

Abstract
Enteroviruses (Picornaviridae family) are a common cause of human illness worldwide and are associated with diverse clinical syndromes, including asymptomatic infection, respiratory illness, gastroenteritis, and meningitis. In this study, we report the identification and complete genome sequence of a novel enterovirus isolated from a case of acute respiratory illness in a Nicaraguan child. Unbiased deep sequencing of nucleic acids from a nose and throat swab sample enabled rapid recovery of the full-genome sequence. Phylogenetic analysis revealed that human enterovirus 109 (EV109) is most closely related to serotypes of human enterovirus species C (HEV-C) in all genomic regions except the 5′ untranslated region (5′ UTR). Bootstrap analysis indicates that the 5′ UTR of EV109 is likely the product of an interspecies recombination event between ancestral members of the HEV-A and HEV-C groups. Overall, the EV109 coding region shares 67 to 72% nucleotide sequence identity with its nearest relatives. EV109 isolates were detected in 5/310 (1.6%) of nose and throat swab samples collected from children in a pediatric cohort study of influenza-like illness in Managua, Nicaragua, between June 2007 and June 2008. Further experimentation is required to more fully characterize the pathogenic role, disease associations, and global distribution of EV109.
The genus Enterovirus (EV) in the family Picornaviridae is a group of related viruses that are associated with a spectrum of disease, ranging from subclinical infections to acute respiratory and gastrointestinal illness to more severe manifestations, such as aseptic meningitis, encephalitis, and acute flaccid paralysis (16, 32). Enteroviruses are small, nonenveloped viruses that share a genomic organization. The RNA genome is a ∼7.5 kb single-stranded, positive-sense, polyadenylated molecule, with a single, long open reading frame flanked by 5′ and 3′ untranslated regions (UTRs). The 5′ UTR is ∼700 nucleotides in length and contains highly structured secondary elements with internal ribosomal entry site (IRES) function. The ∼2,200-amino-acid (aa) polyprotein is cotranslationally processed by viral proteases to yield structural (VP4, VP2, VP3, and VP1) and nonstructural (2A, 2B, 2C, 3A, 3B, 3C, and 3D) proteins (32). Current enterovirus classification is based on the high sequence divergence within the VP1 capsid region, which has been shown to correspond with serotype neutralization (27, 28). Human enterovirus (HEV) types are currently classified into four species, human enterovirus A (HEV-A), HEV-B, HEV-C (including poliovirus), and HEV-D, based on the four phylogenetic clusters observed in comparisons of the coding region sequences. An enterovirus is considered a new type within a species if it possesses <75% nucleotide identity and <85% amino acid identity with known members across the VP1 sequence (27, 30). Molecular identification methods play a crucial role in rapid, sensitive enterovirus diagnostics and have led to the recent discovery of several novel enteroviruses (29, 31, 40, 42, 44). Most approaches target a limited number of conserved regions in the 5′ UTR and VP4-VP2 junction or seek to ascertain serotype information by probing antigenic regions, such as VP1 (5).
Picornavirus RNA-dependent RNA polymerases are highly error prone and lack proofreading ability, resulting in a misincorporation frequency of 1 per 103 to 104 nucleotides (48). The relative infidelity of these polymerases is believed to enable rapid adaptability under selective pressure. Large-impact evolutionary events, such as recombination within and between enterovirus serotypes, also contribute to their evolution and genetic diversity (3, 8, 26, 39) and may lead to changes in disease associations with human enterovirus infections. Human enteroviruses are classified into four species based on coding region sequence phylogeny, and intraspecies recombination events between enteroviruses that are closely related in the coding region are well documented (26, 38, 39). All known enterovirus 5′ UTR sequences, however, cluster into two groups containing either HEV-A and -B sequences or HEV-C and -D sequences. Recent findings have described enterovirus genomes with a coding region that clusters with one species and a 5′ UTR that clusters with a different species, suggesting possible interspecies recombination events (41, 44). Understanding the recombination-driven evolution of HEV-C viruses is of particular public health concern due to the viruses' ability to recombine with vaccine poliovirus, resulting in circulating, highly neurovirulent vaccine-derived polioviruses (17, 21, 34). It is unclear whether recombination events between poliovirus and HEV-C viruses allow for the rapid acquisition of traits that increase pathogenic and circulation potential.
The enterovirus pathogenicity spectrum is related to tissue tropism and is largely determined by cellular receptor usage. Most picornaviruses use receptors from the immunoglobulin superfamily of proteins, such as intracellular adhesion molecule-1 (ICAM-1) or coxsackievirus-adenovirus receptor (CAR) (36). A distinct subgroup of HEV-C viruses, which includes coxsackievirus (CAV) A1, A19, and A22 and enterovirus 104, has not yet been grown successfully in cell culture, and the receptor molecule for this subgroup is unknown (6). HEV-C viruses are believed to be the ancestral source of poliovirus, which resulted from a capsid mutation that caused a cellular receptor switch from ICAM-1 to CD155 (poliovirus receptor [PVR]) (17).
In this study, we report the discovery and characterization of a novel human enterovirus type within species HEV-C, for which we propose the designation human enterovirus 109 (EV109). Sequence analysis reveals considerable nucleotide divergence in the 5′ UTR between EV109 and other HEV-C types, and scanning bootstrap analysis supports the hypothesis that EV109 is the product of an interspecies recombination event with an ancestral member of the HEV-A group. Viral capsid amino acid alignments and homology modeling reveal the predicted three-dimensional arrangement of divergent and conserved residues of EV109 compared with other related enteroviruses. We also report highly similar EV109 isolates within multiple cases of acute pediatric respiratory illness in Managua, Nicaragua.
MATERIALS AND METHODS
Nicaragua Influenza Cohort Study.
The Nicaraguan Influenza Cohort Study is a prospective community-based cohort study of viral respiratory illness in ∼3,800 children aged 2 to 13 years in Managua, Nicaragua (12, 20). Study enrollment began 1 June 2007. At enrollment, participants' families agreed to bring their children to the study health center at the first sign of illness. The study provides medical care free of charge to participants, and data from all medical visits are recorded systematically. Nose and throat swabs are collected from a 25% random sample of patients presenting with influenza-like illness (ILI), as defined by exhibiting fever or history of fever with a cough and/or sore throat, with symptom onset within the previous 4 days. Study participants typically present early in illness, increasing the likelihood that a child will be shedding virus at presentation (92% of participants with ILI presented within 3 days of symptom onset). At collection, nose and throat swabs are placed immediately into a tube containing 3 ml of viral transport medium. Samples are stored at 4°C at the clinical laboratory until they are transported to the Nicaraguan National Virology Laboratory, where they are aliquoted and stored at −80°C.
Specimen nucleic acid extraction, library amplification, and enterovirus PCR.
RNA was extracted from 140 μl of viral transport medium containing the nose/throat swabs using a QIAamp Viral RNA Isolation Kit (Qiagen, Valencia, CA), and cDNA was randomly amplified using a round A/B protocol (45). Specific PCR was performed on cDNA libraries using primers targeting the EV 5′ UTR (22) and the VP1 region (25). PCR was performed using a mixture consisting 17 μl of water, 2.5 μl of 10× Taq buffer, 1 μl of MgCl2 (50 mM), 0.5 μl of the deoxynucleoside triphosphates (dNTPs; 12.5 mM), 0.5 μl of each primer (10 μM), and 0.5 μl of Taq polymerase (Invitrogen) in a final volume of 25 μl. Conditions for the 5′ UTR reaction were 30 cycles of 94°C for 30 s, 50°C for 1 min, and 72°C for 1 min, and conditions for the VP1 reaction were as previously described (25). Amplicons were cloned into plasmid vectors using a TOPO TA Cloning System (Invitrogen) and sequenced on an ABI 3130xl Genetic Analyzer (Applied Biosystems) in both directions using vector primers M13F and M13R and Big Dye terminator sequencing chemistry.
