

Abstract
We report the molecular mechanisms that underlie chemotaxis of macrophages and cell migration of fibroblasts, cells that are essential during the body's innate immune response and during wound repair, respectively. Silencing of phospholipase D1 (PLD1) and PLD2 reduced cell migration (both chemokinesis and chemotaxis) by ∼60% and >80%, respectively; this migration was restored by cell transfection with PLD2 constructs refractory to small interfering RNA (siRNA). Cells overexpressing active phospholipase D1 (PLD1) but, mostly, active PLD2 exhibited cell migration capabilities that were elevated over those elicited by chemoattractants alone. The mechanism for this enhancement is complex. It involves two pathways: one that is dependent on the activity of the lipase (and signals through its product, phosphatidic acid [PA]) and another that involves protein-protein interactions. The first is evidenced by partial abrogation of chemotaxis with lipase activity-defective constructs (PLD2-K758R) and by n-butanol treatment of cells. The second is evidenced by PLD association with the growth factor receptor-bound protein 2 (Grb2) through residue Y169, located within a Src homology 2 (SH2) consensus site. The association Grb2-PLD2 could be visualized by fluorescence microscopy in RAW/LR5 macrophages concentrated in actin-rich membrane ruffles, making possible that Grb2 serves as a docking or intermediary protein. The Grb2/PLD2-mediated chemotaxis process also depends on Grb2's ability to recognize other motility proteins, like the Wiskott-Aldrich syndrome protein (WASP). Cell transfection with WASP, PLD2, and Grb2 constructs yields the highest levels of cell migration response, particularly in a macrophage cell line (RAW/LR5) and only modestly in the fibroblast cell line COS-7. Further, RAW/LR5 macrophages utilize for cell migration an additional pathway that involves S6 kinase (S6K) through PLD2-Y296, known to be phosphorylated by epidermal growth factor receptor (EGFR) kinase. Thus, both fibroblasts and macrophages use activity-dependent and activity-independent signaling mechanisms. However, highly mobile cells like macrophages use all signaling machinery available to them to accomplish their required function in rapid immune response, which sets them apart from fibroblasts, cells normally nonmobile that are only briefly involved in wound healing.
Normalcy of migration is found in the ability of leukocytes to move toward foreign invaders of the body in phagocytic and immunogenic responses. Fibroblasts and endothelial cells migrate to aid wound repair and deposit collagen around the wounded area (32, 41). Migration is also important in embryogenesis and angiogenesis (37). Cellular migration may be involved not only in the normal physiological state of some cells but also in the pathological state, as in metastasis, whereby tumor cells migrate from the initial tumor site into the circulatory system and establish new colonies (32). Before migration can occur, cells must orient themselves to establish a distinct cell front and rear. The orientation of the cell depends on the inflammation site and location of the chemical stimuli or chemoattractants being released (32, 35). A localized enrichment of filamentous actin (F-actin) under the cell membrane occurs shortly after cell stimulation and is responsible for the formation of the lamellipodia and other morphological changes, such as membrane protrusions or ruffles that occur right before chemotaxis (28).
Monocyte chemoattractant protein 1 (MCP-1), macrophage inflammatory protein 1α (MIP-1α), and colony-stimulating factor (CSF-1, also known as macrophage colony-stimulating factor [M-CSF]) are chemoattractants found to increase cell motility of monocytes and macrophages (52). MIP-1α acts through G-protein-coupled cell surface receptors that are highly abundant on those cells (42) and activate the phosphatidylinositol 3-kinase (PI3K) pathway and phospholipase C (PLC) (49). CSF-1 plays essential roles in the ability of monocytes to survive, proliferate, differentiate, and mature (49) and is also a monocyte/macrophage chemoattractant. CSF-1 acts by binding to cell surface receptors (CSF-1R) encoded by the c-fms proto-oncogene (48). Upon the binding of M-CSF, the CSF-1R dimerizes, and the tyrosine kinase domain is activated, resulting in transphosphorylation of the receptor. Grb2 and PI3K bind to two of the four phosphotyrosyl residues created, and the signal is transmitted to the cell interior (8).
Epidermal growth factor (EGF) triggers proliferation and differentiation in fibroblasts (6, 37). EGF also serves other biological functions, such as cell rounding, ruffling, actin cytoskeletal reorganization, filopodium extension, and cell motility (37). Tyrosines that are autophosphorylated at the EGF receptor (EGFR) C terminus upon ligand binding enable binding to Src homology 2 (SH2) and phosphotyrosine binding (PTB) domains (37). Receptor kinase activity, along with at least one of the C-terminal tyrosine autophosphorylation sites, is required for cell movement (9). In addition, PLCγ and protein kinase C (PKC) have been linked to EGF and its ability to enhance cell motility (9, 10).
As for the downstream signaling mechanism of these cell surface receptors, the recognized key players are the small GTPases, Rho, Rac, and cdc42. They are activated during actin cytoskeleton rearrangement and in cellular migration (7). New evidence has implicated other signaling proteins. Phospholipase D (PLD) has been found to play a role in leukocyte chemotaxis and adhesion (35). PLD is also involved in the regulation of essential cellular functions, largely due to the production of second messengers such as phosphatidic acid (PA) and ultimately diacylglycerol (DAG) (2, 11, 21, 22, 25, 38, 47, 53). Once produced, PA is involved in many cellular functions, including cytoskeletal rearrangement, phagocytosis, vesicle trafficking, exocytosis, and neuronal and cardiac stimulation (1, 11, 21, 30, 38). PA mediates chemotaxis, as increasing concentrations of PA enhanced the rate of cell migration of phagocytes (35). In the murine lymphoma cell line EL4, Knoepp et al. found that activated PLD2 promotes phosphorylation of FAK and Akt, leading to cell-substrate adhesion (7, 29). However, while inactivated PLD2 inhibits adhesion, migration, proliferation, and tumor invasion, it does not alter the basal level of FAK and Akt phosphorylation (29). Although PA does play a role in cell migration, the specific mechanisms involved are not completely understood, and more precise structure-function studies are needed.
We have reported earlier an association of PLD2 and Grb2 important for DNA synthesis/cell proliferation at the level of Y179 (13) and Y511 (23). This study uncovers that migrating cells also use PLD2 and Grb2 but through a different mechanism, involving residue Y169 and the presence of the Wiskott-Aldrich syndrome protein (WASP). We also present new evidence implicating PLD2-Y296, known to be phosphorylated by EGFR kinase (24), in a pathway utilizing S6 kinase (S6K). This additional pathway serves to boost the ability of RAW/LR5 cells to migrate more readily than the other type of cell utilized in this study, COS-7 fibroblasts.
MATERIALS AND METHODS
Materials.
Reduced-sodium bicarbonate Dulbecco modified Eagle medium (DMEM) and COS-7 cells were obtained from American Type Culture Collection (ATCC) (Rockville, MD). Human peripheral blood monocytes (HPBMC) and LGM-3 growth medium were obtained from Cambrex Bio Science, Walkersville, Inc. (Walkersville, MD). RAW 264.7/LR5 (RAW/LR5) cells were developed at the laboratory of one of the authors (D.C.). Histopaque-1077 was obtained from Sigma Aldrich (St. Louis, MO). RPMI 1640 (1×) was obtained from Mediatech (Manassas, VA). Lipofectamine and Plus transfection reagents and Opti-MEM were purchased from Invitrogen Co. (Carlsbad, CA). Superfect transfection reagent was obtained from Qiagen (Valencia, CA). Mouse MIP-1α, mouse CSF-1, and human EGF were from PeproTech Inc. (Rocky Hill, NJ). Anti-protein G-agarose, anti-PLD, and anti-myc tag monoclonal antibodies (MAb) were obtained from Millipore (Temecula, CA). Anti-myc mouse monoclonal (IgG2a) antibody-conjugated (AC) agarose beads and antihemagglutinin (anti-HA) mouse monoclonal (IgG2a) antibody-conjugated (AC) agarose beads were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit β-actin antibody and HA-tagged mouse MAb were from Cell Signaling (Danvers, MA). Triton X-100, phalloidin-fluorescein isothiocyanate (FITC) conjugate (conjugate from Amanita phalloides), and sodium citrate solution were purchased from Sigma (St. Louis, MO). Enhanced chemiluminescence (ECL) Western blotting detection reagents and Percoll/RediGrad were purchased from GE Healthcare (Piscataway, NJ).
RAW 264.7/LR5 cell culture and transfection.
LR5 cells, derived from the murine monocyte/macrophage RAW 264.7 cell line (12) (here named RAW/LR5 cells), were resuspended in fresh complete growth medium (CGM) consisting of reduced-sodium bicarbonate DMEM, 20% fetal calf serum (FCS), and 1% gentamicin and transferred to coated tissue culture flasks and maintained at 37°C in 5% CO2. After 2 to 3 passages, 10% newborn calf serum (NCS) was substituted for 20% FCS in the CGM. Cells were fed with fresh CGM every 3 to 4 days and, once confluent, were split at a 1:4 (cell:medium) ratio. The trypan blue exclusion test was used to determine cell viability. Viability of cells prior to transfection was >98%. Cells were serum starved 1 day prior to transfection. Approximately 3 × 106 cells per transfection were lifted from flask surfaces using cell dissociation buffer, diluted with equal volumes of medium and pelleted at 700 rpm for 5 min, and then resuspended in an appropriate volume of CGM. Cells were subcultured in six-well plates, allowed to adhere, and then serum starved 1 day prior to transfection. At the time of transfection, cells were between 80 and 85% confluent. A mixture of SuperFect reagent, plasmid DNA (1 to 5 μg), and CGM (no serum) was incubated for 10 min at room temperature and then added dropwise onto the adherent monolayer of cells to a final volume of 2 ml per well. Cells were incubated with transfection reagent and plasmid for ∼3 h, after which time cells were washed three times with CGM and incubated for an additional 28 h in the presence of CGM prior to experimental use.
COS-7 cell culture and transfection.
