

Abstract
Activation through FcɛRI, a high-affinity IgE-binding receptor, is critical for mast cell function during allergy. The formation of a multimolecular proximal signaling complex nucleated by the adaptor molecules SLP-76 and LAT1 is required for activation through this receptor. Based on previous T-cell studies, current dogma dictates that LAT1 is required for plasma membrane recruitment and function of SLP-76. Unexpectedly, we found that the recruitment and phosphorylation of SLP-76 were preserved in LAT1−/− mast cells and that SLP-76−/− and LAT1−/− mast cells harbored distinct functional and biochemical defects. The LAT1-like molecule LAT2 was responsible for the preserved membrane localization and phosphorylation of SLP-76 in LAT1−/− mast cells. Although LAT2 supported SLP-76 phosphorylation and recruitment to the plasma membrane, LAT2 only partially compensated for LAT1-mediated cell signaling due to its decreased ability to stabilize interactions with phospholipase Cγ (PLCγ). Comparison of SLP-76−/− LAT1−/− and SLP-76−/− mast cells revealed that some functions of LAT1 could occur independently of SLP-76. We propose that while SLP-76 and LAT1 depend on each other for many of their functions, LAT2/SLP-76 interactions and SLP-76-independent LAT1 functions also mediate a positive signaling pathway downstream of FcɛRI in mast cells.
Mast cell activation during allergic inflammation is mediated by the high-affinity immunoglobulin E (IgE)-binding receptor FcɛRI. Cross-linking of FcɛRI on mast cells by IgE/cognate antigen complexes results in the rapid release of a wide array of inflammatory mediators, including vasoactive amines and cytokines/chemokines that give rise to allergic symptoms, ranging in severity from simple urticaria to anaphylactic shock and death (14). As allergy affects ∼30% of the population in developed countries (13), much attention has been placed on studying the signal transduction mechanisms involved in mast cell activation downstream of FcɛRI in hopes of finding novel targets for therapeutic intervention.
Signal transduction downstream of FcɛRI is initiated by the phosphorylation of the immunoreceptor tyrosine-based activation motifs (ITAMs) contained in the signaling components (β and γ chains) of the FcɛRI complex (30, 37). Once phosphorylated, these chains serve as docking sites for several protein tyrosine kinases (PTKs), including Lyn and spleen tyrosine kinase (Syk) (9, 19, 34). Recruitment of Syk to the membrane by FcɛRI results in the phosphorylation of scaffold proteins known as adaptor molecules. Adaptor proteins lack enzymatic activity but instead contain protein-binding domains that are critical for the formation of a multimolecular complex, which orchestrates downstream signaling in a temporal and spatial manner. The adaptor molecules Src homology 2 (SH2) domain-containing leukocyte phosphoprotein of 76 kDa (SLP-76) and linker of activated T cells 1 (LAT1) organize the assembly of a proximal signaling complex downstream of FcɛRI. Failure to form this complex is detrimental to FcɛRI-mediated mast cell function, as demonstrated by the finding that both SLP-76-deficient (22, 29, 41) and LAT1-deficient (25, 31, 32) mast cells display severely diminished degranulation and cytokine/chemokine production following FcɛRI ligation.
Similar proximal signaling complexes are formed downstream of several different ITAM-containing receptors. Much of our understanding of the role of adaptor molecules in signal transduction has come from identification of phosphoproteins during T-cell receptor (TCR)-mediated activation of the human Jurkat T-cell line (1, 33). These studies eventually led to a paradigm describing the sequence of events in the formation of the SLP-76/LAT1 signaling complex. According to this model, SLP-76 is found constitutively bound to Grb2-related adaptor downstream of Shc (GADS) (24) and resides in the cytosol. Upon TCR activation, the tyrosines of membrane-resident LAT1 are phosphorylated and become attachment sites for proteins such as phospholipase Cγ (PLCγ) and GADS (43, 45). SLP-76 is drawn to the membrane through a GADS/LAT1 interaction, which then permits Syk family PTKs to maximally phosphorylate the N-terminal tyrosines of SLP-76 (5, 10). Several lines of evidence support this model whereby a LAT1/SLP-76 module organizes TCR signaling. First, both SLP-76- and LAT1-deficient Jurkat T cells display similar biochemical defects, such as diminished PLCγ and extracellular signal-regulated kinase (ERK) activation (10, 42). Second, T cells in SLP-76−/− and LAT1−/− mice are blocked at the same stage of development (7, 44). Third, SLP-76 can be coimmunoprecipitated with LAT1 but not with LAT1 harboring tyrosine-to-phenylalanine mutations (45). Finally, expression of a fusion protein comprised of the membrane-localizing domain of LAT1 and SLP-76 that forces localization of SLP-76 to the plasma membrane rescues the TCR-induced functional defects of both SLP-76- and LAT1-deficient Jurkat T cells (3). This model implies a mutually dependent relationship between SLP-76 and LAT1, where SLP-76 and LAT1 rely on each other to carry out their roles.
One might suspect that this model for LAT1/SLP-76 function would operate in all other cells that utilize these adaptor molecules for ITAM-containing receptor-mediated signaling. However, the published defects of LAT1-deficient mast cells in FcɛRI-mediated signaling appeared milder than those of SLP-76-deficient mast cells, although a direct comparison has never been reported. In the present study, we show that LAT1-deficient mast cells display distinct functional and biochemical defects compared to SLP-76-deficient mast cells, implying that unlike in T cells, SLP-76 may not depend entirely on LAT1 for its function in mast cells. Surprisingly, the membrane recruitment and phosphorylation of SLP-76 were also preserved in LAT1−/− mast cells. We show that LAT2 (also known as non-T-cell activation linker [NTAL] or linker for activation of B cells [LAB]), which is not expressed in naïve T cells but is expressed in mast cells (15), is responsible for phosphorylation and plasma membrane recruitment of SLP-76 in the absence of LAT1. However, LAT2 cannot support all LAT1/SLP-76-associated functions, such as sustained Ca2+ flux, likely due to decreased stability of the LAT2/SLP-76/PLCγ complex. Comparison of SLP-76−/− LAT1−/− and SLP-76−/− mast cells also revealed that some functions of LAT1 could occur independently of SLP-76. We propose that although SLP-76 and LAT1 are interdependent for many of their functions, LAT2/SLP-76 interactions and SLP-76-independent LAT1 functions mediate positive signaling downstream of FcɛRI in mast cells.
MATERIALS AND METHODS
Mice.
SLP-76−/−, LAT2−/−, and LAT1−/− mice have previously been described (7, 47). LAT1−/− LAT2−/− and SLP-76−/− LAT1−/− mice were obtained by breeding LAT2−/− or SLP-76−/− mice with LAT1−/− mice, followed by interbreeding of LAT2+/− LAT1+/− or SLP-76+/− LAT1−/− mice, respectively. Because of the rare occurrence of adult SLP-76−/− LAT1−/− mice due to perinatal lethality, SLP-76−/− LAT1−/− mast cells were also obtained from mice expressing the tamoxifen-inducible Cre recombinase (T2-Cre) and the SLP-76 exon 2 flanked with loxp sites (SLP-76F) (26). SLP-76+/+ LAT1+/+, SLP-76+/+ LAT1−/−, SLP-76F/− LAT1+/+, and SLP-76F/− LAT1−/− mice expressing T2-Cre and a Cre-inducible ROSA26 promoter-driven yellow fluorescent protein (YFP) reporter gene were treated orally with tamoxifen for 5 days to induce deletion of the gene encoding SLP-76. Mast cells were generated from the bone marrow (BM) of these mice and later sorted for YFP positivity as a marker for successful Cre recombinase induction and gene deletion. Deletion of SLP-76 protein was confirmed by Western blot analysis. All animal care and work were in accordance with national and institutional guidelines and the Institutional Animal Care and Use Committee at the University of Pennsylvania.
Generation of BMMCs.