Sequencing library preparation.
One paired-end, ultradeep-sequencing run was performed using a GAII Sequencer (Illumina) (Fig. 1 ). We utilized one lane of the run for the sample 4327 library. Library generation primers (Table 1) were modified from adapter A and adapter B sequences (Illumina). RNA extracted from sample 4327 nose/throat swab was randomly primed and reverse transcribed using a 14-bp sequence common to the 3′ end of both Illumina adapters attached to a random hexamer (primer 1a). Second-strand synthesis was also primed using primer 1a, followed by PCR amplification using the 14-bp common sequence without the hexamer (primer 1b) for 25 cycles. PCR products of ∼300 bp were purified on a 4% native polyacrylamide gel (30 mM KCl, 1× Tris-borate-EDTA [TBE] buffer, 19:1 acrylamide-bis) run at 4°C, ethanol precipitated, and PCR amplified for 17 additional cycles using a 22-bp-long primer consisting of the 3′ end of Illumina adapter A (primer 2) and the full-length 61-bp Illumina adapter B (primer 4) under the following conditions: 2 cycles of 94°C for 30 s, 40°C for 30 s, and 72°C for 1 min, followed by 15 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min. Amplicons generated with the correct adapter A-adapter B topologies were ∼355 bp and were purified away from adapter A-adapter A amplicons (∼394 bp) and adapter B-adapter B amplicons (∼316 bp) on a 4% native polyacrylamide gel run at 4°C and then ethanol precipitated. An additional 10 cycles of PCR were then performed on the adapter A-adapter B products using primer 2 and the 24 bases from the 5′ end of adapter B (primer 5).
FIG. 1.
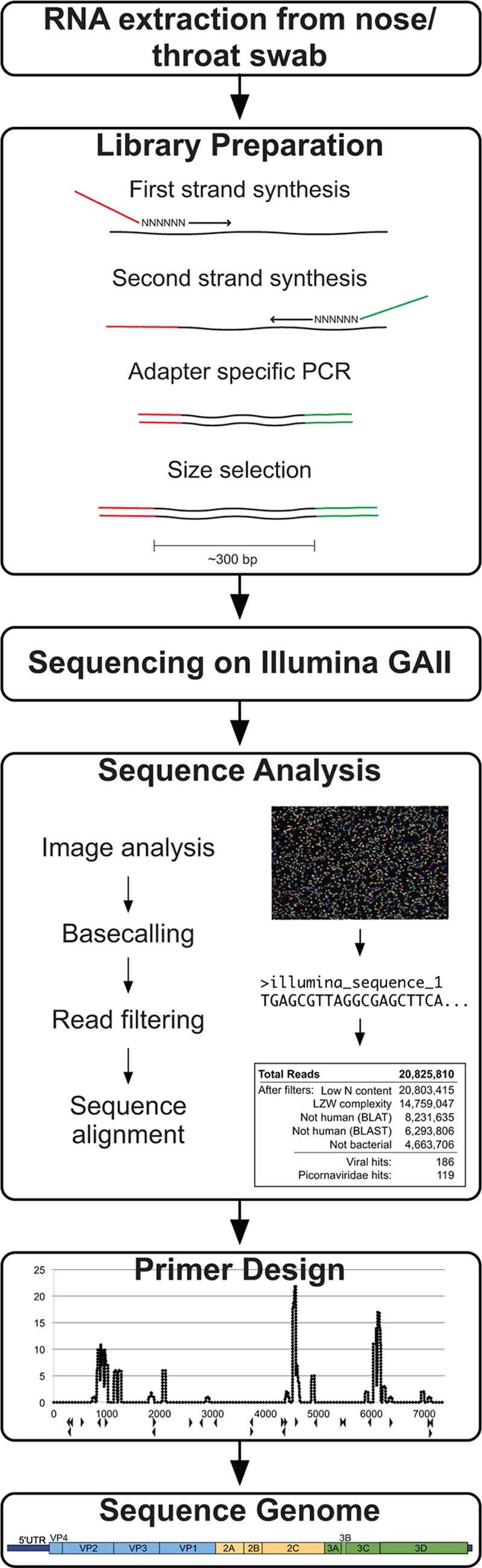
Viral recovery workflow. The method for viral genome recovery begins with RNA extraction from a nose/throat swab of clinical interest. A sequencing library is generated from the nucleic acids using reverse transcription and random-primed amplification to affix known sequencing adapter molecules necessary for the Illumina platform. The Illumina GAII sequencing is performed using the manufacturer's protocol. Sequence analysis includes image analysis and base-calling software, read filtering using quality metrics, and iterative sequence alignments. Reads with similarity to the viral sequence of interest are computationally assembled or used for primer design for subsequent specific amplification of remaining viral regions.
TABLE 1.
Deep-sequencing library preparation primers
| Primer name | Length (bases) | Sequence (5′-3′) |
|---|---|---|
| 1A | 20 | CGCTCTTCCGATCTNNNNNN |
| 1B | 14 | CGCTCTTCCGATCT |
| 2 | 22 | CTACACGACGCTCTTCCGATCT |
| 3 | 58 | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| 4 | 61 | CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT |
| 5 | 24 | CAAGCAGAAGACGGCATACGAGAT |
Deep-sequencing analysis.
The image analysis, base calling, and sequence quality control for the sequencing run were analyzed using the Illumina pipeline (version 1.3.2). A total of 10,412,905 sequencing read pairs, with each pair consisting of 65 nucleotides, were obtained, for a total of 20,825,810 reads and approximately 1.4 gigabases of sequence.
The first six bases of each read, which correspond to the hexamer binding site of the random primer, were trimmed, leaving 61-nucleotide (nt)-long reads. Reads with more than 10% of their bases uncalled (more than six Ns, where N is any nucleotide) were removed. Reads were next subjected to a complexity filter in which sequences with a Lempel-Ziv-Welch (LZW) compression ratio (47) below 0.45 were removed from the data set. The remaining set of higher-complexity reads was aligned to the human genome (University of California, Santa Cruz [UCSC] build 18) using BLAT with the default parameters, and any read with 90% or more of its nucleotides matching identically to the human genome was removed along with its paired end. The set of remaining reads was then aligned to the human genome using a nucleotide BLAST search with an E value of 10−3 and a word size of 20, and any read with 90% or more of its nucleotides matching identically to the human genome was removed along with its paired end. The remaining quality-filtered, nonhuman reads were aligned to Haemophilus influenzae (gi: 148826757), Streptococcus pneumoniae (gi: 116515308), and Porphyromonas gingivalis (gi: 34398108) as sequences related to each of these bacteria were collected during quality validation of the sequence library. The alignment to these bacterial genomes employed nucleotide BLAST with an E value of 10−3 and a word size of 11, and in order to better filter the wide diversity of bacteria, any read with 70% or more of its nucleotides identically matching to one of the bacterial genomes was removed along with its paired end. Finally, the remaining reads were aligned to NCBI's nucleotide (nt) database in an iterative fashion using a nucleotide BLAST search, first with an E value of 10−5 and a word size of 40, then with an E value of 10−3 and a word size of 20, and finally with an E value of 10−3 and a word size of 10. Any query sequence producing alignments to only viral sequences with at least half of its nucleotides matching a known virus was considered viral.
Complete genome sequencing.