COS-7 cultures were established from a frozen stock by washing thawed cells once with prewarmed DMEM and placed in coated tissue culture flasks in CGM. Cells were maintained in 75-cm2 coated tissue culture flasks, and once confluent, cells were trypsinized, counted, and subcultured in six-well culture plates 1 day prior to transfection. Viability was determined by trypan blue exclusion. On the day of transfection, cells were washed twice with warm phosphate-buffered saline (PBS) to remove serum and returned to 37°C and 5% CO2 with 1 ml per well of prewarmed Opti-MEM (serum-free medium). Lipid-DNA complexes were prepared by mixing Lipofectamine reagent, plasmid DNA, Plus reagent, and Opti-MEM and incubating at room temperature for 10 min according to the manufacturer's instructions (Invitrogen). Transfection mixtures were added dropwise to the plated cultures. Cells were incubated in the transfection medium at 37°C and 5% CO2 for 3 h. Transfection medium was replaced with 2 ml CGM (no antibiotics) after being washed once with CGM (no antibiotics). Following transfection, cells were found to be >95% viable by trypan blue exclusion. Cells were allowed to grow for 48 to 55 h prior to harvesting for experimentation.
HL-60 and HeLa cells.
HL-60 cells were grown at 37°C in a 5% CO2 incubator in RPMI 1640 medium supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum, 2 mM l-glutamine, and 5 mg/ml gentamicin, with cell density maintained between 0.1 × 106 and 1.0 × 106/ml. HL-60 cells were induced to differentiate into monocytes (M-HL-60) by incubation with 200 ng/ml phorbol myristate acetate (PMA) for 3 days. HeLa-S3 human cervical carcinoma cells were seeded at 1 × 105 cells/ml on 100-mm Falcon tissue culture dishes and grown in monolayers in F-12 medium supplemented with l-glutamine and 7% FBS. The transfection protocol was similar to that indicated for COS-7 cells.
Chemotaxis.
Cells used in these experiments were between passages 10 and 15. Adherent cells were detached using 0.25% trypsin-EDTA or a nonenzymatic cell dissociation buffer for COS-7 or RAW/LR5 cells, respectively. A hemocytometer was used to count cells, and trypan blue exclusion was used to determine cell viability. Transwell inserts (12-μm pore size and 8-μm pore size for COS-7 and RAW/LR5 cells, respectively) in 20 four-well plates were prewetted with DMEM containing 0.5% bovine serum albumin (BSA). The pore size was based on the fact that COS-7 cells are very large (20 to 25 μm) and a pore size of 12 μm should be adequate to let them pass with difficulty and only when stimulated with the chemoattractant. Conversely, RAW macrophages (12 to 16 μm) are slightly smaller than COS-7 cells, and we chose an 8-μm pore size, as has been successfully used by us in the past (16). Cell concentration was adjusted to 5 × 104 cells per 400 μl of DMEM containing 0.5% BSA per insert. A 600-μl portion of DMEM containing 5% BSA was added to the bottom of the well prior to placing the cells and insert in the well. CSF-1 (1 nM) for RAW/LR5 cells and EGF (3 nM) for COS-7 cells were used as chemoattractants, unless otherwise noted. RAW/LR5 cells were allowed to undergo chemotaxis for up to 2 h, and COS-7 cells underwent chemotaxis for up to 1 h at 37°C and 5% CO2. Unless otherwise indicated, cells were incubated for chemotaxis for times that would avoid saturation of cells in the bottom well, and those times were 45 min for RAW/LR5 and 30 min for COS-7 cells. Afterward, inserts were removed and cells that had migrated to the bottom were fixed with 4% paraformaldehyde. Fixed cells were then allowed to settle and adhere before being counted. Cells were then counted using an inverted microscope at 20×. The average number of cells counted from six fields per well was determined. It should be noted that the chemotaxis assay presented here collects cells in the lower well after migration through a Transwell filter. Even though we have titrated the concentration of BSA in our chemotaxis buffer (0.5% top/5% bottom) to minimize adherence to the underside of the filter, there is a small percentage (8 to 10%) that adheres. However, control experiments have determined that the level of adherent cells remains constant for both controls and chemoattractant-stimulated cells.
Western blotting.
Cells were taken at 28 and 48 h after transfection of RAW/LR5 cells and COS-7 cells, respectively. Lysates were prepared using a lysis buffer containing 5 mM HEPES, pH 7.4, 100 μM sodium orthovanadate, 0.4% Triton X-100, aprotinin, and leupeptin and sonicated twice for 10 s. Sonicates were subjected to SDS-PAGE and proteins from the gel were transferred to polyvinylidene difluoride (PVDF) membranes, blocked with 5% BSA/Tris-buffered saline-Tween 20 (TBS-T) at 4°C (blocking buffer) overnight. Western blotting was carried out using the following dilutions of primary antibodies in blocking buffer: 1:1,000 anti-myc (rabbit or mouse IgG), 1:1,000 anti-HA (rabbit or mouse IgG), 1:1,500 anti-Grb2 (mouse IgG), and 1:3,000 anti-β-actin (rabbit IgG). Washed blots were then incubated in 1:3,000 dilutions of the appropriate secondary antibody-horseradish peroxidase (HRP) conjugate. Amersham's ECL reagent was used to activate the horseradish peroxidase, and X-ray film was exposed to the immunoblots.
Immunofluorescence microscopy.
RAW/LR5 cells transfected with myc- or HA-tagged plasmids were fixed onto 22-mm2 glass coverslips by adding 1 ml of 4% paraformaldehyde to each coverslip for at least 10 min. Paraformaldehyde was aspirated off, and cells were permeabilized with 1 ml 0.5% Triton X-100 in PBS for 10 min. Fixed cells were then blocked with 10% fetal calf serum and 0.1% Triton X-100 in PBS for 4 h at room temperature. Coverslips were then incubated overnight at 4°C in the dark in a 1:1,000 dilution of anti-HA-FITC or anti-myc-FITC IgA conjugate in PBS. Next, coverslips were washed three times for 5 min in 1 ml of PBS. Actin staining was carried out using a 1:200 dilution of phalloidin tetramethyl rhodamine isocyanate (TRITC) in PBS for 2 h at room temperature. Following washing, DAPI (4′,6-diamidino-2-phenylindole), diluted in 1:2,000 PBS, was used to stain cell nuclei by incubating coverslips for 5 min and then once again washing with PBS. Excess liquid was aspirated off, and cells were washed a final time. Air-dried coverslips were then adhered to glass microscope slides (cell side down) using Vectashield mounting medium for fluorescence (Vector Laboratories, Inc.) and were visualized on a Nikon Eclipse 50i inverted microscope with MetaVue software. For fluorescence microscopy with CFP (cerulean)-Grb2 and myc-PLD2, RAW/LR5 cells were cotransfected with yellow fluorescent protein (YFP)-PLD2 and cerulean-Grb2 using FuGENE HD and incubated without or with CSF-1 for 5 min before fixation. Following permeabilization with 0.2% Triton X-100, cells were stained for F-actin using Alexa647-phalloidin. Images were taken using the 60× oil/1.40 phase 3 objective of an Olympus IX71 microscope coupled to a Sensicam cooled charge-coupled-device (CCD) camera.
DNA plasmids (wild type and mutants).
Several PLD2 point mutants have been developed in our laboratory (13, 14). PLD2 mutants Y169F, Y179F, Y296F, and Y511F were prepared from myc-pcDNA3-PLD2 using the following mutagenesis primers: for PLD2-Y169F, 5′-CAGAAATACCTGGAGAATTTTTTAAACTGTCTCTTGACC-3′; for PLD2-Y179F, 5′-TCTTGACCATGTCTTTCTTTCGAAACTACCATGCC-3′; for Y296F, 5′-CTCAAGTGCAGCAGCTTTCGGCAGGCACGGTGG-3′; and for Y511F, 5′-GGCTGGGCAAGGACTTCAGCAATCTTATCACC-3′. All oligonucleotides and their reverse complements were PAGE/high-performance liquid chromatography (HPLC) purified (Integrated DNA Technologies, Coralville, IA). A Grb2-wild-type (WT) plasmid (pcDNA-Xpress-Grb2-WT) was prepared from an expression construct containing the full cDNA of human Grb2 variant 1 cDNA (pCMV6-Grb2; OriGene Technologies, Rockville, MD), which was the template for PCR subcloning. The Grb2 open reading frame (ORF) was PCR amplified using the sense (EcoRI) primer 5′-CACTGAGCAGAATTCAGAATGGAAGCCATGGCC-3′ and the antisense (PspOMI) primer 5′-TTAAATCCAACGGGCCCTCCCACCCCCTAA-3′. The PCR product was EcoRI/PspOMI-digested, purified, and C-terminally subcloned into the EcoRI/NotI-digested Xpress-tagged pcDNA3.1/HisC expression vector (Invitrogen, Carlsbad, CA). The SH2 binding-deficient mutant Grb2-R86K was generated from pcDNA-XpressGrb2-WT using the following mutagenesis primer: Grb2-R86K sense, 5′-GATGGGGCCTTTCTTATAAAAGAGAGTGAGAGCGCTC-3′. The SH3-SH3 binding-deficient double mutant Grb2-P49/206L was generated from pcDNA-XpressGrb2-WT using the following mutagenesis primers: sense primer for Grb2-P49L, 5′-TTAATGGAAAAGACGGCTTCATTCTGAAGAACTACATAGAAATGAAACCAC-3′; sense primer for Grb2-P206L, 5′-GCAGACCGGCATGTTTCTGCGCAATTATGTCACCC-3′. All oligonucleotides and their reverse complements were PAGE/HPLC purified. The S6K DNA plasmid was previously described in reference 19. The WASP DNA plasmid (transfection ready) was from Origen Technology (Rockville, MD) (Homo sapiens Wiskott-Aldrich syndrome-like [WASL], NM_003941.2, in pCMV-XL6 with the SP6 transcriptional promoter).
shRNA Grb2 and rescue plasmid.
