

Abstract
The final step in the egress of herpes simplex virus (HSV) virions requires virion-laden vesicles to bypass cortical actin and fuse with the plasma membrane, releasing virions into the extracellular space. Little is known about the host or viral proteins involved. In the current study, we noted that the conformation of myosin Va (myoVa), a protein known to be involved in melanosome and secretory granule trafficking to the plasma membrane in melanocytes and neuroendocrine cells, respectively, was altered by 4 h after infection with HSV-1 such that an N-terminal epitope expected to be masked in its inactive state was rendered immunoreactive. Wild-type myoVa localized throughout the cytoplasm and to a limited extent in the nuclei of HSV-infected cells. Two different dominant negative myoVa molecules containing cargo-binding domains but lacking the lever arms and actin-binding domains colocalized with markers of the trans-Golgi network (TGN). Expression of dominant negative myoVa isoforms reduced secretion of HSV-1 infectivity into the medium by 50 to 75%, reduced surface expression of glycoproteins B, M, and D, and increased intracellular virus infectivity to levels consistent with increased retention of virions in the cytoplasm. These data suggest that myoVa is activated during HSV-1 infection to help transport virion- and glycoprotein-laden vesicles from the TGN, through the cortical actin, to the plasma membrane. We cannot exclude a role for myoVa in promoting fusion of these vesicles with the inner surface of the plasma membrane. These data also indicate that myoVa is involved in exocytosis in human epithelial cells as well as other cell types.
Herpes simplex virus (HSV) virions, like those of all herpesviruses, comprise a lipid envelope surrounding a layer of proteins called the tegument that covers the surface of the proteinaceous DNA-containing capsid. After assembly in the nucleus, herpes simplex virus nucleocapsids bud through the inner nuclear membrane to obtain an initial virion envelope. In the most widely accepted model of virion egress, the envelopes of nascent virions residing in the perinuclear space then fuse with the outer nuclear membrane, releasing the de-enveloped capsid into the cytoplasm (25). The now cytosolic capsid then buds into a membranous organelle in the cytoplasm to obtain its final envelope. The site of secondary envelopment where the final budding event occurs is believed to contain markers of the trans-Golgi network (TGN) (28) and would be expected to contain the full complement of virion envelope and tegument proteins. Cellular budding machinery would also be expected to be involved, such as that required for multivesicular body formation (1, 4). Other models of virion egress propose that nucleocapsids can exit the nucleus through an expanded nuclear pore (33, 34), or that the original virion envelope derived from the inner nuclear membrane is retained throughout egress (13). In the latter scenario, enveloped virions are incorporated into a vesicle derived from the outer nuclear membrane. This model is not consistent with the observations that a membrane protein encoded by UL34 along with its tegument binding partner encoded by UL31 are present in perinuclear virions but absent from extracellular virions (10, 20).
Regardless of the prior steps involved, all models of virion egress propose a similar final step; namely, that vesicles bearing one or more complete enveloped virions are transported to and fuse with the plasma membrane, releasing the virions into the extracellular space. The extent to which this final exit step contributes to the life cycle is virus and cell type dependent. For example, approximately 10% of total infectivity is secreted into the extracellular space of cultured Vero cells infected with herpes simplex virus 1 (HSV-1), whereas varicella-zoster virus remains almost completely cell associated in cultured MeWo cells (2). Despite general agreement that it occurs, the mechanism by which the virion-laden vesicles are transported to the plasma membrane has not been studied extensively. Myosin Va (myoVa) is a good candidate to mediate such transport because this motor has been shown to be important for the egress of melanosomes (36) and secretory granules (22) by facilitating their transport through the cortical filamentous actin (F-actin) located beneath the plasma membrane. The mechanism underlying this vesicle transport has been described as a “dual transport” process whereby cargoes brought to the cortex via microtubule-associated motors (such as kinesin) are subsequently transferred to cortical F-actin. Egress is then facilitated by myoVa (36) which moves along F-actin by processive (consecutive) “hand-over-hand” steps through a series of ATP hydrolysis reactions (for a review, see references 11 and 29). The motor consists of two heavy chains that dimerize to form duplicate N-terminal head domains (the actin- and nucleotide-binding region) extending into lever arms, a single coiled-coil dimerization tail domain, and two C-terminal globular tail domains (GTDs), which are believed to bind cargo.
The RNA encoding the tail domain of myoVa is alternatively spliced (24, 26), causing variations in different isomers encoded by exons B, D, and F (encoding 3, 27, and 25 amino acids, respectively). Cargo recognition can be regulated by interactions between various effector proteins, including Rab family GTPases and different myoVa exons. Specifically, exon F is required for interaction with Rab27a (35), exon B is necessary to interact with dynein light chain 2 (DLC2) (12, 31), and exon D is essential to bind Rab10 (21). Additionally, Rab11 is known to interact with class V myosins (3). Mutations in the human myoVa gene are the cause of Griscelli syndrome, which can manifest with severe neurological impairment and/or hypopigmentation, depending on the locus of the mutation (15, 18).
Given its known role in vesicle secretion, the current study investigates a potential role for myoVa in the secretion of HSV-1 virions. In the course of these studies, dominant negative myosin Va (DN-myoVa) constructs were used. These contained the exon-encoded portion of the myoVa tail domain as well as the entire (∼400 amino acids) GTD. To address the effects of alternative splicing, and by extension, involvement of different effectors in secretion, we used two DN-myoVa constructs. The melanocyte long tail (MCLT) construct contained exons D and F but lacked exon B, whereas the brain long tail (BRLT) construct contained exon B but not D or F.
MATERIALS AND METHODS
Viruses and cells.
