

Abstract
The development of HIV drugs is an expensive and a lengthy process. In this study, we used drug repositioning, a process whereby a drug approved to treat one condition is used to treat a different condition, to identify clinically approved drugs that have anti-HIV activity. The data presented here show that a combination of two clinically approved drugs, decitabine and gemcitabine, reduced HIV infectivity by 73% at concentrations that had minimal antiviral activity when used individually. Decreased infectivity coincided with a significant increase in mutation frequency and a shift in the HIV mutation spectrum. These results indicate that an increased mutational load is the primary antiviral mechanism for inhibiting the generation of infectious progeny virus from provirus. Similar results were seen when decitabine was used in combination with another ribonucleotide reductase inhibitor. Our results suggest that HIV infectivity can be decreased by combining a nucleoside analog that forms noncanonical base pairs with certain ribonucleotide reductase inhibitors. Such drug combinations are relevant since members of these drug classes are used clinically. Our observations support a model in which increased mutation frequency decreases infectivity through lethal mutagenesis.
There are more than 20 drugs approved for the treatment of HIV infection. However, the efficacy of these drugs is limited by drug resistance, which emerges when drug levels are not high enough to sufficiently inhibit viral replication. While there are currently five classes of HIV therapy, a mutation that confers resistance to one drug often confers resistance to other members of the same drug class. Thus, the emergence of drug resistance limits potential drug therapies, making new anti-HIV therapies essential for successful long-term treatment of HIV infection. However, the development of novel anti-HIV drugs is costly (∼$600 million) and time-consuming (over 12 years) (12). One way to decrease the cost and expedite the development of novel drugs is to use a drug repositioning strategy which involves using drugs that are clinically approved for one condition to treat a different condition (1). Drug repositioning expedites drug development by making use of drugs whose toxicity and pharmacokinetic profiles have already been thoroughly characterized. Such a strategy has been successfully used for the treatment of conditions such as cancer, obesity, and osteoporosis, as well as others (1). For example, zidovudine (AZT), which is clinically approved for the treatment of HIV infection, was originally developed as an anticancer drug (20, 24). Thus, to expedite the development of novel anti-HIV drugs, we examined clinically approved drugs for the ability to inhibit HIV infectivity.
We focused on clinically approved antimetabolites (Table 1) for two reasons. First, none of the current anti-HIV drugs are antimetabolites. Therefore, any compounds identified as having anti-HIV activity would likely offer a new mechanism of action. Second, antimetabolites have been shown to have activity against a wide variety of viruses, such as poliovirus and foot-and-mouth disease virus (FMDV) (34, 35, 37). This antiviral activity is likely attributable to either a reduction in viral replication or an increase in the viral mutation rate. The ability of antimetabolites to reduce replication is likely due to a reduction in deoxynucleoside triphosphate (dNTP) pools, which are required for viral replication (2, 3, 10, 28). Alternatively, alterations of dNTP pools by antimetabolites have been shown to increase the HIV mutation rate, which correlates with a loss of infectivity. This loss of infectivity has been attributed to the process of lethal mutagenesis, a term used to describe the idea that the mutation rate can surpass a threshold beyond which the virus is unable to replicate its genome with enough fidelity to remain infectious. Although an inverse correlation between mutation frequency and infectivity has been shown for a number of viruses, there are few, if any, drugs used clinically that specifically target viral mutation rates.
TABLE 1.
Compounds screened for anti-HIV activitya
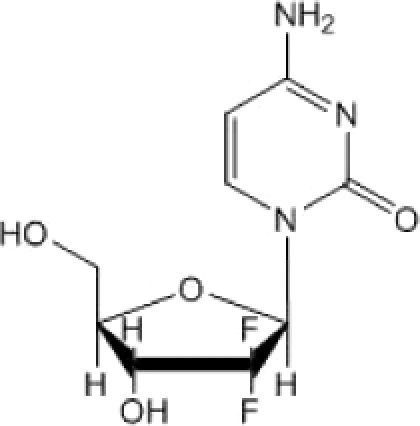 |
All compounds were screened for anti-HIV activity alone or in combination with decitabine using the single-cycle HIV assay shown in Fig. 1.
In this study, we describe the identification of a novel combination therapy for HIV infection composed of two nucleoside analogs that are clinically approved for the treatment of precancerous or cancerous states. The two drugs, decitabine and gemcitabine, significantly decrease HIV infectivity when used individually. However, when used in combination, the drugs worked synergistically to decrease or eliminate HIV infectivity without any detectable effect on cell proliferation. Similar results were seen when decitabine was used in combination with another ribonucleotide reductase inhibitor, hydroxyurea. We provide data that suggest that the combination therapy targets the mutation rate of HIV, a drug target that has yet to be exploited clinically. Importantly, these results reveal a novel therapeutic strategy to inhibit HIV replication. Specifically, we show here that HIV infectivity can be synergistically decreased by combining two classes of compounds, (i) nucleoside analogs that form noncanonical base pairs and (ii) certain ribonucleotide reductase inhibitors. Furthermore, since many of the drugs from each of these drug classes are already clinically approved, it is likely that such a drug combination is clinically relevant to the treatment of HIV infection.
MATERIALS AND METHODS
Cell lines and plasmids.
The 293T cell line was obtained from the American Type Culture Collection. Antibody to mouse heat-stable antigen protein (HSA) was purchased from BD Pharmingen (San Diego, CA). Decitabine was obtained from Moravek (Brea, CA), hydroxyurea was purchased from Sigma (St. Louis, MO), and gemcitabine was from AK Scientific (Mountain View, CA). The CellTiter-Glo cell proliferation kit was purchased from Promega (Madison, WI). The Eppendorf miniprep kit was obtained from Eppendorf (Westbury, NY). The plasmid pNL4-3.HSA.R+.E− was obtained from Nathaniel Landau through the NIH AIDS Research and Reference Reagent program, Division of AIDS, NIAID, NIH (Germantown, MD) (17). Restriction enzymes were purchased from New England Biolabs (Ipswich, MA). U373-MAGI-CXCR4CEM cells were obtained from Michael Emerman through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH (15, 42). pIRES2-EGFP was obtained from Clontech (Mountain View, CA). The pCR-18S plasmid was a kind gift from Mauro Magnani (Universita' Degli Studi Di Urbino) and has been previously described (7).