Bone marrow-derived mast cells (BMMCs) were generated as previously described (20). Briefly, BM cells were obtained from the femurs and tibias of wild-type (WT) or mutant mice. BMMCs were generated by culturing BM cells in mast cell medium (MCM) (RPMI 1640, 15% fetal calf serum [FCS], 100 U/ml penicillin, 100 μg/ml streptomycin, 2.9 mg/ml glutamine, 50 mM 2-mercaptoethanol, 1 mM sodium pyruvate, 1× nonessential amino acids, 10 mM HEPES) containing interleukin 3 (IL-3) (10 ng/ml) and stem cell factor (SCF) (12.5 ng/ml) for 6 to 8 weeks with the medium replenished twice weekly. BMMCs were used when >95% of cells expressed high levels of FcɛRI and c-Kit (CD117), as determined by flow cytometry. Cytokines and culture reagents were purchased from Peprotech (Rocky Hill, NJ) and Invitrogen (Carlsbad, CA), respectively.
FcɛRI-induced inflammatory mediator release.
To measure degranulation, BMMCs were allowed to rest overnight in MCM containing IL-3 (10 ng/ml) and IL-4 (10 ng/ml). The BMMCs were then incubated with 1 μg/ml anti-dinitrophenol (anti-DNP) IgE (clone SPE-7; Sigma-Aldrich, St. Louis, MO) for 2 h at 37°C, washed, and stimulated with various concentrations of dinitrophenol-conjugated human serum albumin (HSA-DNP; Sigma) for 1 h in Tyrode's buffer (130 mM NaCl, 10 mM HEPES, 1 mM MgCl2, 5 mM KCl, 1.4 mM CaCl2, 5.6 mM glucose, 1 mg/ml bovine serum albumin [pH 7.4]). Supernatants were tested for β-hexosaminidase activity by incubating with 1 mM p-nitrophenyl-N-acetyl-ß-d-glucosamide (Sigma-Aldrich) dissolved in 0.1 M citrate buffer (pH 4.5) for 1 h at 37°C and terminating with 0.1 M Na2CO3-NaHCO3 buffer in 96-well flat-bottom plates. Absorbance at 405 nm was read with a plate reader. Total cellular β-hexosaminidase activity was quantified by lysing cells with 1% Triton X-100. Serial dilutions of a known quantity of lysed BMMCs were used as a standard to quantify activity. Percent degranulation was calculated as follows: [(activity of the stimulated BMMCs − activity of unstimulated BMMCs)/(total activity − activity of unstimulated BMMCs)] × 100. To measure cytokine and chemokine production, BMMCs were allowed to rest overnight in MCM containing IL-3 (10 ng/ml), incubated with 1 μg/ml anti-DNP IgE for 2 h at 37°C, washed, and stimulated with various concentrations of HSA-DNP for 24 h in MCM. Supernatants were harvested and tested for the presence of IL-6 and monocyte chemotactic peptide 1 (MCP-1) by enzyme-linked immunosorbent assay (ELISA) (BD Pharmingen, San Diego, CA) according to the manufacturer's protocol.
FcɛRI-induced Ca2+ flux.
BMMCs were sensitized with 1 μg/ml anti-DNP IgE for 2 h at 37°C, washed, and resuspended at 1 × 107 cells/ml in Tyrode's buffer containing 1 mM Probenecid (Sigma-Aldrich) and 3 μg/ml Indo-1 (Invitrogen). The cells were protected from light and incubated at 37°C for 30 min. Indo-1-loaded cells were washed twice and resuspended in warm (37°C) Tyrode's buffer. In some experiments, the cells were resuspended in phosphate-buffered saline (PBS) containing EGTA (10 mM) instead of Tyrode's buffer to remove extracellular Ca2+ immediately prior to activation. The cells were stimulated with HSA-DNP (100 ng/ml), and Ca2+ flux was measured by determining the ratio of fluorescence emitted at λ = 482/398 using an LSR flow cytometer (Becton Dickinson, Franklin Lakes, NJ). In some experiments, thapsigargin (1 μM) was used to inhibit endoplasmic reticulum Ca2+-ATPase and activate store-operated Ca2+ (SOC) channels. Baseline Ca2+ levels were measured for 30 s prior to the addition of HSA-DNP. The sample was collected for a total of 180 s, and ionomycin (1 μg/ml) was added 30 s prior to the end of the assay. Data were analyzed using FlowJo software (TreeStar, Ashland, OR).
Immunoprecipitation and Western blot analysis of mast cell lysates.
BMMCs were presensitized with anti-DNP IgE (1 μg/ml) for 2 h, washed, allowed to rest for 2 h, and stimulated with HSA-DNP (30 ng/ml) for indicated times. The cells were then lysed in 1% Ipegal in Tris-buffered saline with protease/phosphatase inhibitors (protease inhibitor cocktail solution [Roche, Sigma], 1 mM sodium orthovanadate, 50 mM sodium fluoride, 50 mM sodium pyrophosphate, 0.2 mM dichloroisocoumarin, and 1 mM benzamidine), and the proteins were resolved by SDS-PAGE (Bio-Rad Laboratories, Hercules, CA). The phosphorylation of PLCγ1 (Tyr783), PLCγ2 (Tyr1217), ERK1/2 (Thr202/Tyr204), SLP-76 (Tyr128), and Akt (Ser473) were analyzed by Western blotting. Total PLCγ1, p38 mitogen-activated protein kinase (MAPK), and SLP-76 were used as loading controls. All antibodies were from Cell Signaling (Danvers, MA), except for the anti-phospho-Y128 (SLP-76) antibody, which was from eBioscience (San Diego, CA). For immunoprecipitations, BMMCs were presensitized with anti-DNP IgE (1 μg/ml) for 2 h, washed, allowed to rest for 2 h, and stimulated with HSA-DNP (30 ng/ml) for indicated times. The cells were then lysed and incubated for 30 min at 4°C with anti-mouse Ig Trueblot Sepharose beads (Invitrogen) that were preincubated with anti-SLP-76 antibody (1.5 μg/50 μl of beads; eBioscience). The beads were washed extensively in lysis buffer (Tris-buffered saline with 1% Ipegal, 1% Triton X, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 60 mM octyl-β-glucoside, and protease/phosphatase inhibitors), transferred to a separate tube, and boiled with loading buffer. The proteins were resolved by SDS-PAGE, and the presence of PLCγ1, SLP-76, and phosphotyrosine (pY; clone 4G10) was analyzed by Western blotting.
Retroviral transduction of BMMCs.
BM cells were cultured in MCM with IL-3 (10 ng/ml), IL-6 (10 ng/ml), and SCF (50 ng/ml) at 37°C overnight. The BM cells were then centrifuged in 12-well plates containing 1 ml of MCM with Polybrene (4 μg/ml), IL-3, IL-6, SCF, and murine stem cell viral supernatant (MigR1) encoding either green fluorescent protein (GFP)-tagged SLP-76 (GFP-SLP-76), GFP-tagged SLP-76 GADS-binding mutant (SLP-76.G2), GFP-tagged SLP-76 SH2 domain mutant (SLP-76.RK), or LAT1/SLP-76 fusion protein (LAT1-SLP) followed by internal ribosome entry site (IRES)-GFP, or IRES-GFP alone for 90 min at 1,250 × g. The MigR1 constructs have been described previously (36). LAT1-SLP, which is a fusion protein between the first 35 amino acids of LAT1 and full-length SLP-76, and the SLP-76 mutants have been described previously (2, 3, 35). After centrifugation, the plates were incubated for 4 to 6 h without agitation at 37°C. Two milliliters of MCM containing IL-3, IL-6, and SCF was added to each well, and the plates were incubated at 37°C overnight. The same centrifugation procedure was repeated the next day to enhance the efficiency of retroviral transduction into BM cells. BMMCs were then generated from the BM cells and were sorted for GFP-positive mast cells after 4 to 6 weeks. The experiments were performed when CD117+ FcɛRI+ cells represented >95% of BMMC cultures.
Imaging by TIRF microscopy.