Wild-type HSV-1 strain F [HSV-1(F)] and the HSV-1 UL13 deletion virus (R7355) were kindly provided by Bernard Roizman and have been described previously (7, 19). vRR1204 (a kind gift from Richard Roller) contains a lysine-to-alanine mutation at codon 220 in the US3 open reading frame that precludes its kinase activity (23). The K26 virus, kindly provided by Prashant Desai, encodes a VP26 capsid protein fused to green fluorescent protein (GFP) (5). Virus stocks were propagated and titers were determined on Vero cell monolayers, which were maintained in growth medium consisting of Dulbecco's modified Eagle's medium (DMEM) supplemented with 125 U/ml penicillin, 0.125 mg/ml streptomycin, and 10% newborn calf serum. After infection, cells were overlaid with 199V medium (DMEM supplemented with 1% newborn calf serum) and monitored for cytopathic effects. To release intracellular virus, the cells were lysed by freezing and thawing.
Plasmids and transfections.
Dominant negative myosin Va (DN-myoVa) constructs contained the exon-encoded portion of the myoVa tail domain as well as the globular tail domain (GTD). MCLT stands for melanocyte long tail. BRLT stands for brain long tail. Both isoforms were kindly provided by John Hammer III.
The dominant negative myoVa isoforms were excised from their supplied vectors by digestion at flanking Not-I restriction sites and ligated into Not-I digested pcDNA3 (Invitrogen). Monomeric red (mRED) was amplified by PCR analysis such that the amplicon ends contained HindIII/EcoRI sites and a mutation changing the stop codon to alanine. Primers used for this purpose were as follows: Fwd primer, 5′-AAAAAGCTTATGGCCTCCTCCGAGGACG-3′; Rev primer, 5′-AAAGAATTCCCTGCGGCGCCGGTGGAGTGGCGG-3′. The amplicon was ligated into the respective pcDNA3 constructs in frame with the 5′ ends of the MCLT and BRLT open reading frames. The genotypes of plasmid DNAs were verified by DNA sequencing.
For transfections, cells were transfected when they were approximately 90% confluent with approximately 1.6 μg plasmid DNA per well in 12-well dishes or 4.0 μg plasmid DNA per well in 6-well dishes using Lipofectamine 2000 (Invitrogen) according to the directions of the manufacturer.
Immunofluorescence.
At 28.5 h posttransfection, the cells were either mock infected or infected with 5.0 PFU/cell of HSV-1(F) or 10 PFU/cell with HSV-1 K26 as indicated. The cells were washed once with phosphate-buffered saline (PBS) and fixed in 3% paraformaldehyde (PFA) for 15 min, and autofluorescence was quenched by immersion in 50 mM NH4Cl for 15 min. In some experiments, the cells were permeabilized with 0.1% Triton X-100 for 2 min followed by a 10-min block in 10% human serum in PBS. When probing for surface expression, all the steps remained the same, except treatment with Triton X-100 was omitted. Goat anti-myosin Va (1:50; Santa Cruz Biotechnology), rabbit anti-myosin Va (1:50; Santa Cruz Biotechnology), mouse anti-ICP4 (1:100; Rumbaugh-Goodwin Institute for Cancer Research, Plantation, FL), rabbit anti-ICP8 (1:500; a kind gift from Bill Ruyechan), mouse anti-TGN38 (1:100; catalogue no. 610898; BD Transduction Labs), and mouse anti-Golgi 58K (1:100; catalogue no. ab6284; Abcam) primary antibodies were prepared in a 1% bovine serum albumin (BSA)-PBS solution. The mouse anti-gD (DL-6) monoclonal antibody (kindly provided by Roselyn Eisenberg and Gary Cohen) and rabbit anti-gB (kindly provided by David Johnson) were diluted in 1% BSA-PBS (1:100 dilution, or 1:200 in permeabilized cells for gD). The rabbit anti-gM antibody was diluted 1:500 in 1% BSA-PBS. All primary antibodies were incubated with the fixed cells for 30 to 60 min in a humidity chamber held at room temperature. After being washed three times in PBS, the cells were put through a reaction for 30 to 60 min in a humidity chamber with secondary antibodies fluorescein isothiocyanate (FITC)-conjugated anti-goat or FITC-conjugated anti-mouse and/or Texas Red or Cy5-conjugated anti-rabbit IgG (Jackson Laboratories) diluted 1:100 in 1% BSA-PBS. The coverslips were then rinsed three times in PBS and dipped in double-distilled water (ddH2O) before being mounted on glass slides with Mowiol plus 2.5% DABCO (1,4-diazabicyclo[2.2.2]octane) to prevent photobleaching. All digital images except for those used for surface glycoprotein quantification were taken using an IX70 Olympus confocal microscope and were processed with Adobe Photoshop software. Digital images for gD quantification were taken using a Zeiss Axio Imager.M1 fluorescent microscope and analyzed with IP Lab v3.65a software from Scanalytics.
Immunoblot analyses.
Mock-infected and HSV-1(F)-infected HEp-2 cells were washed once in PBS then lysed in 100 μl of 2× SDS-PAGE sample buffer (100 mM Tris-Cl [pH 6.8], 200 mM dithiothreitol, 4% SDS, 20% glycerol, 0.2% bromophenol blue). Lysates were sonicated and boiled for 5 min prior to loading. Samples were separated on a 10% polyacrylamide gel and transferred to a nitrocellulose membrane (30 V for 3.5 h at 4°C). The membrane was blocked in 5% milk-TBST (1× Tris-buffered saline plus 0.1% Tween) for 1 h at room temperature. Primary antibodies were goat anti-myoVa (1:200; Santa Cruz Biotechnology), rabbit anti-ICP8 (1:10,000), and mouse anti-lamin A/C (1:1,000; Santa Cruz Biotechnology). Bound primary antibody was detected by addition of anti-goat or anti-rabbit (1:2,000; Santa Cruz Biotechnology) horseradish peroxidase-conjugated secondary antibody diluted in 5% milk-TBST and visualized by enhanced chemiluminescence (Pierce) followed by exposure to X-ray film (Pierce).