In order to silence endogenous Grb2, we used a short hairpin RNA (shRNA) Grb2 encoding construct pU6+27-shGrb2 from Panomics, Inc. (Fremont, CA), as reported previously (14). Briefly, the human U6 snRNA promoter, the first 27 bp of the U6 snRNA (U6+27), and the shRNA against human Grb2 were confirmed by direct sequencing. The rescue plasmid pcDNA-XpressGrb2Res was generated by introducing seven silent mutations without changing the amino acid sequence of human Grb2 to make XpressGrb2Res resistant to the continuous presence of Grb2 shRNA (nucleotides [nt] 310 to 330: GAT GTG CAG CAC TTC AAG GTT). The sense primer for the mutagenesis was 5′-293CT GTC AAG TTT GGA AAC GAC GTC CAA CAT TTT AAA GTA CTC CGA GAT GGA GCC GGG348-3′ (sites in bold are silently mutated; underlined are the restriction sites AatII and DraI).
All other silencing RNAs.
All double-stranded RNAs (dsRNAs) were from Applied Biosystems (Foster City, CA) as “select validated.” For S6K silencing, we used a dsRNA that targeted exon 1: sense sequence, 5′-CAUGGAACAUUGUGAGAAAtt-3′. For S6K (p70S6 kinase-1) silencing, we used a combination of dsRNAs that targeted exons 4 and 5 (sequences not provided by manufacturer). For PLD2 silencing, we used a dsRNA that targeted exon 15: sense sequence, GGA CUA CAG CAA UCU UAU CTT. For WASP, small interfering RNAs (siRNAs) from Santa Cruz, which consisted of a pool of two target-specific 20- to 25-nucleotide siRNAs designed to knock down gene expression of both WASP and N-WASP [thus named “si-(N)WASP”], were used. The genetic locus of the target is Was (mouse) mapping to XA1.1. A negative control for all silencing was 100 nM siRNA “Neg-siRNA#2,” purchased from Applied Biosystems. This control siRNA is a 19-bp scrambled sequence with 3′ dT overhangs (sequence not disclosed by Ambion) certified not to have significant homology to any known gene sequences from mouse, rat, or human and causes no significant changes in gene expression of transfected cells after 48 h at the same concentration as the dsRNA in the test.
Statistical analyses.
Data are presented as the mean ± standard error of the mean (SEM). The difference between means was assessed by the single-factor analysis of variance (ANOVA) test or by t test, as indicated in the legends of the figures. In either case, a probability of P < 0.05 was considered to indicate a significant difference.
RESULTS
Establishment of RAW/LR5 and COS-7 chemotactic responses.
To investigate the molecular mechanisms that underlie chemotaxis initiated by PLD, we used RAW 264.7/LR5 (“RAW/LR5”) and COS-7 cells. The former are leukocytic (macrophages) and exemplify a key cell in the innate immune response. The latter are fibroblasts and exemplify a key cell in wound healing. Both cell types migrate to injured areas within the body and are important in maintaining homeostasis and the structural integrity of connective tissues. In the laboratory, RAW/LR5 and COS-7 cells provide the additional advantage that they are easily transfectable. In the initial experiments, we wanted to establish the parameters of chemotaxis for these two cell lines. As presented in Fig. 1 A and B, RAW/LR5 cells are chemoattracted to MIP-1α and to CSF-1 (as well as to MCP-1; data not shown). CSF-1, at the concentration of 1 nM for 2 h, was used in subsequent experiments. COS-7 cells are chemoattracted to EGF and to FBS (Fig. 1C and D), albeit with an absolute number slightly lower than those observed for RAW/LR5 macrophages but still very robust. EGF at the concentration of 1 nM for 1 h was used in subsequent experiments. Thus, EGF in addition to its well-known functions as a growth factor or mitogen is a bona fide chemoattractant, or “motogen.”
FIG. 1.

Chemotactic response of RAW/LR5 and COS-7 cells (nontransfected). (A) RAW/LR5 cells migrated toward increasing concentrations of either MIP-1α or CSF-1 in a dose-dependent manner. Cells were incubated in Transwell permeable supports in multiwell plates containing the indicated chemoattractant for a period of 2 h. (B) Time course of RAW/LR5 chemotactic response to 3 nM MIP-1α or 1 nM CSF-1, studied at 30-min increments over a period of 120 min. (C) Dose response of COS-7 chemotactic response to the indicated concentrations of EGF or FBS. The dose response was studied for a period of 1 h. (D) Time course of COS-7 chemotaxis against 3 nM EGF or 7% FBS, studied at 30-min increments over a period of 120 min. Values are the means ± the SEM of results of three independent experiments performed in duplicate.
As the cells considered in this study have small amounts of endogenous PLD, we next investigated if ectopic expression of PLD would have a positive effect on the chemotaxis induced by either CSF-1 or EGF. Figure 2 is a schematic representation of the mutants used in this study. Transfecting either PLD1 or PLD2 constructs led to both an enhancement of chemotaxis of RAW/LR5 macrophages (cells stimulated with CSF-1) and an enhancement of chemokinesis (cells in basal state) (Fig. 3 A). Figure 3 indicates that the effect is more noticeable with PLD2 than with PLD1 (205% versus 163% stimulation due to the PLD alone). A detailed inspection of Fig. 3A indicates that PLD transfection affects both chemokinesis and chemotaxis. First, naturally, cells respond to chemoattractant (compare bars 1 and 9 with 2 and 10). However, just by transfecting the cells with PLD1 or PLD2 (more so with the latter) one can see an elevation of chemokinesis, that is, in the absence of chemoattractant (compare bars 1 and 9 with 5 and 13), that is much further enhanced with the addition of CSF-1 to cells (compare bars 5 and 13 with bars 6 and 14). Thus, it is possible to conclude that PLD affects both random migration and chemotaxis.
FIG. 2.

Sequences of PLD2 and Grb2 mutants used in this study. (A) Scheme of HKD1 (one of the two phospholipase D catalytic sites) domain in PLD1. (B) Scheme of the recognition target for SH2-bearing proteins, a stretch that covers Y296, and the two HKD domains responsible for lipase activity. (C) Amino acid sequence of Grb2 showing the SH2 domain, the two SH3 domains, and the point mutants generated for this study.
FIG. 3.

Silencing and overexpression of phospholipases. (A) Silencing and rescue. Cells were silenced (“si-PLD”) for a total of 4 days, during the last two of which they were (when appropriate) transfected with PLD DNA constructs refractory to siRNA to rescue chemotaxis (“PLD + siPLD”). (B) Typical Western blot, among three total with similar results, of the protein levels after silencing and rescuing. (C) Study of overexpression of PLD2 in four different cell lines (two macrophages and two fibroblasts). Two days prior to chemotaxis experiments cells were either mock transfected (−) or transfected with PLD2-WT (+). Chemotaxis was assayed in Transwell inserts against CSF-1 (for macrophages) or against EGF (for fibroblasts).
Figure 3A demonstrates the effects of PLD1 or PLD2 silencing, and these were confirmed by Western blotting (Fig. 3B). Silencing of PLD1 and, to a greater extent, PLD2 resulted in a profound decrease in chemotaxis. This loss of function was recovered when cells were transfected with a PLD2 construct refractory to siRNA effects. The gain of function with PLD constructs and the loss of function with siRNA experiments unequivocally prove that PLD (PLD2 isoform more so than PLD1 isoform) is essential for chemotaxis. Figure 3C is a study of overexpression of PLD2 in four different cell lines, two macrophages (LR5 and monocytic HL-60 cells) and two fibroblasts (COS-7 and HeLa cells). Chemotaxis was assayed in Transwell inserts against CSF-1 (for macrophages) or against EGF (for fibroblasts) and similar effects of PLD overexpression were detected in all cell lines used. The data indicate that the best models for studying PLD-induced chemotaxis are RAW/LR5 (for macrophages) and COS-7 (for fibroblasts), as the most robust stimulation was observed in those two cell lines. RAW/LR5 cells are a subline of commonly used RAW macrophages that have been used to study CSF-1-induced effects (12, 16). Human cell lines are harder to transfect, and they need to be differentiated using either PMA (as in the case of M-HL-60 cells in Fig. 3C) or dimethyl sulfoxide (DMSO), which might have dramatic effects on signaling in general.
Overexpression of PLD1-WT leads to a 1.7-fold increase in chemotaxis induced by CSF-1 in RAW/LR5 cells (Fig. 4A) or to a 1.5-fold increase in chemotaxis induced by EGF in COS-7 cells (Fig. 4B). Overexpression of PLD2-WT results in greater stimulation levels of chemotaxis: a 2.6-fold increase over CSF-1 alone in RAW/LR5 cells (Fig. 4A) or a 1.9-fold increase over EGF alone in COS-7 cells (Fig. 4B). We should note that transfection efficiency for RAW/LR5 cells with Superfect is 60 to 70% and transfection efficiency for COS-7 with Lipofectamine is 80 to 90%. In either case, the values presented in Fig. 4 are underestimated, and we believe the effect of PLD on chemotaxis might be even greater if the efficiency of transfection were ∼100%.
FIG. 4.