Amplicons derived from specific EV109 PCR primers (Table 2) were gel purified, cloned, and sequenced as described above. Rapid amplification of cDNA ends (RACE) was performed on RNA from sample 4327 to recover the 5′ and 3′ ends of the genome. The 5′ end was captured using an Invitrogen 5′ RACE kit (version 2.0) according to the standard protocol. For 3′ end recovery one round of reverse transcription-PCR (RT-PCR) was employed with a Promega One-Step RT-PCR kit and avian myeloblastosis virus (AMV) reverse transcriptase using primer EV109 7014F and an oligo(dT) primer attached to a known primer B sequence (45), followed by one heminested round of PCR using Taq polymerase (Invitrogen) with primer EV109 7044F and primer B. Genome sequence assembly of PCR amplicons, RACE-derived amplicons, and ultradeep-sequencing reads was generated with the Geneious (version 3.6.1) alignment tool using a 20-bp minimum overlap requiring a 95% overlap identity.
TABLE 2.
Primers used for EV109 sequencing and genome recovery
| Primer name | Length (bases) | Start position (nt) | End position (nt) | Sequence (5′-3′) |
|---|---|---|---|---|
| EV109 5′ RACE 268R | 19 | 268 | 250 | ATGGTGGTACTAGGCTTCC |
| EV109 5′ RACE 274R | 20 | 274 | 255 | CAATCCATGGTGGTACTAGG |
| EV109 5′ RACE 333R | 17 | 333 | 317 | TATTCGGAGCCTCATCG |
| EV109 5′ UTR 492F | 19 | 492 | 510 | CAAACCAGCGTCTGGTAGG |
| EV109 VP4 849R | 21 | 849 | 829 | AATCATCACATCCTTAACTGG |
| EV109 VP2 942F | 20 | 942 | 961 | TACTCAAGAAGCAGCTAACG |
| EV109 VP3 1870F | 20 | 1870 | 1889 | GGGTATGCCTCCATTGTGTTG |
| EV109 VP3 1889R | 21 | 1889 | 1869 | AACACAATGGAGGCATACCC |
| EV109 VP1 2529F | 23 | 2529 | 2551 | GGAGACTGGAGCAACTAGTAAAG |
| EV109 VP1 2769R | 24 | 2769 | 2746 | GGTGAACATTTCCAATTTCCTACG |
| EV109 VP1 3030R | 21 | 3030 | 3010 | GGAAAAGCCGTCATAGAAGTG |
| EV109 2A deg 3662F | 19 | 3662 | 3680 | TCACTGCHGGKGGWGAAGG |
| EV109 2A deg 3688R | 17 | 3688 | 3704 | GCHTTTGCWGACATYAG |
| EV109 2C 4255F | 20 | 4255 | 4274 | ACACTGTTCAACAACATTCG |
| EV109 2C 4307F | 18 | 4307 | 4324 | CCTTGTATGCCAGCGAGG |
| EV109 2C 4324R | 18 | 4324 | 4307 | CCTCGCTGGCATACAAGG |
| EV109 2C 4513F | 17 | 4513 | 4529 | CCCTCTCACTTTGATGG |
| EV109 2C 4916R | 19 | 4916 | 4898 | TTGTCTACAAATTGAATGG |
| EV109 3C deg 5362F | 20 | 5362 | 5381 | GAYTAYGCAGTKGCAATGGC |
| EV109 3C 5452R | 18 | 5452 | 5435 | CTACCCGGTCATAAATGC |
| EV109 3D 5932R | 20 | 5932 | 5913 | CGTCTTTGCTGGGTCTCATC |
| EV109 3D 6286F | 20 | 6286 | 6305 | ATTCTGAACAAAGAAACACG |
| EV109 3′ RACE 7014F | 20 | 7014 | 7033 | GTTCTTGAAACGGTTTTTCC |
| EV109 3′ RACE 7044F | 20 | 7044 | 7063 | ACAATTCCCATACCTAGTGC |
| EV109 3D 7090R | 25 | 7090 | 7066 | CATATTTCTGACATTAGCATAACTGG |
EV109 PCR screening.
Specific EV109 primers targeting the VP1 region were designed from recovered VP1 sequence (EV109 VP1 123F, 5′-GGAGACTGGAGCAACTAGTAAAG-3′; EV109 VP1 363R, 5′-GGTGAACATTTCCAATTTCCTACG-3′). PCRs of 25 μl were performed on cDNA libraries using 17 μl of water, 2.5 μl 10× Taq buffer, 1 μl of MgCl2 (50 mM), 0.5 μl of dNTPs (12.5 mM), 0.5 μl of each primer (10 μM), and 0.5 μl of Taq polymerase (Invitrogen). Conditions for the 5′ UTR reaction were 30 cycles of 94°C for 30 s, 50°C for 1 min, and 72°C for 1 min. To obtain the full-length VP1 sequences and 5′ UTR-VP4 junction from positive samples, PCR was performed using conserved primers flanking these regions, as determined from the full-length 4327 isolate. Amplicons of expected sizes were gel purified, cloned, and sequenced as described above.
Phylogenetic and bootscanning analysis.
Multiple complete and partial genome alignments were constructed using ClustalW (version 2.0.10), and phylogenetic trees were constructed using the neighbor-joining method (100 bootstrap replicates) with Mega software (version 4.0). Bootscanning was performed using the Jukes and Cantor method with RDP3 (23) (window size, 400; step size, 20; 500 replicates).
Comparative protein structure modeling.
EV109 nucleotides 664 to 7281 were translated in the reading frame inferred from all related enteroviruses. The inferred sequences of proteins homologous to the VP1, VP2, VP3, and VP4 proteins of other enteroviruses were extracted for structure modeling of the viral capsid. These sequences were aligned to the sequence and structure of their coxsackievirus A21 (Protein Data Bank [PDB] code 1Z7S) counterparts using the align2d function in MODELLER, version 9v7 (37). Homology models were built using these alignments and the crystallographic structure of the coxsackievirus A21 capsid proteins using the standard automodel routine of MODELLER, version 9v7.
Nucleotide sequence accession numbers.
The completed genome sequence of EV109 isolate 4327 was deposited into GenBank under accession number GQ865517 and reported to the Picornaviridae Study Group (24 August 2009). The sequences of the full-length VP1 regions from four EV109 isolates were deposited in the GenBank under accession numbers GU131224 to GU131227. Sequence read data developed in this study have been deposited in the NCBI Sequence Read Archive under accession number SRA012708.1.
RESULTS
Detection and full-genome sequencing of a new HEV-C type from a nose/throat swab of a patient with influenza-like illness.
We utilized samples collected through the Nicaraguan Influenza Cohort Study, a prospective community-based cohort study of viral respiratory illness in ∼3,800 children aged 2 to 13 years in Managua, Nicaragua (12, 20). For this analysis, nose and throat swab specimens were examined from patients presenting with influenza-like illness (ILI), as defined by exhibiting fever or history of fever with a cough and/or sore throat, with symptom onset in the previous 4 days. First, total RNA was extracted from the swab samples, reverse transcribed to cDNA, and tested by RT-PCR for influenza A and B viruses, parainfluenza viruses 1 to 3, and respiratory syncytial virus. Samples that were negative were then randomly amplified as previously described (45) and tested by PCR for rhinoviruses and enteroviruses using conserved and degenerate picornavirus primers (22, 25). As part of ongoing picornavirus typing studies, 5′ UTR and VP1 amplicons of expected sizes were cloned and sequenced using a capillary electrophoresis sequencer (see Materials and Methods). In one sample, the amplified 5′ UTR and VP1 region had only 79% and 73% BLASTn identity, respectively, to other known enteroviruses.