Design and construction of the HIV vector for mutation detection.
pNL4-3.HSA-R+.E− contains the gene for HSA fused in frame to the nef initiator methionine codon, as well as a frameshift mutation at the 5′ end of env which prevents the production of gp160 (17). The internal ribosome entry site (IRES)-green fluorescent protein (GFP) fragment was PCR amplified from pIRES2-EGFP and subcloned into pCR2.1. This plasmid, as well as pNL4-3.HSA-R+.E−, was restriction digested with XhoI. Following purification, pNL4-3.HSA-R+.E− and the IRES-enhanced GFP (EGFP) fragments were ligated and then transformed using chemically competent DH5α. Restriction digestion and DNA sequencing analysis was used to verify the clones.
Cell culture.
293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal clone 3 (FC3) serum (HyClone, Logan, UT) and penicillin/streptomycin at 37°C in 5% CO2. U373-MAGI-CXCR4CEM cells were maintained at 37°C in 5% CO2 in selection medium composed of DMEM with 10% FC3 serum, 1 μg/ml puromycin, 0.1 mg/ml hygromycin, and 0.2 mg/ml neomycin.
Production of virus stocks.
293T cells (1 × 106) were plated on poly-l-lysine-coated 10-cm culture dishes 24 h before transfection. The cells were then transfected by calcium phosphate coprecipitation with 10 μg of the HIV vector (HIG) and 1 μg of a plasmid encoding the HIV envelope, pNL4-3env (obtained from Eric Freed, NIH). The medium was replaced with 6 ml of DMEM containing 10% FC3 serum and penicillin/streptomycin 24 h after transfection. Virus was harvested 24 h later by filtration of the cell supernatant through a 0.2-μm filter. Viral stocks were used immediately or frozen at −80°C.
Drug treatments and infection.
U373-MAGI-CXCR4CEM cells (6.2 × 104/well) were plated in a 12-well culture dish 24 h prior to drug treatments. Cells were treated with the freshly prepared individual drugs or a combination of the drugs 2 h prior to infection. After the 2-h pretreatment, the viral stock (500 μl) was added to each well to give a final volume of 1 ml and a final drug concentration that is indicated in the figure legends. After infection (24 h), the medium was replaced. Cells were harvested for analysis 48 h after infection.
Cell staining for HSA.
Infected cells were dislodged with trypsin and transferred to a microcentrifuge tube before the samples were centrifuged at 200 × g for 5 min. The supernatant was removed, and the cell pellets were resuspended in 50 μl of phosphate-buffered saline (PBS) containing 2% FC3 serum and phycoerythrin (PE)-conjugated HSA antibody. Cells were incubated with the antibody at 4°C for 15 min. After this incubation, 750 μl of PBS/2% FC3 serum was added and the samples were centrifuged at 300 × g for 5 min. The supernatant was then removed, and the cell pellets were resuspended in PBS containing 2% paraformaldehyde and 2% FC3 serum.
Flow cytometry.
Forward and side scatter gating was done to eliminate nonviable cells. Cells (10,000) were then analyzed for fluorescence at 488 nm and 568 nm. Compensation was adjusted to eliminate carryover of the fluorescent signal. Quadrants were drawn using noninfected cells to determine background levels of fluorescence. Cells expressing both HSA and GFP were used to determine the percentage of infected cells.
Cellular proliferation.
Cell proliferation was examined using the CellTiter-Glo kit from Promega according to the manufacturer's instructions. U373-MAGI-CXCR4CEM cells (4,500/well) were plated in a 96-well dish 24 h prior to drug treatment. Cells were treated with the individual drugs or a combination of the drugs for 24 h, at which point the medium was changed, and 24 h later, proliferation was assessed. Dimethyl sulfoxide (DMSO) was used as a control for the no-drug-treated cells. Twenty percent ethanol was used as a positive control for cellular toxicity. The data were converted to relative cell numbers by setting the value for no-drug-treated cells at 100 for each experiment and then multiplying the data for the other samples by the number used to convert the no-drug-treated cells to 100. This conversion was normalized for differences in luciferase activity among different experiments.
Fluorescence-activated cell sorting (FACS) and sequencing of proviral DNA.
Infected cells were sorted based on the expression of HSA but not GFP (i.e., HSA+, GFP−). A minimum of 5,000 cells was collected from each treatment group. These cells were collected and used to isolate proviral DNA. Genomic DNA was isolated by the High Pure PCR Template kit (Roche). Nested PCR was used to amplify the proviral GFP sequence. The primers used for the first round of PCR were 5′CTGAAGGATGCCCAGAAGG3′ and 5′TGCTTCTAGCCAGGCACAAGC3′. The primers used for the second round of PCR were 5′TTACATGTGTTTAGTCGAGG3′ and 5′GCTACTTGTGATTGCTCCATG3′. The PCR fragment containing GFP was ligated into pCR2.1 and transformed into DH5α. Individual colonies were grown, plasmid DNA was recovered by the Eppendorf miniprep kit, and the GFP gene was sequenced. Two primers were used to obtain the full GFP sequence (5′ATGATTACGCCAAGC3′ and 5′TTCAAGGACGACGGC3′). Any sequences with the same mutations were assumed to be identical, and therefore only mutations from one of these sequences were counted. The number of mutants sequenced was 24 for untreated cells, 38 for cells treated with decitabine, 29 for gemcitabine-treated cells, and 39 for cells treated with both decitabine and gemcitabine.