Chemotactic response of cells transfected with PLD wild-type or lipase-inactive constructs. RAW/LR5 or COS-7 cells were transfected with plasmid constructs containing either wild-type HA-PLD1, wild-type myc-PLD2, or their respective lipase inactive mutations (K→R) in the first and/or second HKD domains. After 2 days of incubation, cells were harvested and used in chemotactic assays of RAW/LR5 cells toward plain buffer (Control) or toward 1 nM CSF-1 (A) or that of COS-7 cells toward plain buffer (Control) or toward 3 nM EGF (B). Another group of transfected cells were harvested for cell lysates that were immunoprecipitated with anti-HA or anti-myc antibodies for PLD1 and PLD2, respectively. The resulting immunocomplex beads were split into two equal sets. The first set was used for Western blot analysis of protein expression (C and D) using anti-β-actin blots to verify similar protein loading, and the second set was used to measure PLD2 activity (E). 100% values represent 2,878 ± 204 dpm/mg for PLD activity. Values are the means ± the SEM of results of at least three independent experiments performed in duplicate. *, differences between mean values for “mock”-transfected (Control), chemoattractant-activated cells and mean values for WT-transfected, chemoattractant-activated cells that were statistically significant (P < 0.05) by ANOVA. #, differences between mean values for chemoattractant-activated control cells and mean values for chemoattractant-activated KR transfectants. (F) Effect of PLD2 inhibition with 1-butanol (n-butanol) or ter-butanol (t-butanol) on cell chemotaxis. Cells were transfected with the indicated PLD constructs (3 μg DNA each). Two days after this, cells were harvested and incubated with the alcohols for 30 min before taking them to Transwell inserts. The figure depicts the averages ± SEM of results of three independent experiments in duplicate. *, differences between mean values with respect to Control-Mock. #, differences between mean values in alcohol treatments and their respective controls.
On the mechanism of PLD-induced chemotaxis.
The most immediate assumption on the mechanism utilized by PLD to mediate chemotaxis is the release of phosphatidic acid (PA), the product of the lipase enzymatic activity on a phospholipid substrate (such as phosphatidylcholine). In order to investigate whether enzymatic activity of PLD is necessary for chemotaxis, we transfected cells with lipase-inactive mutants of PLD1 or PLD2. PLD1-K860R contains a single-point mutation within the second HKD domain of the PLD1 molecule, as does PLD2-K758R for PLD2. PLD2-K444R contains a single-point mutation within the first HKD domain of PLD2. The HKD domains (HXKX4DX6GSXN), or “PLD signature motifs,” are the invariable regions responsible for the enzymatic activity of PLD (13, 22, 30). All the K→R mutants utilized in this study are incapable of synthesizing PA/phosphatidyl butanol (PBut) as the readout for PLD activity (Fig. 4E). There seems to be a correlation between PLD activity and the level of chemotaxis, as lipase-inactive mutants partially failed to elicit chemotaxis and partially negate it in RAW/LR5 cells (compare Fig. 3E with 3A and B). (This is not due to changes in the level of PLD expression, as indicated in the Western blots of Fig. 4C and D, showing that similar protein amounts were loaded per condition.) These data indicate that the lipase activity is only partially required for the enhancement of chemotaxis observed following PLD overexpression. To have a more conclusive idea of the role of phosphatidic acid on the PLD-mediated effect, we used the PA scavenger 1-butanol in chemotaxis assays using wild-type and lipase-deficient mutants, and the results were compared with those obtained in the presence of the tert-butanol (t-butanol) isomer that does not inhibit PLD-induced PA production. As seen in Fig. 4F, n-butanol (but not t-butanol) partially inhibited PLD-induced chemokinesis and chemotaxis, which were further increased in the presence of the “KD” lipase-inactive mutants but never returned completely to the basal levels (Fig. 4F). This indicates that there are two components of PLD-mediated chemotaxis: one that is activity dependent and another that is activity independent. The first one depends on the production of PA, and the second one depends, possibly, on a protein-protein interaction of PLD with signaling elements in the cell. This is discussed in detail in this paper. At any rate, results of Fig. 3 and 4 have indicated that of the two PLD isoforms, it is PLD2 that concedes greater chemokinesis and chemotaxis effect to the cell lines considered, and the rest of this study concentrates on PLD2.
In conclusion, silencing and transfection of PLD2 WT constructs into RAW/LR5 or COS-7 cells lead to a decrease and to an increase, respectively, in chemotaxis. This is also seen with the PLD1 isoform, albeit to a lower extent. The reduced cell migration of the lipase-inactive mutants of PLD2 correlates with a partial decrease in enzymatic activity between these mutants and the PLD2 WT.
Additional requirements for PLD2-mediated chemotaxis.
Our laboratory has reported previously that the tyrosines Y169 and Y179 within the pleckstrin homology (PX) domain of PLD2 form two SH2 motifs capable of offering docking sites for SH2-recognizing signaling proteins (12). We investigated what effect mutations of the tyrosines 169 and 179 would have on chemotaxis. The mutant PLD2-Y169F partially and significantly enhanced CSF-1-led chemotaxis in RAW/LR5 cells (Fig. 5A) or EGF-led chemotaxis in COS-7 cells (Fig. 5B), whereas PLD2-Y179F clearly enhanced chemotaxis at a level similar to that of PLD2-WT. This would set a divergence in signaling. PLD2 could utilize Y169 for chemotaxis, whereas Y179 could be reserved for other PLD2-mediated functions, such as cell proliferation (13). In addition to this region in PLD2, another site that is not within the SH2 consensus motif, Y296, is also important for migration, as shown in Fig. 5B. This led us to conclude that PLD2-mediated chemotaxis is dependent at least on two separate pathways, one relying on an SH2-recognition site (Y169) and the other (Y296) independent of it. We will investigate the Y169-mediated pathway first and will turn our attention to the other later in this study.
FIG. 5.

Chemotactic response of cells transfected with PLD WT and Y→F mutants. RAW/LR5 (A) or COS-7 (B) cells were transfected with plasmid constructs either containing wild-type myc-PLD2 or with indicated Y→F mutations. Procedures are similar to those described in the legend of the previous figure. (A and B) Chemotaxis toward plain buffer (Control) or 1 nM CSF-1 (for RAW/LR5) (A) or toward plain buffer (Control) or 3 nM EGF (for COS-7) (B). (C) Western blot analysis of protein expression. (D) Measurement of PLD2 activity. 100% values represent 3,430 ± 301 dpm/mg for PLD activity. Values are the means ± the SEM of results of at least three independent experiments performed in duplicate. *, differences between means that were statistically significant (P < 0.05) by ANOVA between chemoattractant-activated control cells and chemoattractant-activated WT-transfected cells. #, differences between chemoattractant-activated control cells and chemoattractant-activated YF-transfected cells.
As Y169 is part of the SH2 binding motifs in PLD2's PX domain, it can be hypothesized that chemotaxis is mediated by PLD binding to other partners through these tyrosines once phosphorylated. One such partner is the growth receptor-binding domain 2 protein (Grb2) (13, 14). The Grb2 protein has been found in many types of cancer, possibly lending to the ability of the cells to invade or metastasize (18, 19). We overexpressed Xpress-tagged Grb2 in RAW/LR5 (Fig. 6A) and COS-7 (Fig. 6B) cells, which resulted in greater chemotaxis than levels achieved with CSF-1 or with EGF (2-fold and 1.5-fold, respectively). It is worth noticing that Grb2 alone also caused an increase in chemokinesis (basal or random migration, “control” bars) in cells in the absence of any chemoattractant. This paralleled the effect seen with PLD transfection in basal (chemoattractant-free) cells (Fig. 4 and 5, “control” bars). Thus, we consistently observed that the levels of chemokinesis and chemotaxis induced by Grb2 are similar to those found in PLD2-overexpressing cells (particularly for RAW/LR5).
FIG. 6.

Study of chemotaxis after overexpression of Grb2 and PLD2. Either RAW/LR5 or COS-7 cells were transfected with Grb2 alone or in combination with PLD2. After 2 days of incubation, cells were harvested and used to assay chemotaxis toward plain buffer (“Control”) or 1 nM CSF-1 (for RAW/LR5) (A) or toward plain buffer (“Control”) or 3 nM EGF (for COS-7) (B). Values are the means ± the SEM of three independent experiments performed in duplicate. *, differences between means that were statistically significant (P < 0.05) by ANOVA between chemoattractant-activated control cells and chemoattractant-activated WT transfectants. (C) Patterns of protein expression of the two different constructs used: myc-tagged PLD2 and Xpress-tagged Grb2.
We also observed that Grb2 in combination with PLD2 furthered the chemotaxis induced by either molecule separately. Equal levels of Grb2, whether in the absence or in the presence of PLD2, were demonstrated by immunoblotting (Fig. 6C). We concluded that a potentiating effect of chemotaxis was achieved when both molecules, Grb2 and PLD2, are present. Interestingly, a Grb2-R86K mutant (one that yields a protein incapable of binding to its targets through SH2 motifs) was unable to increase chemotaxis when transfected alone or it was unable to potentiate PLD2-induced chemotaxis, indicating that the presence of intact SH2 motifs on Grb2 is necessary for the observed functional effect.
Next, we affinity purified Grb2 (wild type and R86K mutated, leading to a protein incapable of binding to its targets through SH2 motifs) from Xpress-tagged constructs transfected into COS-7 cells. Additionally, we transfected PLD2 in COS-7 cells and analyzed PLD activity in cell lysates. PLD activity was found to be elevated when Grb2-WT was mixed in the reaction mixture but not when Grb2-R86K was used (Fig. 7A), indicating that Grb2 has a positive enhancing effect on PLD2 activity that is mediated by SH2, as indicated in the previous in vivo experiments.
FIG. 7.

Coimmunoprecipitation and coexpression of Grb2 and PLD2. (A) Affinity-purified Grb2 (wild type or R86K mutated) was mixed with lysates of cells overexpressing PLD2 in vitro, and PLD activity was assayed. (B) Grb2-overexpressing COS-7 cells (Xpress-Grb2 construct) were immunoprecipitated (I.P.) with anti-PLD2 and immunoblotted (W.B.) with anti-Xpress. PLD2-overexpressing COS-7 cells (HA-PLD2 construct) were immunoprecipitated with anti-Grb2 and immunoblotted with anti-HA. (C to J) PLD2 and Grb2 are localized to CSF-1-induced membrane ruffles. Representative images are shown of RAW/LR5 cells cotransfected with YFP-PLD2 (left, red) and cerulean-Grb2 (middle, green) and stained for F-actin (right, blue) with a merged image (far right) incubated without (top row, C to F) or with (bottom row, G to J) CSF-1 for 5 min. Shown are representative fields among five with similar occurrences. Arrows indicate regions of colocalization to ruffles. Scale bar = 5 μm.
