

Abstract
The C-terminal domain (CTD) of the a/Tif32 subunit of budding yeast eukaryotic translation initiation factor 3 (eIF3) interacts with eIF3 subunits j/Hcr1 and b/Prt1 and can bind helices 16 to 18 of 18S rRNA, suggesting proximity to the mRNA entry channel of the 40S subunit. We have identified substitutions in the conserved Lys-Glu-Arg-Arg (KERR) motif and in residues of the nearby box6 element of the a/Tif32 CTD that impair mRNA recruitment by 43S preinitiation complexes (PICs) and confer phenotypes indicating defects in scanning and start codon recognition. The normally dispensable CTD of j/Hcr1 is required for its binding to a/Tif32 and to mitigate the growth defects of these a/Tif32 mutants, indicating physical and functional interactions between these two domains. The a/Tif32 CTD and the j/Hcr1 N-terminal domain (NTD) also interact with the RNA recognition motif (RRM) in b/Prt1, and mutations in both subunits that disrupt their interactions with the RRM increase leaky scanning of an AUG codon. These results, and our demonstration that the extreme CTD of a/Tif32 binds to Rps2 and Rps3, lead us to propose that the a/Tif32 CTD directly stabilizes 43S subunit-mRNA interaction and that the b/Prt1-RRM-j/Hcr1-a/Tif32-CTD module binds near the mRNA entry channel and regulates the transition between scanning-conducive and initiation-competent conformations of the PIC.
Eukaryotic translation initiation factor 3 (eIF3) is a multisubunit protein complex that has been implicated in several steps of the translation initiation pathway (reviewed in reference 19). These steps include recruitment of the eIF2-GTP-Met-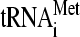 ternary complex (TC) and other eIFs to the small (40S) ribosomal subunit to form the 43S preinitiation complex (PIC), mRNA recruitment by the 43S PIC, and subsequent scanning of the 5′ untranslated region (UTR) for an AUG start codon. The eIF3 in the budding yeast Saccharomyces cerevisiae is composed of only 6 subunits (a/Tif32, b/Prt1, c/Nip1, i/Tif34, g/Tif35, and j/Hcr1), which have homologs in the larger, 13-subunit eIF3 complex in mammals. Yeast eIF3 can be purified with the TC, eIF1, and eIF5 in a ribosome-free assembly called the multifactor complex (MFC) (2), whose formation appears to promote assembly or stability of the 43S PIC and to stimulate scanning and AUG selection (10, 23, 32, 42, 48, 49, 51).
ternary complex (TC) and other eIFs to the small (40S) ribosomal subunit to form the 43S preinitiation complex (PIC), mRNA recruitment by the 43S PIC, and subsequent scanning of the 5′ untranslated region (UTR) for an AUG start codon. The eIF3 in the budding yeast Saccharomyces cerevisiae is composed of only 6 subunits (a/Tif32, b/Prt1, c/Nip1, i/Tif34, g/Tif35, and j/Hcr1), which have homologs in the larger, 13-subunit eIF3 complex in mammals. Yeast eIF3 can be purified with the TC, eIF1, and eIF5 in a ribosome-free assembly called the multifactor complex (MFC) (2), whose formation appears to promote assembly or stability of the 43S PIC and to stimulate scanning and AUG selection (10, 23, 32, 42, 48, 49, 51).
In mammals, there is evidence that eIF3 enhances recruitment of mRNA by interacting directly with eIF4G, the “scaffold” subunit of mRNA cap-binding complex eIF4F, and forming a protein bridge between mRNA and the 43S PIC (24, 25, 35). In budding yeast, direct eIF3-eIF4G interaction has not been detected, and the eIF3-binding domain (25) is not evident in yeast eIF4G. Moreover, depletion of eIF3, but not eIF4G, from yeast cells provokes a strong decrease in the amount of an mRNA (RPL41A) associated with native PICs (23). However, since depletion of eIF3 also reduced the amounts of other MFC components associated with PICs, it remained unclear whether eIF3 acts directly in mRNA recruitment.
In favor of a direct role for eIF3, cross-linking analysis of reconstituted mammalian 48S PICs identified contacts of subunits eIF3a and eIF3d with mRNA residues 8 to 17 nucleotides (nt) upstream of the AUG codon, suggesting that these subunits form an extension of the mRNA exit channel (37). Consistent with this, we found that the N-terminal domain (NTD) of yeast a/Tif32 binds Rps0A, located near the mRNA exit pore, and functionally interacts with sequences 5′ to the regulatory upstream open reading frame 1 (uORF1) in GCN4 mRNA (42). Despite these advances, in vivo evidence supporting a direct role of eIF3 in mRNA recruitment by 43S PICs is lacking.
Recently, there has been progress in elucidating the molecular mechanisms involved in ribosomal scanning and AUG selection. Reconstituted mammalian 43S PICs containing only eIF1, -1A, and -3 and the TC can scan the leader of an unstructured message and form a stable 48S PIC at the 5′-proximal AUG codon (35). eIF1 and -1A are thought to promote scanning by stabilizing an open conformation of the 40S subunit (6, 13, 26, 27), which appears to involve opening the “latch” on the mRNA entry channel formed by helices 18 and 34 of 18S rRNA (33). eIF1A also promotes a mode of TC binding conducive to scanning (39) and seems to prevent full accommodation of Met-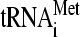 in the P site at non-AUG codons (53). The GTP bound to eIF2 is hydrolyzed, in a manner stimulated by eIF5, but release of phosphate (Pi) from eIF2-GDP-Pi is blocked by eIF1 (1). Entry of AUG into the P site triggers relocation of eIF1 from its binding site on the 40S subunit (27), allowing Pi release (1) and stabilizing the closed, scanning-arrested conformation of the 40S subunit (33).
in the P site at non-AUG codons (53). The GTP bound to eIF2 is hydrolyzed, in a manner stimulated by eIF5, but release of phosphate (Pi) from eIF2-GDP-Pi is blocked by eIF1 (1). Entry of AUG into the P site triggers relocation of eIF1 from its binding site on the 40S subunit (27), allowing Pi release (1) and stabilizing the closed, scanning-arrested conformation of the 40S subunit (33).
Mutations in eIF1 and eIF1A that reduce the stringency of start codon recognition have been isolated by their ability to increase initiation at a UUG codon in his4 alleles lacking the AUG start codon (the Sui− phenotype) (6, 12, 13, 29, 38, 39, 52). eIF1A mutations with the opposite effect of lowering UUG initiation in the presence of a different Sui− mutation (the Ssu− phenotype) were also obtained (13, 39). Previously, we identified Sui− and Ssu− mutations in the N-terminal domain of eIF3 subunit c/Nip1, which alter its contacts with eIF1, -2, and -5, suggesting that integrity of the MFC is important for the accuracy of AUG selection (49).
Several genetic findings also implicate eIF3 in the efficiency of scanning and AUG recognition. The prt1-1 point mutation in b/Prt1 (S518F) (11) impairs translational control of GCN4 mRNA in a manner suggesting a reduced rate of scanning between the short uORFs involved in this control mechanism (30). Disrupting an interaction between a hydrophobic pocket of the noncanonical RNA recognition motif (RRM) in the N terminus of b/Prt1 (henceforth referred to as b/RRM) and a Trp residue in the N-terminal acidic motif of j/Hcr1 (Trp-37) severely reduces the efficiency of initiation at the AUG of uORF1 in GCN4 mRNA, the phenomenon of leaky scanning, implicating the connection between the b/RRM and j/Hcr1 NTD (henceforth referred to as j/NTD) in efficient AUG recognition (10). Similarly, a multiple Ala substitution in RNP1 of the b/RRM evoked leaky scanning of the AUG codon of GCN4 uORF1 (uAUG-1) (32).
Interestingly, besides the b/RRM-j/NTD contact, the b/RRM can simultaneously bind to the j/Hcr1-like domain (HLD) in a/Tif32, and j/Hcr1 also independently binds a/Tif32 (50). This network of interactions involving the b/RRM, a/Tif32-HLD, and j/Hcr1 segments was shown to stabilize an eIF3 subassembly (50), referred to below as the b/RRM-j/Hcr1-a/Tif32-CTD module; however, it was not known whether the a/Tif32 HLD component of this module also participates in AUG recognition or other specific steps of initiation.
In this report, we provide evidence that the evolutionarily conserved KERR motif in the a/Tif32 HLD (hereafter referred to as a/HLD) functions to enhance mRNA recruitment by 43S PICs, processivity of scanning, and the efficiency of AUG recognition. The identification of Ssu− phenotypes for both KERR mutations and replacement of a nearby element (box6) further implicates the a/HLD in promoting the closed, scanning-arrested conformation of the PIC at start codons. Combining these results with our finding that the a/Tif32 CTD binds the 40S proteins Rps3 and Rps2 and the recent evidence that j/Hcr1 promotes AUG recognition and binds Rps2 leads us to propose that the a/HLD is positioned near the 40S mRNA entry channel, where it promotes mRNA binding and, together with j/Hcr1 and the b/RRM, modulates the transition between the open and closed conformations of the PIC during scanning and AUG recognition.
MATERIALS AND METHODS
Construction of yeast strains and plasmids.
The yeast strains and plasmids used in all experiments are listed in Tables 1 and 2, respectively. Yeast strain YAH04 was generated by a genetic cross of H428 and del′32a9A (Table 1) as a haploid ascospore resistant to 3-amino-1,2,4-triazole (3-AT) and geneticin but unable to grow on 5-fluoroorotic acid (5-FOA) and autotrophic for tryptophan. YAH05 was generated by a cross of H428 and H2923 (Table 1) as an ascospore resistant to 3-AT and geneticin, unable to grow on 5-FOA, and autotrophic for tryptophan. To generate strains AY51 and AY52, YAH04 was transformed with pRS-a/TIF32-DS-His and pRS-a/tif32-R731I-His-L, respectively, and the resident TIF32 URA3 plasmid was evicted on 5-FOA. To create strains YSW731 and YSW725, del′32a9A was transformed with pRS-a/tif32-R731I-His-L and pWLCB01, respectively, and the resident TIF32 URA3 plasmid was evicted on 5-FOA. To create strain H3708, H2994 was transformed with YCpTIF32-His-U and then with a PCR fragment containing the tif32Δ::KanMX4 allele amplified from the appropriate deletion mutant from the Saccharomyces Genome Deletion Project (15), purchased from Research Genetics, selecting for resistance to G418. To generate yeast strains H3711, H3714, H3715, and WLCY01 to WLCY03, H3708 was transformed with low-copy-number (lc) LEU2 plasmids harboring the appropriate TIF32-His alleles, and the resident TIF32 URA3 plasmid was evicted on 5-FOA. To create strain WLCY22, primers YLR192C-A and YLR192C-D were used to amplify a PCR product containing the hcr1Δ::KanMX4 allele from strain 6704. H2994 was transformed with the purified DNA fragment by selecting for G418 resistance. Strains WLCY06 to WLCY09, were generated by (i) deleting RPL11B from H3708 by transformation with the HindIII-BamHI fragment containing rpl11bΔ::LEU2 from plasmid pL16b-ΔLeu, selecting for Leu+ clones, and (ii) replacing the resident TIF32 URA3 plasmid with the appropriate single-copy (sc) TRP1 plasmids harboring TIF32-His alleles by counterselection on 5-FOA medium. To generate strain H3774, 1D2-2D was transformed with pRB53, and the resident DED1 URA3 plasmid was evicted on 5-FOA.
TABLE 1.