To recover additional viral sequence from the divergent sample, ultradeep sequencing was performed using the GAII sequencer (Illumina, Inc.) using a random-primed cDNA library derived from RNA from a nose/throat swab from one sample (Fig. 1). The resulting 20.8 million 61-nt reads (10.4 million paired-end reads) were filtered for N content and LZW complexity and aligned to the human genome and three bacterial genomes using BLAT and BLAST, leaving 4.6 million sequencing reads (Fig. 2 A, inset). The remaining reads were aligned to NCBI's nt database in an iterative fashion (see Materials and Methods). A total of 186 reads were identified as viral sequence, and 119 of these reads were tentatively assigned as picornavirus sequence by nucleotide BLAST analysis.
FIG. 2.

Deep-sequencing results and picornavirus genome coverage. (A) Coverage map of deep-sequencing-derived picornavirus reads and their adjusted EV109 genome positions before (perceived coverage) and after (actual coverage) full-genome sequencing. (Inset) Summary of total 61-nt deep-sequencing reads and results from the iterative alignment analysis. (B) Schematic diagram of EV109 genome. The black triangles denote primer positions derived from deep-sequencing reads used to recover the full-length EV109 genome. The black bars denote sequence regions recovered from all five EV109 isolates. The predicted EV109 genome domains are drawn to scale.
Aligning the 119 picornavirus reads to related enterovirus genomes revealed discontinuous regions of high read coverage distributed across the genome, but these were not sufficient to fully assemble the complete genome of the new enterovirus (Fig. 2B). We used read-specific sequence data to design primers to close the remaining genomic gaps (Fig. 2B, arrowheads; Tables 1 and 2). The 3′ end of the genome was recovered using rapid amplification of cDNA ends (3′ RACE), and the 5′ end was recovered using a heminested 5′ RACE strategy. The complete VP1 sequence and genome were submitted to the Picornaviridae Study Group (http://www.picornastudygroup.com), compared to other enterovirus sequences, and named as a new proposed enterovirus type, enterovirus 109 (EV109; GenBank accession number GQ865517).
Genomic description of EV109.
The complete genome of EV109 consists of 7,354 nucleotides, excluding the poly(A) tail. The 5′ UTR contains 663 nucleotides, and the 3′ UTR consists of 73 nucleotides. EV109 features a single open reading frame from base 664 to 7281 that encodes a 2,206-amino-acid polyprotein. The base composition of the full genome is 27.7% A, 23.8% C, 24.1% G, and 24.3% U. To investigate the relationships among EV109 and other members of the Enterovirus genus, we compared the full genome of EV109 with representative members of different enterovirus species and constructed similarity plots (Fig. 3 A). The scanning pairwise nucleotide identity plots suggested that EV109 was most closely related to serotypes of the HEV-C species in all of the genome regions except the 5′ UTR, which was most similar to HEV-A. Overall, EV109 shares only 67 to 72% nucleotide sequence identity with other HEV-C coding regions, including CAV19 (71%), CAV22 (70%), EV104 (72%), and poliovirus 1 (67%). The VP1 capsid subunit region of the EV109 polyprotein is 276 amino acids, and by pairwise similarity, shares 66 to 71% amino acid similarity to that of CAV22, CAV19, and EV104 (64 to 65% nucleotide similarity). A phylogenetic tree constructed from full-length nucleotide sequences using the neighbor-joining method illustrates the four separate enterovirus species clusters and EV109 groups in a subbranch of HEV-C that also contains CAVs 19, 22, and 1 and the recently described EV104 (44) (Fig. 3B). As of this writing, this distinct subgroup of enteroviruses has not been successfully grown in cell culture (6).
FIG. 3.

Relationships between known enteroviruses and EV109 based on full-length genome analysis. (A) Full-genome similarity plot depicting scanning pairwise identity using a 100-nt sliding window evaluated at each nucleotide. The EV109 sequence is compared with a close HEV-C relative, coxsackievirus A19 (CAV19) and more distant HEV-A (EV92) and HEV-D (EV68) serotypes. The conserved enteroviral domains are depicted to scale. The 5′ UTR and VP1 regions are highlighted. (B) Phylogenetic tree constructed from complete enterovirus genomes. The EV109 genome sequenced in this study is depicted with a solid black arrow, coxsackievirus A19 is identified by a red arrow, enterovirus 92 is identified by a purple arrow, and enterovirus 68 is identified by a yellow arrow. ClustalW and MEGA were used for alignments and tree construction, respectively, using the neighbor-joining method and 500 bootstrap replicates.
Several typical picornavirus sequence features are conserved in EV109, including several cis-acting RNA elements, protease catalytic residues, and a nuclear localization sequence (NLS) at the N terminus of the 3D polymerase gene (Fig. 4). Specifically, the 5′ and 3′ UTRs maintain conserved RNA secondary structures with known roles in viral translation and replication, including the X and Y domains of the 3′ UTR that have been shown in related viruses to play a role in minus-strand synthesis (50). The cis-acting replication element (cre) features a stem-loop located in the 2C region with a conserved AAACA motif in the loop (6, 11). The cre hairpin sequence and loop motif are present in EV109 at nucleotide positions 4354 to 4412. The EV109 polyprotein sequence also contains canonical precursor viral protease cleavage sites, as described in other HEV-C viruses (25), and possesses the predicted catalytic triad of residues in both 2Apro and 3Cpro (4, 14, 33). Also conserved in EV109 is an NLS (PNKTKLNPS) near the N terminus of the 3D polymerase sequence that has been shown to direct the 3CDpro of poliovirus and rhinoviruses to the nucleus, where it cleaves host nuclear factors and inhibits cellular RNA transcription (1, 2, 46).
FIG. 4.

Sequence analysis and RNA folding predictions reveal conserved picornavirus features in EV109. RNA secondary structure prediction of the EV109 5′ UTR using pknotsRG (35) and visualized using PseudoViewer, version 3 (7). Previously described type 1 IRES features maintained in EV109 include a 5′ cloverleaf as stem-loop I, a GNRA sequence in stem-loop IV, and a pyrimidine-rich region in stem-loop VI (29). Stem-loops II and III are predicted as one combined stem-loop structure. Other nucleic acid folding software (Mfold and nupack) predicted similar secondary structures (10, 51).
An interspecies recombinant and truncated 5′ UTR.
Intraspecies recombination events occur frequently in enteroviruses, especially within the nonstructural coding region, but interspecies exchanges involving the 5′ UTR have also been observed (3, 8, 15, 26, 33, 38, 39, 41). A phylogenetic tree constructed using enterovirus 5′ UTR sequences (Fig. 5A) was inconsistent with the EV109 complete genome tree (Fig. 3B) because the EV109 5′ UTR did not cluster with its counterparts from other members of HEV-C. EV109 fell outside the two major 5′ UTR clusters and was closely related to EV104 and EV92, which further suggested a recombination event. Tapparel et al. (44) recently described EV104 and reported that its 5′ UTR had also undergone possible recombination. To evaluate the likelihood of genomic recombination, full-length bootscanning analysis was performed with representative strains of HEV-C, HEV-A, and HEV-D (types CAV19, EV92, and EV68, respectively) using EV109 as the query sequence (Fig. 5B). Throughout the 5′ UTR, there was high bootstrap support (>75%) for clustering with EV92 (HEV-A), while the coding region maintained high bootstrap support for CAV19 (HEV-C). The bootscanning analysis revealed a phylogenetic conflict between the 5′ UTR and the downstream coding sequence, which suggests that EV109 arose from an interspecies recombination event preceding the VP4 start codon between an HEV-A- and HEV-C-type enterovirus.
FIG. 5.