Analyses of virion egress.
HeLa cells were transfected as described above. At 15 to 28 h posttransfection, approximately 0.1 PFU HSV-1(F) per cell in 1 ml 199V was added to each well. The cells were incubated for 1 h at 37°C with gentle agitation and subsequently washed three times with Versene (1× PBS and 0.5 mM EDTA) and once with a low-pH buffer (40 mM citric acid [pH 3.0], 10 mM KCl, 135 mM NaCl) to inactivate any residual infectitivity. This overlay was then removed, and 1.5 ml 199V was added to each well. Samples taken at the 0-h postinfection (hpi) time point were collected immediately, and the remaining samples were incubated at 37°C and 5% CO2 for another 20 h. To assess infectivity, parallel cultures of cells and/or overlaying medium was freeze-thawed to release virus, diluted serially, and subjected to a plaque assay on Vero cell monolayers. To check expression of the myoVa constructs, total cell lysates of infected HeLa cells beneath collected supernatants were collected and analyzed by immunoblotting as described above. Rabbit anti-RFP antibody (1 μg/ml; GenScript) was used to detect expression of mRED-DN-myoVa MCLT, mRED-DN-myoVa BRLT, and mRED.
Glycoprotein surface expression analysis.
HEp-2 cells expressing mRED, mRED-DN-myoVa BRLT, or mRED-DN-myoVa MCLT were infected with HSV-1 K26-GFP (MOI = 10 PFU/cell) or mock infected and fixed in 3% PFA at 17 hpi. Fixed cells were immunostained without permeabilization as described above. At least 50 infected cells expressing each construct type were digitally recorded using a Zeiss Imager.M1 Axio fluorescent microscope, and the images were analyzed to quantify surface glycoprotein expression using IP Lab v6.35a software from Scanalytics, Inc.
RESULTS
Immunoreactivity of myosin Va is increased during HSV-1 infection.
The intracellular environment of host cells is highly viscous and not conducive to free diffusion of HSV-1 particles (for a review, see reference 14). Myosin Va (myoVa) is an unconventional actin motor which, through ATP hydrolysis, moves cargo along polar actin fibers toward the plus (or barbed) end. In an effort to determine if HSV-1 utilizes this actin motor, we performed immunofluorescence assays to reveal the localization of myoVa in HSV-1(F)-infected and mock-infected HEp-2 cells. Cells were seeded onto coverslips in a 12-well dish and infected with wild-type HSV-1(F) or were mock infected. At 0, 4, 10, 16, and 22 h postinfection (hpi), cells were washed with PBS and fixed in 3% paraformaldehyde (PFA). Fixed cells were permeabilized in 0.1% Triton X-100, blocked in 10% human serum, and immunostained with goat anti-myoVa and rabbit anti-ICP8 antibodies.
As shown in Fig. 1, the amount of myoVa immunoreactivity increased extensively during the course of infection. This increase was first detectable at 4 hpi, and increased significantly by 16 hpi. MyoVa immunoreactivity localized primarily to the cytoplasm at these time points, although some cells demonstrated substantial intranuclear immunostaining. By 22 hpi, myoVa immunostaining localized primarily to infected nuclei. Similar results were obtained using Vero and HeLa cells (see Fig. S1 in the supplemental material).
FIG. 1.

Increased immunoreactivity of myosin Va during HSV-1 infection. HEp-2 cells were mock infected or infected with 5 PFU HSV-1(F) per cell and fixed at 4, 10, 16, and 22 hpi. Cells were washed with PBS and fixed in 3% paraformaldehyde (PFA) for 15 min and permeabilized with 0.1% Triton X-100. The fixed permeabilized cells were immunostained with goat anti-myoVa and rabbit anti-ICP8 antibodies. Single sections of immunostained cells were digitally collected with an Olympus confocal microscope. Green, anti-myosin Va; red, anti-ICP8.
Steady-state levels of myosin Va protein are unchanged in HSV-infected cells.
To determine whether or not the increased immunoreactivity reflected an increase in steady-state levels of myoVa, we analyzed the amount of myoVa protein in total cell lysates of mock-infected or HSV-1(F)-infected HeLa cells (collected over several time points) by immunoblot assay. We also probed the immunoblot with antibodies to ICP8 and lamin A/C as an infection control and loading control, respectively.
As shown in Fig. 2, similar levels of myoVa were detected in mock-infected and all HSV-1(F)-infected cell lysates collected at five different time points. Since phosphorylation can also play a role in epitope recognition and activation of myosins, we examined by immunofluorescence cells infected with a US3 kinase-dead mutant HSV-1 (23) as well as an HSV-1 mutant lacking the UL13 kinase (19). Fluorescent signals from the myoVa antibody were similar to that of HSV-1(F) in both mutant backgrounds (see Fig. S2 in the supplemental material), indicating that these viral kinases were not responsible for the conformational change in myoVa.
FIG. 2.
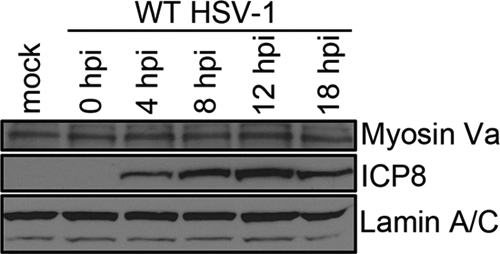
Unchanged steady-state levels of myosin Va protein in HSV-infected cells. Shown is an immunoblot showing levels of myosin Va in mock-infected and HSV-1(F)-infected HEp-2 total cell lysates. Cells were mock infected or infected with 5 PFU per cell HSV-1(F) and lysed in 2× denaturing buffer at 18 hpi, and proteins were separated on a polyacrylamide gel and transferred to nitrocellulose. The blot was probed with a goat anti-myoVa antibody and rabbit anti-ICP8 antibody. Immunoreactivity of goat anti-lamin B antibody served as a loading control.