We hypothesized that the functional increase (chemotaxis) could be due to a protein-protein interaction between the two molecules. Indeed, PLD2 associates with Grb2, as demonstrated by coimmunoprecipitation and immunoblotting using either PLD2 or Grb2 antibodies for immunoprecipitation and Western blotting, respectively, or using Grb2 and PLD2 antibodies for immunoprecipitation and Western blotting, respectively (Fig. 7B). The existence of a PLD2-Grb2 association was also detected by immunofluorescence in COS-7 cells (not shown), but it was more readily apparent in RAW/LR5 macrophages. Figure 7C to J indicate that both PLD2 and Grb2 are localized to CSF-1-induced membrane ruffles. These actin-rich, plasma membrane formations are a hallmark of macrophages and are shown here for the first time to contain both PLD2 and Grb2 in the same location. As cell ruffling is a prelude of chemotaxis (12), Fig. 7C to J tie together the colocalization of Grb2 and PLD2 and the functional effects on chemotaxis.
The results presented in Fig. 6 and 7, taken together, indicate that PLD2 and Grb2 colocalize in the cell, which results in a functional activation of chemotaxis that is mediated by the SH2 domain (in Grb2) and by Y169 and Y179 (in PLD2).
Loss of chemotactic function and rescue by Grb2.
The gain-of-function experiments shown in Fig. 5 and 6 regarding Grb2 were next complemented with loss-of-function designs presented in Fig. 7, utilizing a stable shRNA of Grb2. Figure 8A shows that silencing of endogenous Grb2 after shGrb2 transfection was accomplished (>90% decrease in Grb2 protein in Western blots) in at least two clones (clones 1 and 2) of Sh-Grb2-RAW/LR5 cells (and another two clones for COS-7 cells, clones 3 and 4; not shown). In parallel experiments, also with Grb2 silencing, we consistently observed a significant decrease of chemotaxis of RAW/LR5 and COS-7 cells (Fig. 8B and C, second and third bars). This was more pronounced in the RAW/LR5 cells. Silenced cells were impaired after overexpressing PLD2 (Fig. 8B and C, fourth bar), further indicating the need of functional Grb2 to transduce PLD2-derived migration signals.
FIG. 8.
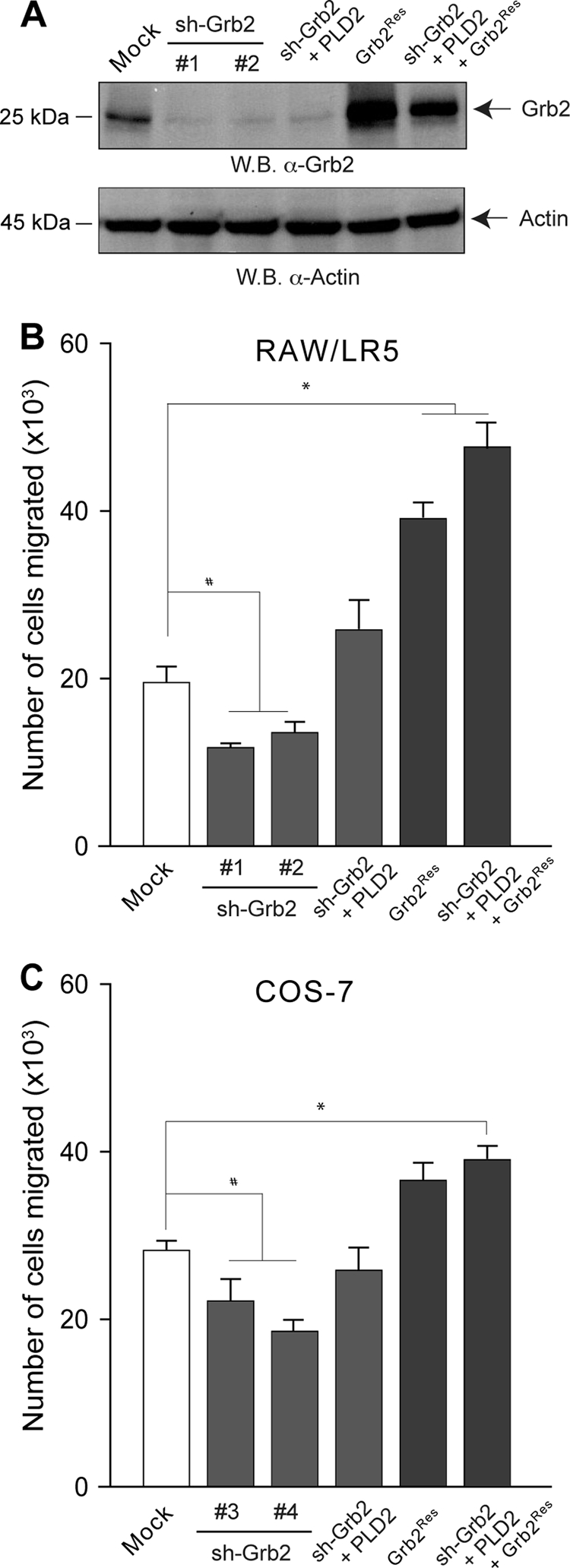
Silencing and rescue of Grb2. (A) Level of depletion of endogenous Grb2 in two different stable clones of RAW/LR5 cells. The panel also shows the level of protein expression after transfection of the Grb2Res plasmid, resistant to Grb2 silencing. For chemotaxis, silenced RAW/LR5 (B) or COS-7 (C) cells were divided into several sets. One set was transfected with PLD2, another with Grb2Res, and a third one with PLD2 and Grb2Res. A separate control set utilized nonsilenced cells that were transfected with Grb2Res alone. Both Grb2-silenced and Grb2-silenced/“rescued” cells were harvested after 4 days and transferred to Transwell inserts for chemotaxis assay toward 1 nM CSF-1 (B) or 3 nM EGF (C). Results are the means ± SEM from results of at least three independent experiments conducted in duplicate. *, values that were statistically significantly different (P < 0.05) above control values by ANOVA; #, values that were statistically significantly different (P < 0.05) below control values by ANOVA.
Since the impediment was not total, and cells still managed to undergo chemotaxis, albeit at a lower number, it suggested that other signaling might exist (in addition to Grb2-derived pathways) for chemotaxis of RAW/LR5 cells, a hypothesis that was tested later in this study. Going back to silencing of Grb2, we next made use of the “rescue” plasmid “Grb2Res,” engineered to be resistant to shGbr2. This plasmid, refractory to shGrb2 action, has an enhancing effect on chemotaxis when transfected alone into cells (Fig. 8B and C, second bar from the right), quantitatively similarly to the results with Grb2-WT in Fig. 6. Importantly, the abolished level of chemotaxis after silencing with shGrb2 demonstrated previously was fully restored with the silencing-resistant Grb2Res rescue plasmid (Fig. 8B and C, last bar on the right; compare to fourth bar), more so in RAW/LR5 cells. These experiments indicate (i) that Grb2 has a significant chemotaxis-potentiating effect on its own and (ii) that for PLD2 to transduce its chemotaxis-inducing signal, it needs a fully functional Grb2 protein.
The SH3 domains of Grb2 are also required for PLD2-mediated signaling.
The results with the Grb2-R86K mutant of Fig. 6 indicate that chemotaxis is hampered by the absence of the SH2 domain. However, given the “coupling” nature of Grb2 we wanted to ascertain if other target proteins might be required to further mediate the PLD2-induced chemotaxis effect. We made use of another Grb2 mutant, GrbP49FL-P206L (referred to here as GrbP49/206L), which has two point mutations, one each in each SH3 domain, that render these two domains incapable of binding to other protein partners. As presented in Fig. 9, Grb2's twin SH3 domains are needed to mediate the observed chemotaxis-enhancing effects due to PLD2. This finding needed to be investigated in more detail.
FIG. 9.

Grb2 SH2 and SH3 domains and chemotaxis. Either RAW/LR5 (A) or COS-7 (B) cells were transfected with Grb2 (WT or the P→L SH3/SH3-binding deficient mutant), with WASP, or with S6K constructs alone or in combination with PLD2. In parallel, some cells were incubated with siRNAs for either N-WASP or S6K. After 2 days of incubation, cells were harvested and used to assay chemotaxis toward 1 nM CSF-1 (for RAW/LR5) (A) or toward 3 nM EGF (for COS-7) (B). (C) PLD2 associates with WASP and N-WASP. COS-7 cells were cotransfected with either myc-PLD2, CFP/YFP-WASP (39), or CFP/YFP-N-WASP (5) as indicated using FuGENE HD (Roche) according to the manufacturer's protocol. Following cell lysis, whole-cell lysates were collected and divided in two sets, one for Western blotting (W.B.) and the other for immunoprecipitation (I.P. GFP) with antibodies against green fluorescent protein (GFP) (Roche) followed by Western blotting with the indicated antibodies.
It is known that the SH3 domains in Grb2 bind target proteins with proline-rich regions. We reasoned that such a target protein(s) in our case at hand has to be important to chemotaxis. One such candidate is WASP. The Wiskott-Aldrich syndrome protein (WASP) is a major regulator of actin assembly and motility in leukocytes. Two important regions in WASP are its poly-proline-rich domain and its carboxyl-terminal verprolin homology/acidic domain (VCA). The latter enables the interaction of WASP with actin (3, 50, 51, 54, 55). For the former we hypothesized that the two SH3 domains of Grb2 could interact with the poly-proline-rich domain of WASP. Dovas et al. (16) have demonstrated that continuous WASP activity is required for podosome formation and function (such as matrix degradation and chemotaxis) in macrophages, and Jones et al. (27) have demonstrated that WASP-deficient macrophages still move but they do not undergo chemotaxis.