Real-time qPCR of late RT products.
MAGI-CXCR4CEM cells (300,000 per well) were plated on a six-well plate. Twenty-four hours later, cells were treated with the indicated drug for 2 h prior to infection with virus that had been treated with DNase I to remove plasmid DNA. As a control, an aliquot of DNase I-treated virus was heat inactivated for 20 min at 100°C. Eighteen hours after infection, cells were harvested with trypsin, washed with PBS, and lysed as previously described (41). Quantitative PCR (qPCR) mixtures contained 1 μl of cell lysate and were set up according to the manufacturer's suggestions (Applied Biosciences). Primers for 18S rRNA were used to normalize sample-to-sample variation. The primers used to detect late reverse transcription (RT) products were 5′TGTGTGCCCGTCTGTTGTGT (forward) and 5′GAGTCCTGCGTCGAGAGAGC (reverse). The primers used to detects 18S rRNA were 5′GTAACCCGTTGAACCCCATT (forward) and 5′CCATCCAATCGGTAGTAGGG (reverse). A standard curve of 10-fold serial dilutions of the single-cycle HIV vector was used to quantitate late RT products in cell lysates, while a similar dilution series was performed using the pCR-18S plasmid. The conditions for amplification were 95°C for 10 min, followed by 40 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s. The amplification efficiencies were similar and within the range of 100%. Gene expression was analyzed using the Pfaffl modification of the ΔΔCT method (36).
Statistical analysis.
Data were analyzed by calculating the mean ± the standard deviation (SD). One-way analysis of variance (ANOVA) with Tukey's posttest was used to assess differences among treatment groups. A P value of <0.05 was considered statistically significant, as indicated by an asterisk.
RESULTS
Identification of decitabine and gemcitabine as potent anti-HIV compounds.
To examine clinically approved drugs for the ability to decrease HIV infectivity, we used an assay that enables the detection of HIV-infected cells through the expression of two marker proteins, HSA and GFP. The assay, shown in Fig. 1, uses an HIV construct that has a mutated Env gene which limits the virus to one round of replication.
FIG. 1.

Single-cycle HIV assay used to assess infectivity and mutation frequency. HIV is produced in 293T cells by transfection of two plasmid constructs: the envelope-deficient HIV vector contains two marker genes (those for HSA and GFP) that are used to assess infectivity and mutation frequency. The second plasmid encodes the HIV envelope. At 48 h after transfection, the supernatant containing virus is passed through a 0.2-μm filter and added to target cells that have been pretreated with drugs for 2 h. The target cells are harvested 48 h after infection and stained with PE-conjugated HSA antibody. Flow cytometry is used to examine the percentage of cells expressing HSA and GFP.
293T cells are used to produce virus, which is then used to infect target cells that express the HIV receptor (CD4) and coreceptor (CXCR4). The target cells are either drug or solvent (DMSO) treated, and then expression of the target genes is assessed by flow cytometry to determine the percentage of cells that have been infected. Approved anti-HIV drugs were used to validate the assay. For example, treatment of MAGI cells with AZT showed a dose response with a 50% effective concentration (EC50) of approximately 100 nM (data not shown). This assay was used to screen a number of drugs (Table 1), three of which showed potent anti-HIV activity, including decitabine and gemcitabine. Figure 2 shows the abilities of decitabine and gemcitabine to decrease HIV infectivity in a concentration-dependent manner. As shown in Fig. 2a, decitabine, a nucleoside analog, significantly decreased HIV infectivity at low concentrations (50% inhibitory concentration, ∼200 nM). Figure 2b shows that gemcitabine, a potent ribonucleotide reductase inhibitor, also decreased HIV infectivity at low concentrations (EC50, ∼8 nM).
FIG. 2.

Effects of decitabine and gemcitabine on HIV infectivity. (a) The single-cycle HIV assay was used to assess viral infectivity as shown in Fig. 1. Decitabine leads to a concentration-dependent decrease in HIV infectivity, as measured by expression of HSA and GFP by flow cytometry. The data were normalized by setting the untreated control cells (ND, no drug) to 100% infectivity and comparing all of the other treatment groups to this value. (b) Dose response of HIV infectivity to gemcitabine. The data shown are means ± SDs. Each data point is from three independent experiments.
Decitabine is used clinically for its ability to inhibit DNA methylation (30-32), while gemcitabine is used for its ability to inhibit ribonucleotide reductase (29), the enzyme required for reduction of nucleoside diphosphates to deoxynucleoside diphosphates. Since decitabine and gemcitabine have two distinct activities, they may decrease HIV infectivity by two different mechanisms. If gemcitabine and decitabine inhibit infectivity by two separate mechanisms, then they may act additively or synergistically when used in combination. To observe synergy, we examined concentrations of decitabine and gemcitabine that, when used alone, had little, if any, effect on infectivity. These concentrations were then combined to see if, when used together, the two drugs would inhibit infectivity in an additive or synergistic manner. In doing so, we found that the combination of 50 nM decitabine and 5 nM gemcitabine was significantly more effective at decreasing infectivity than was each drug used individually (Fig. 3 a). In fact, the combination of decitabine and gemcitabine decreased infectivity by 73% whereas the individual drugs did not dramatically decrease infectivity. Furthermore, the drug combination showed a dose-dependent decrease in infectivity when the ratio of decitabine to gemcitabine was kept constant (Fig. 3b).
FIG. 3.