Yeast strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| H428a | MATaleu2-3,112 ura3-52 hcr1::KanMX3 | 32 |
| H2923 | MATα leu2-3,112 ura3-52 trp1Δ prt1::hisG gcn2::hisG pRS316-PRT1 (PRT1 URA3) | 30 |
| del′32a9Aa | MATα leu2-3,112 ura3-52 trp1Δ tif32::hisG gcn2::hisG YCp-a/TIF32-His-U (TIF32 URA3) | 42 |
| YAH04a | MATaleu2-3,112 ura3-52 trp1Δ GCN2 hcr1::KanMX3 tif32::hisG YCp-a/TIF32-His-U (TIF32-His URA3) | This study |
| YAH05a | MATaleu2-3,112 ura3-52 GCN2 hcr1::KanMX3 prt1::hisG pRS316-PRT1 (PRT1 URA3) | This study |
| AY51a | MATaleu2-3,112 ura3-52 trp1Δ GCN2 hcr1::KanMX3 tif32::hisG pRS-a/TIF32 DS-His (TIF32-His LEU2) | This study |
| AY52a | MATaleu2-3,112 ura3-52 trp1Δ GCN2 hcr1::KanMX3 tif32::hisG pRS-a/tif32-R731I-His (tif32-His-R731I LEU2) | This study |
| YSW731a | MATα leu2-3,112 ura3-52 trp1Δ tif32::hisG gcn2::hisG pRS-a/tif32-R731I-His-L (tif32-His-R731I LEU2) | This study |
| YSW725a | MATα leu2-3,112 ura3-52 trp1Δ tif32::hisG gcn2::hisG pRS-a/tif32-H725P-His-L (tif32-His-H725P LEU2) | This study |
| H2994b | MATaura3-52 trp1-63 leu2-3,112 his4-301(AUU) | 49 |
| H3708b | MATaura3-52 trp1-63 leu2-3,112 his4-301(AUU) tif32Δ::KanMX4 YCp-a/TIF32-His-U (TIF32-His URA3) | This study |
| H3711b | MATaura3-52 trp1-63 leu2-3,112 his4-301(AUU) tif32Δ::KanMX4 pRS-a/tif32-box2-His-L (tif32-His-box2 LEU2) | This study |
| H3714b | MATaura3-52 trp1-63 leu2-3,112 his4-301(AUU) tif32Δ::KanMX4 pRS-a/tif32-box5-His-L (tif32-His-box5 LEU2) | This study |
| H3715b | MATaura3-52 trp1-63 leu2-3,112 his4-301(AUU) tif32Δ::KanMX4 pRS-a/tif32-box6-His-L (tif32-His-box6 LEU2) | This study |
| WLCY01b | MATaura3-52 trp1-63 leu2-3,112 his4-301(AUU) tif32Δ::KanMX4 pRS-a/TIF32-His-L (TIF32-His LEU2) | This study |
| WLCY02b | MATaura3-52 trp1-63 leu2-3,112 his4-301(AUU) tif32Δ::KanMX4 pWLCB01 (tif32-His-H725P LEU2) | This study |
| WLCY03b | MATaura3-52 trp1-63 leu2-3,112 his4-301(AUU) tif32Δ::KanMX4 pRS-a/tif32-R731I-His-L (tif32-His-R731I LEU2) | This study |
| WLCY06b | MATaura3-52 trp1-63 leu2-3,112 his4-301(AUU) tif32Δ::KanMX4 rpl11bΔ::LEU2 pWLCB02 (TIF32-His TRP1) | This study |
| WLCY07b | MATaura3-52 trp1-63 leu2-3,112 his4-301(AUU) tif32Δ::KanMX4 rpl11bΔ::LEU2 pWLCB03 (tif32-His-H725P TRP1) | This study |
| WLCY08b | MATaura3-52 trp1-63 leu2-3,112 his4-301(AUU) tif32Δ::KanMX4 rpl11bΔ::LEU2 pWLCB04 (tif32-His-R731I TRP1) | This study |
| WLCY09b | MATaura3-52 trp1-63 leu2-3,112 his4-301(AUU) tif32Δ::KanMX4 rpl11bΔ::LEU2 pWLCB05 (tif32-His-box6 TRP1) | This study |
| 1D2-2D | MAT ura3-1 trp1-1 his3-11,15 leu2-3,112 can1-100 ade2-1 ded1::HISMX6 (DED1 URA3) | 22 |
| H3774 | MAT ura3-1 trp1-1 his3-11,15 leu2-3,112 can1-100 ade2-1 ded1::HISMX6 pRB53(ded1-57 LEU2) | This study |
| 6704 | MATahcr1::KanMX4 his3-Δ1 leu2-Δ0 ura3-Δ0 met15-Δ0 | Research Genetics |
| WLCY22b | MATaura3-52 trp1-63 leu2-3,112 his4-301(AUU) hcr1Δ::KanMX4 | This study |
| H2879c | MATaleu2-3,112 ura3-52 | 30 |
| H3675c | MATaleu2-3,112 ura3-52 hcr1Δ | 32 |
Isogenic strains.
Isogenic strains.
Isogenic strains.
TABLE 2.
Plasmids used in this study
| Plasmid | Description | Source or reference |
|---|---|---|
| pRS-b/PRT1-HisXS | PRT1-His in lc LEU2 plasmid, from pRS315 | 10 |
| YEplac195 | hc URA3 vector | 16 |
| YEp-j/HCR1-DS | HCR1 coding region flanked by BamHI and NcoI sites, respectively, in hc LEU2 plasmid, from YEplac181 | 10 |
| YEp-j/HCR1-DS-U | HCR1 coding region flanked by BamHI and NcoI sites, respectively, in hc URA3 plasmid, from YEplac195 | 10 |
| YEp-j/hcr1-NTD | hcr1-NTD [1-135] in hc LEU2 plasmid, from YEplac181 | 10 |
| YEp-j/hcr1-NTD-U | hcr1-NTD [1-135] in hc URA3 plasmid, from YEplac195 | This study |
| YEp-j/hcr1-CTD | hcr1-CTD [136-265] in hc LEU2 plasmid, from YEplac181 | 10 |
| YEp-j/hcr1-CTD-U | hcr1-CTD [136-265] in hc URA3 plasmid, from YEplac195 | This study |
| YEp-j/hcr1-R215I-DS | hcr1-R215I in hc LEU2 plasmid, from YEplac181 | This study |
| YEp-j/hcr1-R215I-U | hcr1-R215I in hc URA3 plasmid, from YEplac195 | This study |
| YEp-j/hcr1-box6 | hcr1-box6 in hc LEU2 plasmid, from YEplac181 | This study |
| YEp-j/hcr1-box6-U | hcr1-box6 in hc URA3 plasmid, from YEplac195 | This study |
| YEp-j/hcr1-box6-R215I | hcr1-box6-R215I in hc LEU2 plasmid, from YEplac181 | This study |
| YEp-j/hcr1-box6-R215I-U | hcr1-box6-R215I in hc URA3 plasmid, from YEplac195 | This study |
| YCp-j/HCR1-DS-U | HCR1 in lc URA3 plasmid, from YCplac33 | 10 |
| YCp-j/HCR1-DS-L | HCR1 in lc LEU2 plasmid, from YCplac111 | This study |
| YCp-j/hcr1-R215I-U | hcr1-R215I in lc URA3 plasmid, from YCplac33 | This study |
| pRS-a/TIF32-His-L | TIF32-His in lc LEU2 plasmid, from pRS315 | 48 |
| pRS-a/tif32-Box9-His-L | tif32-box9-His, encoding 11 Ala substitutions in box9 (aa 721 to 732) in lc LEU2 plasmid, from pRS315 | This study |
| pRS-a/tif32-R731I-His-L | tif32-R731I-His in lc LEU2 plasmid, from pRS315 | This study |
| pWLCB01 | tif32-H725P-His in lc LEU2 plasmid, from pRS315 | This study |
| pGEX-5X-3 | Cloning vector for GST fusions | 40 |
| pGEX-b/PRT1-RRM | Encodes GST-b/prt1-RRM [1-136] fusion, from pGEX-5X-3 | 50 |
| pT7-a/TIF32 | TIF32 ORF under T7 promoter | 3 |
| pT7-HLD | Encodes TIF32[483-786] under T7 promoter | This study |
| pT7-HLD-box6 | Encodes tif32-box6[483-786] under T7 promoter | This study |
| pT7-HLD-box9 | Encodes tif32-box9[483-786] under T7 promoter | This study |
| pT7-HLD-R731I | Encodes tif32-R731I[483-786] under T7 promoter | This study |
| pGEX-j/HCR1 | Encodes GST-j/HCR1 fusion, from pGEX-5X-3 | 50 |
| pGEX-j/hcr1-NTD | Encodes GST-j/hcr1-NTD [1-135] fusion, from pGEX-5X-3 | 10 |
| pGEX-j/hcr1-CTD | Encodes GST-j/hcr1-CTD [136-265] fusion, from pGEX-5X-3 | 10 |
| pGEX-j/hcr1-BOX6 | Encodes GST-j/hcr1-box6 fusion, from pGEX-5X-3 | This study |
| pGEX-j/hcr1-BOX9 | Encodes GST-j/hcr1-box9 fusion, from pGEX-5X-3 | 10 |
| pGEX-j/hcr1-R215I | Encodes GST-j/hcr1-R215I fusion, from pGEX-5X-3 | This study |
| YEp-j/HCR1-W | HCR1 in hc TRP1 plasmid, from YEplac112 | 46 |
| YEp-j/hcr1-R215I-DS-W | hcr1-R215I in hc TRP1 plasmid, from YEplac112 | This study |
| pGBK-T7-RPS2 | RPS2 ORF in pGBKT7, TRP1 (Clontech) | 47 |
| pGAD-T7-RPS3 | RPS3 ORF in pGADT7, LEU2 (Clontech) | 47 |
| pGBK-T7-RPS0e | RPS0A ORF (lacking intron) in GBKT7, TRP1 (Clontech) | This study |
| pGEX-RPS2 | Encodes GST-RPS2 fusion, from pGEX-5X-3 | 10 |
| pGEX-a/TIF32-Δ4 | Encodes GST-a/TIF32-Δ4[790-964] fusion, from pGEX-5X-3 | 48 |
| pRS-a/tif32-box6-His-L | tif32-box6-His in lc LEU2 plasmid, from pRS315 | This study |
| YEp tif32-box + 9-His-L | tif32-box9-His in hc LEU2 plasmid, from YEplac181 | This study |
| YCplac22 | Single-copy TRP1 vector | 16 |
| p4280/YCpSUI3-S264Y-W | SUI3-S264Y in single-copy TRP1 plasmid, from YCplac22 | 49 |
| p4281/YCpSUI5-G31R-W | SUI5-G31R/TIF5-G31R in single-copy TRP1 plasmid, from YCplac22 | 49 |
| pAS57 | tif11Δ125-153 in hc TRP1 plasmid, from YEplac112 | This study |
| pM226 | Derivative of pM199; elongated uORF1 extends into the GCN4-lacZ coding region | 18 |
| p180 | GCN4-lacZ with WT leader in single-copy URA3 plasmid, from YCp50 | 20 |
| p227 | GCN4-lacZ with point mutations in all 4 uAUGs in single-copy URA3 plasmid, from YCp50 | 28 |
| p209 | GCN4-lacZ with point mutations in uAUGs 2 to 4 in single-copy URA3 plasmid, from YCp50 | 17 |
| p367 | HIS4-lacZ in single-copy URA3 plasmid | 9 |
| p391 | HIS4-UUG-lacZ, with TTG replacing ATG start codon, in single-copy URA3 plasmid | 9 |
| p4642/pJM753 | L0LUC reporter in single-copy URA3 plasmid | 4 |
| p4645/pJM261 | L2LUC reporter in single-copy URA3 plasmid | 4 |
| pWLCB06 | GCN4-lacZ lacking all uORFs, with hairpin insertion between nt +481 and +482 in single-copy URA3 plasmid, from YCp50 | This study |
| pWLCB07 | GCN4-lacZ lacking all uORFs, with hairpin insertion between nt +481 and +482 in single-copy URA3 plasmid, from YCp50 | This study |
Yeast biochemical methods.
Glutathione S-transferase (GST) pulldown experiments with GST fusions and in vitro-synthesized 35S-labeled polypeptides (Table 2 lists vector descriptions) were conducted as follows. Individual GST fusion proteins were expressed in Escherichia coli, immobilized on glutathione-Sepharose beads, and incubated with 10 μl of 35S-labeled potential binding partners at 4°C for 2 h. The beads were washed 3 times with 1 ml of phosphate-buffered saline, and bound proteins were separated by SDS-PAGE. Gels were first stained with Gelcode Blue Stain Reagent (Pierce) and then subjected to autoradiography. Nickel chelation chromatography of eIF3 complexes containing His8-tagged a/Tif32 or b/Prt1 from yeast whole-cell extracts (WCEs) and Western blot analysis were conducted as described in detail previously (31). In short, WCEs were incubated with 4 μl of 50% Ni-nitrilotriacetic acid (NTA)-silica resin (Qiagen) suspended in 200 μl of buffer A for 2 h at 4°C, followed by washing and elution. For assaying expression of GCN4-lacZ reporters, β-galactosidase activities in WCEs were measured as described previously (18). For assaying luciferase reporters, cells were disrupted with glass beads in 50 mM Tris-HCl (pH 7.5), 1 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride hydrochloride (AEBSF), and WCEs were assayed for luciferase activities using the luminescence reader Monolight 3010 (BD Biosciences) according to the supplier's protocol (Promega).
Analysis of polysome profiles and native 43S/48S PICs.
For polysome analysis, strains were grown in yeast extract-peptone-dextrose (YPD) at 30°C, shifted to 36°C, and cultured for 6 h (A600, ∼1). Cycloheximide was added (50 μg/ml) 5 min prior to harvesting, and WCEs were prepared in breaking buffer (20 mM Tris-HCl, pH 7.5, 50 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol [DTT], 5 mM NaF, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1× Complete Protease Inhibitor Mix tablets without EDTA [Roche]). Five A260 units of WCEs was separated by velocity sedimentation on a 4.5 to 45% sucrose gradient by centrifugation at 39,000 rpm for 2.5 h in an SW41Ti rotor (Beckman). Gradient fractions were collected and scanned at 254 nm to visualize ribosomal species.