GFP-SLP-76 microclusters were visualized as previously described (35). Delta T4 dishes (Bioptechs, Butler, PA) were coated with 0.01% poly-l-lysine (Sigma) plus 10 μg/ml anti-IgE or isotype control antibody (BD Pharmingen). IgE-preincubated GFP-SLP-76-expressing BMMCs were deposited onto dishes maintained at 37°C in Tyrode's buffer. Samples were excited using a 488-nm laser line (Coherent), and visualized by a 60× 1.45-numerical-aperture (NA) total internal reflection fluorescence (TIRF) objective fitted to an inverted microscope system (Olympus IX71; Olympus, Center Valley, PA) equipped with a charge-coupled device (CCD) camera (Hamamatsu, Bridgewater, NJ). Images were recorded and analyzed using HCImage software (Hamamatsu), while background fluorescence was equalized using a Blackman filter.
Statistical analysis.
Statistical significance was calculated by t test or paired t test, as indicated in the figure legends, using Microsoft Excel computer software.
RESULTS
The function of SLP-76 is partially independent of LAT1 in mast cells.
Models generated from T-cell studies predict that the recruitment of SLP-76 from the cytosol to the plasma membrane by phosphorylated LAT1 is critical for SLP-76 function. Thus, SLP-76- and LAT1-deficient T cells are expected to elicit similar functional and biochemical defects. To test this notion in mast cells, BMMCs were generated from wild-type (WT) mice and mice lacking SLP-76 or LAT1. As described previously (29, 31), WT, SLP-76−/−, and LAT1−/− BMMCs were phenotypically similar and expressed equivalent levels of CD117 and FcɛRI (data not shown). Upon stimulation through FcɛRI, WT BMMCs exhibited robust degranulation and produced IL-6 and MCP-1. Each of these mast cell functions was dramatically decreased in both SLP-76−/− and LAT1−/− BMMCs (Fig. 1 A to C). However, a direct comparison of SLP-76−/− and LAT1−/− BMMCs revealed that SLP-76−/− BMMCs were significantly more defective than LAT1−/− BMMCs in both degranulation and MCP-1 production (Fig. 1A to C). Importantly, SLP-76- and LAT1-deficient BMMCs degranulated and produced MCP-1 to a similar extent when proximal signaling was bypassed by stimulation with a phorbol ester (data not shown). These data demonstrate that SLP-76−/− mast cells are functionally more defective than LAT1−/− mast cells.
FIG. 1.

SLP-76−/− BMMCs are significantly more defective than LAT1−/− BMMCs are. (A) WT, LAT1−/− (LAT1 knockout [KO]), and SLP-76−/− (SLP KO) BMMCs were preincubated with anti-DNP IgE (1 μg/ml) and stimulated with various concentrations of HSA-DNP. (B and C) Cell-free culture supernatants were analyzed for degranulation and MCP-1 production (B) or IL-6 production (C). The right-hand plots were generated by averaging four independent experiments normalized to WT levels stimulated with 30 ng/ml or 10 ng/ml HSA-DNP. Results are expressed as means plus standard errors of the means (SEMs) (error bars). N.S., not statistically significant.
Activation of PLCγ by SLP-76 and LAT1 leads to a subsequent rise in intracellular Ca2+ that is critical for mast cell function. To examine whether SLP-76−/− and LAT1−/− mast cells differed in activation of the PLCγ pathway, the phosphorylation of PLCγ was examined. Compared to WT BMMCs, the inducible phosphorylation of PLCγ1 and PLCγ2 was markedly but similarly diminished in SLP-76−/− and LAT1−/− BMMCs following FcɛRI-mediated stimulation (Fig. 2A). However, the ability of SLP-76−/− and LAT1−/− BMMCs to mobilize intracellular Ca2+ following FcɛRI-mediated activation appeared to differ. For the first 15 to 30 s following FcɛRI activation, SLP-76−/− BMMCs exhibited a marked delay in Ca2+ flux, while the initial upstroke of Ca2+ flux was almost completely intact in LAT1−/− BMMCs. Despite these differences, intracellular Ca2+ levels of SLP-76−/− and LAT1−/− BMMCs eventually converged to a similarly depressed level compared to WT mast cells.
FIG. 2.
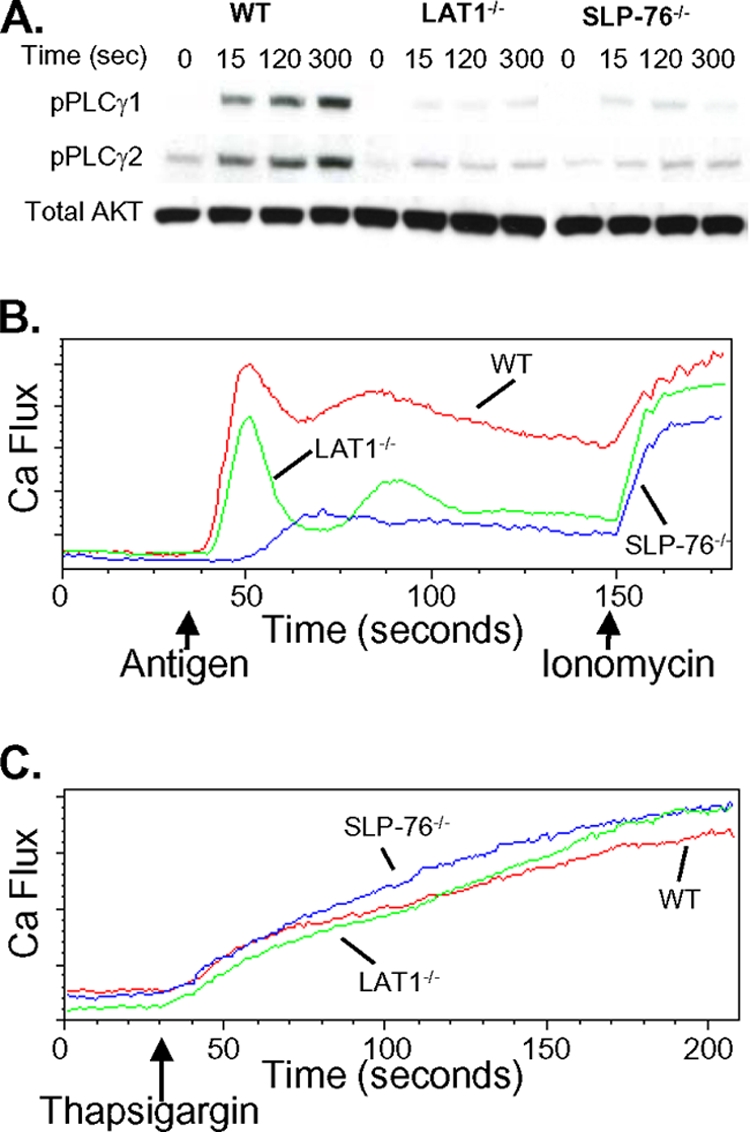
FcɛRI-mediated Ca2+ flux is qualitatively different between SLP-76−/− and LAT1−/− BMMCs. (A) WT, LAT1−/−, and SLP-76−/− BMMCs were preincubated with anti-DNP IgE (1 μg/ml) and stimulated with HSA-DNP (30 ng/ml) for the indicated times (in seconds). Cell lysates were analyzed for phosphorylated PLCγ1 (pPLCγ1) and PLCγ2 (pPLCγ2) by Western blotting. Total Akt served as a loading control. (B and C) WT, LAT1−/−, and SLP-76−/− BMMCs were monitored for elevations in intracellular Ca2+ by flow cytometry. The arrows indicate the time when the stimulus (HSA-DNP, ionomycin, or thapsigargin) was added to the mast cells. The results are representative of at least three independent experiments.
SOC channels respond to depletion of internal endoplasmic reticulum (ER) stores of Ca2+ and allow entry of extracellular Ca2+ to maintain high intracellular Ca2+ concentrations following FcɛRI-mediated stimulation (39). Thus, the inability of LAT1−/− mast cells to sustain Ca2+ flux despite a normal initial Ca2+ upstroke raised the possibility that LAT1−/− mast cells may harbor defects in SOC channels. However, WT, SLP-76−/−, and LAT1−/− BMMCs displayed an equivalent rise in intracellular Ca2+ following depletion of ER Ca2+ stores by thapsigargin (Fig. 2C), suggesting that SOC channels function normally in LAT1−/− mast cells.