The EV109 5′ UTR region reveals evidence of an ancestral recombination event. (A) Phylogenetic tree constructed from 5′ UTR regions of 74 annotated human enteroviruses. A black arrow denotes EV109. Also highlighted are CAV19 (red arrow), EV92 (blue arrow), and EV68 (yellow arrow), which are used in the subsequent bootscanning analysis. ClustalW and MEGA were used for alignments and tree construction, respectively, using the neighbor-joining method and 500 bootstrap replicates. (B) Bootscanning analysis of EV109. Bootscanning analysis was performed with other serotypes of HEV-A (EV92), HEV-C (CAV19), and HEV-D (EV68) using a word size of 400 and step size of 20. (C) Sequence alignment depicting the 5′ UTR-VP4 junction site. The EV109 sequence is compared to representative members of HEV-A and HEV-C.
Enterovirus 5′ UTRs maintain a common structural organization that includes a 5′-end cloverleaf structure and several pseudoknots that function as an internal ribosomal entry site (IRES) required for translation of the viral polyprotein. The IRES of EV109 (Fig. 4) features several required motifs, such as a GNRA consensus sequence in stem-loop IV and a pyrimidine-rich region in stem-loop VI, and is followed by a hypervariable region preceding the VP4 start codon (33). The average 5′ UTR length of human enteroviruses is 740 nt. Pairwise alignments to other HEV-A and HEV-C 5′ UTR-VP4 junctions revealed a truncated hypervariable region in the 3′ end of the EV109 5′ UTR (Fig. 5C), resulting in a shorter than average 633-nt 5′ UTR. In spite of this shortened sequence, RNA folding algorithms (Mfold and NuPack) predicted that the EV109 5′ UTR maintains the canonical enterovirus cloverleaf and IRES structures, including required sequence motifs (Fig. 4).
EV109 capsid homology modeling.
The nonenveloped picornavirus capsid proteins are subject to the diversifying effects of host immunologic pressure. To gain additional insight into the viral capsid diversity of EV109, we examined the position-specific amino acid conservation of the four structural protein sequences (VP4, VP2, VP3, and VP1) of EV109 compared to other HEV-C relatives. To perform this analysis, the structural protein amino acid sequences were aligned and scored with a position-specific scoring matrix (PSSM) for amino acid position-specific conservation. The PSSM scores were then mapped onto the three-dimensional viral pentamer crystal structure of coxsackievirus A21 (PDB code 1Z7S), revealing positions of amino acid diversity throughout the enterovirus capsid pentamer (Fig. 6). Nonconserved EV109 residues (as denoted by a negative PSSM score) are located both along protrusions and within the capsid canyons on the external pentamer surface but are not aggregated within one particular external location (Fig. 6A and B). Nonconserved residues are also located on the internal capsid surface and predominantly cluster along the edges of the five tetrameric units that make up the pentamer (Fig. 6C).
FIG. 6.
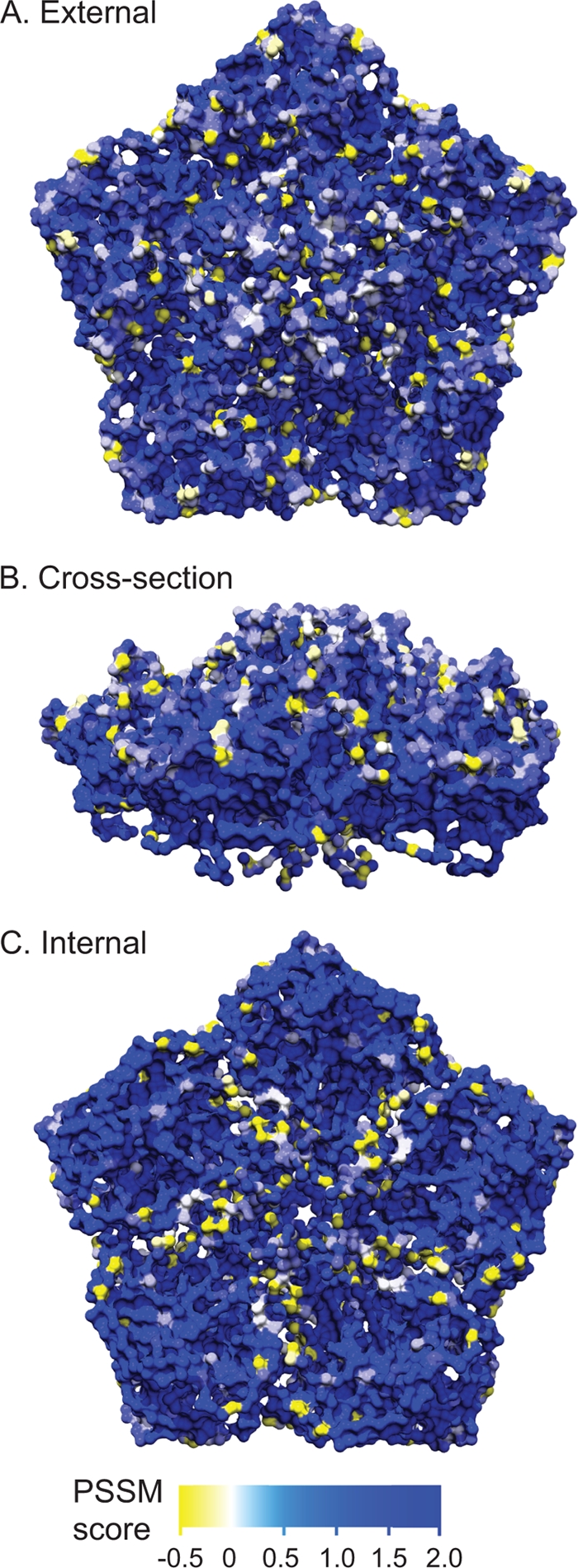
Amino acid conservation on the enterovirus capsid pentamer subunit. Amino acid alignment PSSM score of EV109 compared to other HEV-C capsid sequences and mapped on the pentamer crystal structure of coxsackievirus A21. Residues shaded in blue have a higher PSSM score and are more conserved in EV109, whereas yellow residues have a negative PSSM and are nonconserved. A scale bar is given below panel C. (A) External pentamer view. (B) Cross-sectional view. (C) Internal pentamer view.
Identification of additional EV109 isolates.
EV109-specific PCR primers for the VP1 region were designed and used to screen a total of 310 ILI samples from the Nicaraguan pediatric cohort in a blinded fashion. Four additional EV109-positive samples were detected (1.6% detection rate). Additional PCR primers were then designed and employed to recover the full-length VP1 sequence and a 350-bp region spanning the 5′ UTR hypervariable and VP4 junction region from each sample. The five viruses shared >95% nucleotide and >94% amino acid pairwise identity across the full-length VP1 region and >95% nucleotide pairwise identity in the 5′ UTR-VP4 junction. This high relatedness in multiple regions (as denoted by black bars on Fig. 2B) suggested that all five isolates, indeed, belong to our newly characterized enterovirus type.
Unmasking the clinical status and demographic information of the five samples revealed symptom onset dates between 14 January 2008 and 23 April 2008 (Table 3). In addition to ILI symptoms, three of five patients exhibited gastroenteritis symptoms, including abdominal pain or vomiting. One patient was transferred to the National Pediatric Reference Hospital. All five cases originated from separate households within a 2.5-km2 area of northwest Managua and involved female patients.
TABLE 3.