Effects of decitabine (Dec) and/or gemcitabine (Gem) on viral infectivity, cell number, and mutation frequency. (a) Infectivity was assessed by the single-cycle HIV assay as described in Fig. 1. The untreated cells were set at 100%, and all other treatment groups were compared to this control value. Each column represents the mean ± the SD from at least three independent experiments. An asterisk indicates a statistically significant difference (P < 0.05). (b) Dose-response curve showing the inhibitory effect of the combination of decitabine and gemcitabine at different concentrations. (c) Effect of decitabine and/or gemcitabine on relative cell number. Relative cell number was assessed as described in Materials and Methods. The untreated control cells were set at 100, and all of the other treatment groups were compared to this control value. Each column represents the mean ± the SD from at least three independent experiments. (d) Mutation frequencies assessed by the single-cycle HIV assay. Mutation frequencies were determined by flow cytometry. The following equation was used: Mutation frequency = [(% HSA+ GFP− cells) + (% HSA− GFP+ cells)]/[(% HSA+ GFP− cells) + (% HSA− GFP+) + (HSA+ GFP+ cells)]. For each experiment, the untreated cells were set at 1 and all of the other drug treatments were compared to this control value. (e) Mutation frequency upon treatment with ddA, a chain terminator that inhibits reverse transcriptase. (f) Inhibition of infectivity mediated by ddA. The data represent the mean ± the SD from at least three independent experiments. The asterisk indicates a statistically significant difference (P < 0.05).
Decitabine and gemcitabine are not cytotoxic at concentrations that have antiviral activity.
Toxicity caused by drug treatment could nonspecifically decrease HIV infectivity. To determine if the apparent decrease in infectivity was due to toxicity, the effect of decitabine and gemcitabine on cell proliferation was examined. Figure 3c shows that at the concentrations indicated, cell proliferation, as determined by intracellular ATP levels, was not significantly different than that of untreated control cells. Importantly, cell proliferation was not dramatically affected at the concentrations used in the infectivity assay. Thus, the loss of infectivity was not due to toxicity.
The combination of decitabine and gemcitabine increases HIV mutation frequency.
Gemcitabine is a nucleoside analog that inhibits ribonucleotide reductase (18), while decitabine is a nucleoside analog that is used clinically for its ability to inhibit DNA methylation (30, 33). Ribonucleotide reductase inhibitors can induce dNTP imbalances, which are known to alter the fidelity of reverse transcriptase (4). Also, nucleosides that form noncanonical base pairs could be incorporated into viral DNA and subsequently induce mutations through noncanonical base pair formation (16, 25). Based upon these observations, we hypothesized that the combination of decitabine and gemcitabine (Fig. 3a and b) may elevate mutation frequencies. To test this hypothesis, we used the assay outlined in Fig. 1 to assess the HIV mutation frequency in the presence of decitabine and gemcitabine. The two target genes, those for HSA and GFP, present in the HIV plasmid were used to simultaneously detect HIV infectivity and mutation frequency. To determine mutation frequency, the flow data were divided into four quadrants based on the expression of GFP and/or HSA. Cells infected with the wild-type HIV construct were expected to express both GFP and HSA, whereas cells infected with a mutant HIV construct express either one marker gene or neither marker gene. Cells that express only the gene for HSA (HSA+ GFP−) are cells that have been infected with HIV that carries a mutation that has abrogated the expression of GFP. Similarly, expression of GFP only (HSA− GFP+) indicates that a mutation has abrogated the expression of HSA. Therefore, cells infected with mutant HIV were detected as cells expressing either HSA or GFP, but not both. The relative mutation frequency was then calculated as a fraction by dividing the percentage of cells infected with mutant virus by the total percentage of cells infected. To normalize for variation in viral titers among different experiments, untreated cells were defined as having a mutation frequency of 1. The results shown in Fig. 3d demonstrate that neither 50 nM decitabine nor 5 nM gemcitabine increased the mutation frequency; however, when these compounds were used in combination at the same concentrations, there was a 3.4-fold increase in the relative mutation frequency. This finding is in contrast to that observed with dideoxyadenosine (ddA), a chain terminator that inhibits the infectivity of HIV (Fig. 3e and f) and is not known to affect its mutation frequency.
To assess whether the drug combination was mutagenic to the host genome, we used the CHO/HGPRT (hypoxanthine-guanine phosphoribosyltransferase) assay (21) to examined the genotoxic potential of decitabine and gemcitabine. Previous results have shown that decitabine is not toxic by the HGPRT assay, and our results (data not shown) confirm this finding and indicate that the combination of decitabine with gemcitabine is not genotoxic.
Decitabine alters the HIV mutation spectrum.
The results in Fig. 3d demonstrate that the combination of decitabine and gemcitabine increased the HIV mutation frequency. Decitabine is a nucleoside analog that can be incorporated into DNA in place of dCTP. Although there is no evidence that decitabine is mutagenic in humans, there is some evidence that decitabine could be mutagenic in some systems, such a transgenic mouse model where an increase in C:G→G:C transversions in the lacZ transgene was reported (23). Similarly, exposure of HIV type 1 with 5-azacytidine, the ribonucleoside analog of decitabine, increased C:G→G:C mutations (9). The model of the mutagenic lesions identified suggests that decitabine can undergo a ring-opening step, followed by hydrolysis to give a structure that is able to base pair with cytosine (23). Thus, decitabine is incorporated in place of dCTP but behaves chemically as a guanosine by base pairing with cytosine. Incorporation of decitabine during plus-strand DNA synthesis would generally not result in a mutation, as it would be removed from the proviral DNA by the host DNA repair machinery (39). Thus, we examined the mutation spectrum of HIV exposed to decitabine, gemcitabine, or a combination thereof. To do this, cells infected with mutant HIV were identified through the single-cycle HIV assay (Fig. 1), followed by FACS analysis to isolate the HSA+ GFP− cell population. Genomic DNA was isolated from these cells, and the GFP gene was PCR amplified and sequenced to examine the mutational spectrum. There was a dramatic change in the mutation spectrum of sequenced HIV from cells treated with decitabine or a combination of decitabine and gemcitabine compared to that of HIV from untreated cells (Fig. 4 and data not shown). Specifically, decitabine dramatically increased G-to-C mutations, with G-to-C mutations accounting for 38% of all mutations. Conversely, no G-to-C mutations were identified in either the untreated or gemcitabine-treated cells. This abundance of G-to-C mutations supports the notion that decitabine may base pair with cytosine, leading to an increase in G-to-C transversions (23). Cells treated with both decitabine and gemcitabine appear to have a modest increase in G-to-C mutations compared to untreated cells. Although gemcitabine is a ribonucleotide reductase inhibitor, the data do not suggest that it alters dNTP pools in a way that would increase the incorporation of decitabine during viral DNA synthesis since there was no increase in G-to-C mutations in cells treated with both drugs compared to those treated with decitabine alone.