A myosin Va epitope is exposed preferentially in HSV-infected cells.
The goat polyclonal antibody used as described above is directed against (sc-17707; Santa Cruz Biotechnology) an epitope within the N-terminal motor domain of myoVa (amino acids 117 to 137). This epitope is located near an N-terminal region that interacts with a C-terminal domain to hold the myosin molecule in the folded (inactive) conformation. We therefore asked whether a second myoVa antibody binding to an epitope that would be exposed in both the folded (inactive) and extended (active) conformations would produce similar levels of fluorescent intensity upon immunostaining mock- and HSV-infected cells. To address this possibility, HeLa cells were either mock infected or HSV-1(F) infected and coimmunostained with the original goat polyclonal antibody to myoVa and a second polyclonal antibody made in rabbits that binds to amino acids 981 to 1070 (sc-25726; Santa Cruz Biotechnology) in the myoVa tail dimerization domain. The cells were also immunostained with a mouse anti-ICP4 antibody to mark infected cell nuclei. The results are shown in Fig. 3.
FIG. 3.

A myosin Va epitope is exposed preferentially in HSV-infected cells. HeLa cells were mock infected or infected with HSV-1(F) (MOI = 5 PFU per cell). At 16 hpi the cells were washed in PBS, fixed in 3% PFA for 15 min, and permeabilized with 0.1% Triton X-100. Fixed cells were immunostained with rabbit anti-myosin Va (top rows) or goat anti-myosin Va (bottom row). Mouse antibody to ICP4 was used to mark infected cell nuclei. Red, myoVa with epitope in tail region; green, myoVa with epitope in N-terminal region; blue, ICP4.
Examination of the immunostained cells by confocal microscopy revealed that the tail-specific myoVa antibody similarly stained mock-infected and HSV-infected cells (red), whereas the previous myoVa antibody recognizing the N-terminal epitope (green) preferentially stained HSV-infected cells (also see Fig. 1). These data, taken together with data shown in Fig. 2, suggest that the increase in myoVa immunoreactivity results from the unmasking of the myoVa epitope rather than an increase in protein level. We speculate that this unfolding reflects activation of myoVa in HSV-infected cells.
DN-myoVa colocalizes primarily with the TGN in HSV-1(F)-infected cells.
Because HSV-1 virions are thought to transit into or near the trans-Golgi network (TGN) on their way to the extracellular space (28), we hypothesized that expression of a dominant negative myoVa motor would block HSV-1 virion secretion by inhibiting virion-laden TGN vesicles from passing through cortical F-actin. One prediction of this hypothesis is that dominant negative myoVa would localize at the TGN because the globular domain would bind to these vesicles and the absence of the actin-binding domain would preclude movement to the plasma membrane. To test this prediction, we transfected HeLa cells with the mRED-DN-myoVa constructs and compared localization of mRED-DN-myoVa to TGN and Golgi markers. The results are presented in Fig. 4.
FIG. 4.
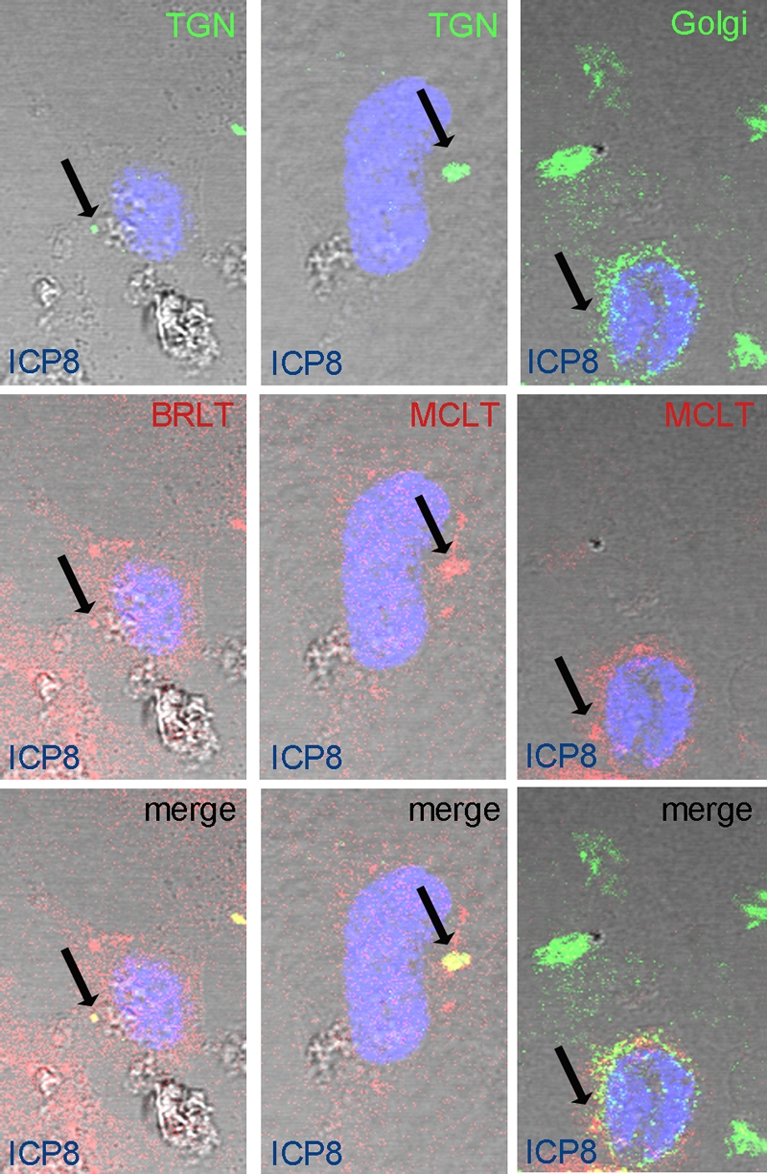
DN-MyoVa colocalizes with TGN but not Golgi markers in HSV-infected cells. HeLa cells were transfected with mRED-DN-myoVa BRLT (left column) or MCLT (center and right columns). At 23 h posttransfection the cells were infected with HSV-1(F) at an MOI of 5 PFU per cell. At 16 hpi the cells were washed with PBS, fixed in 3% PFA for 15 min, and permeabilized with 0.1% Triton X-100. Fixed cells were immunostained with mouse anti-TGN (top row, first and second columns) or mouse anti-Golgi markers (top row, right column). Rabbit anti-ICP8 was used to mark the infected cell nuclei. Red, DN-myoVa; green, TGN and Golgi; blue, ICP8. Arrows indicate sites of colocalization of the respective markers.