For analysis of native 43S PICs, cells were cultured as described above and cross-linked with 1% formaldehyde prior to being harvested. WCEs were prepared in breaking buffer, and 25 A260 units was separated by velocity sedimentation on a 7.5 to 30% sucrose gradient by centrifugation at 41,000 rpm for 5 h in an SW41Ti rotor. Fractions (0.7 ml) were collected, precipitated by adding 1.7 volumes of 100% ethanol, resuspended in SDS loading buffer, and resolved by SDS-PAGE, followed by Western blotting with antibodies against the relevant eIFs. For analysis of 48S PICs, total RNA was isolated from 0.3 ml of gradient fractions, prepared as described above by hot-phenol extraction, and resuspended in 50 μl of diethyl pyrocarbonate (DEPC)-treated H2O. Two microliters of RNA was subjected to reverse transcription with SuperScript III reverse transcriptase (Invitrogen). Aliquots of cDNA were diluted 10-fold, and PCR amplifications were performed on 2 μl of diluted cDNA in 20-μl reaction mixtures prepared with the Brilliant II SYBR green qPCR Master Mix (Stratagene) and primers for RPL41A (0.3 μM) or 18S rRNA (0.4 μM) using the Mx3000P system (Stratagene).
RESULTS
Substitutions in the a/HLD impair general translation initiation.
To identify functionally important residues in the C-terminal portion of the a/HLD, we introduced Ala substitutions in consecutive blocks of 10 (or in one instance 11) residues between amino acids (aa) 642 and 791 (dubbed boxes 1 to 15 [Fig. 1 A]). The mutations were generated in plasmid-borne TIF32-His, encoding His8-tagged a/Tif32, and introduced into a tif32Δ strain by plasmid shuffling (5). Except for box9, none of these mutations was lethal, allowing eviction of the resident TIF32+ URA3 plasmid on medium containing 5-FOA, and only three of the mutations produced detectable slow-growth (Slg−) phenotypes: box2, -5, and -6 (summarized in Fig. 1A). The tif32-box9 allele was lethal whether present on an lc or high-copy-number (hc) plasmid (data not shown). We also subjected the TIF32 CTD to random mutagenesis and identified three mutant alleles with temperature-sensitive (Ts−) phenotypes, only one of which produced a single-amino-acid substitution (H725P). The other two Ts− mutants contained C-terminal truncations of the Tif32 CTD and were not studied further.
FIG. 1.

a/HLD substitutions impair cell growth. (A) Schematic representation of a/Tif32, indicating N- and C-terminal halves of the j/Hcr1-like domain (HLD). The CTD of a/Tif32 subjected to mutagenesis is indicated as a bar and string of ovals (boxes 1 to 15) representing 10 consecutive residues replaced by alanines. Shades of gray indicate the degree of Slg− phenotype, and black indicates lethality. A sequence alignment of the C-terminal portion of the a/HLD with j/Hcr1 (residues 168 to 222) is shown, indicating the nature of tif32 mutations box6, box9, H725P, and R731I above and the KERR motif below. S.c., S. cerevisiae. Asterisks, amino acids conserved in all sequences; colons, homologous substitutions; periods, nonhomologous substitutions; boldface, box6 region and KERR motif conserved residues. (B) ClustalW2 was used to align regions of j/Hcr1 and the a/HLD using sequences from the indicated species (K.lactis, Kluyveromyces lactis; H.sapiens, Homo sapiens; M.musculus, Mus musculus; B.taurus, Bos taurus; S.pombe, Schizosaccharomyces pombe; A.thaliana, Arabidopsis thaliana) with the following accession numbers (in the order shown, from the top): NP_013293, Q6CMJ8, O75822, Q66JS6, Q0VCU8, NP_009635, CAA21076, Q9LD55, and Q14152. (C) Phenotypes of tif32 a/HLD mutants. Serial dilutions of GCN2 his4-301 tif32Δ strains harboring lc plasmids with TIF32+-His (WLCY01), tif32-H725P-His (WLCY02), tif32-R731I-His (WLCY03), or tif32-box6-His (H3715) were spotted on synthetic dextrose minimal medium supplemented with histidine, tryptophan, and uracil (SD+HWU) and incubated at the indicated temperatures for 3 days.
Interestingly, the histidine residue replaced by the H725P mutation is located in the region altered by the lethal Ala replacement of box9 and is only 6 residues N-terminal to the R731I substitution produced by the rpg1-1 allele described previously (44) (Fig. 1A). These mutations all replace a region of sequence conservation among a/Tif32 homologs, designated the KERR motif after its conserved residues (K721-X5-E727-R728-X2-R731) (10), which is also conserved in the C-terminal region of j/Hcr1 (Fig. 1A and B). Based on the identification of multiple substitutions in the KERR motif, and of box6 only 20 residues upstream, which impaired the essential function of a/Tif32, we selected these mutations for further analysis.
The two KERR mutations, H725P and R731I, impair growth only at elevated temperatures (33°C and 36°C), whereas the box6 mutation confers Slg− at all temperatures (Fig. 1C). All three mutations provoke substantial (>3-fold) reductions in the polysome/monosome (P/M) ratio after incubation at 36°C (Fig. 2 A), indicating marked reductions in rates of translation initiation in vivo. Importantly, none of the mutations, including lethal box9, reduced the steady-state level of a/Tif32 protein (Fig. 2B, right and left).
FIG. 2.

a/HLD substitutions impair translation initiation. (A) Polysome profiles of the strains shown in Fig. 1C cultured in YPD medium at 30°C and shifted to 36°C for 6 h, with cycloheximide added just prior to harvesting. WCEs were separated by velocity sedimentation through 4.5 to 45% sucrose gradient centrifugation, and fractions were collected while scanning at 254 nm to visualize ribosomal species and determine P/M ratios. (B) Western analysis of a/Tif32 proteins. (Left) WCEs were prepared from the strains in panel A after being cultured in YPD at 30°C or after being shifted to 36°C for 6 h. Aliquots (2.5, 5, and 10 μl in successive lanes) were separated by SDS-PAGE and subjected to Western analysis with monoclonal antibodies against His8 epitope or polyclonal antibodies against Gcd6. The amounts of His8-a/Tif32 were normalized to the Gcd6 amounts measured in the same lanes, and the resulting ratios were normalized to those measured in WT cells (set to 1.0). The mean and standard errors calculated from replicate determinations are plotted in the histogram below. (Right) Western analysis of WCEs from transformants of TIF32+ strain H2994 harboring lc WT TIF32-His or lc or hc tif32-box9-His and cultured in synthetic complete medium lacking uracil at 30°C. (Because of its lethality, expression of His8-a/Tif32-box9 was examined in cells containing TIF32+).
a/HLD substitutions impair 48S PIC assembly.
To determine whether the tif32 mutations affect association of eIF3 or other components of the MFC with native 40S subunits, we employed formaldehyde cross-linking of living cells to stabilize native 43S/48S PICs. WCEs were separated by sedimentation through sucrose gradients, and the gradient fractions were subjected to Western analysis with antibodies against MFC components and the 40S subunit protein Rps22. The Western signals in the 40S fractions were quantified and normalized to Rps22 levels, and the mean eIF/40S ratio for each MFC component was determined from replicate experiments. After growth at 36°C, neither H725P nor R731I produced a reduction in the eIF/40S ratios compared to the wild type (WT) (Fig. 3 A and B), suggesting that the rate-limiting defects conferred by these mutations occur at a step following 43S PIC assembly. The box6 mutation leads to reductions of from 25 to 50% in the eIF/40S ratios for MFC constituents, including subunits of eIF3, eIF2γ, eIF5, and possibly eIF1, but does not reduce the level of 40S-bound eIF1A, which is not an MFC component (Fig. 3C). These findings suggest that a portion of the initiation defect in this mutant involves a reduced rate of formation, or decreased stability, of 43S PICs.
FIG. 3.

a/Tif32 KERR substitutions diminish native 48S PICs containing RPL41A mRNA. (A to C) Strains shown in Fig. 1C were grown in YPD at 30°C and shifted to 36°C for 6 h, and the cells were cross-linked with HCHO for 1 h prior to being harvested. WCEs were sedimented through 7.5 to 30% sucrose gradients, and fractions were subjected to Western analysis with antibodies against the indicated proteins. The amounts of each factor in the 40S fractions (boxed) were normalized for the Rps22 level, and the ratios of the eIF/40S levels in the mutant to those in the WT were plotted in the adjoining histograms (means ± standard errors [SE]; n = 3). (D) (Left) rpl11bΔ strains isogenic to those in panels A to C were cultured and cross-linked as described above. Total RNA was extracted from each fraction, and the amounts of 18S rRNA and RPL41A mRNA were measured by real-time quantitative PCR (qPCR). The amounts of mRNA were calculated as 2−CT × 10−7 for RPL41A mRNA and 2−CT × 10−4 for 18S rRNA. (Right) The ratio of RPL41A mRNA in fractions 10 to 12 to 18S rRNA in fractions 9 to 11 was calculated for each mutant and normalized to the corresponding value for WT (means ± SE; n = 4). Student's t test indicated that the value for each mutant differed significantly from that for the WT (P < 0.01).
To examine the effects of the KERR and box6 substitutions on 48S PIC assembly, we measured the amounts of RPL41A mRNA associated with native 48S complexes in extracts of cross-linked cells. These experiments were conducted using isogenic strains lacking one of the genes encoding the 60S protein Rpl11 (RPL11B), as the reduced level of 60S subunits, and attendant reduced rate of 60S subunit joining, produced by rpl11BΔ increases the concentration of 48S PICs (23). This accentuates the peak of 48S-associated RPL41A mRNA relative to the free RPL41A mRNP and facilitates quantification of the 48S species. Using real-time quantitative reverse transcription (RT)-PCR to assay RPL41A mRNA and 18S rRNA in each fraction, we found that all three mutations significantly reduced the mean RPL41A mRNA/18S rRNA ratio by 40 to 50% in three replicate experiments (Fig. 3D). Although the total amount of RPL41A mRNA was lower in the mutants, there was an obvious shift in the distribution of RPL41A mRNA toward free mRNP, indicating a substantial underutilized pool of free mRNP. It seems possible that the reduced amounts of total RPL41A mRNA in the mutants results from increased degradation due to its decreased recruitment by 43S PICs, leading to an underestimate of the actual defect in 48S assembly. Marked reductions in the RPL41A mRNA/18S rRNA ratio, coupled with a relative accumulation of free RPL41A mRNPs, were also observed for all three tif32 mutations in the RPL11 background (data not shown). Hence, we conclude that the tif32 mutations impair the assembly or stability of 48S PICs. The decrease in 48S abundance provoked by box6 could arise partly from its effect on 43S assembly (Fig. 3C), while the KERR mutations seem to impair a function specifically required for mRNA recruitment.
a/HLD mutations impair GCN4 translational induction and increase leaky scanning of uAUG-1.
We next examined the tif32 mutations for defects in the efficiency of scanning and AUG recognition, using GCN4 translational control as a sensitive genetic reporter of this process (21). GCN4 encodes a transcriptional activator of amino acid biosynthetic genes whose translation is derepressed by amino acid starvation when formation of TC is reduced by phosphorylation of eIF2α by kinase Gcn2. This regulation is mediated by the four uORFs in the GCN4 mRNA leader. Under nonstarvation conditions, ribosomes scan from the cap and translate uORF1, and the fraction that resumes scanning will rebind the TC; reinitiate at uORF2, -3, or -4; and subsequently dissociate from the mRNA, failing to translate GCN4. When the TC level drops under starvation conditions, a portion the subunits rescanning from uORF1 do not rebind the TC until after bypassing uORF2 to -4, and reinitiate at GCN4 instead. Thus, translation of uORF1, and the ability of ribosomes to reinitiate downstream following termination at uORF1, is crucial for induction of GCN4 translation in starved cells.
Mutations that impair derepression of GCN4 translation confer sensitivity to inhibitors of amino acid biosynthesis, including sulfometuron (SM), indicating a Gcn− phenotype (21). Interestingly, all three tif32 mutations confer sensitivity to SM at permissive (30°C) and semipermissive (33°C) temperatures (Fig. 4 A) and produce dramatic reductions in derepression of a GCN4-lacZ reporter containing all four uORFs in cells treated with SM at 33°C (Fig. 4B, construct i). Only box6 reduced expression of the construct lacking all four uORFs (Fig. 4B, ii), but the magnitude of the effect, 10 to 20%, was far less than that observed for the uORF-containing construct. Hence, the tif32 mutations impair derepression of GCN4 translation.
FIG. 4.

a/HLD mutations impair derepression of GCN4 translation and increase leaky scanning of uAUG-1. (A) a/HLD mutants have Gcn− phenotypes. Serial dilutions of the strains in Fig. 1C were spotted on SD+HWU and SD+HWU containing 0.5 μg/ml SM and incubated at 30°C, 33°C, or 36°C for 3 days. (B) a/HLD mutations alter expression of GCN4-lacZ reporters. Shown are the strains in Fig. 1C harboring GCN4-lacZ reporter plasmid p180 (i), p227 (ii), pM226 (iii), or p209 (iv), containing the 5′ UTR configurations shown schematically, with Xs indicating AUG mutations in uORFs. Transformants were grown in SD+HWU (−) or SD+HWU containing 0.5 μg/ml SM (+), as indicated in the left-hand column, at 33°C for 6 h, and β-galactosidase activity (in nmol of o-nitrophenyl-β-d-galactopyranoside cleaved per min per mg) was assayed in WCEs. The means and SE from four independent transformants are reported, along with the means expressed as a percentage of the corresponding WT value. The values in the column “box6 norm.” are the results from column “box6” normalized to correct for the different expression of construct ii (without SM) in box6 versus WT cells.