FIG. 4.

Decitabine alters the HIV mutation spectrum. Proviral GFP was sequenced from GFP− HSA+ cells. Each number within a wedge represents a specific mutation, with the value indicating the prevalence of this mutation in terms of the percentage of total mutations. Number of mutations sequenced: no drug, 36; decitabine (Dec), 82; gemcitabine (Gem), 48; decitabine plus gemcitabine, 63.
Thus far, the data suggest that decitabine and gemcitabine can reduce viral infectivity by elevating the viral mutation rate. Since the drug combination inhibits HIV infectivity synergistically, we hypothesized that other ribonucleotide reductase inhibitors could be used in combination with decitabine to synergistically inhibit HIV infectivity. To test this hypothesis, we examined the antiviral activity of other ribonucleotide reductase inhibitors. When decitabine was used in combination with hydroxyurea, a synergistic decrease in HIV infectivity similar to that seen with decitabine and gemcitabine was seen (Fig. 5 a). Additionally, this decrease was not attributed to cellular toxicity (Fig. 5b). Although ribonucleotide reductase inhibitors such as luteolin, didox, trimidox, and guanazole were also used in combination with decitabine, none of these were found to synergistically decrease infectivity data not shown.
FIG. 5.

Decitabine (Dec) and hydroxyurea (HU) significantly decrease viral infectivity at nontoxic concentrations. (a) Infectivity was assessed by the single-cycle HIV assay. The untreated cells were set at 100%, and all other treatment groups were compared to this control value. Each column represents the mean ± the SD from at least three independent experiments. ND, no drug. (b) Relative number of target cells after treatment with solvent (DMSO), decitabine, hydroxyurea, or a combination of decitabine and hydroxyurea. The untreated cells were set at 100 and used as a comparison for all of the other treatment groups. Each column represents the mean ± the SD from at least three independent experiments.
Lack of correlation between the decitabine/gemcitabine infectivity loss and reduction in viral DNA synthesis.
The data presented here indicate that provirus made in the presence of decitabine and gemcitabine is mutated and contributes to their antiviral activity. However, since both compounds are nucleoside analogs, it is possible that the primary antiviral mechanism is inhibition of RT. Although their structures do not indicate that decitabine and gemcitabine would act as chain terminators, there have been reports that gemcitabine acts as a masked chain terminator (22). Therefore, real-time qPCR was used to examine the effect of a combination of 50 nM decitabine and 5 nM gemcitabine on the synthesis of late viral DNA products. As shown in Fig. 6 a, this combination of decitabine and gemcitabine decreased late RT products compared to those in untreated cells. However, the corresponding loss of infectivity cannot be attributed solely to inhibition of RT (Fig. 6b). In contrast, cells treated with 200 nM AZT, a well-characterized chain terminator, decreased late RT products by approximately 70% (Fig. 6a), which strongly correlates with the observed loss of infectivity (Fig. 6b).
FIG. 6.

Lack of correlation between decitabine (Dec)- and -gemcitabine (Gem)-mediated infectivity loss and inhibition of RT. (a) Late products of RT were examined by real-time qPCR as described in Materials and Methods. Cells were treated with DMSO as a control (ND), 50 nM decitabine plus 5 nM gemcitabine, or 200 nM AZT. Heat-inactivated virus was used as a control to account for amplification of any plasmid carried over in the viral supernatant taken from transfected cells. Data are expressed as the mean ± the SD from three separate experiments. The single asterisk indicates that the combination treatment is significantly different from the untreated control (ND), while the double asterisks indicate that AZT treatment is significantly different from both the untreated cells and the combination treatment (Dec + Gem). Statistical significance was determined by one-way ANOVA with Tukey's posttest, and a P value of <0.05 was considered significant. (b) Decreased infectivity in cells treated with 50 nM decitabine plus 5 nM gemcitabine or with 200 nM AZT. The single-cycle HIV assay was done in parallel with qPCR to examine the relationship between infectivity and inhibition of RT products.
DISCUSSION
Recent setbacks in the development of an AIDS vaccine, as well as the prevalence of drug-resistant HIV, emphasize the need for new approaches to anti-HIV therapies. To decrease the cost of and time required for the development of novel drugs to treat HIV infection, a number of clinically approved drugs were screened for the ability to inhibit HIV infectivity, including nucleoside analogs and antimetabolites. Since HIV reverse transcriptase is error prone, the virus is highly susceptible to mutations caused by alterations in dNTP pools or by the incorporation of nucleoside analogs that form noncanonical base pairs (4, 16, 25, 27). The infectivity of HIV and other RNA viruses is significantly reduced with even a modest increase in the mutation rate (19, 38). This finding has led to the design of novel nucleoside analogs, including 5-hydroxy-2′-deoxycytidine and 5-aza-5,6-dihydro-2′-deoxycytidine (KP-1212), that are incorporated into viral DNA during replication (16). Although these compounds effectively increase the viral mutation rate, none have been shown to be potent or safe enough to be used clinically.
