. Nakazawa, T. Osonoe, H. Kikuchi, T. Shiba, Y. Sakaki, and M. Hattori. 2001. Genome sequence of an industrial microorganism Streptomyces avermitilis: deducing the ability of producing secondary metabolites. Proc. Natl. Acad. Sci. U. S. A. 98:12215-12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palenik, B., and F. M. M. Morel. 1990. Amino acid utilization by marine phytoplankton: a novel mechanism. Limnol. Oceanogr. 35:260-269. [Google Scholar]
- 32.Pfaffl, M. W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Porsby, C. H., K. F. Nielsen, and L. Gram. 2008. Phaeobacter and Ruegeria species of the Roseobacter clade colonize separate niches in a Danish turbot (Scophthalmus maximus)-rearing farm and antagonize Vibrio anguillarum under different growth conditions. Appl. Environ. Microbiol. 74:7356-7364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sambrock, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 35.Sanchez-Amat, A., P. Lucas-Elio, E. Fernández, J. C. Garcia-Borrón, and F. Solano. 2001. Molecular cloning and functional characterization of a unique multipotent polyphenol oxidase from Marinomonas mediterranea. Biochim. Biophys. Acta 1547:104-116. [DOI] [PubMed] [Google Scholar]
- 36.Serganov, A., and D. J. Patel. 2009. Amino acid recognition and gene regulation by riboswitches. Biochim. Biophys. Acta 1789:592-611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sikora, L., and G. A. Marzluf. 1982. Regulation of L-amino acid oxidase and of D-amino acid oxidase in Neurospora crassa. Mol. Gen. Genet. 186:33-39. [DOI] [PubMed] [Google Scholar]
- 38.Solano, F., E. García, E. Pérez de Egea, and A. Sanchez-Amat. 1997. Isolation and characterization of strain MMB-1 (CECT 4803), a novel melanogenic marine bacterium. Appl. Environ. Microbiol. 63:3499-3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Solano, F., P. Lucas-Elío, E. Fernández, and A. Sanchez-Amat. 2000. Marinomonas mediterranea MMB-1 transposon mutagenesis: isolation of a multipotent polyphenol oxidase mutant. J. Bacteriol. 182:3754-3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stelzer, S., S. Egan, M. R. Larsen, D. H. Bartlett, and S. Kjelleberg. 2006. Unravelling the role of the ToxR-like transcriptional regulator WmpR in the marine antifouling bacterium Pseudoalteromonas tunicata. Microbiology 152:1385-1394. [DOI] [PubMed] [Google Scholar]
- 41.Takahashi, E., M. Furui, H. Seko, and T. Shibatani. 1997. d-Methionine preparation from racemic methionines by Proteus vulgaris IAM 12003 with asymmetric degrading activity. Appl. Microbiol. Biotechnol. 47:173-179. [DOI] [PubMed] [Google Scholar]
- 42.Tong, H., W. Chen, W. Shi, F. Qi, and X. Dong. 2008. SO-LAAO, a novel l-amino acid oxidase that enables Streptococcus oligofermentans to outcompete Streptococcus mutans by generating H2O2 from peptone. J. Bacteriol. 190:4716-4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Varadi, M., N. Adanyi, E. E. Szabo, and N. Trummer. 1999. Determination of the ratio of D- and L-amino acids in brewing by an immobilised amino acid oxidase enzyme reactor coupled to amperometric detection. Biosens. Bioelectron. 14:335-340. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.