At high levels of PMT or in stacked images, the mRED and DNmyoVa constructs localized throughout the cytoplasm, making it difficult to discern the predominant localization of the proteins. At lower PMT settings, image slices taken with a confocal microscope showed that the mRED-DN-myoVa isoforms concentrated within cytoplasmic puncta that were not present in the mRED-expressing control cells. Some of these puncta colocalized with sites containing substantial immunostaining with the TGN38 marker. We did not see substantial colocalization between cytoplasmic foci bearing high concentrations of mRED-DN-myoVa isoforms and markers of the Golgi apparatus, although some overlap of the immunostaining was apparent even at low PMT settings. We found similar results in HEp-2 cells (see Fig. S3 in the supplemental material). Similar localization of DN-myoVa was observed in uninfected Hep2 cells (data not shown). The data indicate that the DN-myoVa isoforms localize primarily at the TGN and to a much lesser extent at the Golgi apparatus.
Myosin Va is required for efficient secretion of HSV-1 infectivity but not for production of intracellular infectivity.
To test our hypothesis that DN-myoVa impacts virion secretion, we transfected HeLa cells with the mRED-DN-myoVa isoforms and mRED control constructs and infected the cells the next day with HSV-1(F) at an MOI of 0.1 PFU per cell. After virus adsorption for 1 h at 37°C, the cells were washed three times with PBS and once with a low-pH buffer to neutralize any remaining extracellular infectivity. At this time and again 20 h later, we collected the 199V overlay medium containing secreted virions as well as total cell lysates of the infected cell monolayers beneath the 199V. At the same time points, the remaining cultures containing intracellular virus and medium overlay were subjected to freeze-thawing to release intracellular infectivity. The infectivity was then measured by plaque assay. The expression of the dominant negative and control constructs were monitored in parallel by immunoblot analysis.
As shown in Fig. 5A, the levels of the MCLT and BRLT isoforms were detected as high-molecular-weight bands (Mr of approximately 130,000) that reacted with rabbit anti-RFP antibody, indicating fusion of the fluorescent tag to the construct. MCLT ran slightly slower than BRLT, which was due to the additional exons contained in the MCLT transcript. A band indicative of mRED was detected in lanes containing lysates of the control cells.
FIG. 5.

(A) Transient expression of mRED-DN-myoVa and mRED control constructs in HeLa cells. HeLa cells were transfected with expression vectors for mRED-DN-myoVa-BRLT, mRED-DN-myoVa-MCLT, or mRED alone. Total cell lysates were collected at 0 and 20 hpi, separated on a polyacrylamide gel, and transferred to nitrocellulose. The blot was probed with a rabbit anti-RFP antibody. Signals from goat anti-lamin B antibody served as loading controls. (B) Myosin Va is required for efficient secretion of HSV-1 virions but not for intracellular production of infectivity. HeLa cells were transfected with expression vectors for mRED-DN-myoVa-BRLT, mRED-DN-myoVa-MCLT, or mRED alone. At 26 h posttransfection, cells were infected with HSV-1(F) at an MOI of 0.1. The resulting infectivity was determined by titration on Vero cells. The graph represents averages of data collected from three separate experiments.
At 0 hpi almost no residual HSV-1 infectivity was detected inasmuch as the highest titer was 1 PFU per cell (data not shown). As shown in Fig. 5B, at 20 hpi, secreted infectivity from the mRED-DN-myoVa BRLT- and MCLT-transfected cells was lower than the level of infectivity released from cells transfected with the mRED control vector (approximately 51% and 25% of the control, respectively). In contrast, the intracellular infectivity in the BRLT- and MCLT-expressing cells was higher than the infectivity from mRED-expressing control cells (approximately 183% and 172% of the control, respectively). Table S1 in the supplemental material lists the amount of infectivity determined in three separate trials. We interpret these data to indicate that the DN-myoVa blocks release of HSV-1 virions but not the accumulation of infectious virus within cells.
Expression of DN-myoVa results in decreased glycoprotein presentation on the surfaces of HSV-infected cells.
