

Abstract
Nevirapine forms the mainstay of our efforts to curtail the pediatric AIDS epidemic through prevention of mother-to-child transmission of HIV-1. A key limitation, however, is the rapid selection of HIV-1 strains resistant to nevirapine following the administration of a single dose. This rapid selection of resistance suggests that nevirapine-resistant strains preexist in HIV-1 patients and may adversely affect outcomes of treatment. The frequencies of nevirapine-resistant strains in vivo, however, remain poorly estimated, possibly because they exist as a minority below current assay detection limits. Here, we employ stochastic simulations and a mathematical model to estimate the frequencies of strains carrying different combinations of the common nevirapine resistance mutations K103N, V106A, Y181C, Y188C, and G190A in chronically infected HIV-1 patients naïve to nevirapine. We estimate the relative fitness of mutant strains from an independent analysis of previous competitive growth assays. We predict that single mutants are likely to preexist in patients at frequencies (∼0.01% to 0.001%) near or below current assay detection limits (>0.01%), emphasizing the need for more-sensitive assays. The existence of double mutants is subject to large stochastic variations. Triple and higher mutants are predicted not to exist. Our estimates are robust to variations in the recombination rate, cellular superinfection frequency, and the effective population size. Thus, with 107 to 108 infected cells in HIV-1 patients, even when undetected, nevirapine-resistant genomes may exist in substantial numbers and compromise efforts to prevent mother-to-child transmission of HIV-1, accelerate the failure of subsequent antiretroviral treatments, and facilitate the transmission of drug resistance.
Mother-to-child transmission of HIV infection has triggered a pediatric AIDS epidemic in many parts of the world. In 2008, 2.1 million children under the age of 15 years were living with HIV infection worldwide, 430,000 children were newly infected, and 280,000 children died due to HIV/AIDS (62). Most HIV infections in children occur due to the transmission of the virus from the mothers during pregnancy or delivery or subsequently through breast-feeding (9). Nevirapine (NVP), a nonnucleoside reverse transcriptase inhibitor (NNRTI), forms the mainstay of our efforts to prevent mother-to-child transmission of HIV-1 infection in the developing world. A single dose of NVP (SDNVP) to the mother at the onset of labor and to the infant within 72 h of birth lowered the risk of mother-to-child transmission by 41% (22). A key limitation of SDNVP, however, is the rapid selection of NVP resistance mutations (15). For instance, after 6 to 8 weeks of SDNVP, 87% of mothers and infants were found to harbor NVP resistance mutations by an allele-specific assay (34). NVP resistance mutations decay with time following their selection after SDNVP but may persist in some patients for several years (15, 17, 34, 44). NVP resistance mutations may also be archived in latently infected cells (64), which have half-lives of ∼44 months (55). The rapid selection and persistence of NVP resistance mutations following SDNVP may compromise the efficacy of SDNVP during subsequent pregnancies, may adversely impact outcomes of subsequent antiretroviral therapies containing NNRTIs, and may result in the transmission of drug-resistant strains of HIV-1 (18, 27, 33, 36, 38, 60).
HIV exists in infected individuals as a quasispecies, a collection of related yet distinct viral genomes (7, 57). Specific mutations in the viral genome are responsible for resistance to current antiretroviral drugs (6, 25). Thus, genomes carrying drug resistance mutations may exist as part of the viral quasispecies in infected individuals prior to the onset of therapy (31, 41). Mutations typically carry a fitness penalty (3). Consequently, drug-resistant genomes may exist as a minority, with the fitter, susceptible genomes dominant in the quasispecies (19, 24, 39, 56). Resistant genomes have a fitness advantage in the presence of drugs and, consequently, are rapidly selected following the onset of therapy, increasing the risk of the failure of therapy (19, 24, 39, 56). The rapid selection of NVP resistance mutations following SDNVP implies that these mutations may exist as a minority quasispecies in treatment-naïve individuals. Indeed, the risk of failure of NNRTI-based antiretroviral therapy increased substantially when initiated after administration of SDNVP (27, 33, 60). In particular, among 60 women who started NVP-based antiretroviral therapy within 6 months of receiving either SDNVP or placebo, 41.7% of the women who received SDNVP and none who received placebo had virologic failure (P < 0.001) (33).