In this study, we identify a novel strategy for HIV infection treatment that takes advantage of a drug target that has yet to be exploited clinically—the HIV mutation rate. Specifically, we show that HIV infectivity can be synergistically decreased by combining two classes of compounds, (i) a nucleoside analog that forms noncanonical base pairs and (ii) a ribonucleotide reductase inhibitor. Another significant finding was that the antiviral activity of the drug combination can be attributed to an increase in the HIV mutation rate. The most potent drug combination characterized consists of decitabine and gemcitabine, two clinically approved drugs used in cancer treatment. The data shown here demonstrate that when used in combination, decitabine and gemcitabine appear to synergistically decrease HIV infectivity at concentrations well below those used in cancer therapy (14, 30). Furthermore, a combination of decitabine and gemcitabine increased the HIV mutation frequency by 3.4-fold (Fig. 3d) compared to that in untreated cells and this increase in the mutation frequency correlated with a 73% reduction in infectivity (Fig. 3a). Although the increase in the mutation frequency was modest, similar increases have been found to be sufficient to eliminate the infectivity or viability of other viruses, such as FMDV, poliovirus, and HIV (19, 38). The hypothesis that combination drug therapy induces viral mutations was further supported by sequencing data, which showed a striking difference between the mutation spectra of viruses exposed to combination drug therapy and unexposed viruses. A model is proposed in Fig. 7 to account for the observed changes in the mutation spectrum when decitabine is incorporated into viral DNA. These findings suggest that combination drug therapy decreases infectivity by increasing the mutation rate. This is further supported by data showing that similar antiviral effects were observed when decitabine was combined with a different ribonucleotide reductase inhibitor, hydroxyurea. Hydroxyurea has previously been shown to have anti-HIV activity and acts synergistically with chain terminators such as dideoxyinosine (11, 26, 28). Although hydroxyurea has been used clinically to treat HIV infection, its use is not favored because of its adverse effects. While it was expected that other ribonucleotide reductase inhibitors would have the same antiviral effects as that seen with hydroxyurea and gemcitabine, this was not supported by our data. This may be related to the specific dNTP pools that are altered by each of the different compounds or cellular processes that compensate for alterations in dNTP pools.
FIG. 7.

Model of how decitabine acts as a viral mutagen to induce G-to-C mutations during minus-strand viral DNA synthesis. Positive-stranded viral RNA is reverse transcribed into double-stranded viral DNA. Decitabine (D*) is incorporated in place of dCTP and base pairs with cytosine. Incorporation of decitabine into the minus strand of viral DNA leads to a C-to-G transversion mutation in the positive strand. This results in fixation of the mutation. In contrast, incorporation of decitabine into the positive strand results in D*-to-G mispairing. Decitabine would likely be excised during host DNA repair and replaced with a cytosine. In this proposed mechanism, decitabine would primarily act as a viral mutagen during minus-strand viral DNA synthesis.
As a clinical therapy, lethal mutagenesis has been met with hesitation, primarily because of concerns related to the cytotoxicity of the compounds, the high concentrations of mutagen required, and the potential side effects associated with the long-term use of these compounds (25). However, the concentrations of decitabine and gemcitabine used here are well below the concentrations used in cancer therapy (31, 40), suggesting that any adverse effects of these drugs should also be minimized. Although our data indicate that decitabine can act as a mutagenic nucleoside to inhibit HIV replication, clinically, decitabine is not known to be mutagenic. Instead, decitabine is thought to work by covalently binding to DNA methyltransferase I, irreversibly inhibiting the enzyme and decreasing DNA methylation (8, 30). Since the concentrations of decitabine used here are well below those used in cancer treatment (31), it seems unlikely that decitabine, as part of an anti-HIV therapy regimen, would introduce mutations into genomic DNA, given the presence of host DNA repair mechanisms. This model is supported by our data, which did not show an increase in C-to-G mutations, the mutations expected if decitabine were not excised from the viral DNA during integration. Instead, our data suggest that decitabine is removed after integration by the host DNA repair machinery and that the G-to-C mutations are due to the guanosines that replace decitabine in the minus strand viral DNA. Furthermore, decitabine was not genotoxic, as determined with the HGPRT assay by us (data not shown) and others (32). Additionally, decitabine was not mutagenic to male Fisher rats that were treated with decitabine for 1 year (6). Despite this evidence that decitabine is not mutagenic to the human genome, there was one report that decitabine was mutagenic in a transgenic mouse model (23).
Although it is possible that the combination of decitabine and gemcitabine decreases infectivity by more than one mechanism, an additional antiviral activity would not negate the data presented, which show that completion of RT in the presence of decitabine and gemcitabine significantly alters the mutation spectrum. Although decitabine is known to affect the methylation of cellular genes, it is unlikely that this activity contributes to the decrease in infectivity shown here (5). Similarly, there is no evidence that decitabine acts as a chain terminator to inhibit replication. In fact, while the combination of decitabine and gemcitabine modestly inhibited viral DNA synthesis, the decrease in RT products was not enough to account for the loss of infectivity. Since mutagens are not expected to act as chain terminators, no significant decrease in viral DNA would be expected from compounds acting as lethal mutagens. In fact, previous studies have demonstrated that even when mutagens eliminate infectivity, there are still detectable levels of viral RNA (13). This is likely due to the production of defective viruses that are not infectious and can interfere with the infectivity of viruses that were not lethally mutagenized, a process known as lethal defection (13).
The combination therapy we have identified in this study has many advantages, including that (i) it has a low effective concentration of each drug due to synergy, which decreases concerns of host toxicity; (ii) it uses a nucleoside analog that can be incorporated into the HIV genome but is unlikely to be incorporated into the host genome or, if incorporated, may be efficiently excised from the human genome; and (iii) both of the drugs used are already approved for human clinical use for conditions other than HIV infection, which should shorten the time and decrease the cost associated with drug development. Given these advantages, the combination of decitabine and gemcitabine may offer a novel treatment option for HIV-infected individuals. More importantly, this study reveals a novel treatment strategy of combining a nucleoside analog and a ribonucleotide reductase inhibitor to decrease infectivity through an increase in the HIV mutation rate.
Acknowledgments
Support for these studies was from a Center for Drug Design funding agreement and NIH grants GM56615 and T32 CA009138.