Western analysis of WCEs from cells induced with SM revealed no significant reduction in eIF2α phosphorylation in any of the mutants (data not shown). This suggested that the mutations impair one or more steps in the GCN4 reinitiation mechanism, which could include decreased recognition of uORF1 by “leaky scanning” of its start codon (uAUG-1), loss of posttermination 40S subunits at the uORF1 stop codon, or a reduced rate of scanning by reinitiating ribosomes between uORF1 and uORF4. Analysis of a construct containing a single elongated version of uORF1, which permits GCN4-lacZ expression only by leaky scanning of uAUG-1, revealed an ∼2.6-fold increase in GCN4-lacZ expression in the H725P and R731I mutants (Fig. 4B, iii). A smaller but significant increase of 1.9-fold was observed for box6 after normalizing for its effect on the expression of GCN4-lacZ lacking a uORF (Fig. 4B, iii, box6 norm.). Thus, all three tif32 mutations moderately increased leaky scanning of uAUG-1.
Analysis of a construct containing only WT uORF1 at its normal location, which allows a high level of reinitiation at GCN4, revealed increases in expression of 1.1- to 1.7-fold (Fig. 4B, iv). These results eliminate the possibility that the mutations reduce the number of posttermination 40S subunits that resume scanning following termination at uORF1, a defect observed previously for truncation of the a/Tif32 NTD (42), as this defect would decrease expression of the solitary-uORF1 construct. In fact, R731I confers a significant increase in expression of this solitary-uORF1 construct (P < 0.01) that is too large to be explained solely by the amount of leaky scanning of uAUG-1 that occurs in this strain (Fig. 4B, 660 − 390 = 270 units, from row 6, versus 27 − 10 = 17 units, from row 5). Hence, R731I might also reduce the rate of scanning by reinitiating 40S subunits, as this would increase the number of subunits that rebind the TC before reaching GCN4 and thereby increase GCN4-lacZ expression from the solitary-uORF1 construct (row 6).
Evidence for scanning defects in tif32 box6 and KERR mutants.
We also examined the effects of tif32 mutations on the expression of two luciferase constructs designed to reveal defects in the processivity of ribosomal scanning, as they differ greatly in 5′ UTR lengths at 43 nt (L0-LUC) and 1,177 nt (L2-LUC) (4). Consistent with previous findings (4), the ded1-57 mutation, affecting DEAD box RNA helicase (Ded1) (8), has a greater effect on expression of the reporter with the long 5′ UTR, lowering the ratio of L2-LUC to L0-LUC expression by 45% after cells were shifted to 36°C. Interestingly, all three tif32 mutations have effects similar to those of the ded1-57 mutation. While it is conceivable that a longer 5′ UTR would reduce the efficiency of 43S PIC attachment, we think it is more likely that the tif32 mutations disproportionately reduce expression of the L2-LUC reporter because they diminish the processivity of ribosomal scanning (Fig. 5 A).
FIG. 5.

Evidence that a/HLD substitutions confer scanning defects in vivo. (A) Effects of a/HLD mutations on expression of LUC reporters with 5′ UTRs of different lengths. The yeast strains in Fig. 1C and H3774 (ded1-57), harboring the plasmid-borne L0LUC or L2LUC reporters under the control of the GAL1 promoter with the indicated 5′ UTR lengths, were grown in SD supplemented with adenine, histidine, and tryptophan at 30°C to an A600 of ∼0.6, shifted to the same medium containing galactose instead of glucose, and incubated at 36°C for 6 h. Luciferase activities were assayed in WCEs, and the means and SE from 6 independent transformants for L2LUC were normalized to the corresponding values for L0LUC. (B) The yeast strains from panel A were transformed with GCN4-lacZ reporter plasmid p227 (i), pWCB07 (ii), or pWCB06 (iii), indicated schematically as in Fig. 4B, except that constructs ii and iii contained the indicated sequences inserted 21 nt 5′ of the GCN4 AUG codon, with complementary bases underlined. The cells were cultured in SD+HW at 33°C for 6 h, and β-galactosidase activities were assayed in WCEs of four independent transformants. Means ± SE (n = 4) and activities as percentages of WT values are indicated. The values in the columns “box6 norm.” and “ded1 norm.” are the results from columns “box6” and “ded1-57” normalized to correct for the different expression levels of construct i in box6 or ded1-57 versus WT cells.
We also examined the effects of the tif32 mutations on the inhibitory effect of a stem-loop structure inserted in the 5′ UTR of the GCN4-lacZ construct lacking a uORF, which diminished translation in WT cells by a factor of ∼8 (Fig. 5B, WT, iii versus i). Whereas the H725P and R731I mutations did not affect sensitivity to the stem-loop, box6 increased the inhibitory effect 4-fold (Fig. 5B, iii, WT versus box6 norm.). Importantly, box6 had a substantially smaller effect (∼40% reduction) on a construct harboring an insertion with alterations eliminating 2 of the 7 predicted base pairs of the stem-loop (Fig. 5B, ii, WT versus box6 norm.), which dramatically reduced the predicted minimum free energy of the inserted hairpin structure (ΔH) and inhibitory effect of the insertion in WT cells (Fig. 5B, WT, ii versus iii). Interestingly, the ded1-57 mutation also greatly increased sensitivity to the more stable stem-loop, by a factor of ∼5, after normalizing for its effect on expression of the parental construct lacking uORF. Thus, in the several different assays shown in Fig. 4 and 5, the H725P, R731I, and box6 mutations exhibited defects in scanning and efficient AUG selection, with H725P and R731I showing similar effects and differing in some respects from box6.
a/HLD substitutions suppress the enhanced UUG initiation conferred by dominant Sui− mutations.
Previously, we showed that eIF1A mutations that exhibit Gcn− phenotypes and increase leaky scanning of GCN4 uORF1, like the tif32 a/HLD mutants, also exhibit Ssu− phenotypes, suppressing initiation at UUG start codons in Sui− mutants (13). The Sui− phenotype is manifested in strains containing the his4-301 allele, which lacks the ATG start codon and confers histidine auxotrophy (His− phenotype). By increasing initiation at the third (UUG) codon in his4-301 mRNA, Sui− mutations allow growth on medium lacking histidine (−His). As shown previously, the dominant Sui− allele SUI5 (encoding the eIF5-G31R mutant) allowed TIF32+ his4-301 cells to grow in −His medium (Fig. 6 A, rows 1 and 2). Interestingly, the His+ phenotype of SUI5 was diminished by the H725P and R731I mutations at 30°C (Fig. 6A, rows 4 and 6 versus 2), suggesting that these tif32 mutations have Ssu− phenotypes at the permissive growth temperature. The box6 mutation is synthetically lethal with SUI5, and H725P and R731I cells are barely viable at 33°C in the presence of SUI5 (data not shown). Given that SUI5 reduces the efficiency of AUG initiation, as well as increasing the frequency of UUG initiation (26), these observations might be explained by proposing that the combined deleterious effects of SUI5 and the tif32 mutations on AUG recognition greatly reduce cell viability.
FIG. 6.

a/HLD mutations suppress UUG initiation in Sui− mutants. (A) Suppression of His+/Sui− phenotypes of SUI5. Serial dilutions of his4-301 strains in Fig. 1C harboring empty vector or a SUI5 plasmid were grown at 30°C on SD+H for 3 days or SD for 7 days. (B and C) Suppression of increased UUG/AUG initiation ratio. Strains from Fig. 1C carrying empty vector or the SUI5, SUI3-2, or tif11Δ125-153 plasmid and harboring HIS4-lacZ reporter plasmids with an AUG or UUG start codon were grown in SD+H medium and assayed for β-galactosidase activity in WCEs. The mean ratios and SE of UUG versus AUG reporter expression from six independent transformants are shown, with the fold increases relative to the WT for the vector transformants of each strain in the column “vector” and the fold increases relative to vector for each plasmid-borne Sui− allele in each strain in the remaining three columns.
To confirm the Ssu− phenotypes of H725P and R731I mutants, we measured the expression of matched HIS4-lacZ reporters containing AUG or UUG start codons. Indeed, all three tif32 mutations completely eliminated the ability of SUI5 and two other dominant Sui− mutations affecting eIF2β (SUI3-2) or eIF1A (tif11Δ125-153) to increase the efficiency of UUG initiation. Thus, in the TIF32+ strain, the dominant Sui− mutations increased the UUG/AUG initiation ratio by factors of 2.5 to 5 but had little effect on the UUG/AUG ratio in the tif32 mutants (Fig. 6B and C). Interestingly, the tif32 mutations by themselves produced an ∼2-fold increase in the UUG/AUG ratio in cells bearing an empty vector, suggesting that, besides being Ssu−, they also confer a moderate Sui− phenotype in otherwise WT cells (Fig. 6B and C, vector). Examination of the absolute expression levels of the UUG and AUG reporters (data not shown) revealed that the H725P and R731I mutations lowered the expression of both reporters but had a greater effect on AUG than on UUG initiation (increasing the UUG/AUG ratio) in otherwise WT cells and had a greater effect on UUG than AUG (reducing the UUG/AUG ratio) in the SUI5 background. Together, these findings indicate that the tif32 mutations impair one or more mechanisms that ensure accurate selection of AUG start codons, increasing the relative frequency of UUG initiation on their own but completely masking the effects of Sui− alterations in eIF2, eIF5, and eIF1A on the UUG/AUG ratio.
tif32 KERR mutations reduce j/Hcr1 association with eIF3 in a manner contributing to their growth phenotypes.
In view of our findings that the a/Tif32 KERR substitutions do not affect MFC binding to native 40S subunits (Fig. 3A and B), we wondered whether they might alter intersubunit interactions within eIF3 as a way of impairing its functions in mRNA recruitment, scanning, or AUG recognition in the 48S PIC. Accordingly, we measured the copurification of MFC components with His8-tagged a/Tif32 in nickel chelation chromatography of WCEs. Following growth at 36°C, the H725P, R731I, and box6 mutants all displayed significant reductions in copurifying j/Hcr1 (relative to the yield of His8-a/Tif32), with little additional effect on other MFC components, except for an ∼50% reduction in the amount of eIF2 associated with the complex (Fig. 7 A and B).
FIG. 7.

a/HLD substitutions weaken interactions of eIF3 with j/Hcr1 and eIF2. (A) Strains described in Fig. 1C were cultured in YPD medium at 30°C and shifted to 36°C for 6 h. WCEs were incubated with Ni-NTA-silica resin, and bound proteins were eluted and subjected to Western blot analysis with antibodies against the His8 epitope (for a/Tif32) or against the factors listed on the right. Three percent of input WCE (In), 15% and 30% of the eluate (1× and 2×), and 3% of flowthrough (FT) were analyzed in successive lanes. (B) Western signals for each factor in the eluates from panel A were normalized to that of His8-a/Tif32, and the ratio of each tif32 mutant to WT was plotted (means ± SE; n = 3).
It was shown previously that overexpressing j/Hcr1 from an hc plasmid partially suppressed the Ts− phenotype of tif32-R731I (46). As all three substitutions in the a/HLD reduce j/Hcr1 association with eIF3, we asked whether the growth defects conferred by H725P and box6 are also suppressible by hc HCR1. Interestingly, hc HCR1 partially suppressed the Ts− phenotype of H725P, as well as R731I, but not the Slg− phenotype of the box6 mutant (Fig. 8 A). Consistent with this, hc HCR1 increased the yield of j/Hcr1 copurifying with His8-tagged a/Tif32 from H725P and R731I cells, but not from box6 or WT cells (Fig. 8B to E, cf. lanes 2 to 3 versus 6 to 7 in the blots and adjoining histograms). These findings suggest that the initiation defects conferred by H725P and R731I can be partially corrected by restoring j/Hcr1 association with eIF3 in the PIC.
FIG. 8.

Overexpression of j/Hcr1 partially suppresses the Ts− phenotypes of tif32 KERR mutants and restores j/Hcr1 association with eIF3. (A) Serial dilutions of the strains from Fig. 1C harboring an hc empty vector or hc HCR1 plasmid were spotted on SD+HWU and incubated at 30°C or 36°C for 3 days. (B to E) Nickel chelation chromatography of WCEs from strains in panel A was conducted as for Fig. 7A. Western signals were quantified, and the ratios of normalized values for hc HCR1 versus empty vector are plotted in the histograms (means ± SE; n = 3).
The fact that the H725P and R731I mutations reduce the amounts of j/Hcr1 and eIF2γ that copurify with His8-a/Tif32 (Fig. 7A and B), but not the amounts bound to 40S subunits in vivo, can be explained by proposing that the mutations impair only a subset of the interactions that stabilize j/Hcr1 and eIF2 association with eIF3, eIF5, and eIF1 in the MFC. Loss of these interactions is sufficient to allow j/Hcr1 and eIF2 to dissociate from eIF3 when the factors are free of the ribosome during affinity purification in vitro, but not to provoke j/Hcr1 and eIF2 dissociation from native PICs in vivo, owing to their independent interactions with the 40S subunit and the ability of the ribosome to bridge association among MFC components (47, 48).