To determine if secretion of cargo other than virions required myoVa, we asked whether or not the mRED-DN-myoVa constructs could preclude viral glycoprotein transport to the cell surface. We therefore transfected HEp-2 cells with the dominant negative BRLT and MCLT isoforms or the mRED control vector. At 24 h posttransfection, the cells were mock infected or infected with 10 PFU per cell of HSV-1 K26-GFP, a virus that encodes a VP26 capsid protein fused to GFP. The cells were subjected to analysis by indirect immunofluorescence as indicated above except that they were fixed with PFA in the absence of detergent. To monitor glycoprotein expression at the cell surface, we put these unpermeabilized cells through a reaction with DL6 monoclonal antibody that recognizes a luminal epitope of HSV-1 envelope protein gD, gB antibody, or gM antibody and examined the cells by fluorescent microscopy. The results are presented in Fig. 6.
FIG. 6.

(A) Quantification of gB, gM, and gD surface expression in the presence of dominant negative myoVa. HEp-2 cells were transfected with mRED-DN-myoVa-BRLT, mRED-DN-myoVa-MCLT, or mRED control vector. Transfected cells were infected with HSV-1 K26-GFP (MOI = 10 PFU per cell) or mock infected at 26 h posttransfection. At 15.5 hpi the cells were washed with PBS, fixed in 3% PFA for 15 min, and blocked in 10% human serum. The fixed, unpermeabilized cells were immunostained with gD, gB, or gM antibody. K26-infected cells expressing the mRED control, mRED-DN-myoVa BRLT, or mRED-DN-myoVa MCLT were identified at random by intrinsic red and GFP fluorescence, and the amount of glycoprotein-specific fluorescence was quantified using a Zeiss Imager.M1 Axio fluorescent microscope and IP Lab v3.65a software. At least 50 infected cells expressing each construct were analyzed. Mean fluorescence is the sum of measured fluorescence divided by the area. The mRED-DN-myoVa BRLT- and MCLT-expressing cells were determined by Student's t test (P < 0.05) to have less surface expression of gB, gM, and gD compared to that of mRED control cells. (B) Expression of DN-myoVa decreases glycoprotein immunoreactivity on the surfaces of HSV-infected HEp-2 cells. Example of K26-infected cells transfected with DN-myoVa or the mRED control demonstrating decreased immunoreactivity with gD antibody in DN-myoVa-expressing cells. White arrows indicate cells of interest. Similar results were seen for gM and gB staining (data not shown). Green, HSV-1; blue, gD; red, DN-myoVa or mRED control.
Cells infected with HSV-1 K26-GFP were indicated by the presence of intrinsic bright GFP-mediated fluorescence in their nuclei as described previously (5). Uninfected cells did not display notable gD-specific fluorescence, as expected. In contrast, large amounts of glycoprotein-specific immunofluorescence were noted in unpermeabilized cells infected with the K26-GFP virus and expressing the mRED control. In infected cells expressing the mRED-DN-myoVa constructs, considerably less glycoprotein-specific immunoreactivity was detected on the cell surface.
Glycoprotein surface expression was quantified in infected cells expressing mRED or the mRED-DN-myoVa constructs by analysis of digital images of at least 50 infected cells (i.e., exhibiting GFP fluorescence) expressing each mRED or a mRED-DN-myoVa construct using IP Lab v3.65a software. The quantification data are presented in Fig. 6A and indicate significantly less surface expression of gB, gM, and gD on cells expressing either mRED-DN-myoVa construct compared to those expressing mRED alone (P < 0.05 by Student's t test). To demonstrate that DN-myoVa expression did not preclude expression we repeated this experiment under similar conditions but permeabilized the cells with detergent and probed with the gD antibody. We found readily detectable levels of gD (see Fig. S4 in the supplemental material), gB, and gM (data not shown) within the permeabilized infected mRED-DN-myoVa-expressing cells. Taken together, the data suggest that myoVa plays a role in surface expression of viral glycoproteins.
DISCUSSION
MyoVa has been implicated in transport of secretory granules and melanosomes to the cell surfaces of neuroendocrine cells (6, 22) and melanocytes, respectively (36). Through the use of two different dominant negative myosin Va (DN-myoVa) constructs lacking the actin-binding domain but containing the exon-encoded portion of the myoVa tail domain and the C-terminal cargo-binding domain, we have now shown that myoVa plays a role in exocytosis of HSV virions and glycoproteins in two different epithelial cell lines (specifically HEp-2 and HeLa cells).
HSV alters the conformation of myoVa as indicated by a substantial increase in immunostaining with a myoVa-specific polyclonal antibody starting at about 4 hpi with HSV-1 (Fig. 1). According to the manufacturer, this antibody recognizes an epitope located between amino acids 260 to 310. Previous work has shown that myoVa exists in two conformations, folded or extended (32), and that the folded (inactive) conformation is facilitated by an interaction between the C-terminal globular tail domain (GTD) and amino acids 117 to 137 of the N-terminal motor domain (27). Based on this information, we postulate that the epitope between amino acids 260 to 310 is masked in the nonactive conformation as in uninfected cells, whereas the myoVa antibody preferentially binds to the active (extended) myoVa in infected cells. Thus, the increased fluorescence observed in infected cells stained with this antibody suggests that myoVa activity is augmented during HSV-1 infection. This activation might play to the advantage of the virus inasmuch as substantial transport activity is likely required to support virion secretion.
In a previous study, treatment of HSV-infected cells with BDM (2,3-butanedione monoxime) decreased secretion of infectivity into the medium by 20- to 50-fold and decreased production of intracellular virus by 2-fold. BDM is a potent inhibitor of myosin II but also affects calcium flux and many other proteins that might affect HSV egress (17, 30). The current study specifically implicates myoVa in virus secretion inasmuch as expression of dominant negative myoVa reduced secretion of infectivity up to 4-fold while titration of extracellular plus intracellular infectivity revealed slightly elevated levels. This observation indicates that myoVa is involved in transporting infectious virions to the extracellular space, whereas earlier steps in the virion assembly pathway in which infectious virus is generated do not require myoVa. A role in the final egress step of the virion egress pathway is consistent with myoVa's role in the final exocytic step allowing transport of melanosomes (36) and secretory granules (22) into the extracellular space.