Membrane recruitment and phosphorylation of SLP-76 are crucial for the function of SLP-76, a process that is dependent on LAT1 in T cells. As SLP-76−/− and LAT1−/− BMMCs showed functionally and biochemically distinct activation profiles after FcɛRI stimulation, we questioned whether SLP-76 membrane recruitment and phosphorylation were dependent on LAT1 in mast cells. To measure SLP-76 phosphorylation in mast cells, an antibody that specifically recognizes the phosphorylated form of Y128 of SLP-76 was used. In addition, we immunoprecipitated SLP-76 and looked for all tyrosine phosphorylation of SLP-76, since at least two other tyrosines known to be phosphorylated (Y112 and Y145) contribute greatly to SLP-76 function. By using both methods, SLP-76 phosphorylation was similar in LAT1−/− BMMCs compared to WT BMMCs at all time points tested (Fig. 3A and B). Because SLP-76 phosphorylation was intact, we next sought to determine whether SLP-76 was recruited to signaling microclusters in LAT1−/− BMMCs following FcɛRI engagement. Normal SLP-76 recruitment to membrane microclusters was observed in LAT1−/− BMMCs by fluorescence microscopy (Fig. 3C). These data suggest that activation of SLP-76 could occur independently of LAT1 in mast cells.
FIG. 3.

LAT2 can support SLP-76 phosphorylation and membrane recruitment in the absence of LAT1. (A and B) WT, LAT1−/−, LAT2−/−, and LAT1−/− LAT2−/− (double knockout [DKO]) BMMCs were preincubated with anti-DNP IgE (1 μg/ml) and stimulated with HSA-DNP (30 ng/ml) for the indicated time. Cell lysates were analyzed for phosphorylated Y128 of SLP-76 (pY128 SLP) with a pY128-specific antibody and total SLP-76 (A) or immunoprecipitated (IP) with anti-SLP-76 antibody and immunoblotted (IB) for phosphotyrosine (pY) and total SLP-76 (B). (C) Anti-DNP IgE-presensitized GFP-SLP-76-transduced WT, LAT1−/−, LAT2−/−, and LAT1−/− LAT2−/− BMMCs were deposited onto anti-IgE antibody-coated coverslips, and GFP-SLP-76 clustering at the plasma membrane was monitored by TIRF microscopy. Twenty cells were visualized per treatment, and images are representative of four independent experiments. (D and E) WT, LAT1−/−, LAT2−/−, LAT1−/− LAT2−/−, and SLP-76−/− BMMCs were preincubated with anti-DNP IgE and stimulated with various concentrations of HSA-DNP. Cell-free culture supernatants were analyzed for degranulation (D) and MCP-1 production (E). One representative of at least three independent experiments is shown for each panel. (F) WT, SLP-76.G2 (G2) mutant, or SLP-76.RK (RK) mutant GFP-SLP-76-transduced WT or LAT1−/− BMMCs were preincubated with anti-DNP IgE (1 μg/ml) and stimulated with HSA-DNP (30 ng/ml) for the indicated time. Cell lysates were analyzed for phosphorylated Y128 of SLP-76 (pY128 SLP) and total SLP-76. One representative of two independent experiments is shown.
LAT2 is responsible for localization/phosphorylation of SLP-76 and residual function in LAT1−/− mast cells.
In contrast to T cells, mast cells express another LAT1-like molecule known as LAT2. LAT2 is a membrane-bound adaptor molecule containing five tyrosines that are putative binding sites for Grb2 (4) and GADS (15), thereby making it potentially suitable for SLP-76 localization to the plasma membrane. To test whether LAT2 compensated for LAT1 deficiency in mast cells, BMMCs were generated from WT, LAT1−/−, LAT2−/−, and LAT1−/− LAT2−/− mice. The level of SLP-76 phosphorylation was similar among WT, LAT1−/−, and LAT2−/− BMMCs following FcɛRI stimulation (Fig. 3A and B). However, in the absence of both LAT1 and LAT2, SLP-76 phosphorylation was markedly decreased (Fig. 3A and B). Similarly, membrane recruitment of SLP-76 was observed in WT, LAT1−/−, and LAT2−/− BMMCs but was severely diminished in LAT1−/− LAT2−/− BMMCs (Fig. 3C). Next, the function of LAT1−/− LAT2−/− BMMCs was compared to SLP-76−/− BMMCs. Although LAT1−/− BMMCs were more functional than SLP-76−/− BMMCs, LAT1−/− LAT2−/− and SLP-76−/− BMMCs were equally defective in degranulation and MCP-1 production (Fig. 3D and E). These data suggest that LAT2 is responsible for the localization and phosphorylation of SLP-76 and for the residual function in LAT1−/− mast cells. Of note, the increased degranulation response of the LAT2−/− BMMCs is consistent with previous reports demonstrating that LAT2−/− BMMCs are hyperfunctional due to an increased amount of LAT1 in the plasma membrane (23, 40).
To gain insight into the molecular mechanism by which LAT2 interacted with SLP-76, we tested the ability of two mutant SLP-76 molecules to be phosphorylated in BMMCs following FcɛRI cross-linking. BMMCs were retrovirally transduced with either GFP-tagged forms of WT SLP-76, SLP-76 with a mutation of the GADS-binding domain (SLP-76.G2), or SLP-76 with a mutation in the SH2 domain (SLP-76.RK). Upon FcɛRI activation, equal phosphorylation of both WT SLP-76 and SLP-76.RK was observed, suggesting that the SH2 domain of SLP-76 was not necessary for SLP-76 phosphorylation. In contrast, phosphorylation of SLP-76.G2 was severely diminished, suggesting that the GADS-binding domain of SLP-76 is critical for SLP-76 phosphorylation (Fig. 3F). Similarly, phosphorylation of SLP-76.G2 was not seen in LAT1−/− BMMCs (Fig. 3F), suggesting that LAT2 allowed SLP-76 phosphorylation to occur, potentially through a LAT2/GADS/SLP-76 interaction.
LAT2 mediates the initial upstroke of intracellular Ca2+ flux in LAT1−/− mast cells.
The rise in intracellular Ca2+ levels during the initial 15 to 30 s following FcɛRI activation is delayed in SLP-76−/− mast cells, but not in LAT1−/− mast cells. To test whether this rapid rise in intracellular Ca2+ preserved in LAT1−/− mast cells was mediated by LAT2, Ca2+ flux profiles were examined in WT, LAT1−/−, SLP-76−/−, and LAT1−/− LAT2−/− BMMCs. While LAT1−/− BMMCs exhibited a quick rise in intracellular Ca2+ immediately following FcɛRI stimulation, Ca2+ flux in both SLP-76−/− and LAT1−/− LAT2−/− BMMCs was similarly delayed (Fig. 4A). This observation suggests that LAT2 can activate the PLCγ pathway and induce Ca2+ flux in mast cells. This seems discordant, however, with our observation that LAT1−/− and SLP-76−/− mast cells show an equivalent defect in PLCγ phosphorylation (Fig. 2A). It is noted though that this analysis was performed 30 s after TCR stimulation, and it is possible that the activation of PLCγ by LAT2/SLP-76 was too transient to be detected using our Western blot approach. Thus, to examine the effect of LAT1 and SLP-76 deficiency on very early PLCγ activation, we examined Ca2+ flux in mast cells in the absence of extracellular Ca2+. This restricts the source of Ca2+ to intracellular stores only and is a reflection of the amount of inositol trisphosphate (IP3) produced due to PLCγ activation. Upon chelation of extracellular Ca2+, we found that LAT1−/− mast cells did display a rapid rise and then a decrease in intracellular Ca2+, whereas SLP-76−/− mast cells failed to demonstrate a similar early rise in intracellular Ca2+ levels (Fig. 4B). These data support the notion that LAT2 can support a rapid but transient activation of PLCγ.
FIG. 4.