Clinical and demographic information from five EV109-positive patients, including patient age, date of onset of illness, and symptomsa
| Patient no. | Accession no. of 5′ UTR-VP4 junction | Accession no. of full-length VP1 | Age (yr) | Date of onset | Symptoms |
|---|---|---|---|---|---|
| 4733.01 | GU265819 | GU131227 | 3 | 14 January 2008 | Fever, sore throat, lymphadenopathy, cough, coryza, abdominal pain |
| 4323.04 | GU265817 | GU131225 | 3 | 27 January 2008 | Fever, headache, sore throat, lymphadenopathy, cough, coryza, vomitingc |
| 4327.01 | GQ865517b | GQ865517b | 3 | 23 February 2008 | Fever, sore/red throat, cough, coryza |
| 1578.01 | GU265816 | GU131226 | 11 | 15 April 2008 | Fever, cough, coryza, vomiting |
| 2751.01 | GU265818 | GU131224 | 11 | 23 April 2008 | Fever, sore/red throat, coryza |
All patients were female, and all specimens were from nose/throat swabs.
Full-length genome.
Patient was transferred to a hospital.
DISCUSSION
In this study we report the discovery and full genome sequence of a novel enterovirus isolated from a case of acute pediatric respiratory illness in Nicaragua, and we propose the name enterovirus 109, according to Picornaviridae Study Group naming conventions. EV109 was detected as part of respiratory virus surveillance studies of pediatric ILI patients who are participants in a long-term community-based cohort study in Managua (20). EV109 is a member of the HEV-C species and is most closely related to CAV19, CAV22, CAV1, and EV104 (Fig. 3), a distinct subgroup within HEV-C that has yet to be grown in cell culture (6). The VP1 sequence of EV109 shared <72% amino acid and <66% nucleotide identity with its relatives, fulfilling current classification requirements for designating new enterovirus types.
This study demonstrated the utility of deep sequencing as a strategy to quickly sequence the full-length genome of a novel virus too divergent to be easily recovered by other methods. Recovering the genome of a previously uncharacterized virus has been historically accomplished by screening bacterial or phage libraries, primer walking, and/or PCR using degenerate primers. In the best cases, recovery of the genome is a straightforward iterative trial-and-error process. However, several factors, such as the degree of identity to known viruses and the copy number of the genome present in the sample, can make this process highly inefficient, time-consuming, and costly. In a case where the presence of the viral target is vanishingly small (<1:100,000), ultradeep sequencing can provide a set of distributed reads that can then be used to rapidly close the genome, even in the context of a host organism that lacks a reference genome (19). Even when the entire genome cannot be assembled from the primary sequencing reads, as was the case for EV109, this strategy yields an overall cost savings in terms of supplies, labor, and time, all of which will improve as sequencing output per run continues to grow and cost decreases. The sequence data presented here represent one-eighth of an Illumina GAII sequencing run (one lane on an eight-lane flow cell) and cost $1,000. Thus, the cost per recovered viral read (211 total, out of 20 million) is $4.74, which we note is less than the cost of a conventional PCR primer (25-mer, at $0.28/base). While colony screening, degenerate PCR, and primer-walking strategies still have their places in novel genome recovery, the time when these techniques are rendered completely obsolete by ultradeep sequencing is clearly on the horizon. It is notable that of the complete sequencing data set, only 211 reads out of 20.8 million (0.001%) actually originated from the EV109 genome (Fig. 2A). Given the nonsterile location of the sample (nose and throat swab) and the unbiased nucleic acid extraction, it may not be surprising that so few reads could be obtained. In this regard, deep-sequencing technologies that produce less depth (fewer than 1 million reads) may have missed this species completely. Regardless, using our alignment parameters, more reads were identified with the picornavirus family than all other virus families combined. Furthermore, the use of paired-end sequencing greatly diminished the possibility of obtaining spurious hits.
The coding region of EV109 is most closely related to HEV-C species, but the 5′ UTR is not closely related to either the HEV-C or HEV-A phylogenetic groups, suggesting ancestral recombination with a divergent species outside the two major groups (Fig. 5). The observed recombination product between different HEV species is consistent with previous reports that interspecies recombination involving the 5′ UTR can occur in enteroviruses, as reported by Smura et al., who describe an enterovirus genome closely related to HEV-A types except for the 5′ UTR region, which clusters with HEV-C and -D sequences (41). The recombination evidence in EV109 is similar to the recombination event reported by Tapparel et al. in EV104 (44). The existence of recombinant enteroviruses has practical implications for diagnostic strategies that attempt to type enteroviruses using solely the 5′ UTR sequence and highlights the importance of obtaining VP1 sequence for definitive identification. The 663-nt 5′ UTR of EV109 is shorter than average by virtue of a truncated hypervariable region between the maintained IRES secondary structure (Fig. 4) and the start codon, indicating that the hypervariable region is deletion tolerant. It has been suggested by poliovirus studies (43) that this unstructured variable stretch plays a role as a spacer element preceding the authentic translation start codon of internal initiation and that upstream AUG codons, including two in stem-loop VI at bases 578 and 584 of EV109, may allow ribosome docking preceding transfer to the downstream AUG at base 664. We speculate that, beyond its role as a spacer element, the hypervariable region may also be a hot spot for recombination between enterovirus species.
A functional enterovirus with a recombinant 5′ UTR would be required to maintain the necessary known interactions with both viral proteins, such as 2B, 2BC, 3A, and 3D, and host proteins, such as polypyrimidine tract-binding protein and poly(rC) binding protein, which are important for viral translation and RNA synthesis (24, 50). To conserve interaction with viral proteins, such a recombination event in the 5′ UTR may well have provoked compensatory mutations in the viral coding region. However, the specific sequence motifs necessary for specific viral protein-nucleotide interactions are currently poorly defined, limiting our ability to recognize them. Structural features may also play a functional role, as demonstrated by 3CD-cloverleaf interaction experiments that alter sequence but maintain structure (49). Chimeric polioviruses possessing heterologous 5′ UTRs from coxsackievirus or rhinovirus have exhibited growth deficiencies in cell culture and attenuated neuropathogenesis in poliomyelitis mouse models though they share ∼70% nucleotide identity across the 5′ UTRs (13, 18). Other investigators have observed complementary mutations in 3A/3AB and 3C/3CD that alleviate cell-type-specific growth defects in enteroviruses with incompatible UTRs; although the mutations were confined to the 3A and 3C regions, homogenous mutations were not identified at defined sites (9). Future site-directed mutagenesis studies using EV109 could elucidate the selected coding region mutations needed to maintain UTR-viral protein interactions within the context of a natural recombination event.
A total of five EV109 isolates were detected in nose/throat swabs from five children in separate households between January and April 2008. We screened 310 nose/throat swab samples by PCR for EV109 and obtained a detection rate of 1.6%. In addition to presenting with acute influenza-like illness (fever or history of fever with a cough and/or sore throat), three of the five patients also presented with gastrointestinal symptoms (Table 3). EV109 is genetically similar to other known pathogenic enteroviruses, and though present findings are suggestive, human disease causation has not yet been formally established. Each of the five isolates reported here was negative by RT-PCR testing for a number of additional respiratory viruses, including influenza A and B viruses, respiratory syncytial virus, and human parainfluenza viruses 1, 2, and 3 (data not shown). Deep sequencing did not reveal a high number of reads aligning to other viral families, with the exception of reads with identity to human endogenous retroviruses. Other low-level viral reads observed in the sequencing library are likely false positives derived from spurious alignments. These findings are consistent with a pathogenetic role in the illnesses described here, but further experimentation is required to formally establish such a role. Our findings should make possible development of serologic as well as genomic testing, which would allow (i) documentation of seroconversion during illness and (ii) estimation of seroprevalence in other populations. Additionally, given the wide variety of enterovirus-associated diseases, which include ILI, gastroenteritis, encephalitis, aseptic meningitis, and acute flaccid paralysis (16, 32), more expansive screening of both asymptomatic and symptomatic subjects with other clinical syndromes will be required to establish the full spectrum of EV109's disease associations.