We thank K. Pankiewicz and R. Vince for stimulating discussions and valuable feedback and I. Dorweiler and J. Chauhan for technical assistance and stimulating discussions.
We have no competing financial interests.
Footnotes
Published ahead of print on 7 July 2010.
REFERENCES
- 1.Ashburn, T. T., and K. B. Thor. 2004. Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 3:673-683. [DOI] [PubMed] [Google Scholar]
- 2.Baba, M., R. Pauwels, J. Balzarini, P. Herdewijn, E. De Clercq, and J. Desmyter. 1987. Ribavirin antagonizes inhibitory effects of pyrimidine 2′,3′-deoxynucleosides but enhances inhibitory effects of purine 2′,3′-dideoxynucleosides on replication of human immunodeficiency virus in vitro. Antimicrob. Agents Chemother. 31:1613-1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balzarini, J., D. A. Cooney, M. Dalal, G.-J. Kang, J. E. Cupp, E. De Clercq, S. Broder, and D. G. Johns. 1987. 2′,3′-Dideoxycytidine: regulation of its metabolism and anti-retroviral potency by natural pyrimidine nucleosides and by inhibitors of pyrimidine nucleotide synthesis. Mol. Pharmacol. 32:798-806. [PubMed] [Google Scholar]
- 4.Bebenek, K., J. D. Roberts, and T. A. Kunkel. 1992. The effects of dNTP pool imbalances on frameshift fidelity during DNA replication. J. Biol. Chem. 267:3589-3596. [PubMed] [Google Scholar]
- 5.Bednarik, D. P., J. A. Cook, and P. M. Pitha. 1990. Inactivation of the HIV LTR by DNA CpG methylation: evidence for a role in latency. EMBO J. 9:1157-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carr, B. I., S. Rahbar, Y. Asmeron, A. Riggs, and C. D. Winberg. 1988. Carcinogenicity and haemoglobin synthesis induction by cytidine analogues. Br. J. Cancer 57:395-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casabianca, A., C. Orlandi, A. Fraternale, and M. Magnani. 2004. Development of a real-time PCR assay using SYBR Green I for provirus load quantification in a murine model of AIDS. J. Clin. Microbiol. 42:4361-4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christman, J. K. 2002. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 21:5483-5495. [DOI] [PubMed] [Google Scholar]
- 9.Dapp, M. J., C. L. Clouser, S. Patterson, and L. M. Mansky. 2009. 5-Azacytidine can induce lethal mutagenesis in human immunodeficiency virus type 1. J. Virol. 83:11950-11958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao, W.-Y., D. G. Johns, M. Tanaka, and H. Mitsuya. 1999. Suppression of replication of multidrug-resistant HIV type 1 variants by combinations of thymidylate synthase inhibitors with zidovudine or stavudine. Mol. Pharmacol. 55:535-540. [PubMed] [Google Scholar]
- 11.Gao, W. Y., A. Cara, R. C. Gallo, and F. Lori. 1993. Low levels of deoxynucleotides in peripheral blood lymphocytes: a strategy to inhibit human immunodeficiency virus type 1 replication. Proc. Natl. Acad. Sci. U. S. A. 90:8925-8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garg, S., R. Kandarapu, K. Vermani, K. R. Tambwekar, A. Garg, D. P. Waller, and L. J. Zaneveld. 2003. Development pharmaceutics of microbicide formulations. Part I: preformulation considerations and challenges. AIDS Patient Care STDS 17:17-32. [DOI] [PubMed] [Google Scholar]
- 13.Grande-Pérez, A., E. Lazaro, P. R. Lowenstein, E. Domingo, and S. C. Manrubia. 2005. Suppression of viral infectivity through lethal defection. Proc. Natl. Acad. Sci. U. S. A. 102:4448-4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grunewald, R., H. Kantarjian, M. Du, K. Faucher, P. Tarassoff, and W. Plunkett. 1992. Gemcitabine in leukemia: a phase I clinical, plasma, and cellular pharmacology study. J. Clin. Oncol. 10:406-413. [DOI] [PubMed] [Google Scholar]
- 15.Harrington, R. D., and A. P. Geballe. 1993. Cofactor requirement for human immunodeficiency virus type 1 entry into a CD4-expressing human cell line. J. Virol. 67:5939-5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris, K. S., W. Brabant, S. Styrchak, A. Gall, and R. Daifuku. 2005. KP-1212/1461, a nucleoside designed for the treatment of HIV by viral mutagenesis. Antiviral Res. 67:1-9. [DOI] [PubMed] [Google Scholar]
- 17.He, J., S. Choe, R. Walker, P. Di Marzio, D. O. Morgan, and N. R. Landau. 1995. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 69:6705-6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heinemann, V., Y.-Z. Xu, S. Chubb, A. Sen, L. W. Hertel, G. B. Grindey, and W. Plunkett. 1990. Inhibition of ribonucleotide reduction in CCRF-CEM cells by 2′,2′-difluorodeoxycytidine. Mol. Pharmacol. 38:567-572. [PubMed] [Google Scholar]
- 19.Holland, J. J., E. Domingo, J. C. De La Torre, and D. A. Steinhauer. 1990. Mutation frequencies at defined single codon sites in vesicular stomatitis virus and poliovirus can be increased only slightly by chemical mutagenesis. J. Virol. 64:3960-3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horwitz, J. P., J. Chua, and M. Noel. 1964. Nucleosides. V. The monomesylates of 1-(2′-deoxy-β- d-lyxofuranosyl)thymine. J. Org. Chem. 29:2076-2078. [Google Scholar]