Cooperation of SLP-76 and LAT1 is important for FcɛRI-mediated signaling and function. (A) WT, LAT1−/−, LAT1−/− LAT2−/−, and SLP-76−/− BMMCs were preincubated with anti-DNP IgE (1 μg/ml) and monitored for elevations in intracellular Ca2+ by flow cytometry after stimulation in Tyrode's buffer. (B) WT, LAT1−/−, and SLP-76−/− BMMCs were preincubated with anti-DNP IgE (1 μg/ml) and monitored for elevations in intracellular Ca2+ by flow cytometry after stimulation in PBS containing EGTA (10 mM). The arrows indicate the time when the stimulus (HSA-DNP or ionomycin) was added to the mast cells. (C) WT, LAT1−/−, and SLP-76−/− BMMCs were preincubated with anti-DNP IgE and stimulated with HSA-DNP (30 ng/ml) for the indicated time. Cell lysates were immunoprecipitated with anti-SLP-76 antibody and blotted for total SLP-76 and PLCγ1. (D) WT, SLP-76−/−, or LAT1−/− BMMCs were retrovirally transduced with vector alone (MIGR) or full-length SLP-76 fused to the extracellular domain of LAT1 (LAT1-SLP), preincubated with anti-DNP IgE, and stimulated with various concentrations of HSA-DNP. Cell-free culture supernatants were analyzed for degranulation. Results are representative of at least two independent experiments.
We reasoned that the inability of LAT2 to sustain PLCγ activation might be due to the lack of a PLCγ-binding tyrosine in LAT2. The PLCγ-binding tyrosine of LAT1 has been shown to be crucial for the function of LAT1 (32), and thus, cooperative stabilization of PLCγ by LAT1 and SLP-76 may be important for prolonged PLCγ activation. LAT2 lacks this putative PLCγ-binding tyrosine motif (18) and therefore may not be able to assist SLP-76 in stabilizing PLCγ at the plasma membrane. To test this possibility, PLCγ binding to SLP-76 was measured in WT and LAT1−/− BMMCs. Basal and FcɛRI-induced binding of PLCγ to SLP-76 was greatly reduced in LAT1−/− BMMCs compared to WT BMMCs (Fig. 4C), suggesting that LAT2 alone cannot stabilize the SLP-76/PLCγ interaction.
Localization of SLP-76 can be forced to the plasma membrane by fusing the transmembrane portion of LAT1 to full-length SLP-76 (LAT1-SLP). Expression of LAT1-SLP rescues the function of both SLP-76- and LAT1-deficient Jurkat T cells (3). This protein mimics the LAT2/SLP-76 interaction, since it allows for SLP-76 localization to the plasma membrane without cooperative binding of PLCγ. Since the LAT2/SLP-76 interaction was insufficient for full activation of mast cells, we reasoned that unlike Jurkat T cells, LAT1-SLP might not be able to rescue the defect afforded by LAT1 deficiency in mast cells. To test this notion, SLP-76−/− and LAT1−/− BMMCs were retrovirally transduced with LAT1-SLP and analyzed for FcɛRI-mediated degranulation responses. Expression of LAT1-SLP rescued the degranulation defect of SLP-76−/− BMMCs, but not LAT1−/− BMMCs (Fig. 4D). Thus, localization of SLP-76 to the plasma membrane is insufficient for full activation of mast cells in the absence of LAT1.
SLP-76 and LAT1 independently contribute to phosphorylation of ERK.
Although SLP-76−/− mast cells are severely defective in FcɛRI-mediated mast cell activation, some residual Ca2+ flux is still observed (Fig. 2A). Moreover, ERK phosphorylation is entirely intact in SLP-76−/− mast cells (41). LAT1 may direct Ca2+ flux and ERK phosphorylation independently of SLP-76 by localizing PLCγ and Grb2/SOS (SOS stands for son of sevenless), respectively. To test this notion, BMMCs from SLP-76−/− LAT1−/− mice were generated and compared to LAT1−/− and SLP-76−/− BMMCs. LAT1−/− and SLP-76−/− BMMCs displayed decreased Ca2+ flux upon FcɛRI stimulation compared to WT BMMCs (Fig. 5A). SLP-76−/− LAT1−/− BMMCs exhibited a further decrease in Ca2+ flux (Fig. 5A), suggesting that LAT1 contributed to Ca2+ flux independently of SLP-76. Next, ERK phosphorylation was examined in FcɛRI-stimulated BMMCs. Similar to previous observations (41, 47), ERK phosphorylation remained largely intact in SLP-76- and LAT1-deficient BMMCs compared to WT BMMCs (Fig. 5C). In contrast, ERK phosphorylation was markedly decreased in SLP-76−/− LAT1−/− BMMCs. Only one SLP-76−/− LAT1−/− mouse could be obtained for these experiments due to high perinatal lethality of these mice. Thus, to confirm these results, SLP-76−/− LAT1−/− BMMCs were also obtained by using LAT1−/− mice with conditionally deleted SLP-76 (SLP-76F/−). Similar results were obtained from experiments using these mast cells (Fig. 5B and D). Surprisingly, LAT1−/− LAT2−/− mast cells exhibited near-normal ERK phosphorylation after FcɛRI cross-linking (Fig. 5E) despite the markedly decreased ability of these mast cells to localize and phosphorylate SLP-76. This suggests that only a small amount of SLP-76 phosphorylation might be necessary for ERK activation in mast cells. Together, these findings suggest that although SLP-76 and LAT1 depend on each other for many of their functions, SLP-76-independent LAT1-dependent and LAT1-independent SLP-76-dependent signals also contribute to FcɛRI-mediated activation of mast cells.
FIG. 5.

SLP-76 and LAT1 contribute independently to Ca2+ flux and ERK phosphorylation. (A and B) WT, LAT1−/−, SLP-76−/−, and SLP-76−/− LAT1−/− BMMCs (A) or YFP-positive SLP-76+/+ LAT1+/+, SLP-76+/+ LAT1−/−, SLP-76F/− LAT1+/+, and SLP-76F/− LAT1−/− BMMCs (B) were preincubated with anti-DNP IgE (1 μg/ml) and monitored for elevations in intracellular Ca2+ by flow cytometry. The arrows indicate the time when the stimulus (HSA-DNP or ionomycin) was added to the mast cells. (C and D) WT, LAT1−/−, SLP-76−/−, and SLP-76−/− LAT1−/− BMMCs (C) or YFP-positive SLP-76+/+ LAT1+/+, SLP-76+/+ LAT1−/−, SLP-76F/− LAT1+/+, and SLP-76F/− LAT1−/− BMMCs (D) were preincubated with anti-DNP IgE and stimulated with HSA-DNP (30 ng/ml) for the indicated time. Cell lysates were analyzed for phosphorylated ERK (pERK), total p38 MAPK, or total PLCγ1 by Western blotting. (E) WT, LAT1−/−, LAT2−/−, and LAT2/LAT1 DKO BMMCs were preincubated with anti-DNP IgE (1 μg/ml) and stimulated with HSA-DNP (30 ng/ml) for the indicated time. Cell lysates were analyzed for phosphorylated ERK and total p38 MAPK. Total p38 MAPK or PLCγ1 served as loading controls. Results are representative of at least two independent experiments.
DISCUSSION
We demonstrate herein that LAT1−/− BMMCs display milder functional and biochemical defects than SLP-76−/− BMMCs do. Moreover, localization and phosphorylation of SLP-76 in LAT1−/− BMMCs were largely normal, which were mediated by LAT2 in LAT1−/− mast cells. However, LAT2 failed to fully support LAT1-mediated signaling due to diminished stability of the SLP-76/PLCγ interaction. SLP-76 and LAT1 also independently contribute to mast cell activation, as SLP-76−/− LAT1−/− BMMCs showed a further decrease in Ca2+ flux and impaired ERK phosphorylation compared to SLP-76−/− or LAT1−/− BMMCs. Collectively, these data establish cooperative and independent roles for these adaptor molecules in their contribution to FcɛRI-mediated mast cell activation (Fig. 6).
FIG. 6.