Given that the tif32 mutations alter the j/Hcr1-like region of a/Tif32, we explored what regions in overexpressed j/Hcr1 are required to suppress the growth defects of the KERR mutants. The N-terminal 135 residues of j/Hcr1 are necessary and sufficient to complement the Slg− phenotype in hcr1Δ TIF32+ cells (10), and consistent with this, eliminating the j/Hcr1 NTD (j/NTD) in the hcr1-CTD allele eliminates the ability of hc HCR1 to suppress the tif32 KERR mutants containing chromosomal HCR1+ (Table 3, column 5). Interestingly, although eliminating the j/Hcr1 CTD (j/CTD) in the hcr1-NTD allele has no effect on complementation of hcr1Δ (10), it destroys the suppressor activity of hc HCR1 (Table 3, column 3). As noted above, the j/CTD contains sequences similar to box6 and the box9/KERR motif of a/Tif32 (Fig. 1A; see Fig. 10A). Introducing the 10-Ala substitution of box6 into j/Hcr1 by the hcr1-box6 allele also abolished the ability of hc HCR1 to suppress the tif32 KERR mutants without affecting complementation of the hcr1Δ mutant (Table 3, column 4). Thus, both the critical NTD and the dispensable CTD of j/Hcr1, including the box6 element, are required for the ability of overexpressed j/Hcr1 to boost translation initiation in the tif32 KERR mutants.
TABLE 3.
Effects of HCR1 mutations on suppression of the Ts− phenotypes of tif32-R731I and tif32-H725P mutantsa
| Strain | Growth |
||||
|---|---|---|---|---|---|
| Empty vector | hc HCR1 | hc hcr1-NTD | hc hcr1-box6 | hc hcr1-CTD | |
| tif32-R731I HCR1 | − | + | − | − | − |
| tif32-H725P HCR1 | − | + | − | − | − |
| TIF32 hcr1Δ | − | + | + | + | − |
tif32Δ HCR1+ strains YSW731 and YSW725 harboring pRS-a/tif32-R731I-His-L (tif32-R731I HCR1) and pWLCB01 (tif32-H725P HCR1), respectively, or hcr1Δ strain H428 (TIF32 hcr1Δ), were transformed with YEplac195 (Empty vector), YEp-j/HCR1-DS-U (Hc HCR1), YEp-j/hcr1-NTD-U (Hc hcr1-NTD), YEp-j/hcr1-box6-U (Hc hcr1-box6), and YEp-j/hcr1-CTD-U (Hc hcr1-CTD) and examined for growth at 36°C as described in the legend to Fig. 8A, and the results are summarized qualitatively (+, growth; −, no growth).
FIG. 10.

Effects of box6 and KERR substitutions in j/CTD and a/HLD on binary interactions in the b/RRM-j/Hcr1-a/Tif32-CTD module and leaky scanning of GCN4 uAUG-1. (A) Schematic of j/Hcr1 showing the positions of the N-terminal acidic (nta) and KERR (kerr) motifs, with arrows delimiting minimal binding domains for the indicated proteins. Shown below are sequence alignments of the box6 and KERR segments of j/Hcr1 and a/Tif32, indicating identical (*) or conserved (:) positions, the KERR residues (underlined), and substitutions present in the indicated hcr1 mutants. Lowercase letters indicate j/Hcr1 residues that were subjected to site-directed mutagenesis. (B) Summary of molecular interactions of components of the b/RRM-j/Hcr1-a/Tif32-CTD module. The shaded rectangles represent the b/RRM, a/HLD, and full-length j/Hcr1. The solid arrow indicates interaction between the NTA motif of j/Hcr1 and helix α1 and loop L5 of the b/RRM, whose structural determinants are known. The dashed arrows depict other interactions that were mapped previously or determined in this study. The results presented here establish the roles of box6 and KERR residues in the a/HLD in binding to the b/RRM and of the equivalent residues in j/CTD in binding a/Tif32 regions flanking the a/HLD. (C) Both the NTD and CTD of j/Hcr1 are required for binding to a/Tif32 in vitro. GST fusions to full-length j/Hcr1 (lane 3), the j/NTD (lane 4) or j/CTD (lane 5), or GST alone (lane 2), were tested for binding to full-length 35S-a/TIF32 in pulldown assays. The GST fusions visualized by Coomassie blue staining (top) and the 35S-a/TIF32 visualized by autoradiography (bottom) in the bound fractions are shown in lanes 2 to 5. Lane 1 contains 10% of the input 35S-a/TIF32. (D) The KERR motif and box6 of j/Hcr1 are critical for its binding to a/Tif32. Same as panel C, except that GST fusions to full-length j/Hcr1 (lane 3) or its box6 (lane 4), box9 (lane 5), or R215I (lane 6) mutant derivatives were examined. (E) Substitutions in the KERR motif and box6 in the a/HLD strongly reduce its binding to the b/RRM. A GST fusion to the b/RRM (aa 1 to 136) or GST alone was tested for binding to the indicated WT and mutant derivatives of 35S-a/HLD (aa 490 to 790). (F) Leaky-scanning phenotypes of substitutions in box6 or KERR residues of j/Hcr1 or a/Tif32. (Top) Transformants of hcr1Δ strain H428 bearing plasmid YEp-j/HCR1, YEp-j/hcr1-NTD, YEp-j/hcr1-R215I, YEp-j/hcr1-Box6, or YEp-j/hcr1-Box6-R215I and containing GCN4-lacZ reporter plasmid pM226 were analyzed for β-galactosidase activities as for Fig. 4B (iii). Mean values and standard errors from 6 or more measurements of three transformants containing pM226 are shown, along with activities in the hcr1 strains normalized to that in the HCR1 strain. All values were normalized to correct for any differences among the strains in expression of the reporter lacking uORF on p227. (Bottom) Same as above, but using AY51 (TIF32-His) or AY52 (tif32-R731I-His) transformants bearing YEp-j/HCR1-W or YEp-j/hcr1-R215I-DS-W, respectively.
Having found that suppression of the tif32 KERR mutations by hc WT HCR1 is associated with increased interaction of j/Hcr1 with eIF3, we examined whether the loss of dosage suppression by hc HCR1 conferred by different hcr1 mutations is associated with diminished interaction of the cognate mutant j/Hcr1 proteins with eIF3. In fact, we showed recently that destroying the specific contact between the b/Prt1 RRM and j/Hcr1 by site-specific mutations abolished association of j/Hcr1 with His8-tagged b/Prt1 and other MFC components in WCEs (10). Hence, as expected, removing the entire NTD (in construct hcr1-CTD) also completely eliminated j/HCR1 from the MFC (Fig. 9 A, lanes 14 and 15 versus 6 and 7 and adjoining histogram). Likewise, eliminating j/CTD in j/hcr1-NTD, substituting box6, or altering the KERR motif with a substitution (R215I) equivalent to R731I in a/Tif32 all had similar effects of impairing association of the mutant j/Hcr1 proteins with His8-b/Prt1 (Fig. 9A, lanes 10 and 11 versus 6 and 7). The results in Table 3 and Fig. 9 together indicate that both the NTD and CTD of j/Hcr1 are required for its tight binding to eIF3 and the ability to suppress a/Tif32 KERR substitutions when overexpressed.
FIG. 9.

The NTD, box6, and KERR motif in the CTD of j/Hcr1 are required for its association with eIF3. (A) WCEs were prepared from a transformant of YAH05 (prt1Δ hcr1Δ pRS316-PRT1 [PRT1 URA3]) containing plasmid YEp-j/HCR1-DS (lanes 1 to 4) or from YAH05 derivatives lacking pRS316-PRT1 and containing pRS-b/PRT1-His and YEp-j/HCR1-DS-U (lanes 5 to 8), pRS-b/PRT1-His and YEp-j/hcr1-NTD-U (lanes 9 to 12), pRS-b/PRT1-His and YEp-j/hcr1-CTD-U (lanes 13 to 16), or pRS-b/PRT1-His and YEp-j/hcr1-box6-U (lanes 17 to 20), cultured in SD at 30°C. Nickel chelation chromatography and Western blot analysis were conducted as for Fig. 7A, except that 5% of input WCEs (In), 30% (E1) or 60% (E2) of the eluates, and 5% of flowthrough (FT) was loaded. Mean Western signals were normalized to those of b/Prt1-His8 and plotted as percentages of the corresponding values calculated for the HCR1+ strain. (B) Same as panel A, except that WCEs were prepared from a YAH05 transformant harboring YCp-j/HCR1-DS-L (lanes 1 to 4) or YAH05 derivatives lacking pRS316-PRT1 and containing pRS-b/PRT1-His and YCp-j/HCR1-DS-U (lanes 5 to 8) or pRS-b/PRT1-His and YCp-j/hcr1-R215I-U (lanes 9 to 12).
Interestingly, all of the j/Hcr1 mutations we examined reduced the amount of eIF2γ copurifying with His8-b/Prt1 (Fig. 9), similar to our findings above that KERR and box6 substitutions in a/Tif32 reduce copurification of both j/Hcr1 and eIF2γ with His8-a/Tif32. We showed previously that the extreme CTD of a/Tif32 is critical for eIF2 and j/Hcr1 association with eIF3 in the MFC in vivo (48). Perhaps binding of j/Hcr1 to the extreme CTD of a/Tif32 is required to “prime” the conformation of this domain for stable interaction with eIF2. In this view, loss of eIF2 from the MFC caused by mutations in j/Hcr1 or the a/HLD is an indirect consequence of impaired j/Hcr1 association with the a/Tif32 CTD.
We showed recently that overexpressing the three subunits of eIF2 and 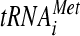 , the macromolecular components of the TC, partially suppresses the growth defect of hcr1Δ cells (10), consistent with the contribution of j/Hcr1 to proper association of the TC with the MFC. In contrast, we found that overexpressing the components of the TC does not suppress the growth defects of the a/HLD substitutions (data not shown), suggesting that weakened interaction of the TC with the MFC is not the rate-limiting defect in these tif32 mutants. As discussed below, this is consistent with the fact that the a/HLD substitutions impair certain aspects of initiation that are unaffected by elimination of j/Hcr1.
, the macromolecular components of the TC, partially suppresses the growth defect of hcr1Δ cells (10), consistent with the contribution of j/Hcr1 to proper association of the TC with the MFC. In contrast, we found that overexpressing the components of the TC does not suppress the growth defects of the a/HLD substitutions (data not shown), suggesting that weakened interaction of the TC with the MFC is not the rate-limiting defect in these tif32 mutants. As discussed below, this is consistent with the fact that the a/HLD substitutions impair certain aspects of initiation that are unaffected by elimination of j/Hcr1.
Mutations in both the NTD and CTD of j/Hcr1 exacerbate the growth defects of a/HLD substitutions.
In addition to the fact that overexpressing j/Hcr1 partially suppresses the Ts− phenotype of the tif32-R731I HCR1+ mutant, it was shown previously that deleting HCR1 exacerbates the effect of this tif32 mutation on cell growth (46), producing a stronger Slg− phenotype at 30°C than was displayed by either single mutant (Table 4, rows 1 and 2 and columns 1 and 2). We observed the same synthetic sick interaction on combining hcr1Δ with the tif32-box6 or tif32-H725P mutation (Table 4, rows 3 and 4 and columns 1 and 2). As might be expected, absence of the critical NTD in the hcr1-CTD allele also produced synthetic sick interactions with tif32-R731I and tif32-box6 (Table 4, rows 1 to 3 and column 3 versus 1). Importantly, both the absence of the normally dispensable CTD in hcr1-NTD and the hcr1-box6 mutation likewise exacerbated the Slg− phenotypes of the tif32 mutations (Table 4, columns 5 and 6 versus column 1). Moreover, the KERR mutation hcr1-R215I further exacerbated the effect of hcr1-box6, as revealed in the hcr1-box6-R215I tif32 triple mutants (Table 4, columns 4 and 7 versus column 1).
TABLE 4.
Synthetic sick phenotypes of tif32 hcr1 double mutantsa
| Transformant | Growthb |
||||||
|---|---|---|---|---|---|---|---|
| WT | None (vector) | hcr1-CTD | hcr1-R215I | hcr1-box6 | hcr1-NTD | hcr1-box6-R215I | |
| TIF32 | 8+ | 5+ | 6+ | 8+ | 8+ | 8+ | 8+ |
| tif32-R731I | 4+ | 1+ | 1+ | 4+ | 3+ | 2+ | 1+ |
| tif32-box6 | 5+ | 2+ | 2+ | 5+ | 2+ | 3+ | 2+ |
| tif32-H725P | 5+ | 1+ | ND | 5+ | 3+ | ND | 1+ |
Transformants of tif32Δ hcr1Δ strain YAH04 harboring combinations of pRS-a/TIF32-His-L (TIF32), pRS-a/tif32-R731I-His-L (tif32-R731I), pRS-a/tif32-box6-His-L (tif32-box6), and pWLCB01 (tif32-H725P) with YEp-j/HCR1-DS-U (WT), YEplac195 (None), YEp-j/hcr1-CTD-U (hcr1-CTD), YEp-j/hcr1-R215I-U (hcr1-R215I), YEp-j/hcr1-box6-U (hcr1-box6), YEp-j/hcr1-NTD-U (hcr1-NTD), and YEp-j/hcr1-box6-R215I-U (hcr1-box6-R215I), from which the original covering plasmid carrying TIF32 URA3 was evicted on 5-FOA, were examined for growth at 30°C (for tif32-R731I and tif32-H725P) or 37°C (for tif32-box6), and the results are summarized qualitatively. Note that the tif32 mutant alleles confer a Slg− phenotype at 30°C in this strain background that is not evident in the strains shown in Fig. 1.