These studies clearly indicate the key importance of WASP in chemotaxis. Figure 9A and B (middle group of bars) confirm the results of those two studies of both LR5 and COS-7 cells, as silencing of (N-)WASP decreased chemotaxis in both the presence and absence of PLD2. The figure also indicates (bars to the right) that overexpression of WASP in combination with Grb2 and PLD2 yields the highest level of chemotaxis, particularly in LR5 cells. As our laboratory has shown previously, PLD2 forms a complex with Grb2 through the SH2 domain. What is left to demonstrate is the interaction of PLD2 with WASP. This was confirmed by immunoprecipitation and Western blotting analysis (Fig. 9C). The figure shows that PLD2 associates with WASP and N-WASP. Thus, we propose that Grb2, a docking protein that is known to serve as a nexus for other signaling proteins (Sos in the ERK pathway, etc.), could bind to PLD2 (at the Y169 and Y179 sites) through its only SH2 domain and to WASP (at the proline-rich region) through its two SH3 domains and that this is a step required for chemotaxis.
Of note, Fig. 9 also shows that RAW/LR5 (but not COS-7) cells were still able to undergo chemotaxis in the presence of silencing WASP plus PLD2, a result that is in agreement with Fig. 8, in that PLD2 overcame partially the silencing of Grb2 only in RAW/LR5 cells. We hypothesized that in addition to Grb2, other signaling pathways downstream of PLD2 must exist in RAW/LR5 chemotaxis. Other authors and our own laboratory (4, 20) have previously pointed at the importance of the ribosomal S6 kinase (S6K) protein in neutrophil motility. We therefore followed this lead and tested whether or not S6K would also be implicated in macrophages or fibroblasts.
As presented in Fig. 10A and B, overexpression of S6K and its silencing in RAW/LR5 cells elevated and abrogated cell motility, respectively. As to what upstream signaling event(s) S6K would be using that does not involve Y169/Grb2/WASP, we followed our recent report (24) that a crucial residue, Y296, lies at the crux of PLD2 regulation by phosphorylation-dephosphorylation. Y296 is phosphorylated by EGF receptor (EGFR) kinase, and we hypothesized that the S6K pathway would involve Y296. Indeed, as presented in Fig. 10A and B, PLD2-Y296F fails to induce cell migration and the combination of S6K/PLD2-Y296F is incapable of restoring the normal enhancing effect of PLD2-WT on chemotaxis, particularly, and again, in RAW/LR5 macrophages. Contrarily, the combination of S6K/PLD2-Y296F does fully restore cell migration in COS-7 cells to the level of that of the PLD2-WT control. Thus, as the dependence on Grb2 is consistently more pronounced in RAW/LR5 cells than in COS-7 cells, in all the experimental conditions we have assayed, it is evident that these cells use all molecules/pathways available to them during chemotaxis.
FIG. 10.
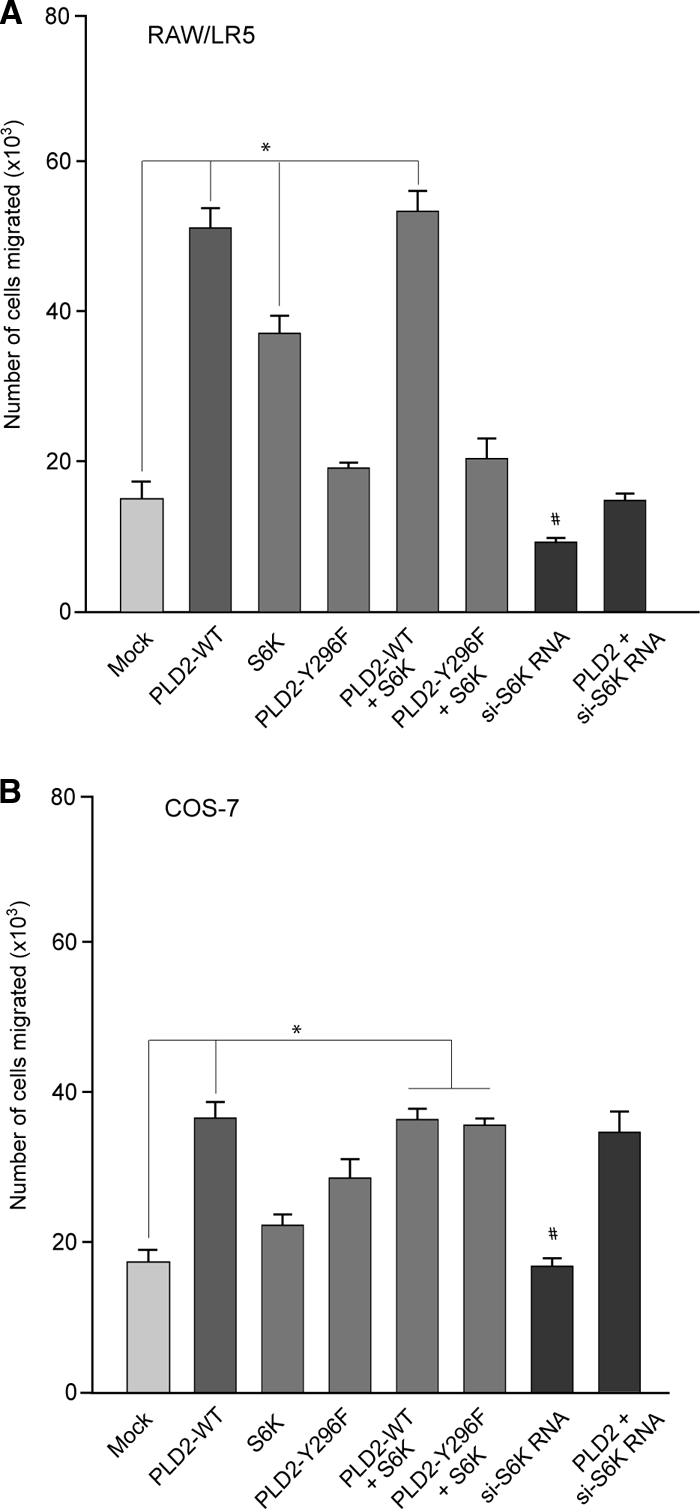
S6K and chemotaxis of RAW/LR5 or COS-7 cells. Cells were transfected with PLD2 (wt or Y296F), S6K or a combination of these plasmids. Cells were also silenced for 3 days with si-S6K RNAs. Values are the means ± the SEM of results of at least three independent experiments performed in duplicate. *, values that were statistically significantly different (P < 0.05) above control values by ANOVA; #, values that were statistically significantly different (P < 0.05) below control values by ANOVA.
DISCUSSION
We have demonstrated in this study that PLD has the capability to increase cell migration in two different cell types: macrophages and fibroblasts. These increases are in addition to the levels achieved by stimulation with known chemoattractants (CSF-1 in the case of macrophages) or motogens (EGF in the case of fibroblasts). We have studied the mechanism of PLD-induced chemotaxis and found that the enzymatic activity of the lipase needs to be intact. This is in agreement with the findings of Mazie et al. (43), who demonstrated that EGF triggers the activation of PLD in MDCK cells. PA, the product of PLD activation, is a critical mediator of EGF-dependent cell motility. Whereas the activity of the enzyme is necessary for chemotaxis, as demonstrated in this and previous studies, PLD activity alone is not sufficient for chemotaxis, as our data on the mutant PLD2-Y179F, which displays an unaltered PLD2 activity, indicated that it failed to increase chemotaxis. We are providing evidence for the participation of the docking protein Grb2 in mediating PLD2-induced chemotaxis.
As seen throughout this study, a specific chemokine that is used against a specific cell line is enough to cause a cell to migrate. Macrophages respond to CSF-1 and MIP-1α. Fibroblasts, known for their ability to synthesize collagen and maintain the structural integrity of connective tissue, have been found to respond to EGF during wound healing. Fibroblasts, by nature, are slower migratory cells, which may explain why EGF has less of an effect on their migration (37) but one that is still robust. In order to investigate the molecular mechanism(s) by which chemotaxis occurs in these two cell types, we used overexpression to identify signals that modulate the rate of chemotaxis. We have found that overexpression of PLD, particularly that of the isoform PLD2, enhanced a cell's ability to undergo chemotaxis. We have also demonstrated that a protein-protein interaction between PLD2 and Grb2 enhanced chemotaxis. Thus, PLD, signaling through Grb2, is a key regulator of the functionality of the cell types studied. This may play an important role in facilitating wound healing and innate defense capabilities of our body.
We have seen that PLD is able to affect other signaling pathways that regulate chemotaxis independently of PLD activity and that the downstream pathways differ for macrophages and fibroblasts. In a rapidly dividing cell like COS-7, recruitment of the Grb2/Sos complex could lead to the activation of Ras effectors and subsequent phosphorylation of MEK through Raf (12, 15) and mitosis. A parallel signaling pathway, through PI3K and, in particular, through S6K, could be triggered for migration. Conversely, in highly motile macrophages, the Grb2/Ras/MEK pathway is switched to serve as a function of motility.
As shown here, Grb2 has been identified as a regulator of PLD2 during chemotaxis, and PLD2-Y169 is needed in order for Grb2 to interact with PLD2. All throughout this study it becomes apparent that in RAW/LR5 cells PLD2 has two different mechanisms to regulate chemotaxis: one dependent and one independent of Grb2. For example, while the Grb2 effect is absolutely dependent on residue R86 (see Fig. 6A), overexpression of PLD2 partially overcomes the negative effect of Grb2-R86K. This is also evident in Fig. 8B where overexpression of PLD2 partially overcomes the inhibitory effect elicited by Grb2 downregulation. These two examples illustrate the Grb2-independent effect of PLD2 on chemotaxis in RAW/LR5 cells. On the other hand, it is clear that the PLD2 effect is potentiated by the expression of a functional Grb2.