Contribution of proximal signaling complexes downstream of FcɛRI to Ca2+ flux and ERK activation. Upon mast cell activation through FcɛRI, both LAT1 and LAT2 recruit SLP-76 to the plasma membrane and allow for SLP-76 phosphorylation. This occurs through the GADS-binding domain of SLP-76, implicating the involvement of GADS in this process. Three distinct molecular complexes involving PLCγ contribute to its activation: LAT1/SLP-76, LAT2/SLP-76, and LAT1 alone. Because LAT1, but not LAT2, cooperatively binds to PLCγ with SLP-76, LAT1/SLP-76 contributes most to increasing intracellular Ca2+ levels. LAT1 and SLP-76 both cooperatively and independently lead to ERK phosphorylation. Thus, deletion of both LAT1 and SLP-76 is required for disruption of the ERK signaling pathway. SLP-76-independent LAT1-dependent ERK activation may involve localization of Grb2/SOS by LAT1. The upstream molecule(s) involved in LAT1/2-independent SLP-76 activation leading to ERK phosphorylation is unknown at this time.
In contrast to the milder defects in degranulation and MCP-1 production by LAT1−/− BMMCs, IL-6 production was equally depressed in SLP-76- and LAT1-deficient BMMCs. While we do not know the exact mechanism that contributes to this finding, it is possible that the activation threshold for IL-6 production is higher than that of MCP-1 or degranulation. This notion is supported by a previous study demonstrating that strong signals from FcɛRI preferentially lead to IL-6 production, while weaker signals lead to MCP-1 production (12). Thus, the weak signal transduced by LAT1−/− BMMCs may be sufficient for some MCP-1 production, whereas it is insufficient for IL-6 production.
SLP-76 localization and phosphorylation were greatly diminished in LAT1−/− LAT2−/− BMMCs but intact in LAT1−/− BMMCs, suggesting that LAT2 is as efficient as LAT1 in drawing SLP-76 to the membrane. It is important to note, however, that the phosphorylation of SLP-76 was not completely lost in LAT1−/− LAT2−/− BMMCs. One potential explanation for this is that some SLP-76 phosphorylation may occur independently of its recruitment to the plasma membrane. This is concordant with results seen in the T-cell compartment where LAT1 deficiency results in a significant reduction, but not an absolute elimination, of SLP-76 phosphorylation (10), and the complementary finding that a mutant variant of SLP-76 that cannot bind to GADS (and hence, LAT1) is also phosphorylated to a limited extent in T cells (27). This likely results from some SLP-76 interaction with activated Syk family kinases in the cell, leading to phosphorylation without localization to the plasma membrane. In addition, we have found that the phosphorylation of SLP-76 with a mutation in the GADS-binding site is further decreased in LAT1/LAT2 double knockout (DKO) mast cells (data not shown), suggesting that another protein that interacts with SLP-76 through GADS could also be involved in the phosphorylation of SLP-76. For example, another LAT1 family member, such as linker for activation of X cells (LAX) (48), might allow for some phosphorylation of SLP-76. However, it is clear that for optimal SLP-76 phosphorylation to occur, membrane localization through LAT1 or LAT2 is required.
The recruitment of SLP-76 to LAT2 likely brings SLP-76-bound PLCγ to the plasma membrane and may drive the quick upstroke in Ca2+ flux immediately following FcɛRI stimulation. Indeed, an indirect association of LAT2 and PLCγ involving an unidentified cytosolic adaptor protein has been reported (16). Although LAT2 supports initial Ca2+ flux, it is insufficient to sustain Ca2+ flux at WT levels. Consistent with this observation, PLCγ phosphorylation is largely diminished in LAT1−/− BMMCs, suggesting that the LAT2/SLP-76 interaction is insufficient for optimal PLCγ phosphorylation. This finding may be explained by the lack of the PLCγ-binding tyrosine in LAT2. Unlike LAT1, LAT2 cannot cooperatively bind to PLCγ with SLP-76, and hence, the localization of PLCγ can be sustained only temporarily, which is reflected in the decreased association of PLCγ with SLP-76 in the presence of LAT2 alone. The levels of PLCγ phosphorylation in LAT1−/− BMMCs and SLP-76−/− BMMCs are indistinguishable 30 s after FcɛRI stimulation, suggesting that PLCγ activation cannot be sustained in the absence of LAT1. Thus, despite having the ability to localize SLP-76, LAT2 differs from LAT1 in its ability to mediate FcɛRI-mediated positive signals.
Although FcɛRI-mediated signaling is severely defective in SLP-76−/− mast cells, some biochemical signaling pathways are retained. For example, the phosphorylation of ERK is unaltered in SLP-76−/− mast cells. ERK activation involves a cascade of phosphorylation events mediated by a series of kinases beginning with the activation of Ras GTPase. In mast cells, the activation of Ras can be mediated by at least two distinct guanine nucleotide exchange factors, RasGRP (8) and son of sevenless (SOS) (17). Activation of both factors potentially involves LAT1 and SLP-76, since RasGRP is activated by diacylglycerol (DAG)/Ca2+ following PLCγ activation and Grb2-associated SOS is localized by LAT1. However, the proximal signaling pathways leading to activation of Ras in mast cells have been unclear, as mast cells lacking either SLP-76 or LAT1 display intact phosphorylation of ERK (11, 41, 47). Because multiple signaling pathways contribute to Ras activation and the threshold for ERK phosphorylation is low, blockade of more than one pathway may be necessary to observe any reduction in ERK phosphorylation. Indeed, our data suggest that LAT1 and SLP-76 independently contribute to ERK activation and that ERK phosphorylation is decreased only when both SLP-76 and LAT1 are lacking in mast cells. The SLP-76-independent LAT1-dependent ERK phosphorylation is likely due to binding of Grb2/SOS to LAT1, which has been shown to occur independently of SLP-76 (42). Surprisingly, LAT1−/− LAT2−/− mast cells exhibited near-normal ERK phosphorylation after FcɛRI cross-linking, despite the decreased abilities of these mast cells to localize and phosphorylate SLP-76. This result suggests that LAT2/LAT1-independent SLP-76-dependent function also contributes to ERK phosphorylation.
Optimal PLCγ activation requires the cooperative stabilization of PLCγ by LAT1 and SLP-76. However, SLP-76 and LAT1 can also contribute independently to Ca2+ immobilization, as SLP-76−/− LAT1−/− BMMCs exhibit a further decrease in Ca2+ flux compared to mast cells lacking either SLP-76 or LAT1. It appears that the PLCγ-binding tyrosine of LAT1 has some ability to localize PLCγ without SLP-76. Alternatively, it is possible that another SLP-76-like molecule compensates in the absence of SLP-76. Mast cells express the SLP-76-related protein cytokine-dependent hematopoietic cell linker (Clnk), which has the ability to localize PLCγ through its phosphorylated tyrosine. Although Clnk−/− mast cells have been shown to degranulate normally compared to WT controls (38), a similar scenario to LAT2/LAT1 may exist where the positive signaling effects of Clnk may be apparent only in the absence of SLP-76. Thus, SLP-76−/− Clnk−/− mast cells may show additional defects over SLP-76−/− mast cells and phenocopy SLP-76−/− LAT1−/− mast cells.
Not all signals downstream of FcɛRI require SLP-76, LAT1, and LAT2. The phosphorylation of Akt is unaltered in SLP-76−/− mast cells as well as SLP-76−/− LAT1−/− mast cells (data not shown), suggesting that these adaptor molecules do not contribute to Akt activation. Akt activation likely occurs through an entirely distinct signaling pathway involving Fyn and the adaptor protein Gab2 (28). Accordingly, SLP-76−/− Fyn−/− mast cells but not SLP-76−/− mast cells demonstrate decreased Akt phosphorylation (21).
It is unclear what role LAT2 plays in FcɛRI-mediated signaling in WT mast cells expressing LAT1. In initial studies, LAT2 was thought to be a negative regulator of mast cell activation, as LAT2−/− mast cells displayed heightened activation compared to WT controls (40, 47); however, this effect was explained by LAT1 and LAT2 competing for sites in the plasma membrane and for phosphorylation by Syk. Nevertheless, expression of LAT2 would be expected to negatively impact mast cell activation, given that LAT2 is less effective than LAT1 in mediating a positive signal downstream of FcɛRI. It is possible that the regulation of the LAT1/LAT2 ratio within mast cells plays a role in modulating mast cell activation. Decreased expression of LAT1 or increased LAT2 expression (decreased LAT1/LAT2 ratio) could make mast cells less responsive to FcɛRI cross-linking stimuli. Such changes in the expression pattern of LAT1/LAT2 have been reported to occur in TCR-activated T cells (46) and cytokine-stimulated NK cells (6). Although resting T cells do not express LAT2, inducible LAT2 expression by activated T cells appears to negatively regulate their function, as aged LAT2−/− mice develop a T-cell-mediated autoimmune syndrome (46). It is thus conceivable that similar regulation of LAT1/LAT2 ratios in mast cells could also alter their activation threshold.