It should be noted that EV109 was successfully detected using conserved 5′ UTR primers first derived by Lönnrot et al. in 1999 (22), but subsequent testing in other studies has not detected EV109 or has perhaps failed to detect it due to the 5′ UTR recombination. Although the global distribution of EV109 is currently unknown, this study describes the first novel picornavirus isolated in Nicaragua. Given the scarcity of dedicated molecular diagnostic testing and the large percentage of undiagnosed cases of respiratory illness in developing settings, our findings underscore the importance of conducting molecular diagnostic and surveillance studies in tropical, developing regions.
Acknowledgments
We thank the children and families participating in the Nicaragua Influenza Cohort Study and the staff at the Health Center Sócrates Flores Vivas and the Nicaraguan National Virology Laboratory, in particular, Roger López, Cristhiam Cerpas, Carolina Flores, Heyri Roa, Moises Navarro, and Patricia Castillo. We are grateful to J. Graham Ruby for helpful advice and editing and to Lucille Huang for technical support.
This work was supported in part by a grant from the Pacific Southwest Regional Center of Excellence, NIH grant U54-AI65359, the Pediatric Dengue Vaccine Initiative (VE-1 to E.H.), the Howard Hughes Medical Institute, the Doris Duke Foundation, and the Packard Foundation. Joseph DeRisi and Don Ganem are supported by the Howard Hughes Medical Institute.
Footnotes
Published ahead of print on 30 June 2010.
The authors have paid a fee to allow immediate free access to this article.
REFERENCES
- 1.Aminev, A. G., S. P. Amineva, and A. C. Palmenberg. 2003. Encephalomyocarditis virus (EMCV) proteins 2A and 3BCD localize to nuclei and inhibit cellular mRNA transcription but not rRNA transcription. Virus Res. 95:59-73. [DOI] [PubMed] [Google Scholar]
- 2.Amineva, S. P., A. G. Aminev, A. C. Palmenberg, and J. E. Gern. 2004. Rhinovirus 3C protease precursors 3CD and 3CD′ localize to the nuclei of infected cells. J. Gen. Virol. 85:2969-2979. [DOI] [PubMed] [Google Scholar]
- 3.Andersson, P., K. Edman, and A. M. Lindberg. 2002. Molecular analysis of the echovirus 18 prototype: evidence of interserotypic recombination with echovirus 9. Virus Res. 85:71-83. [DOI] [PubMed] [Google Scholar]
- 4.Bazan, J. F., and R. J. Fletterick. 1988. Viral cysteine proteases are homologous to the trypsin-like family of serine proteases: structural and functional implications. Proc. Natl. Acad. Sci. U. S. A. 85:7872-7876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bolanaki, E., C. Kottaridi, P. Markoulatos, L. Margaritis, and T. Katsorchis. 2005. A comparative amplification of five different genomic regions on Coxsackie A and B viruses. Implications in clinical diagnostics. Mol. Cell. Probes 19:127-135. [DOI] [PubMed] [Google Scholar]
- 6.Brown, B., M. S. Oberste, K. Maher, and M. A. Pallansch. 2003. Complete genomic sequencing shows that polioviruses and members of human enterovirus species C are closely related in the noncapsid coding region. J. Virol. 77:8973-8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Byun, Y., and K. Han. 2009. PseudoViewer3: generating planar drawings of large-scale RNA structures with pseudoknots. Bioinformatics 25:1435-1437. [DOI] [PubMed] [Google Scholar]
- 8.Chevaliez, S., A. Szendröi, V. Caro, J. Balanant, S. Guillot, G. Berencsi, and F. Delpeyroux. 2004. Molecular comparison of echovirus 11 strains circulating in Europe during an epidemic of multisystem hemorrhagic disease of infants indicates that evolution generally occurs by recombination. Virology 325:56-70. [DOI] [PubMed] [Google Scholar]
- 9.de Sessions, P. F., E. Dobrikova, and M. Gromeier. 2007. Genetic adaptation to untranslated region-mediated enterovirus growth deficits by mutations in the nonstructural proteins 3AB and 3CD. J. Virol. 81:8396-8405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dirks, R. M., and N. A. Pierce. 2003. A partition function algorithm for nucleic acid secondary structure including pseudoknots. J. Comput. Chem. 24:1664-1677. [DOI] [PubMed] [Google Scholar]
- 11.Goodfellow, I., Y. Chaudhry, A. Richardson, J. Meredith, J. W. Almond, W. Barclay, and D. J. Evans. 2000. Identification of a cis-acting replication element within the poliovirus coding region. J. Virol. 74:4590-4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gordon, A., O. Ortega, G. Kuan, A. Reingold, S. Saborio, A. Balmaseda, and E. Harris. 2009. Prevalence and seasonality of influenza-like illness in children, Nicaragua, 2005-2007. Emerg. Infect. Dis. 15:408-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gromeier, M., L. Alexander, and E. Wimmer. 1996. Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proc. Natl. Acad. Sci. U. S. A. 93:2370-2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hämmerle, T., C. U. Hellen, and E. Wimmer. 1991. Site-directed mutagenesis of the putative catalytic triad of poliovirus 3C proteinase. J. Biol. Chem. 266:5412-5416. [PubMed] [Google Scholar]
- 15.Huang, T., W. Wang, M. Bessaud, P. Ren, J. Sheng, H. Yan, J. Zhang, X. Lin, Y. Wang, F. Delpeyroux, and V. Deubel. 2009. Evidence of recombination and genetic diversity in human rhinoviruses in children with acute respiratory infection. PLoS One. 4:e6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacques, J., H. Moret, D. Minette, N. Leveque, N. Jovenin, G. Deslee, F. Lebargy, J. Motte, and L. Andreoletti. 2008. Epidemiological, molecular, and clinical features of enterovirus respiratory infections in French children between 1999 and 2005. J. Clin. Microbiol. 46:206-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang, P., J. A. J. Faase, H. Toyoda, A. Paul, E. Wimmer, and A. E. Gorbalenya. 2007. Evidence for emergence of diverse polioviruses from C-cluster coxsackie A viruses and implications for global poliovirus eradication. Proc. Natl. Acad. Sci. U. S. A. 104:9457-9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson, V. H., and B. L. Semler. 1988. Defined recombinants of poliovirus and coxsackievirus: sequence-specific deletions and functional substitutions in the 5′-noncoding regions of viral RNAs. Virology 162:47-57. [DOI] [PubMed] [Google Scholar]
- 19.Kistler, A., A. Gancz, S. Clubb, P. Skewes-Cox, K. Fischer, K. Sorber, C. Chiu, A. Lublin, S. Mechani, Y. Farnoushi, A. Greninger, C. Wen, S. Karlene, D. Ganem, and J. DeRisi. 2008. Recovery of divergent avian bornaviruses from cases of proventricular dilatation disease: identification of a candidate etiologic agent. Virology J. 5:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuan, G., A. Gordon, W. Aviles, O. Ortega, S. N. Hammond, D. Elizondo, A. Nunez, J. Coloma, A. Balmaseda, and E. Harris. 2009. The Nicaraguan Pediatric Dengue Cohort Study: study design, methods, use of information technology, and extension to other infectious diseases. Am. J. Epidemiol. 170:120-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu, H., D. Zheng, L. Zhang, M. S. Oberste, M. A. Pallansch, and O. M. Kew. 2000. Molecular evolution of a type 1 wild-vaccine poliovirus recombinant during widespread circulation in China. J. Virol. 74:11153-11161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lönnrot, M., M. Sjöroos, K. Salminen, M. Maaronen, T. Hyypiä, and H. Hyöty. 1999. Diagnosis of enterovirus and rhinovirus infections by RT-PCR and time-resolved fluorometry with lanthanide chelate labeled probes. J. Med. Virol. 59:378-384. [PubMed] [Google Scholar]