- 21.Hsie, A. W., D. A. Casciano, D. B. Couch, D. F. Krahn, J. P. O'Neill, and B. L. Whitfield. 1981. The use of Chinese hamster ovary cells to quantify specific locus mutation and to determine mutagenicity of chemicals. A report of the gene-tox program. Mutat. Res. 86:193-214. [DOI] [PubMed] [Google Scholar]
- 22.Huang, P., S. Chubb, L. W. Hertel, G. B. Grindey, and W. Plunkett. 1991. Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res. 51:6110-6117. [PubMed] [Google Scholar]
- 23.Jackson-Grusby, L., P. W. Laird, S. N. Magge, B. J. Moeller, and R. Jaenisch. 1997. Mutagenicity of 5-aza-2′-deoxycytidine is mediated by the mammalian DNA methyltransferase. Proc. Natl. Acad. Sci. U. S. A. 94:4681-4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin, T.-S., and W. H. Prusoff. 1978. Synthesis and biological activity of several amino analogues of thymidine. J. Med. Chem. 21:109-112. [PubMed] [Google Scholar]
- 25.Loeb, L. A., J. M. Essigmann, F. Kazazi, J. Zhang, K. D. Rose, and J. I. Mullins. 1999. Lethal mutagenesis of HIV with mutagenic nucleoside analogs. Proc. Natl. Acad. Sci. U. S. A. 96:1492-1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lori, F., A. Malykh, A. Cara, D. Sun, J. N. Weinstein, J. Lisziewicz, and R. C. Gallo. 1994. Hydroxyurea as an inhibitor of human immunodeficiency virus-type 1 replication. Science 266:801-805. [DOI] [PubMed] [Google Scholar]
- 27.Martinez, M. A., J.-P. Vartanian, and S. Wain-Hobson. 1994. Hypermutagenesis of RNA using human immunodeficiency virus type 1 and biased dNTP concentrations. Proc. Natl. Acad. Sci. U. S. A. 91:11787-11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meyerhans, A., J.-P. Vartanian, C. Hultgren, U. Plikat, A. Karlsson, L. Wang, S. Eriksson, and S. Wain-Hobson. 1994. Restriction and enhancement of human immunodeficiency virus type 1 replication by modulation of intracellular deoxynucleoside triphosphate pools. J. Virol. 68:535-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mini, E., S. Nobili, B. Caciagli, I. Landini, and T. Mazzei. 2006. Cellular pharmacology of gemcitabine. Ann. Oncol. 17:v7-v12. [DOI] [PubMed] [Google Scholar]
- 30.Momparler, R. L. 2005. Pharmacology of 5-aza-2′-deoxycytidine (decitabine). Semin. Hematol. 42:S9-S16. [DOI] [PubMed] [Google Scholar]
- 31.Momparler, R. L., D. Y. Bouffard, L. F. Momparler, J. Dionne, K. Belanger, and J. Ayoub. 1997. Pilot phase I-II study on 5-aza-2′-deoxycytidine (Decitabine) in patients with metastatic lung cancer. Anticancer Drugs 8:358-368. [DOI] [PubMed] [Google Scholar]
- 32.Momparler, R. L., J. Samson, L. F. Momparler, and G. E. Rivard. 1984. Cell cycle effects and cellular pharmacology of 5-aza-2′-deoxycytidine. Cancer Chemother. Pharmacol. 13:191-194. [DOI] [PubMed] [Google Scholar]
- 33.Oki, Y., E. Aoki, and J. P. Issa. 2007. Decitabine—bedside to bench. Crit. Rev. Oncol. Hematol. 61:140-152. [DOI] [PubMed] [Google Scholar]
- 34.Pariente, N., A. Airaksinen, and E. Domingo. 2003. Mutagenesis versus inhibition in the efficiency of extinction of foot-and-mouth disease virus. J. Virol. 77:7131-7138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pariente, N., S. Sierra, P. R. Lowenstein, and E. Domingo. 2001. Efficient virus extinction by combinations of a mutagen and antiviral inhibitors. J. Virol. 75:9723-9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pfaffl, M. W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruiz-Jarabo, C. M., C. Ly, E. Domingo, and J. C. Torre. 2003. Lethal mutagenesis of the prototypic arenavirus lymphocytic choriomeningitis virus (LCMV). Virology 308:37-47. [DOI] [PubMed] [Google Scholar]
- 38.Sierra, S., M. Davila, P. R. Lowenstein, and E. Domingo. 2000. Response of foot-and-mouth disease virus to increased mutagenesis: influence of viral load and fitness in loss of infectivity. J. Virol. 74:8316-8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Snyder, R. D., and P. J. Lachmann. 1989. Differential effects of 5-azacytidine and 5-azadeoxycytidine on cytotoxicity, DNA-strand breaking and repair of X-ray-induced DNA damage in HeLa cells. Mutat. Res. 226:185-190. [DOI] [PubMed] [Google Scholar]
- 40.Sugiyama, E., N. Kaniwa, S. Ryang Kim, R. Kikura-Hanajiri, R. Hasegawa, K. Maekawa, Y. Saito, S. Ozawa, J. Sawada, N. Kamatani, J. Furuse, H. Ishii, T. Yoshida, H. Ueno, T. Okusaka, and N. Saijo. 2007. Pharmacokinetics of gemcitabine in Japanese cancer patients: the impact of a cytidine deaminase polymorphism. J. Clin. Oncol. 25:32-42. [DOI] [PubMed] [Google Scholar]
- 41.Victoria, J. G., D. J. Lee, B. R. McDougall, and W. E. Robinson. 2003. Replication kinetics for divergent type 1 human immunodeficiency viruses using quantitative SYBR Green I real-time polymerase chain reaction. AIDS Res. Hum. Retroviruses 19:865-874. [DOI] [PubMed] [Google Scholar]
- 42.Vodicka, M. A., W. C. Goh, L. I. Wu, M. E. Rogel, S. R. Bartz, V. L. Schweickart, C. J. Raport, and M. Emerman. 1997. Indicator cell lines for detection of primary strains of human and simian immunodeficiency viruses. Virology 233:193-198. [DOI] [PubMed] [Google Scholar]