In sum, we have demonstrated that redundancy of function exists between the related adaptor molecules LAT1 and LAT2 in the activation of SLP-76. Although SLP-76 and LAT1/LAT2 mainly exert their functions through cooperation, each of these molecules can also contribute individually to FcɛRI-mediated positive signals. Further investigation is required to clarify the precise role of each of these adaptor molecules and to establish relevance to FcɛRI-stimulated mast cell functions.
Acknowledgments
We thank members of the Koretzky and Kambayashi laboratories for helpful discussions and Rebecca May, Kim Nichols, and Justina Stadanlick for careful reading of our manuscript.
This work was supported by grants from the Sandler Program for Asthma Research and from the National Institutes of Health.
Footnotes
Published ahead of print on 6 July 2010.
REFERENCES
- 1.Abraham, R. T., and A. Weiss. 2004. Jurkat T cells and development of the T-cell receptor signalling paradigm. Nat. Rev. Immunol. 4:301-308. [DOI] [PubMed] [Google Scholar]
- 2.Baker, R. G., C. J. Hsu, D. Lee, M. S. Jordan, J. S. Maltzman, D. A. Hammer, T. Baumgart, and G. A. Koretzky. 2009. The adapter protein SLP-76 mediates “outside-in” integrin signaling and function in T cells. Mol. Cell. Biol. 29:5578-5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boerth, N. J., J. J. Sadler, D. E. Bauer, J. L. Clements, S. M. Gheith, and G. A. Koretzky. 2000. Recruitment of SLP-76 to the membrane and glycolipid-enriched membrane microdomains replaces the requirement for linker for activation of T cells in T cell receptor signaling. J. Exp. Med. 192:1047-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brdicka, T., M. Imrich, P. Angelisova, N. Brdickova, O. Horvath, J. Spicka, I. Hilgert, P. Luskova, P. Draber, P. Novak, N. Engels, J. Wienands, L. Simeoni, J. Osterreicher, E. Aguado, M. Malissen, B. Schraven, and V. Horejsi. 2002. Non-T cell activation linker (NTAL): a transmembrane adaptor protein involved in immunoreceptor signaling. J. Exp. Med. 196:1617-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bubeck Wardenburg, J., C. Fu, J. K. Jackman, H. Flotow, S. E. Wilkinson, D. H. Williams, R. Johnson, G. Kong, A. C. Chan, and P. R. Findell. 1996. Phosphorylation of SLP-76 by the ZAP-70 protein-tyrosine kinase is required for T-cell receptor function. J. Biol. Chem. 271:19641-19644. [DOI] [PubMed] [Google Scholar]
- 6.Chiesa, S., M. Mingueneau, N. Fuseri, B. Malissen, D. H. Raulet, M. Malissen, E. Vivier, and E. Tomasello. 2006. Multiplicity and plasticity of natural killer cell signaling pathways. Blood 107:2364-2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clements, J. L., S. E. Ross-Barta, L. T. Tygrett, T. J. Waldschmidt, and G. A. Koretzky. 1998. SLP-76 expression is restricted to hemopoietic cells of monocyte, granulocyte, and T lymphocyte lineage and is regulated during T cell maturation and activation. J. Immunol. 161:3880-3889. [PubMed] [Google Scholar]
- 8.Ebinu, J. O., S. L. Stang, C. Teixeira, D. A. Bottorff, J. Hooton, P. M. Blumberg, M. Barry, R. C. Bleakley, H. L. Ostergaard, and J. C. Stone. 2000. RasGRP links T-cell receptor signaling to Ras. Blood 95:3199-3203. [PubMed] [Google Scholar]
- 9.El-Hillal, O., T. Kurosaki, H. Yamamura, J. P. Kinet, and A. M. Scharenberg. 1997. syk kinase activation by a src kinase-initiated activation loop phosphorylation chain reaction. Proc. Natl. Acad. Sci. U. S. A. 94:1919-1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finco, T. S., T. Kadlecek, W. Zhang, L. E. Samelson, and A. Weiss. 1998. LAT1 is required for TCR-mediated activation of PLCgamma1 and the Ras pathway. Immunity 9:617-626. [DOI] [PubMed] [Google Scholar]
- 11.Gomez, G., C. Gonzalez-Espinosa, S. Odom, G. Baez, M. E. Cid, J. J. Ryan, and J. Rivera. 2005. Impaired FcepsilonRI-dependent gene expression and defective eicosanoid and cytokine production as a consequence of Fyn deficiency in mast cells. J. Immunol. 175:7602-7610. [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez-Espinosa, C., S. Odom, A. Olivera, J. P. Hobson, M. E. Martinez, A. Oliveira-Dos-Santos, L. Barra, S. Spiegel, J. M. Penninger, and J. Rivera. 2003. Preferential signaling and induction of allergy-promoting lymphokines upon weak stimulation of the high affinity IgE receptor on mast cells. J. Exp. Med. 197:1453-1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gould, H. J., and B. J. Sutton. 2008. IgE in allergy and asthma today. Nat. Rev. Immunol. 8:205-217. [DOI] [PubMed] [Google Scholar]
- 14.Hines, C. 2002. The diverse effects of mast cell mediators. Clin. Rev. Allergy Immunol. 22:149-160. [DOI] [PubMed] [Google Scholar]
- 15.Iwaki, S., B. M. Jensen, and A. M. Gilfillan. 2007. Ntal/Lab/LAT2. Int. J. Biochem. Cell Biol. 39:868-873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iwaki, S., J. Spicka, C. Tkaczyk, B. M. Jensen, Y. Furumoto, N. Charles, M. Kovarova, J. Rivera, V. Horejsi, D. D. Metcalfe, and A. M. Gilfillan. 2008. Kit- and Fc epsilonRI-induced differential phosphorylation of the transmembrane adaptor molecule NTAL/LAB/LAT2 allows flexibility in its scaffolding function in mast cells. Cell Signal. 20:195-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jabril-Cuenod, B., C. Zhang, A. M. Scharenberg, R. Paolini, R. Numerof, M. A. Beaven, and J. P. Kinet. 1996. Syk-dependent phosphorylation of Shc. A potential link between FcepsilonRI and the Ras/mitogen-activated protein kinase signaling pathway through SOS and Grb2. J. Biol. Chem. 271:16268-16272. [DOI] [PubMed] [Google Scholar]
- 18.Janssen, E., M. Zhu, B. Craven, and W. Zhang. 2004. Linker for activation of B cells: a functional equivalent of a mutant linker for activation of T cells deficient in phospholipase C-gamma1 binding. J. Immunol. 172:6810-6819. [DOI] [PubMed] [Google Scholar]
- 19.Jouvin, M. H., M. Adamczewski, R. Numerof, O. Letourneur, A. Valle, and J. P. Kinet. 1994. Differential control of the tyrosine kinases Lyn and Syk by the two signaling chains of the high affinity immunoglobulin E receptor. J. Biol. Chem. 269:5918-5925. [PubMed] [Google Scholar]
- 20.Kambayashi, T., J. D. Baranski, R. G. Baker, T. Zou, E. J. Allenspach, J. E. Shoag, P. L. Jones, and G. A. Koretzky. 2008. Indirect involvement of allergen-captured mast cells in antigen presentation. Blood 111:1489-1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kambayashi, T., D. F. Larosa, M. A. Silverman, and G. A. Koretzky. 2009. Cooperation of adapter molecules in proximal signaling cascades during allergic inflammation. Immunol. Rev. 232:99-114. [DOI] [PubMed] [Google Scholar]