8+, WT growth; 1+, the lowest growth rate observed; 2+ to 7+, different degrees of growth discernible between these two extremes.
The foregoing results indicate that, even though the j/CTD is dispensable for growth in WT cells, it is critically required, together with the j/NTD, when the a/HLD is compromised and that both the box6 and KERR elements in j/Hcr1 contribute to its ability to complement tif32 mutations. As shown in Table 3, both the j/CTD and j/NTD are also required for the ability of overexpressed j/Hcr1 to suppress tif32 KERR mutations. These findings imply that the NTD and CTD of j/Hcr1 either functionally substitute for an activity of the a/HLD or enhance the ability of the a/HLD to carry out its function when impaired by KERR or box6 substitutions.
Substitutions in the KERR and box6 elements in both a/Tif32 and j/Hcr1 destabilize the b/RRM-j/Hcr1-a/Tif32-CTD module.
Previously, we showed that both j/Hcr1 and a/Tif32 can interact directly with the RRM domain in b/Prt1, so that the b/RRM can bridge a/Tif32-j/Hcr1 interaction (50), and we showed recently that the NTD of j/Hcr1 mediates its direct interaction with the b/RRM (10). a/Tif32 and j/Hcr1 can also interact directly, but the domains involved were not determined, except that regions both N terminal and C terminal to the a/HLD contribute to the interaction (50) (summarized in Fig. 10 B). To gain further insights into the consequences of a/HLD and j/Hcr1 mutations for eIF3 integrity, we examined their effects on interactions between different components of the b/RRM-j/Hcr1-a/Tif32-CTD module.
As observed previously (50), in vitro-translated 35S-labeled a/Tif32 interacts specifically with GST-j/Hcr1 in a pulldown assay (Fig. 10C, lanes 1 to 3). Absence of the CTD in the GST-j/Hcr1-NTD, or absence of the NTD in GST-j/Hcr1-CTD, essentially eliminates the interaction with a/Tif32 (Fig. 10C, lanes 3 to 5). Consistent with this, substitutions in box6, box9, or R215I in the j/CTD strongly impair GST-j/Hcr1 binding to a/Tif32 (Fig. 10D). Thus, whereas the j/NTD is critical for binding the b/RRM (10), the j/CTD, including its box6 and KERR elements, is more crucial for j/Hcr1-a/Tif32 interaction.
Consistent with previous findings that the a/HLD is not required for j/Hcr1-a/Tif32 interaction (50), we observed no effect of box6 or KERR substitutions in a/TIF32 on its binding to j/Hcr1 in vitro (data not shown). Interestingly, however, these substitutions impair binding of the a/HLD to a GST-b/Prt1-RRM fusion (Fig. 10E), indicating that the box6 and KERR elements in the a/HLD are crucial for its direct binding to the b/RRM. Together, the binding data in Fig. 10 indicate that substitutions in the a/HLD and j/CTD impair distinct interactions in the b/RRM-j/Hcr1-a/Tif32-CTD module. These destabilizing effects could be a contributing factor in the synthetic sick interactions produced by combining tif32 box6 or KERR mutations with mutations affecting the NTD or CTD of j/Hcr1.
It is intriguing that the a/HLD substitutions impair j/Hcr1 association with eIF3 (Fig. 7A), even though, as described above, they do not affect direct j/Hcr1-a/Tif32 interaction but disrupt binding of the a/HLD to the b/RRM instead. To explain this finding, it could be proposed that the a/HLD-b/RRM interaction stabilizes the conformation of the b/RRM competent for binding the j/NTD and that the tif32 KERR mutations disrupt this indirect contribution of the a/HLD to j/Hcr1-b/RRM interaction. The fact that the c/Nip1 subunit of eIF3 stabilizes interaction between a/Tif32 and b/Prt1 by binding to both of these subunits (48) could explain why association of b/Prt1 with eIF3 is not reduced by the a/Tif32 KERR substitutions (Fig. 7A and B), despite the fact that they weaken the a/HLD-b/RRM contact.
Integrity of the b/RRM-j/Hcr1-a/Tif32-CTD module is required to prevent leaky scanning.
It was of interest to determine whether destabilizing the b/RRM-j/Hcr1-a/Tif32-CTD module in different ways would impair the same steps in translation initiation. This possibility is supported by our previous finding that impairing the j/NTD-b/RRM interaction leads to increased leaky scanning of the elongated version of GCN4 uORF1, the same phenotype described here for the box6 and KERR substitutions in the a/HLD. Thus, 4- to 5-fold increases in leaky scanning were observed for substitutions in the b/RRM, including the RNP1 motif (32) and the j/NTD interaction surface in alpha helix 1 and loop 5 of the b/RRM, and an ∼8-fold increase occurred with elimination of the j/NTD by the hcr1-CTD mutation (10). Hence, we investigated whether mutations in the j/CTD affecting its direct interaction with a/Tif32 would likewise increase leaky scanning.
Eliminating the j/CTD (in hcr1-NTD) or replacing its box6 or KERR elements produced only small increases of 1.5- to 2-fold in leaky scanning, much less than the ∼8-fold increase observed on eliminating the j/NTD (in hcr1-CTD) (10). Interestingly, combining the box6 and R215I substitutions in the hcr1-box6 R215 double mutant increased leaky scanning by a factor of ∼4 (Fig. 10F). Given that the j/CTD is largely dispensable for WT recognition of GCN4 uAUG-1, it is possible that the j/hcr1-box6 R215I mutant protein interferes with the function of the a/HLD in a way that reduces recognition of uAUG-1. Combining hcr1-R215I with tif32-R731I in a double mutant did not produce a statistically significant increase in leaky scanning compared to that seen for each single mutant (Fig. 10F), consistent with the possibility that hcr1-R215I increases leaky scanning by interfering with the function of the a/HLD. In any event, it seems clear that the j/CTD and its interaction with a/Tif32 is less important than the j/NTD-b/RRM interaction for efficient AUG recognition.
Given that the a/HLD substitutions reduce mRNA recruitment by native 43S complexes (Fig. 3D), we wondered if disrupting the b/RRM-j/Hcr1 interaction would also impair this step of PIC assembly. At odds with this possibility, there is no defect in RPL41A mRNA binding to native 43S complexes in hcr1Δ cells (data not shown). Consistent with this, we found previously that replacing RNP1 of the b/RRM had no effect on 40S binding of RPL41A mRNA (32). Thus, the function of the a/HLD in mRNA recruitment does not require an intact b/RRM-j/NTD interaction. A similar conclusion holds for the role of the a/HLD in suppressing UUG initiation (Ssu− phenotype), as we found that hcr1Δ does not significantly reduce the elevated UUG/AUG initiation ratio conferred by the dominant Sui− alleles SUI5, SUI3-2, and tif11Δ-125-153 (data not shown). Together, these findings indicate that disrupting different components of the b/RRM-j/Hcr1-a/Tif32-CTD module can have differential effects on individual steps of the initiation pathway.
The extreme CTD of a/Tif32 interacts with the 40S proteins Rps2 and Rps3.
We showed previously that the extreme CTD of a/Tif32 (residues 791 to 964) can bind directly to helices 16 to 18 of 18S rRNA (47). This interaction could implicate a/Tif32 in regulating the connection between h16 and Rps3 on the solvent side of the 40S subunit that stabilizes the open conformation of the latch of the mRNA channel (33). Consistent with this possibility, we found that the a/Tif32 CTD also interacts specifically with Rps3. Thus, a GST fusion to the a/Tif32 CTD (aa 791 to 964) binds to 35S-Rps3, but not to 35S-Rps0 (Fig. 11 A). (Rps0 was previously shown to interact with a/Tif32 NTD [47].) Remarkably, the GST-a/Tif32-CTD fusion also binds to Rps2 (Fig. 11A), which is situated near the mRNA entry channel adjacent to Rps3 on the solvent side of the 40S subunit (Fig. 11C, top). The a/Tif32 CTD does not bind to Rps23 (Fig. 11A), which resides on the interface side of the mRNA entry channel (41). Interaction of a/Tif32 with Rps2 was confirmed in pulldown assays using GST-Rps2 and in vitro-translated a/Tif32 (Fig. 11B). Interestingly, we found recently that the CTD of j/Hcr1 also binds to Rps2 in vitro, in a manner impaired by mutations in its KERR motif (10). Thus, it seems very likely that the entire b/RRM-j/Hcr1-a/Tif32-CTD module is situated near the mRNA entry channel pore.
FIG. 11.

a/Tif32 CTD interacts with Rps2 and Rps3 in vitro: a hypothetical model for binding of a/Tif32 CTD near the mRNA entry channel pore of the 40S subunit. (A and B) The extreme CTD of a/Tif32 interacts with Rps2 and Rps3 in vitro. (A) A GST fusion to the a/Tif32-CTD (aa 791 to 964) or GST alone was tested for binding to 35S-labeled full-length Rps2, Rps3, Rps0, or Rps23, as shown in Fig. 10C. (B) Same as panel A, except that Rps2 fused to GST was examined for binding to 35S-a/Tif32. (C) Hypothetical location of eIF3 on the solvent side of the S. cerevisiae 40S subunit based on cryo-EM reconstruction (adapted from reference 47). The 40S subunit is shown from the solvent side, with RNA segments in yellow and proteins in green. The positions of Rps2, Rps3, helices 16 and 18 of 18S rRNA, the a/HLD and CTD of a/Tif32, the b/RRM, and j/Hcr1 are highlighted in color and/or boldface. The mRNA exit channel is indicated by an arrow. The blue lines represent mRNA. The positions of Rps2 and Rps3 were modified according to reference 43.
DISCUSSION
In this study, we showed that substitutions in the KERR motif (H725P and R731I) and Ala substitution for nearby residues 692 to 701 (box6) in the a/HLD impair its essential function in translation initiation, at least partly by reducing the recruitment of RPL41A mRNA to native 40S subunits in vivo. We reported previously that depletion of the entire eIF3 complex from cells impaired RPL41A mRNA recruitment in vivo; however, TC binding to native 40S subunits also was reduced (23). Because base pairing of 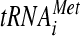 with AUG stabilizes 48S PICs (27, 36), a large portion of the defect in mRNA recruitment in cells depleted of eIF3 could be secondary to reduced TC recruitment. As the tif32 KERR substitutions reduce 40S binding of RPL41A mRNA without diminishing 43S complexes, our results provide the strongest evidence yet that eIF3, and a/Tif32 in particular, has a direct role in mRNA recruitment by 43S complexes in vivo.
with AUG stabilizes 48S PICs (27, 36), a large portion of the defect in mRNA recruitment in cells depleted of eIF3 could be secondary to reduced TC recruitment. As the tif32 KERR substitutions reduce 40S binding of RPL41A mRNA without diminishing 43S complexes, our results provide the strongest evidence yet that eIF3, and a/Tif32 in particular, has a direct role in mRNA recruitment by 43S complexes in vivo.
In addition to impairing mRNA recruitment, replacing the KERR motif or box6 in a/Tif32 produces phenotypes indicating reduced efficiency of scanning or AUG recognition. First, these mutations preferentially decrease translation of a luciferase reporter with a long 5′ UTR, resembling a substitution in DEAD box helicase Ded1 (4), suggesting that the a/HLD enhances scanning processivity. The box6 mutation also resembles the ded1-57 mutation in reducing the ability to scan through a stem-loop structure inserted in the GCN4 mRNA leader ∼400 nt from the 5′ end.
Second, the KERR and box substitutions strongly impair the derepression of GCN4 translation (Gcn− phenotype) without affecting eIF2α phosphorylation, indicating that they prevent reinitiating ribosomes from bypassing the inhibitory uORF2 to -4 in cells with reduced TC levels. We ruled out the possibility that posttermination 40S subunits cannot resume scanning following translation of uORF1 but observed a moderate increase in leaky scanning of uAUG-1 in the KERR and box6 mutants. The R731I KERR mutation also seems to reduce the rate of scanning by posttermination 40S subunits between uORF1 and -4. It seems unlikely, however, that the magnitude of these two defects can account for the nearly complete block to induction of GCN4 translation observed in these mutants. Hence, we presume that the KERR and box6 substitutions produce additional defects that depend on the presence of 48S PICs at uAUG-2 to -4 in order to be expressed and thus cannot be detected with GCN4-lacZ reporters containing uORF1 alone.