To address the effects of alternative splicing, we used two DN-myoVa constructs. The melanocyte long tail (MCLT) construct contained exons D and F but lacked exon B, whereas the brain long tail (BRLT) construct contained exon B but not D or F. Because both constructs affected virion secretion similarly, we conclude that regions of myoVa required for interaction with Rab27a (encoded by exon F) (35), dynein light chain 2 (encoded by exon B) (12, 31), and Rab10 (encoded by exon D) (21) were dispensable for the dominant negative effects. Thus, either these effectors were not involved in myoVa-dependent virion secretion or their involvement was redundant.
It was of interest to us that surface expression of at least gB, gD, and gM at the cell surface was significantly (P < 0.05) reduced by the expression of DN myoVa isoforms, suggesting that vesicles bearing viral glycoproteins also utilize myoVa to facilitate their transport to the plasma membrane. This suggests that the final step of virion transport is functionally similar to that of glycoprotein transport to the cell surface, at least in the cell types tested. The observation that the dominant negative proteins colocalized with TGN markers suggest that myoVa mediates primarily transport from the TGN to the plasma membrane. This makes sense in light of the fact that HSV virions are known to transit near or through the TGN on their way to the extracellular space (28).
We also noted substantial amounts of myoVa in the nuclei of infected cells, especially at late times after infection (Fig. 1). MyoVa in the nucleus has also been observed in cells infected with pseudorabies virus, a herpesvirus of swine (8). While it is possible that myoVa plays a role in active intranuclear movement of capsids (9), the production of substantial intracellular infectivity in cells expressing the DN-myoVa suggests very minor or no effect on transport before the final secretion step when virions are released from the plasma membrane.
The current work supports a model in which myoVa is activated during HSV-1 infection to enhance the transport of secretory vesicles bearing virions and viral glycoproteins past cortical actin to eventually fuse with the plasma membrane. The study does not rule out roles for other nonmuscle myosins and does not exclude other roles for myoVa in the final secretion step. For example, the observation that myosin IIa plays a role in stabilizing the fusion pore that links the interior of the secretory vesicle to the extracellular space (16) would conceivably also enhance virion secretion. From a heuristic standpoint, defining the roles of nonmuscle myosins in virion secretion is expected to be a useful approach to study how these proteins perform their functions in uninfected as well as infected cells.
Supplementary Material
Acknowledgments
We thank Roselyn Eisenberg and Gary Cohen for the antibody to gD, David Johnson for the antibody to gB, Bill Ruyechan for the ICP8 antibody, Prashant Desai for the K26 virus, and John Hammer III for the dominant negative myoVa constructs.
These studies were supported by grants R01 AI52341 and T32 AI007618 from the National Institutes of Health.
Footnotes
Published ahead of print on 14 July 2010.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Calistri, A., P. Sette, C. Salata, E. Cancellotti, C. Forghieri, A. Comin, H. Gottlinger, G. Campadelli-Fiume, G. Palu, and C. Parolin. 2007. Intracellular trafficking and maturation of herpes simplex virus type 1 gB and virus egress require functional biogenesis of multivesicular bodies. J. Virol. 81:11468-11478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carpenter, J. E., E. P. Henderson, and C. Grose. 2009. Enumeration of an extremely high particle-to-PFU ratio for varicella-zoster virus. J. Virol. 83:6917-6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Correia, S. S., S. Bassani, T. C. Brown, M. F. Lise, D. S. Backos, A. El-Husseini, M. Passafaro, and J. A. Esteban. 2008. Motor protein-dependent transport of AMPA receptors into spines during long-term potentiation. Nat. Neurosci. 11:457-466. [DOI] [PubMed] [Google Scholar]
- 4.Crump, C. M., C. Yates, and T. Minson. 2007. Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J. Virol. 81:7380-7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desai, P., and S. Person. 1998. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J. Virol. 72:7563-7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eichler, T. W., T. Kogel, N. V. Bukoreshtliev, and H. H. Gerdes. 2006. The role of myosin Va in secretory granule trafficking and exocytosis. Biochem. Soc. Trans. 34:671-674. [DOI] [PubMed] [Google Scholar]
- 7.Ejercito, P. M., E. D. Kieff, and B. Roizman. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J. Gen. Virol. 2:357-364. [DOI] [PubMed] [Google Scholar]
- 8.Feierbach, B., S. Piccinotti, M. Bisher, W. Denk, and L. W. Enquist. 2006. Alpha-herpesvirus infection induces the formation of nuclear actin filaments. PLoS Pathog. 2:e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forest, T., S. Barnard, and J. D. Baines. 2005. Active intranuclear movement of herpesvirus capsids. Nat. Cell Biol. 7:429-431. [DOI] [PubMed] [Google Scholar]
- 10.Fuchs, W., B. G. Klupp, H. Granzow, N. Osterrieder, and T. C. Mettenleiter. 2002. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J. Virol. 76:364-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hammer, J. A., III, and X. Wu. 2007. Slip sliding away with myosin V. Proc. Natl. Acad. Sci. U. S. A. 104:5255-5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hódi, Z., A. L. Nemeth, L. Radnai, C. Hetenyi, K. Schlett, A. Bodor, A. Perczel, and L. Nyitray. 2006. Alternatively spliced exon B of myosin Va is essential for binding the tail-associated light chain shared by dynein. Biochemistry 45:12582-12595. [DOI] [PubMed] [Google Scholar]