Knowledge of the frequencies of minority NVP resistance mutations is essential for accurate assessment of the risk of mother-to-child transmission, the outcomes of NNRTI-based antiretroviral therapies, and the epidemiology of the transmission of drug resistance (38). Indeed, substantial efforts are ongoing to assess the frequencies and the prevalence of minority NVP resistance mutations in HIV-infected individuals using novel, highly sensitive detection techniques (14, 16, 23, 44). For instance, in 50 South African women without any detectable NVP resistance mutations before SDNVP, 10 were detected with the common K103N NVP resistance mutation following SDNVP, using a standard population-based sequencing analysis which detects mutations present in frequencies of >20%, whereas when a sensitive real-time reverse transcription-PCR (RT-PCR) technique with an assay detection limit of ∼0.1% was used, an additional 16 women were found to harbor the K103N mutation (23). Recent advances in detection techniques have thus provided improved estimates of the frequencies and the prevalence of drug resistance mutations. Yet, current techniques may not be able to assess fully the impact of drug resistance mutations. The population of infected cells within an HIV-1 patient is ∼107 to 108 (20). Thus, even with the best assay sensitivities of ∼0.01% (43), genomes that infect fewer than 103 to 104 cells would remain undetected and yet may have a substantial impact on treatment outcome. Alternative ways of estimating the frequencies of minority NVP resistance mutations are therefore necessary. Here, we employ detailed simulations of the within-host evolution of HIV-1 and a mathematical model of HIV-1 dynamics to estimate the frequencies of NVP resistance mutations in a chronically infected individual naïve to antiretroviral therapy.
MATERIALS AND METHODS
Estimation of the relative fitness of genomes by analysis of competitive growth assays.
We consider the experiments of Collins et al. (8), who performed competitive growth assays in which cells were exposed at low multiplicity of infection (MOI) simultaneously to the wild-type virus and a mutant, i, and the time evolution of the ratio of the mutant and wild-type viruses was measured. The experiments proceeded over a short duration (∼6 days), when viral populations grow predominantly by replication; mutation and recombination are negligible. The time evolutions of the populations of the wild-type and mutant virions are then governed by the following equations:
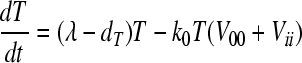 |
(1a) |
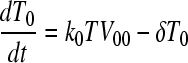 |
(1b) |
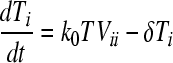 |
(1c) |
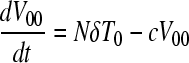 |
(1d) |
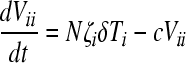 |
(1e) |
Here, uninfected cells, T, proliferate in proportion to their abundance with a rate constant, λ, and die with a rate constant, dT. Uninfected cells are also infected by wild-type virions, V00, and mutant virions, Vii, with the second-order infection rate constant k0. We denote by T0 and Ti cells infected with the wild-type and the mutant genomes, respectively. The latter cells die with a first-order rate constant, δ. Cells infected with the wild type produce N progeny virions per cell, the viral burst size. Cells infected with the mutant produce a smaller number of virions, Nζi, where ζi (0 ≤ ζi ≤ 1) is the relative fitness of genome i. Free virions are lost with the first-order rate constant c.
We solve the above equations using parameter values representative of HIV-1 infection in vitro (2, 58, 61): λ = 0.624 day−1, dT = 0.018 day−1, δ = 1.44 day−1, c = 3.5 day−1, N = 103, and k0 = 10−8 day−1. At time t = 0, 5 × 105 uninfected cells were exposed to virions (MOI = 0.001), of which an experimentally determined fraction contained mutant genomes (8). The ratio Vii/V00 yields the mutant/wild-type ratio measured experimentally. We fit model predictions of the time evolution of Vii/V00 to the experimental data using ζi as an adjustable parameter for each of the viable NVP-resistant mutants. The fits were performed using the Levenberg-Marquardt algorithm in MATLAB.
Simulations of viral evolution. (i) Representation and initialization.
We consider a fixed population, C, of uninfected cells exposed to a pool of V virions. Each virion consists of two RNA genomes represented by bit-strings of length L = 5 each. The five positions represent the NVP resistance loci 103, 106, 181, 188, and 190. Infection begins with a pool of identical homozygous virions, where each position has the wild-type allele.
(ii) Infection.
We let infections occur in discrete generations. In any generation, each cell is infected by M virions drawn from the viral pool, where M follows a predetermined distribution of the frequency of multiple infections of cells (see below). Virions are chosen for infection randomly from the viral pool.
(iii) Reverse transcription.