The box6 and KERR substitutions in a/Tif32 also affect the accuracy of start codon recognition, as they completely suppress the ability of dominant Sui− mutations in eIF5, eIF2β, and eIF1A to increase the UUG/AUG ratio—a pronounced Ssu− phenotype. Previously, we described eIF1A mutations that resemble the a/HLD substitutions in conferring both increased leaky scanning of GCN4 uAUG-1 and Ssu− phenotypes (13) and proposed that such mutations destabilize the closed, scanning-incompatible conformation of the 48S PIC with AUG or UUG in the P site. The decreased ratio of UUG to AUG initiation conferred by Ssu− mutations implies that the defect in accessing the closed conformation is more pronounced at UUG than at AUG. Thus, the a/HLD substitutions could likewise destabilize the closed PIC conformation as a means of reducing recognition of uAUG-1 in otherwise WT cells and of UUG start codons in Sui− mutants, such as SUI5. Examining the matched HIS4-lacZ reporters revealed that, in otherwise WT cells, the a/HLD KERR substitutions reduce initiation at both AUG and UUG but have a greater effect at AUG, producing a weak Sui− phenotype. This trend is reversed in SUI5 and other Sui− mutants, where the a/HLD mutations have a greater effect on UUG than on AUG initiation, conferring the Ssu− phenotype. Thus, it appears that the a/HLD substitutions impede the transition from the open to the closed conformation in a complex way, diminishing the greater probability of the transition at AUG in otherwise wild-type cells and overriding the effect of SUI5 and other Sui− mutations in boosting the transition at UUG codons.
The proposal that the a/HLD mutations destabilize the closed PIC conformation as the means of reducing start codon recognition is consistent with the fact that a/Tif32 interacts with components of the 40S subunit located near the mRNA entry channel pore on the solvent side of the 40S subunit. Thus, the extreme CTD of a/Tif32 can bind an rRNA segment containing h16-h18 (47), and we found here that it also binds Rps2 and Rps3 in vitro. Hydroxyl radical cleavage mapping of mammalian eIF3 in the 48S PIC is also consistent with the idea that a segment of eIF3 interacts with h16 (37). Interestingly, cryo-electron microscopy (EM) analysis suggests that Rps3 plays a key role in opening and closing the mRNA entry channel latch, stabilizing the closed position by interacting with h34 but interacting with h16 on the solvent side of the 40S subunit to promote the open-latch conformation (33). It is tempting to consider that the a/Tif32 CTD, by interacting with Rps3, h16, or h18, can modulate the mRNA entry channel latch as a way of influencing the transition from open to closed PIC conformations. If so, then tif32 a/HLD mutations could reduce the efficiency of uAUG-1 selection and of UUG selection in Sui− mutants by destabilizing the interaction of Rps3 with h34, or the h18-h34 connection, to impede latch closing on start codon recognition. The proximity of the a/Tif32-CTD to Rps2 and Rps3 at the mRNA entry channel pore might also underlie the ability of the a/HLD to promote mRNA recruitment, serving as an extension of the mRNA binding channel in the manner suggested for mammalian eIF3a and eIF3d (37) and yeast a/Tif32-NTD (42).
Interestingly, the tif32 a/HLD mutations also produce phenotypes suggesting the opposite effect of destabilizing the open, scanning-compatible conformation of the PIC, as they appear to reduce the processivity of scanning. To account for the seemingly opposite effects of the tif32 a/HLD mutations on scanning and start codon selection, it could be proposed that they reduce the rate or extent of switching in both directions between the fully open and fully closed conformations of the PIC.
The a/Tif32 KERR and box6 substitutions weaken interactions of j/Hcr1 with the eIF3 complex in WCEs. Both the growth defects and reduced j/Hcr1 association with eIF3 are partially rescued by overexpressing j/Hcr1, suggesting that the a/HLD mutations disrupt a critical function of a/Tif32 in a manner that can be mitigated by restoring j/Hcr1 association with eIF3. This conclusion is consistent with our finding that the hcr1-box6 mutation, or eliminating the entire CTD or NTD of j/Hcr1, impairs j/Hcr1 association with eIF3 and also destroys suppression of the a/Tif32 KERR mutants by overexpressed j/Hcr1. Given that the a/Tif32 KERR substitutions impair its direct binding to the b/RRM, one possibility is that partial suppression of these tif32 mutations by hc HCR1 involves the ability of j/Hcr1 to bridge interaction between a/Tif32 and the b/Prt1 RRM and to stabilize the b/RRM-j/Hcr1-a/Tif32-CTD module in mutant eIF3. This mechanism could also account for our finding that the growth defects conferred by the a/Tif32 KERR mutations are exacerbated by mutations affecting either the NTD or CTD of j/Hcr1 in cells lacking WT j/Hcr1.
An alternative explanation for the genetic interactions could be that j/Hcr1 and the a/HLD have overlapping functions in one or more steps of initiation. In this view, overexpressing WT j/Hcr1 increases its ability to functionally compensate for a/HLD defects in a manner dependent on both the NTD and CTD of j/Hcr1. Likewise, mutating either domain of j/Hcr1 would prevent it from complementing a/HLD defects in cells lacking WT j/Hcr1. According to this model, the j/CTD is required for robust growth only in the presence of a/HLD mutations, because their shared function can be fully executed by the wild-type a/HLD.
One line of evidence consistent with the model of overlapping functions is that increased leaky scanning is a phenotype conferred by mutations affecting either the a/HLD or j/Hcr1. Because we did not observe a significant additive effect on leaky scanning on combining equivalent KERR substitutions in the a/HLD and the j/CTD, it is possible that these related segments act coordinately and that each single KERR substitution disrupts most of the activity they provide as a unit. Considering that the strongest effects on leaky scanning have been observed by eliminating the j/NTD-b/RRM interaction (10), and given our finding that a/HLD substitutions weaken j/Hcr1 association with eIF3, it could be argued that the leaky-scanning phenotype of the a/HLD mutations is an indirect consequence of reduced interaction of j/Hcr1 with the b/RRM. This might be unlikely, however, as we found that overexpressing WT j/Hcr1 did not mitigate the leaky-scanning defect in tif32-R731I cells (data not shown). Hence, we favor the idea that both the j/NTD-b/RRM interaction and the box6/KERR elements of the a/HLD and j/CTD make independent contributions to efficient AUG recognition and to blocking of leaky scanning. As the CTD of j/Hcr1 also interacts with Rps2 (10), it is highly likely that the entire b/Prt1-RRM-j/Hcr1-a/Tif32-CTD module is positioned near the mRNA entry channel by the multiple interactions of the j/CTD and a/Tif32 CTD with Rps2, Rps3, and h16-h18 (Fig. 11C). Accordingly, the leaky-scanning phenotypes of mutations in the b/RRM, j/Hcr1, or a/HLD might all involve destabilizing the closed conformation of the PIC at AUG codons.
Other findings indicate that the a/HLD and j/Hcr1 have distinct functions in translation initiation. Although related in sequence, the results presented here and elsewhere indicate clearly that they make different interactions within the b/Prt1-RRM-j/Hcr1-a/Tif32-CTD module. In addition, whereas a/HLD substitutions impair mRNA recruitment by 43S complexes and confer Ssu− phenotypes, neither defect was observed in hcr1Δ cells. The fact that hcr1Δ reduces recognition of uAUG-1 (10) but does not suppress UUG initiation in Sui− mutants seems to indicate that eliminating j/Hcr1 destabilizes the closed conformation equally with UUG or AUG in the P site so as not to alter the UUG/AUG initiation ratio in Sui− mutants. The fact that the tif32 mutations exert this effect preferentially at UUG start codons suggests a difference between j/Hcr1 and the a/HLD in how they regulate access to the closed PIC conformation. The fact that overexpressing TC partially suppresses the growth defect in hcr1Δ, but not in the tif32 mutant cells, suggests that the defects in mRNA recruitment and/or start codon selection conferred by the a/HLD substitutions (but lacking in hcr1Δ cells) contribute more than the impaired eIF2 association with the MFC to the overall reduction in translation rates in the tif32 mutants.
Another function that distinguishes the a/HLD from j/Hcr1 is the role of j/Hcr1 in the final stage of 40S ribosome biogenesis occurring in the cytoplasm (45). It could be proposed that the strong leaky-scanning phenotype conferred by hcr1Δ (10) results indirectly from the decreased levels of 40S subunits present in this mutant. To evaluate this possibility, we measured leaky scanning of GCN4 uAUG-1 (in the manner described in the legend to Fig. 4B) in three mutants harboring deletions of one of two duplicate genes encoding a 40S subunit protein, RPS14A, RPS18A, or RPS23A, all of which confer reductions in 40S subunit levels at least as great as those found in hcr1Δ cells (7, 34, 45). We found that all three rps mutants displayed moderate (20 to 40%) reductions, rather than an increase, in leaky scanning (data not shown). Hence, the leaky-scanning phenotype of hcr1Δ most likely reflects a direct role for j/Hcr1 in promoting start codon recognition.
Hydroxyl radical mapping data position the CTD of mammalian eIF3j in the 40S mRNA entry channel and A site on the interface side of the 40S subunit, and it was shown that eIF3j antagonizes 40S subunit binding by mRNA in a manner overcome by the bound TC. It was proposed that by interacting with the decoding center, the eIF3j CTD would coordinate binding of mRNA with initiation factors and modulate 40S subunit-mRNA interactions during scanning and AUG selection (14). Consistent with this, the j/Hcr1 CTD can bind in vitro to Rps23 (10), which is located near the A site and mRNA entry channel on the interface side of the 40S subunit (41). Given that interactions of the j/CTD with both Rps2 and Rps23 require its KERR motif (10), it seems plausible that the j/CTD “toggles” between 40S binding sites on Rps23 (on the interface side) and Rps2 (on the solvent side) in performing its proposed roles in regulating PIC assembly and scanning. In contrast, the a/Tif32 CTD binds to Rps2 and Rps3, but not Rps23, suggesting that the function(s) of a/Tif32 shared with, or modulated by, j/Hcr1 is executed from the solvent side of the 40S subunit.
Acknowledgments
We are grateful to the members of the Hinnebusch, Valášek, Dever, and Krásný laboratories for helpful comments. We thank Jean Fringer and Tom Dever for the rps mutants.
This work was supported in part by the Intramural Research Program of the NIH (A.G.H.) and by The Wellcome Trusts grant 076456/Z/05/Z, NIH research grant R01 TW007271 funded by Fogarty International Center, Czech Science Foundation 305/10/0335, and Inst. Research Concept AV0Z50200510 to L.V.
Footnotes
Published ahead of print on 28 June 2010.