- 13.Johnson, D. C., and P. G. Spear. 1982. Monensin inhibits the processing of herpes simplex virus glycoproteins, their transport to the cell surface, and the egress of virions from infected cells. J. Virol. 43:1101-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lyman, M. G., and L. W. Enquist. 2009. Herpesvirus interactions with the host cytoskeleton. J. Virol. 83:2058-2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ménasché, G., C. H. Ho, O. Sanal, J. Feldmann, I. Tezcan, F. Ersoy, A. Houdusse, A. Fischer, and B. G. de Saint. 2003. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J. Clin. Invest. 112:450-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neco, P., C. Fernandez-Peruchena, S. Navas, L. M. Gutierrez, G. A. de Toledo, and E. Ales. 2008. Myosin II contributes to fusion pore expansion during exocytosis. J. Biol. Chem. 283:10949-10957. [DOI] [PubMed] [Google Scholar]
- 17.Ostap, E. M. 2002. 2,3-Butanedione monoxime (BDM) as a myosin inhibitor. J. Muscle Res. Cell Motil. 23:305-308. [DOI] [PubMed] [Google Scholar]
- 18.Pastural, E., F. J. Barrat, R. Dufourcq-Lagelouse, S. Certain, O. Sanal, N. Jabado, R. Seger, C. Griscelli, A. Fischer, and B. G. de Saint. 1997. Griscelli disease maps to chromosome 15q21 and is associated with mutations in the myosin-Va gene. Nat. Genet. 16:289-292. [DOI] [PubMed] [Google Scholar]
- 19.Purves, F. C., and B. Roizman. 1992. The UL13 gene of herpes simplex virus 1 encodes the functions for posttranslational processing associated with phosphorylation of the regulatory protein alpha 22. Proc. Natl. Acad. Sci. U. S. A. 89:7310-7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reynolds, A. E., E. G. Wills, R. J. Roller, B. J. Ryckman, and J. D. Baines. 2002. Ultrastructural localization of the HSV-1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 76:8939-8952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roland, J. T., L. A. Lapierre, and J. R. Goldenring. 2009. Alternative splicing in class V myosins determines association with Rab10. J. Biol. Chem. 284:1213-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rudolf, R., T. Kogel, S. A. Kuznetsov, T. Salm, O. Schlicker, A. Hellwig, J. A. Hammer III, and H. H. Gerdes. 2003. Myosin Va facilitates the distribution of secretory granules in the F-actin rich cortex of PC12 cells. J. Cell Sci. 116:1339-1348. [DOI] [PubMed] [Google Scholar]
- 23.Ryckman, B. J., and R. J. Roller. 2004. Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J. Virol. 78:399-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seperack, P. K., J. A. Mercer, M. C. Strobel, N. G. Copeland, and N. A. Jenkins. 1995. Retroviral sequences located within an intron of the dilute gene alter dilute expression in a tissue-specific manner. EMBO J. 14:2326-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stackpole, C. W. 1969. Herpes-type virus of the frog renal adenocarcinoma. I. Virus development in tumor transplants maintained at low temperature. J. Virol. 4:75-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strobel, M. C., P. K. Seperack, N. G. Copeland, and N. A. Jenkins. 1990. Molecular analysis of two mouse dilute locus deletion mutations: spontaneous dilute lethal20J and radiation-induced dilute prenatal lethal Aa2 alleles. Mol. Cell. Biol. 10:501-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thirumurugan, K., T. Sakamoto, J. A. Hammer, J. R. Sellers, and P. J. Knight. 2006. The cargo-binding domain regulates structure and activity of myosin 5. Nature 442:212-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turcotte, S., J. Letellier, and R. Lippe. 2005. Herpes simplex virus type 1 capsids transit by the trans-Golgi network, where viral glycoproteins accumulate independently of capsid egress. J. Virol. 79:8847-8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vale, R. D. 2003. Myosin V motor proteins: marching stepwise towards a mechanism. J. Cell Biol. 163:445-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Leeuwen, H., G. Elliott, and P. O'Hare. 2002. Evidence of a role for nonmuscle myosin II in herpes simplex virus type 1 egress. J. Virol. 76:3471-3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wagner, W., E. Fodor, A. Ginsburg, and J. A. Hammer. 2006. The binding of DYNLL2 to myosin Va requires alternatively spliced exon B and stabilizes a portion of the myosin's coiled-coil domain. Biochemistry 45:11564-11577. [DOI] [PubMed] [Google Scholar]
- 32.Wang, F., K. Thirumurugan, W. F. Stafford, J. A. Hammer, P. J. Knight, and J. R. Sellers. 2004. Regulated conformation of myosin V. J. Biol. Chem. 279:2333-2336. [DOI] [PubMed] [Google Scholar]
- 33.Wild, P., M. Engels, C. Senn, K. Tobler, U. Ziegler, E. M. Schraner, E. Loepfe, M. Ackermann, M. Mueller, and P. Walther. 2005. Impairment of nuclear pores in bovine herpesvirus 1-infected MDBK cells. J. Virol. 79:1071-1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wild, P., C. Senn, C. L. Manera, E. Sutter, E. M. Schraner, K. Tobler, M. Ackermann, U. Ziegler, M. S. Lucas, and A. Kaech. 2009. Exploring the nuclear envelope of herpes simplex virus 1-infected cells by high-resolution microscopy. J. Virol. 83:408-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu, X. S., K. Rao, H. Zhang, F. Wang, J. R. Sellers, L. E. Matesic, N. G. Copeland, N. A. Jenkins, and J. A. Hammer III. 2002. Identification of an organelle receptor for myosin-Va. Nat. Cell Biol. 4:271-278. [DOI] [PubMed] [Google Scholar]
- 36.Wu, X., B. Bowers, K. Rao, Q. Wei, and J. A. Hammer III. 1998. Visualization of melanosome dynamics within wild-type and dilute melanocytes suggests a paradigm for myosin V function in vivo. J. Cell Biol. 143:1899-1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.