- 23.Martin, D. P., C. Williamson, and D. Posada. 2005. RDP2: recombination detection and analysis from sequence alignments. Bioinformatics 21:260-262. [DOI] [PubMed] [Google Scholar]
- 24.Martinez-Salas, E., A. Pacheco, P. Serrano, and N. Fernandez. 2008. New insights into internal ribosome entry site elements relevant for viral gene expression. J. Gen. Virol. 89:611-626. [DOI] [PubMed] [Google Scholar]
- 25.Nix, W. A., M. S. Oberste, and M. A. Pallansch. 2006. Sensitive, Seminested PCR amplification of VP1 sequences for direct identification of all enterovirus serotypes from original clinical specimens. J. Clin. Microbiol. 44:2698-2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Norder, H., L. Bjerregaard, and L. O. Magnius. 2002. Open reading frame sequence of an Asian enterovirus 73 strain reveals that the prototype from California is recombinant. J. Gen. Virol. 83:1721-1728. [DOI] [PubMed] [Google Scholar]
- 27.Oberste, M. S., K. Maher, M. R. Flemister, G. Marchetti, D. R. Kilpatrick, and M. A. Pallansch. 2000. Comparison of classic and molecular approaches for the identification of untypeable enteroviruses. J. Clin. Microbiol. 38:1170-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oberste, M. S., K. Maher, D. R. Kilpatrick, and M. A. Pallansch. 1999. Molecular evolution of the human enteroviruses: correlation of serotype with VP1 sequence and application to picornavirus classification. J. Virol. 73:1941-1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oberste, M. S., K. Maher, W. A. Nix, S. M. Michele, M. Uddin, D. Schnurr, S. al-Busaidy, C. Akoua-Koffi, and M. A. Pallansch. 2007. Molecular identification of 13 new enterovirus types, EV79-88, EV97, and EV100-101, members of the species human enterovirus B. Virus Res. 128:34-42. [DOI] [PubMed] [Google Scholar]
- 30.Oberste, M. S., S. M. Michele, K. Maher, D. Schnurr, D. Cisterna, N. Junttila, M. Uddin, J. Chomel, C. Lau, W. Ridha, S. al-Busaidy, H. Norder, L. O. Magnius, and M. A. Pallansch. 2004. Molecular identification and characterization of two proposed new enterovirus serotypes, EV74 and EV75. J. Gen. Virol. 85:3205-3212. [DOI] [PubMed] [Google Scholar]
- 31.Oberste, M. S., D. Schnurr, K. Maher, S. al-Busaidy, and M. A. Pallansch. 2001. Molecular identification of new picornaviruses and characterization of a proposed enterovirus 73 serotype. J. Gen. Virol. 82:409-416. [DOI] [PubMed] [Google Scholar]
- 32.Pallansch, M. A., and R. Roos. 2007. Enteroviruses: Polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses, pp. 839-893. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 33.Racaniello, V. 2007. Picornavirdae: the viruses and their replication, pp. 795-838. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 34.Rakoto-Andrianarivelo, M., S. Guillot, J. Iber, J. Balanant, B. Blondel, F. Riquet, J. Martin, O. Kew, B. Randriamanalina, L. Razafinimpiasa, D. Rousset, and F. Delpeyroux. 2007. Co-circulation and evolution of polioviruses and species C enteroviruses in a district of Madagascar. PLoS Pathog. 3:e191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reeder, J., P. Steffen, and R. Giegerich. 2007. pknotsRG: RNA pseudoknot folding including near-optimal structures and sliding windows. Nucleic Acids Res. 35:W320-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rossmann, M. G., Y. He, and R. J. Kuhn. 2002. Picornavirus-receptor interactions. Trends Microbiol. 10:324-331. [DOI] [PubMed] [Google Scholar]
- 37.Sali, A., and T. L. Blundell. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234:779-815. [DOI] [PubMed] [Google Scholar]
- 38.Santti, J., T. Hyypia, L. Kinnunen, and M. Salminen. 1999. Evidence of recombination among enteroviruses. J. Virol. 73:8741-8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simmonds, P., and J. Welch. 2006. Frequency and Dynamics of Recombination within Different Species of Human Enteroviruses. J. Virol. 80:483-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smura, T., S. Blomqvist, T. Hovi, and M. Roivainen. 2009. The complete genome sequences for a novel enterovirus type, enterovirus 96, reflect multiple recombinations. Arch. Virol. 154:1157-1161. [DOI] [PubMed] [Google Scholar]
- 41.Smura, T., S. Blomqvist, A. Paananen, T. Vuorinen, Z. Sobotova, V. Bubovica, O. Ivanova, T. Hovi, and M. Roivainen. 2007. Enterovirus surveillance reveals proposed new serotypes and provides new insight into enterovirus 5′-untranslated region evolution. J. Gen. Virol. 88:2520-2526. [DOI] [PubMed] [Google Scholar]
- 42.Smura, T. P., N. Junttila, S. Blomqvist, H. Norder, S. Kaijalainen, A. Paananen, L. O. Magnius, T. Hovi, and M. Roivainen. 2007. Enterovirus 94, a proposed new serotype in human enterovirus species D. J. Gen. Virol. 88:849-858. [DOI] [PubMed] [Google Scholar]
- 43.Stewart, S. R., and B. L. Semler. 1997. RNA determinants of picornavirus cap-independent translation initiation. Semin. Virol. 8:242-255. [Google Scholar]
- 44.Tapparel, C., T. Junier, D. Gerlach, S. V. Belle, L. Turin, S. Cordey, K. Mühlemann, N. Regamey, J. Aubert, P. M. Soccal, P. Eigenmann, E. Zdobnov, and L. Kaiser. 2009. New respiratory enterovirus and recombinant rhinoviruses among circulating picornaviruses. Emerg. Infect. Dis. 15:719-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang, D., A. Urisman, Y. Liu, M. Springer, T. G. Ksiazek, D. D. Erdman, E. R. Mardis, M. Hickenbotham, V. Magrini, J. Eldred, J. P. Latreille, R. K. Wilson, D. Ganem, and J. L. DeRisi. 2003. Viral discovery and sequence recovery using DNA microarrays. PLoS Biol. 1:E2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weidman, M. K., R. Sharma, S. Raychaudhuri, P. Kundu, W. Tsai, and A. Dasgupta. 2003. The interaction of cytoplasmic RNA viruses with the nucleus. Virus Res. 95:75-85. [DOI] [PubMed] [Google Scholar]
- 47.Welch, T. 1984. A technique for high performance data compression. Computer 17:8-19. [Google Scholar]
- 48.Wimmer, E., C. U. T. Hellen, and X. Cao. 1993. Genetics of poliovirus. Genetics 27:353-436. [DOI] [PubMed] [Google Scholar]
- 49.Zell, R., K. Sidigi, E. Bucci, A. Stelzner, and M. Görlach. 2002. Determinants of the recognition of enteroviral cloverleaf RNA by coxsackievirus B3 proteinase 3C. RNA 8:188-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zoll, J., H. A. Heus, F. J. M. van Kuppeveld, and W. J. G. Melchers. 2009. The structure-function relationship of the enterovirus 3′-UTR. Virus Res. 139:209-216. [DOI] [PubMed] [Google Scholar]
- 51.Zuker, M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31:3406-3415. [DOI] [PMC free article] [PubMed] [Google Scholar]