- 22.Kettner, A., V. Pivniouk, L. Kumar, H. Falet, J. S. Lee, R. Mulligan, and R. S. Geha. 2003. Structural requirements of SLP-76 in signaling via the high-affinity immunoglobulin E receptor (Fc epsilon RI) in mast cells. Mol. Cell. Biol. 23:2395-2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koonpaew, S., E. Janssen, M. Zhu, and W. Zhang. 2004. The importance of three membrane-distal tyrosines in the adaptor protein NTAL/LAB. J. Biol. Chem. 279:11229-11235. [DOI] [PubMed] [Google Scholar]
- 24.Liu, S. K., N. Fang, G. A. Koretzky, and C. J. McGlade. 1999. The hematopoietic-specific adaptor protein gads functions in T-cell signaling via interactions with the SLP-76 and LAT1 adaptors. Curr. Biol. 9:67-75. [DOI] [PubMed] [Google Scholar]
- 25.Malbec, O., M. Malissen, I. Isnardi, R. Lesourne, A. M. Mura, W. H. Fridman, B. Malissen, and M. Daeron. 2004. Linker for activation of T cells integrates positive and negative signaling in mast cells. J. Immunol. 173:5086-5094. [DOI] [PubMed] [Google Scholar]
- 26.Maltzman, J. S., L. Kovoor, J. L. Clements, and G. A. Koretzky. 2005. Conditional deletion reveals a cell-autonomous requirement of SLP-76 for thymocyte selection. J. Exp. Med. 202:893-900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Myung, P. S., G. S. Derimanov, M. S. Jordan, J. A. Punt, Q. H. Liu, B. A. Judd, E. E. Meyers, C. D. Sigmund, B. D. Freedman, and G. A. Koretzky. 2001. Differential requirement for SLP-76 domains in T cell development and function. Immunity 15:1011-1026. [DOI] [PubMed] [Google Scholar]
- 28.Parravicini, V., M. Gadina, M. Kovarova, S. Odom, C. Gonzalez-Espinosa, Y. Furumoto, S. Saitoh, L. E. Samelson, J. J. O'Shea, and J. Rivera. 2002. Fyn kinase initiates complementary signals required for IgE-dependent mast cell degranulation. Nat. Immunol. 3:741-748. [DOI] [PubMed] [Google Scholar]
- 29.Pivniouk, V. I., T. R. Martin, J. M. Lu-Kuo, H. R. Katz, H. C. Oettgen, and R. S. Geha. 1999. SLP-76 deficiency impairs signaling via the high-affinity IgE receptor in mast cells. J. Clin. Invest. 103:1737-1743. [PMC free article] [PubMed] [Google Scholar]
- 30.Pribluda, V. S., C. Pribluda, and H. Metzger. 1994. Transphosphorylation as the mechanism by which the high-affinity receptor for IgE is phosphorylated upon aggregation. Proc. Natl. Acad. Sci. U. S. A. 91:11246-11250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saitoh, S., R. Arudchandran, T. S. Manetz, W. Zhang, C. L. Sommers, P. E. Love, J. Rivera, and L. E. Samelson. 2000. LAT1 is essential for Fc(epsilon)RI-mediated mast cell activation. Immunity 12:525-535. [DOI] [PubMed] [Google Scholar]
- 32.Saitoh, S., S. Odom, G. Gomez, C. L. Sommers, H. A. Young, J. Rivera, and L. E. Samelson. 2003. The four distal tyrosines are required for LAT1-dependent signaling in FcepsilonRI-mediated mast cell activation. J. Exp. Med. 198:831-843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Samelson, L. E. 2002. Signal transduction mediated by the T cell antigen receptor: the role of adapter proteins. Annu. Rev. Immunol. 20:371-394. [DOI] [PubMed] [Google Scholar]
- 34.Shiue, L., J. Green, O. M. Green, J. L. Karas, J. P. Morgenstern, M. K. Ram, M. K. Taylor, M. J. Zoller, L. D. Zydowsky, J. B. Bolen, et al. 1995. Interaction of p72syk with the gamma and beta subunits of the high-affinity receptor for immunoglobulin E, Fc epsilon RI. Mol. Cell. Biol. 15:272-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Silverman, M. A., J. Shoag, J. Wu, and G. A. Koretzky. 2006. Disruption of SLP-76 interaction with Gads inhibits dynamic clustering of SLP-76 and FcepsilonRI signaling in mast cells. Mol. Cell. Biol. 26:1826-1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singer, A. L., S. C. Bunnell, A. E. Obstfeld, M. S. Jordan, J. N. Wu, P. S. Myung, L. E. Samelson, and G. A. Koretzky. 2004. Roles of the proline-rich domain in SLP-76 subcellular localization and T cell function. J. Biol. Chem. 279:15481-15490. [DOI] [PubMed] [Google Scholar]
- 37.Turner, H., and J. P. Kinet. 1999. Signalling through the high-affinity IgE receptor Fc epsilonRI. Nature 402:B24-B30. [DOI] [PubMed] [Google Scholar]
- 38.Utting, O., B. J. Sedgmen, T. H. Watts, X. Shi, R. Rottapel, A. Iulianella, D. Lohnes, and A. Veillette. 2004. Immune functions in mice lacking Clnk, an SLP-76-related adaptor expressed in a subset of immune cells. Mol. Cell. Biol. 24:6067-6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vig, M., and J. P. Kinet. 2007. The long and arduous road to CRAC. Cell Calcium 42:157-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Volna, P., P. Lebduska, L. Draberova, S. Simova, P. Heneberg, M. Boubelik, V. Bugajev, B. Malissen, B. S. Wilson, V. Horejsi, M. Malissen, and P. Draber. 2004. Negative regulation of mast cell signaling and function by the adaptor LAB/NTAL. J. Exp. Med. 200:1001-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu, J. N., M. S. Jordan, M. A. Silverman, E. J. Peterson, and G. A. Koretzky. 2004. Differential requirement for adapter proteins Src homology 2 domain-containing leukocyte phosphoprotein of 76 kDa and adhesion- and degranulation-promoting adapter protein in FcepsilonRI signaling and mast cell function. J. Immunol. 172:6768-6774. [DOI] [PubMed] [Google Scholar]
- 42.Yablonski, D., M. R. Kuhne, T. Kadlecek, and A. Weiss. 1998. Uncoupling of nonreceptor tyrosine kinases from PLC-gamma1 in an SLP-76-deficient T cell. Science 281:413-416. [DOI] [PubMed] [Google Scholar]
- 43.Zhang, W., J. Sloan-Lancaster, J. Kitchen, R. P. Trible, and L. E. Samelson. 1998. LAT1: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell 92:83-92. [DOI] [PubMed] [Google Scholar]
- 44.Zhang, W., C. L. Sommers, D. N. Burshtyn, C. C. Stebbins, J. B. DeJarnette, R. P. Trible, A. Grinberg, H. C. Tsay, H. M. Jacobs, C. M. Kessler, E. O. Long, P. E. Love, and L. E. Samelson. 1999. Essential role of LAT1 in T cell development. Immunity 10:323-332. [DOI] [PubMed] [Google Scholar]
- 45.Zhang, W., R. P. Trible, M. Zhu, S. K. Liu, C. J. McGlade, and L. E. Samelson. 2000. Association of Grb2, Gads, and phospholipase C-gamma 1 with phosphorylated LAT1 tyrosine residues. Effect of LAT1 tyrosine mutations on T cell antigen receptor-mediated signaling. J. Biol. Chem. 275:23355-23361. [DOI] [PubMed] [Google Scholar]
- 46.Zhu, M., S. Koonpaew, Y. Liu, S. Shen, T. Denning, I. Dzhagalov, I. Rhee, and W. Zhang. 2006. Negative regulation of T cell activation and autoimmunity by the transmembrane adaptor protein LAB. Immunity 25:757-768. [DOI] [PubMed] [Google Scholar]
- 47.Zhu, M., Y. Liu, S. Koonpaew, O. Granillo, and W. Zhang. 2004. Positive and negative regulation of FcepsilonRI-mediated signaling by the adaptor protein LAB/NTAL. J. Exp. Med. 200:991-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu, M., I. Rhee, Y. Liu, and W. Zhang. 2006. Negative regulation of Fc epsilonRI-mediated signaling and mast cell function by the adaptor protein LAX. J. Biol. Chem. 281:18408-18413. [DOI] [PubMed] [Google Scholar]