With respect to PLD2-Grb2 interaction, and as indicated, Y169 plays a key role for Grb2 to interact with PLD2, whereas another tyrosine, Y179, is more relevant to DNA synthesis/cell proliferation (13). Grb2 has multiple functions associated with embryogenesis, cancer, regulation of the cytoskeleton, cell differentiation, and DNA synthesis (14, 40). Grb2 is a cytoplasmic protein and is able to transmit signals into the cytoplasm (45). When Grb2 is stimulated with EGF, cytoplasmic Grb relocates to the plasma membrane. The Grb2/Sos complex leads to activation of Ras, but it may also lead to activation of another GTPase, Rac, through which macrophage chemotaxis functions (26). PLD was found to be elevated in cells that have been transformed by several oncogenes, including v-Src, v-Ras, v-Raf, and v-Fps, leading to the belief that there is a chronic turnover of PLD-dependent phosphatidylcholine (44). Increased amounts of Grb2 have been correlated with formation of tumors in the liver of mice as well as in human breast cancer cells (46), possibly leading to the ability to metastasize.
Both cell lines studied were able to undergo chemotaxis through a permeable membrane in response to CSF-1 or EGF. For the first time, we have shown that PLD1 and PLD2 are able to augment the chemotactic responses of these cells to CSF-1 and EGF. Transfection of the PLD2 WT plasmid causes a 3-fold increase in chemotaxis rate over that of the mock-transfected cells in the two cell lines studied. The potency could be described as PLD2 > PLD1. In RAW/LR5 cells, transfection of PLD2 WT increased cell migration over that of mock-transfected COS-7 and RAW/LR5 cells.
In summary (Fig. 11), the experiments presented in this paper indicate that PLD2 has a potentiating effect of basal cell migration that is likely due to membrane ruffling, that PLD2 has a potentiating effect on chemoattractant-induced cell migration, that the chemotaxis effect is mediated by PLD2's enzymatic activity and by interaction with other signaling proteins independently of its activity, that there are Grb2-dependent and independent mechanisms for PLD2 to transduce its chemotaxis-inducing signal, and that Grb2 could bind PLD2 (at the Y169 site) through its only SH2 domain and to WASP (at the proline-rich region) through its two SH3 domains. Lastly, RAW/LR5 macrophages are capable of using other signaling routes to accomplish an elevated migratory response to chemoattractants, particularly S6K. Residue Y296 is phosphorylated by EGF receptor (EGFR) kinase, and we believe that the S6K pathway, as it pertains to cell migration, involves Y296 in early signaling. In the end, the data presented here indicate that highly mobile cells like macrophages use all signaling machinery available to them to accomplish their required function in rapid immune response, which sets them apart from fibroblasts, cells normally nonmobile that rarely become migratory (as when they are involved in wound healing).
FIG. 11.

Proposed model for the participation of PLD in chemotaxis. Based on results of this study, along with those of other authors, we propose the participation of PLD and Grb2 in cell chemotaxis involving three major pathways. First, the product of PLD activity, PA, binds to target protein mTOR, S6K, or Sos (17, 36, 56). S6K then stimulates actin polymerization (4). Data in the present study indicate that Y169 is involved in lipase activity (PA production), leading to chemotaxis. Second, PLD can bind to either Grb2 or Sos (5, 12), with Y179 involved in a PLD2-Grb2 protein-protein interaction that leads to a downstream activation of mitogen-activated protein kinase (MAPK). MAPK can cross talk to S6K and provide positive feedback (34) to enhance migration. S6K is mainly implicated in migration of RAW/LR5 macrophages, and that is through the residue Y296. This residue is phosphorylated by EGFR kinase (24). Third, PLD and PA can directly interact with actin (31, 33) or indirectly, as this study indicates, through WASP.
Acknowledgments
We thank Mauricio Di Fulvio for construction of the shGrb2 rescue plasmid and for helpful suggestions on this study.
This work has been supported by the National Institutes of Health grants HL056653 (J.G.-C.) and GM071828 (D.C.).
Footnotes
Published ahead of print on 20 July 2010.
REFERENCES
- 1.Ahn, B., S. Y. Sim, E. H. Kim, K. S. Choi, T. K. Kwon, Y. H. Lee, J. Chang, M. Kim, Y. Jo, and D. S. Min. 2003. Transmodulation between phospholipase D and c-Src enhances cell proliferation. Mol. Cell. Biol. 23:3103-3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banno, Y. 2002. Regulation and possible role of mammalian phospholipase D in cellular functions. J. Biochem. 131:301-306. [DOI] [PubMed] [Google Scholar]
- 3.Benesch, S., S. Lommel, A. Steffen, T. E. Stradal, N. Scaplehorn, M. Way, J. Wehland, and K. Rottner. 2002. Phosphatidylinositol 4,5-biphosphate (PIP2)-induced vesicle movement depends on N-WASP and involves Nck, WIP, and Grb2. J. Biol. Chem. 277:37771-37776. [DOI] [PubMed] [Google Scholar]
- 4.Berven, L. A., F. S. Willard, and M. F. Crouch. 2004. Role of the p70(S6K) pathway in regulating the actin cytoskeleton and cell migration. Exp. Cell Res. 296:183-195. [DOI] [PubMed] [Google Scholar]
- 5.Cammer, M., J. C. Gevrey, M. Lorenz, A. Dovas, J. Condeelis, and D. Cox. 2009. The mechanism of CSF-1-induced Wiskott-Aldrich syndrome protein activation in vivo: a role for phosphatidylinositol 3-kinase and Cdc42. J. Biol. Chem. 284:23302-23311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carpenter, G., and S. Cohen. 1990. Epidermal growth factor. J. Biol. Chem. 265:7709-7712. [PubMed] [Google Scholar]
- 7.Chae, Y. C., J. W. Kim, K. L. Kim, H. W. Kim, H. Y. Lee, W. D. Heo, T. Meyer, P. Suh, and S. H. Ryu. 2008. Phospholipase D activity regulates integrin-mediated cell spreading and migration by inducting GTP-Rac translocation to the plasma membrane. Mol. Biol. Cell 19:3111-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, H. E., S. Chang, T. Trub, and B. G. Neel. 1996. Regulation of colony-stimulating factor 1 receptor signaling by the SH2 domain-containing tyrosine phosphatase SHPTP1. Mol. Cell. Biol. 16:3685-3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, P., K. Gupta, and A. Wells. 1994. Cell movement elicited by epidermal growth factor receptor requires kinase and autophosphorylation but is separable from mitogenesis. J. Cell Biol. 124:547-555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen, P., H. Xie, M. C. Seka, K. Gupta, and A. Wells. 1994. Epidermal growth factor receptor-mediated cell motility: phospholipse C activity is required, but mitogen-activated protein kinase activity is not sufficient for induced cell movement. J. Cell Biol. 127:847-857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi, W. S., T. Hiragun, J. H. Lee, Y. M. Kim, H. Kim, A. Chahdi, E. Her, J. W. Han, and M. A. Beaven. 2004. Activation of RBL-2H3 mast cells is dependent on tyrosine phosphorylation of phospholipase D2 by fyn and fgr. Mol. Cell. Biol. 24:6980-6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cox, D., P. Chang, Q. Zhang, P. G. Reddy, G. M. Bokoch, and S. Greenberg. 1997. Requirements for both Rac1 and Cdc42 in membrane ruffling and phagocytosis in leukocytes. J. Exp. Med. 186:1487-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Fulvio, M., N. Lehman, X. Lin, I. Lopez, and J. Gomez-Cambronero. 2006. The elucidation of novel SH2 binding sites on PLD2. Oncogene 25:3032-3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Fulvio, M., K. Frondorf, K. Henkels, N. Lehman, and J. Gomez-Cambronero. 2007. The Grb2/PLD2 interaction is essential for lipase activity, intracellular localization and signaling, in response to EGF. J. Mol. Biol. 367:814-824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Fulvio, M., K. Frondorf, and J. Gomez-Cambronero. 2008. Mutation of Y179 on phospholipase D2 (PLD2) upregulates DNA synthesis in a PI3K-and Akt-dependent manner. Cell. Signal. 20:176-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dovas, A., J. C. Gevrey, A. Grossi, H. Park, W. Abou-Kheir, and D. Cox. 2009. Regulation of podosome dynamics by WASp phosphorylation: implication in matrix degradation and chemotaxis in macrophages. J. Cell Sci. 122:3873-3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fang, Y., M. Vilella-Bach, R. Bachmann, A. Flanigan, and J. Chen. 2001. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 294:1942-1945. [DOI] [PubMed] [Google Scholar]
- 18.Galliher-Beckley, A., and W. Schiemann. 2008. Grb2 binding to Tyr284 in TβR-II is essential for mammary tumor growth and metastasis stimulated by TGF-β. Carcinogenesis 29:244-251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giubellino, A., Y. Gao, S. Lee, M.-J. Lee, J. Vasselli, S. Medepalli, J. Trepel, T. Burke, and P. Bottaro. 2007. Inhibition of tumor metastasis by a growth factor receptor bound protein 2 Src homology 2 domain-binding antagonist. Cancer Res. 67:6012-6016. [DOI] [PubMed] [Google Scholar]
- 20.Gomez-Cambronero, J., J. Horn, C. C. Paul, and M. A. Baumann. 2003. GM-CSF is a chemoattractant cytokine for neutrophils: involvement of the ribosomal p70S6K signaling pathway. J. Immunol. 171:6846-6855. [DOI] [PubMed] [Google Scholar]