REFERENCES
- 1.Algire, M. A., D. Maag, and J. R. Lorsch. 2005. Pi release from eIF2, not GTP hydrolysis, is the step controlled by start-site selection during eukaryotic translation initiation. Mol. Cell 20:251-262. [DOI] [PubMed] [Google Scholar]
- 2.Asano, K., J. Clayton, A. Shalev, and A. G. Hinnebusch. 2000. A multifactor complex of eukaryotic initiation factors eIF1, eIF2, eIF3, eIF5, and initiator tRNAMet is an important translation initiation intermediate in vivo. Genes Dev. 14:2534-2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asano, K., L. Phan, J. Anderson, and A. G. Hinnebusch. 1998. Complex formation by all five homologues of mammalian translation initiation factor 3 subunits from yeast Saccharomyces cerevisiae. J. Biol. Chem. 273:18573-18585. [DOI] [PubMed] [Google Scholar]
- 4.Berthelot, K., M. Muldoon, L. Rajkowitsch, J. Hughes, and J. E. McCarthy. 2004. Dynamics and processivity of 40S ribosome scanning on mRNA in yeast. Mol. Microbiol. 51:987-1001. [DOI] [PubMed] [Google Scholar]
- 5.Boeke, J. D., J. Trueheart, G. Natsoulis, and G. R. Fink. 1987. 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol. 154:164-175. [DOI] [PubMed] [Google Scholar]
- 6.Cheung, Y. N., D. Maag, S. F. Mitchell, C. A. Fekete, M. A. Algire, J. E. Takacs, N. Shirokikh, T. Pestova, J. R. Lorsch, and A. G. Hinnebusch. 2007. Dissociation of eIF1 from the 40S ribosomal subunit is a key step in start codon selection in vivo. Genes Dev. 21:1217-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiocchetti, A., J. Zhou, H. Zhu, T. Karl, O. Haubenreisser, M. Rinnerthaler, G. Heeren, K. Oender, J. Bauer, H. Hintner, M. Breitenbach, and L. Breitenbach-Koller. 2007. Ribosomal proteins Rpl10 and Rps6 are potent regulators of yeast replicative life span. Exp. Gerontol. 42:275-286. [DOI] [PubMed] [Google Scholar]
- 8.Chuang, R. Y., P. L. Weaver, Z. Liu, and T. H. Chang. 1997. Requirement of the DEAD-Box protein ded1p for messenger RNA translation. Science 275:1468-1471. [DOI] [PubMed] [Google Scholar]
- 9.Donahue, T. F., and A. M. Cigan. 1988. Genetic selection for mutations that reduce or abolish ribosomal recognition of the HIS4 translational initiator region. Mol. Cell. Biol. 8:2955-2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elantak, L., S. Wagner, A. Herrmannova, M. Karaskova, E. Rutkai, P. Lukavsky, and L. Valasek. 2010. The indispensable N-terminal half of eIF3j/HCR1 cooperates with its structurally conserved binding partner eIF3b/PRT1-RRM and with eIF1A in stringent AUG selection. J. Mol. Biol. 396:1097-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evans, D. R. H., C. Rasmussen, P. J. Hanic-Joyce, G. C. Johnston, R. A. Singer, and C. A. Barnes. 1995. Mutational analysis of the Prt1 protein subunit of yeast translation initiation factor 3. Mol. Cell. Biol. 15:4525-4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fekete, C. A., D. J. Applefield, S. A. Blakely, N. Shirokikh, T. Pestova, J. R. Lorsch, and A. G. Hinnebusch. 2005. The eIF1A C-terminal domain promotes initiation complex assembly, scanning and AUG selection in vivo. EMBO J. 24:3588-3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fekete, C. A., S. F. Mitchell, V. A. Cherkasova, D. Applefield, M. A. Algire, D. Maag, A. Saini, J. R. Lorsch, and A. G. Hinnebusch. 2007. N- and C-terminal residues of eIF1A have opposing effects on the fidelity of start codon selection. EMBO 26:1602-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fraser, C. S., K. E. Berry, J. W. Hershey, and J. A. Doudna. 2007. eIF3j is located in the decoding center of the human 40S ribosomal subunit. Mol. Cell. 26:811-819. [DOI] [PubMed] [Google Scholar]
- 15.Giaever, G., A. M. Chu, L. Ni, C. Connelly, L. Riles, S. Veronneau, S. Dow, A. Lucau-Danila, K. Anderson, B. Andre, A. P. Arkin, A. Astromoff, M. El-Bakkoury, R. Bangham, R. Benito, S. Brachat, S. Campanaro, M. Curtiss, K. Davis, A. Deutschbauer, K. D. Entian, P. Flaherty, F. Foury, D. J. Garfinkel, M. Gerstein, D. Gotte, U. Guldener, J. H. Hegemann, S. Hempel, Z. Herman, D. F. Jaramillo, D. E. Kelly, S. L. Kelly, P. Kotter, D. LaBonte, D. C. Lamb, N. Lan, H. Liang, H. Liao, L. Liu, C. Luo, M. Lussier, R. Mao, P. Menard, S. L. Ooi, J. L. Revuelta, C. J. Roberts, M. Rose, P. Ross-Macdonald, B. Scherens, G. Schimmack, B. Shafer, D. D. Shoemaker, S. Sookhai-Mahadeo, R. K. Storms, J. N. Strathern, G. Valle, M. Voet, G. Volckaert, C. Y. Wang, T. R. Ward, J. Wilhelmy, E. A. Winzeler, Y. Yang, G. Yen, E. Youngman, K. Yu, H. Bussey, J. D. Boeke, M. Snyder, P. Philippsen, R. W. Davis, and M. Johnston. 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418:387-391. [DOI] [PubMed] [Google Scholar]
- 16.Gietz, R. D., and A. Sugino. 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74:527-534. [DOI] [PubMed] [Google Scholar]
- 17.Grant, C. M., and A. G. Hinnebusch. 1994. Effect of sequence context at stop codons on efficiency of reinitiation in GCN4 translational control. Mol. Cell. Biol. 14:606-618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grant, C. M., P. F. Miller, and A. G. Hinnebusch. 1994. Requirements for intercistronic distance and level of eIF-2 activity in reinitiation on GCN4 mRNA varies with the downstream cistron. Mol. Cell. Biol. 14:2616-2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hinnebusch, A. G. 2006. eIF3: a versatile scaffold for translation initiation complexes. Trends Biochem. Sci. 31:553-562. [DOI] [PubMed] [Google Scholar]
- 20.Hinnebusch, A. G. 1985. A hierarchy of trans-acting factors modulate translation of an activator of amino acid biosynthetic genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 5:2349-2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hinnebusch, A. G. 2005. Translational regulation of GCN4 and the general amino acid control of yeast. Annu. Rev. Microbiol. 59:407-450. [DOI] [PubMed] [Google Scholar]
- 22.Iost, I., M. Dreyfus, and P. Linder. 1999. Ded1p, a DEAD-box protein required for translation initiation in Saccharomyces cerevisiae, is an RNA helicase. J. Biol. Chem. 274:17677-17683. [DOI] [PubMed] [Google Scholar]
- 23.Jivotovskaya, A. V., L. Valasek, A. G. Hinnebusch, and K. H. Nielsen. 2006. Eukaryotic translation initiation factor 3 (eIF3) and eIF2 can promote mRNA binding to 40S subunits independently of eIF4G in yeast. Mol. Cell. Biol. 26:1355-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koromilas, A. E., A. Lazaris-Karatzas, and N. Sonenberg. 1992. mRNAs containing extensive secondary structure in their 5′ non-coding region translate efficiently in cells overexpressing initiation factor eIF-4E. EMBO J. 11:4153-4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LeFebvre, A. K., N. L. Korneeva, M. Trutschl, U. Cvek, R. D. Duzan, C. A. Bradley, J. W. Hershey, and R. E. Rhoads. 2006. Translation initiation factor eIF4G-1 binds to eIF3 through the eIF3e subunit. J. Biol. Chem. 281:22917-22932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maag, D., M. A. Algire, and J. R. Lorsch. 2006. Communication between eukaryotic translation initiation factors 5 and 1A within the ribosomal pre-initiation complex plays a role in start site selection. J. Mol. Biol. 356:724-737. [DOI] [PubMed] [Google Scholar]
- 27.Maag, D., C. A. Fekete, Z. Gryczynski, and J. R. Lorsch. 2005. A conformational change in the eukaryotic translation preinitiation complex and release of eIF1 signal recognition of the start codon. Mol. Cell. 17:265-275. [DOI] [PubMed] [Google Scholar]
- 28.Mueller, P. P., and A. G. Hinnebusch. 1986. Multiple upstream AUG codons mediate translational control of GCN4. Cell 45:201-207. [DOI] [PubMed] [Google Scholar]
- 29.Nanda, J. S., Y. N. Cheung, J. E. Takacs, P. Martin-Marcos, A. K. Saini, A. G. Hinnebusch, and J. R. Lorsch. 2009. eIF1 controls multiple steps in start codon recognition during eukaryotic translation initiation. J. Mol. Biol. 394:268-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nielsen, K. H., B. Szamecz, L. Valasek, A. Jivotovskaya, B. S. Shin, and A. G. Hinnebusch. 2004. Functions of eIF3 downstream of 48S assembly impact AUG recognition and GCN4 translational control. EMBO J. 23:1166-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nielsen, K. H., and L. Valasek. 2007. In vivo deletion analysis of the architecture of a multiprotein complex of translation initiation factors. Methods Enzymol. 431:15-32. [DOI] [PubMed] [Google Scholar]
- 32.Nielsen, K. H., L. Valasek, C. Sykes, A. Jivotovskaya, and A. G. Hinnebusch. 2006. Interaction of the RNP1 motif in PRT1 with HCR1 promotes 40S binding of eukaryotic initiation factor 3 in yeast. Mol. Cell. Biol. 26:2984-2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Passmore, L. A., T. M. Schmeing, D. Maag, D. J. Applefield, M. G. Acker, M. A. Algire, J. R. Lorsch, and V. Ramakrishnan. 2007. The eukaryotic translation initiation factors eIF1 and eIF1A induce an open conformation of the 40S ribosome. Mol. Cell 26:41-50. [DOI] [PubMed] [Google Scholar]
- 34.Paulovich, A. G., J. R. Thompson, J. C. Larkin, Z. Li, and J. L. Woolford, Jr. 1993. Molecular genetics of cryptopleurine resistance in Saccharomyces cerevisiae: expression of a ribosomal protein gene family. Genetics 135:719-730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pestova, T. V., and V. G. Kolupaeva. 2002. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev. 16:2906-2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peterson, D. T., W. C. Merrick, and B. Safer. 1979. Binding and release of radiolabeled eukaryotic initiation factors 2 and 3 during 80 S initiation complex formation. J. Biol. Chem. 254:2509-2519. [PubMed] [Google Scholar]
- 37.Pisarev, A. V., V. G. Kolupaeva, M. M. Yusupov, C. U. Hellen, and T. V. Pestova. 2008. Ribosomal position and contacts of mRNA in eukaryotic translation initiation complexes. EMBO J. 27:1609-1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reibarkh, M., Y. Yamamoto, C. R. Singh, F. del Rio, A. Fahmy, B. Lee, R. E. Luna, M. Ii, G. Wagner, and K. Asano. 2008. Eukaryotic initiation factor (eIF) 1 carries two distinct eIF5-binding faces important for multifactor assembly and AUG selection. J. Biol. Chem. 283:1094-1103. [DOI] [PubMed] [Google Scholar]
- 39.Saini, A. K., J. S. Nanda, J. R. Lorsch, and A. G. Hinnebusch. 2010. Regulatory elements in eIF1A control the fidelity of start codon selection by modulating tRNA (i) (Met) binding to the ribosome. Genes Dev. 24:97-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith, D. B., and K. S. Johnson. 1988. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 67:31-40. [DOI] [PubMed] [Google Scholar]
- 41.Spahn, C. M., R. Beckmann, N. Eswar, P. A. Penczek, A. Sali, G. Blobel, and J. Frank. 2001. Structure of the 80S ribosome from Saccharomyces cerevisiae—tRNA ribosome and subunit-subunit interactions. Cell 107:373-386. [DOI] [PubMed] [Google Scholar]
- 42.Szamecz, B., E. Rutkai, L. Cuchalova, V. Munzarova, A. Herrmannova, K. H. Nielsen, L. Burela, A. G. Hinnebusch, and L. Valasek. 2008. eIF3a cooperates with sequences 5′ of uORF1 to promote resumption of scanning by post-termination ribosomes for reinitiation on GCN4 mRNA. Genes Dev. 22:2414-2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor, D. J., B. Devkota, A. D. Huang, M. Topf, E. Narayanan, A. Sali, S. C. Harvey, and J. Frank. 2009. Comprehensive molecular structure of the eukaryotic ribosome. Structure 17:1591-1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valášek, L. 1998. Characterization of RPG1, the large subunit of the yeast S. cerevisiae initiation translation factor 3 (eIF3). Ph.D. thesis. University of Vienna, Vienna, Austria.
- 45.Valášek, L., J. Hašek, K. H. Nielsen, and A. G. Hinnebusch. 2001. Dual function of eIF3j/Hcr1p in processing 20 S pre-rRNA and translation initiation. J. Biol. Chem. 276:43351-43360. [DOI] [PubMed] [Google Scholar]
- 46.Valášek, L., J. Hašek, H. Trachsel, E. M. Imre, and H. Ruis. 1999. The Saccharomyces cerevisiae HCRI gene encoding a homologue of the p35 subunit of human translation eukaryotic initiation factor 3 (eIF3) is a high copy suppressor of a temperature-sensitive mutation in the Rpg1p subunit of yeast eIF3. J. Biol. Chem. 274:27567-27572. [DOI] [PubMed] [Google Scholar]
- 47.Valášek, L., A. Mathew, B. S. Shin, K. H. Nielsen, B. Szamecz, and A. G. Hinnebusch. 2003. The Yeast eIF3 subunits TIF32/a and NIP1/c and eIF5 make critical connections with the 40S ribosome in vivo. Genes Dev. 17:786-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Valášek, L., K. H. Nielsen, and A. G. Hinnebusch. 2002. Direct eIF2-eIF3 contact in the multifactor complex is important for translation initiation in vivo. EMBO J. 21:5886-5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Valasek, L., K. H. Nielsen, F. Zhang, C. A. Fekete, and A. G. Hinnebusch. 2004. Interactions of eukaryotic translation initiation factor 3 (eIF3) subunit NIP1/c with eIF1 and eIF5 promote preinitiation complex assembly and regulate start codon selection. Mol. Cell. Biol. 24:9437-9455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Valášek, L., L. Phan, L. W. Schoenfeld, V. Valásková, and A. G. Hinnebusch. 2001. Related eIF3 subunits TIF32 and HCR1 interact with an RNA recognition motif in PRT1 required for eIF3 integrity and ribosome binding. EMBO J. 20:891-904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamamoto, Y., C. R. Singh, A. Marintchev, N. S. Hall, E. M. Hannig, G. Wagner, and K. Asano. 2005. The eukaryotic initiation factor (eIF) 5 HEAT domain mediates multifactor assembly and scanning with distinct interfaces to eIF1, eIF2, eIF3, and eIF4G. Proc. Natl. Acad. Sci. U. S. A. 102:16164-16169. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
52.Yoon, H. J., and T. F. Donahue. 1992. The sui1 suppressor locus in Saccharomyces cerevisiae encodes a translation factor that functions during
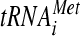 recognition of the start codon. Mol. Cell. Biol. 12:248-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
recognition of the start codon. Mol. Cell. Biol. 12:248-260. [DOI] [PMC free article] [PubMed] [Google Scholar] - 53.Yu, Y., A. Marintchev, V. G. Kolupaeva, A. Unbehaun, T. Veryasova, S. C. Lai, P. Hong, G. Wagner, C. U. Hellen, and T. V. Pestova. 2009. Position of eukaryotic translation initiation factor eIF1A on the 40S ribosomal subunit mapped by directed hydroxyl radical probing. Nucleic Acids Res. 37:5167-5182. [DOI] [PMC free article] [PubMed] [Google Scholar]