- 21.Gomez-Cambronero, J., M. Di Fulvio, and K. Knapek. 2007. Understanding phospholipase D (PLD) using leukocytes: PLD involvement in cell chemotaxis and adhesion. J. Leukoc. Biol. 82:272-281. [DOI] [PubMed] [Google Scholar]
- 22.Henage, L. G., J. H. Exton, and H. A. Brown. 2006. Kinetic analysis of a mammalian phospholipase D. Allosteric modulation by monomeric GTPases, protein kinase C, and polyphosphoinositides. J. Biol. Chem. 281:3408-3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henkels, K. M., S. Short, H.-J. Peng, M. Di Fulvio, and J. Gomez-Cambronero. 2009. PLD2 has both enzymatic and cell proliferation-inducing capabilities, that are differentially regulated by phosphorylation and dephosphorylation. Biochem. Biophys. Res. Commun. 389:224-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henkels, K. M., H.-J. Peng, K. Frondorf, and J. Gomez-Cambronero. 2010. A comprehensive model that explains the regulation of phospholipase D2 (PLD2) activity by phosphorylation-dephosphorylation. Mol. Cell. Biol. 30:2251-2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horn, J., I. Lopez, M. W. Miller, and J. Gomez-Cambronero. 2005. The uncovering of a novel regulatory mechanism for PLD2: formation of a ternary complex with protein tyrosine phosphatase PTP1B and growth factor receptor-bound protein Grb2. Biochem. Biophys. Res. Commun. 332:58-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Innocenti, M., P. Tenca, E. Frittoli, M. Faretta, A. Tocchetti, P. Paolo Di Fiore, and G. Scita. 2002. Mechanisms through which Sos-1 coordinates the activation of ras and rac. J. Cell Biol. 156:125-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones, G. E., D. Zicha, G. A. Dunn, M. Blundell, and A. Thrasher. 2002. Restoration of podosomes and chemotaxis in Wiskott-Aldrich syndrome macrophages following induced expression of WASp. Int. J. Biochem. Cell Biol. 34:806-815. [DOI] [PubMed] [Google Scholar]
- 28.Jones, G. E. 2000. Cellular signaling in macrophage migration and chemotaxis. J. Leukoc. Biol. 68:593-602. [PubMed] [Google Scholar]
- 29.Knoepp, S. M., M. S. Chahal, Y. Xie, Z. Zhang, D. J. Brauner, M. A. Hallman, S. A. Robinson, S. Han, M. Imai, S. Tomlinson, and K. E. Meier. 2008. Effects of active and inactive phospholipase D2 on signal transduction, adhesion, migration, invasion, and metastasis, in EL4 lymphoma cells. Mol. Pharmacol. 74:574-584. [DOI] [PubMed] [Google Scholar]
- 30.Kook, S., and J. H. Exton. 2005. Identification of interaction sites of protein kinase Ca on phospholipase D1. Cell. Signal. 17:1423-1432. [DOI] [PubMed] [Google Scholar]
- 31.Kusner, D. J., J. A. Barton, K. K. Wen, X. Wang, P. A. Rubenstein, and S. S. Iyer. 2002. Regulation of phospholipase D activity by actin. Actin exerts bidirectional modulation of mammalian phospholipase D activity in a polymerization-dependent, isoform-specific manner. J. Biol. Chem. 277:50683-50692. [DOI] [PubMed] [Google Scholar]
- 32.Lauffenburger, D. A., and F. F. Horwitz. 1996. Cell migration: a physically integrated molecular process. Cell 84:359-369. [DOI] [PubMed] [Google Scholar]
- 33.Lee, S., J. B. Park, J. H. Kim, Y. Kim, J. H. Kim, K. J. Shin, J. S. Lee, S. H. Ha, P. G. Suh, and S. H. Ryu. 2001. Actin directly interacts with phospholipase d, inhibiting its activity. J. Biol. Chem. 276:28252-28260. [DOI] [PubMed] [Google Scholar]
- 34.Lehman, J. A., and J. Gomez-Cambronero. 2002. Molecular crosstalk between p70S6K and MAPK cell signaling pathways. Biochem. Biophys. Res. Commun. 293:463-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lehman, N., M. Di Fulvio, N. McCray, I. Campos, F. Tabatabaian, and J. Gomez-Cambronero. 2006. Phagocyte cell migration is mediated by phospholipases PLD1 and PLD2. Blood 108:3564-3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lehman, N., B. Ledford, M. Di Fulvio, K. Frondorf, L. C. McPhail, and J. Gomez-Cambronero. 2007. Phospholipase D2-derived phosphatidic acid binds to and activates ribosomal p70 S6 kinase independently of mTOR. FASEB J. 21:1075-1087. [DOI] [PubMed] [Google Scholar]
- 37.Li, J., M. Lin, G. J. Wiepz, A. G. Guadarrama, and P. J. Bertics. 1999. Integrin-mediated migration of murine B82L fibroblasts is dependent on the expression of an intact epidermal growth factor receptor. J. Biol. Chem. 274:11209-11219. [DOI] [PubMed] [Google Scholar]
- 38.Liscovitch, M., M. Czarny, G. Fiucci, and X. Tang. 2000. Phospholipase D: molecular and cell biology of a novel gene family. Biochem. J. 345:401-415. [PMC free article] [PubMed] [Google Scholar]
- 39.Lorenz, M., H. Yamaguchi, Y. Wang, R. H. Singer, and J. Condeelis. 2004. Imaging sites of N-wasp activity in lamellipodia and invadopodia of carcinoma cells. Curr. Biol. 14:697-703. [DOI] [PubMed] [Google Scholar]
- 40.Lowenstein, E. J., R. J. Daly, A. G. Batzer, W. Li, B. Margolis, R. Lammers, A. Ullrich, E. Y. Skolnik, D. Bar-Sagi, and J. Schlessing. 1992. The SH2 and SH3 domain-containing protein Grb2 links receptor tyrosine kinases to ras signaling. Cell 70:431-442. [DOI] [PubMed] [Google Scholar]
- 41.Matthay, M. A., J. Thiery, F. Lafont, M. F. Stampfer, and B. Boyer. 1993. Transient effect of epidermal growth factor on the motility of an immortalized mammary epithelial cell line. J. Cell Sci. 106:869-878. [DOI] [PubMed] [Google Scholar]
- 42.Maurer, M., and E. von Stebut. 2004. Macrophage inflammatory protein. Int. J. Biochem. Cell Biol. 36:1882-1886. [DOI] [PubMed] [Google Scholar]
- 43.Mazie, A. R., J. K. Spix, E. R. Block, H. B. Achebe, and J. K. Klarlund. 2006. Epithelial cell motility is triggered by activation of the EGF receptor through phosphatidic acid signaling. J. Cell Sci. 119:1645-1654. [DOI] [PubMed] [Google Scholar]
- 44.Min, D. S., T. K. Kwon, W. Park, J. Chang, S. Park, B. Ahn, Z. Ryoo, Y. H. Lee, Y. S. Lee, D. Rhie, S. Yoon, S. J. Hahn, M. Kim, and Y. Jo. 2001. Neoplastic transformation and tumorigenesis associated with overexpression of phospholipase D isozymes in cultured murine fibroblasts. Carcinogenesis 22:1641-1647. [DOI] [PubMed] [Google Scholar]
- 45.Morimatsu, M., H. Takagi, K. G. Ota, R. Iwamoto, T. Yanagida, and Y. Sako. 2007. Multiple-state reactions between the epidermal growth factor receptor and Grb2 as observed by using single-molecule analysis. Proc. Natl. Acad. Sci. U. S. A. 104:18013-18018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ogura, K., T. Shiga, M. Yokochi, S. Yuzawa, T. R. Burke, Jr., and F. Inagaki. 2008. Solution structure of the Grb2 SH2 domain complexed with a high-affinity inhibitor. J. Biomol. NMR 42:197-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saito, M., and J. Kanfer. 1975. Phosphatidohydrolase activity in a solubilized preparation from rat brain particulate fraction. Arch. Biochem. Biophys. 169:318-323. [DOI] [PubMed] [Google Scholar]
- 48.Sasmono, R. T., D. Oceandy, J. W. Pollard, W. Tong, P. Pavli, B. J. Wainwright, M. C. Ostrowski, S. R. Himes, and D. A. Hume. 2003. A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood 101:1155-1163. [DOI] [PubMed] [Google Scholar]
- 49.Schönlau, F., C. Schlesiger, J. Ehrehen, S. Grabbe, C. Sorg, and C. Sunderkötter. 2003. Monocyte and macrophage functions in M-CSF-deficient op/op mice during experimental leishmaniasis. J. Leukoc. Biol. 73:564-573. [DOI] [PubMed] [Google Scholar]
- 50.Snapper, S. B., P. Meelu, D. Nguyen, B. M. Stockton, P. Bozza, F. W. Alt, F. S. Rosen, U. H. von Andrian, and C. Klein. 2005. WASP deficiency leads to global defects of directed leukocyte migration in vitro and in vivo. J. Leukoc. Biol. 77:993-998. [DOI] [PubMed] [Google Scholar]
- 51.Takenawa, T., and S. Suetsugu. 2007. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat. Rev. Mol. Cell Biol. 8:37-48. [DOI] [PubMed] [Google Scholar]
- 52.Taub, D. D., P. Proost, W. J. Murphy, M. Anver, D. L. Longo, J. V. Damme, and J. J. Oppenheim. 1994. Monocyte chemotactic protein-1 (MCP-1), -2, and -3 are chemotactic for human T lymphocytes. J. Clin. Invest. 95:1370-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weernink, P. A. O., M. López de Jesús, and M. Schmidt. 2007. Phospholipase D signaling: orchestration by PIP2 and small GTPases. Naunyn-Schmiedebergs Arch. Pharmacol. 374:399-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weiner, O. D., M. C. Rentel, A. Ott, G. E. Brown, M. Jedrychowski, M. B. Yaffe, S. P. Gygi, L. C. Cantley, H. R. Bourne, and M. W. Kirschner. 2006. Hem-1 complexes are essential for Rac activation, actin polymerization, and myosin regulation during neutrophil chemotaxis. PLoS Biol. 4:e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang, H., U. Y. Schaff, C. E. Green, H. Chen, M. R. Sarantos, Y. Hu, D. Wara, S. I. Simon, and C. A. Lowell. 2006. Impaired integrin-dependent function in Wiskott-Aldrich syndrome protein-deficient murine and human neutrophils. Immunity 25:285-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao, C., G. Du, K. Skowronek, M. A. Frohman, and D. Bar-Sagi. 2007. Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by Sos. Nat. Cell Biol. 9:706-712. [DOI] [PubMed] [Google Scholar]