Following infection, reverse transcription converts the viral RNA in each infecting virion to proviral DNA of length L. Here, mutation and recombination introduce genomic variations. We implement recombination first as follows. At the first position, we choose the nucleotide from one of the two strands with a probability of 0.5. At the next position, we choose the nucleotide from the same strand if an even number of crossovers, or template switches, occurs between the two positions. If l is the number of nucleotides between positions 1 and 2, then the probability that even crossovers occurs between the two positions is Peven(l) = exp(−ρl)cosh(ρl), where ρ is the recombination rate expressed as the number of crossovers per site per replication (61). For instance, positions 1 and 2 here correspond to amino acid positions 103 and 106, respectively. The number of nucleotides, l, between these positions is assumed to be 9, three times the corresponding amino acid separation. We generate a random number from a uniform distribution between 0 and 1. If the number is smaller than Peven(l), then we choose the nucleotide at the second position from the same strand as the first. If not, we choose the nucleotide from the other strand. We repeat this process for all the five positions and generate a recombinant genome. Next, we mutate the recombinant genome at each position with a probability, μ, per site per replication and obtain the resulting proviral DNA. We repeat the process for all virions infecting cells.
(iv) Viral production.
The proviral DNAs are then transcribed into viral RNAs, which are assorted randomly into pairs and released as new virions. We assume that the production of virions from a cell depends on the relative fitness of genomes infecting the cell. We consider a cell infected with M proviral DNA, numbered j = 1, 2,…, M, with fitness ζj. The cell then produces Pζ progeny virions, where 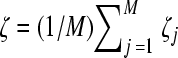 is the average fitness of the proviral DNA. Viral RNAs for inclusion in the progeny virions are picked with a probability equal to the relative fitness of genomes within the cell. Thus, provirus j is chosen with a probability
is the average fitness of the proviral DNA. Viral RNAs for inclusion in the progeny virions are picked with a probability equal to the relative fitness of genomes within the cell. Thus, provirus j is chosen with a probability 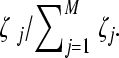 This process of viral production is repeated for all cells. In effect, we ensure that within a cell, fitter genomes are chosen more frequently than less-fit genomes during viral production and that across cells, fitter virions are produced in larger numbers. The progeny virions form the viral pool for infection of the next generation of uninfected cells.
This process of viral production is repeated for all cells. In effect, we ensure that within a cell, fitter genomes are chosen more frequently than less-fit genomes during viral production and that across cells, fitter virions are produced in larger numbers. The progeny virions form the viral pool for infection of the next generation of uninfected cells.
(v) Calculations of viral diversity and genome frequencies.
In each generation, we compute the average diversity, 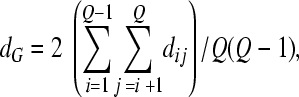 of the Q proviruses present in that generation and their average divergence from the founder sequence,
of the Q proviruses present in that generation and their average divergence from the founder sequence, 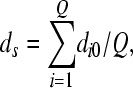 where dij is the Hamming distance per position between genomes i and j and 0 represents the founder sequence. The Hamming distance between two genomes is the number of positions at which the two genomes differ. We also compute the frequency of each of the S mutant genomes among the proviral DNA,
where dij is the Hamming distance per position between genomes i and j and 0 represents the founder sequence. The Hamming distance between two genomes is the number of positions at which the two genomes differ. We also compute the frequency of each of the S mutant genomes among the proviral DNA,  , where Qi is the number of proviral DNAs of type i∈(0, 1, 2,…, S), and among the viral RNAs,
, where Qi is the number of proviral DNAs of type i∈(0, 1, 2,…, S), and among the viral RNAs, 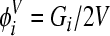 , where Gi is the number of viral RNA genomes of type i and V is the total viral population. We average the frequencies over 1,000 generations after viral diversity reaches equilibrium (see text). Furthermore, 600 realizations of the infection process are averaged to obtain the expected values of φiC and φiV.
, where Gi is the number of viral RNA genomes of type i and V is the total viral population. We average the frequencies over 1,000 generations after viral diversity reaches equilibrium (see text). Furthermore, 600 realizations of the infection process are averaged to obtain the expected values of φiC and φiV.
The simulations are implemented using a computer program written in C++.
(vi) Simulation parameters.
We employ the following parameter values. Each infected cell may produce ∼102 to 104 virions (5, 20, 21), only a few of which may be infectious (10, 11, 45). Recent studies estimate the median basic reproductive ratio of HIV-1 in vivo to be 6 to 8, indicating that each infected cell would produce at least 6 to 8 virions that successfully infect target cells (49, 59). We therefore let each cell produce a maximum of P = 10 infectious progeny virions and let the number decrease as the fitness of genomes infecting the cell decreases (see above). We fix the substitution rate at μ = 3 × 10−5 substitutions per site per replication, which is approximately the rate found by Mansky and Temin (29, 35), and we fix the recombination rate at ρ = 8.3 × 10−4 crossovers per site per replication (32, 61). We employ the relative fitness, ζi, of each of the S resistant genomes obtained from our analysis of growth competition assays (see Fig. 2).
Estimates of the within-host effective population size of HIV-1 vary over a wide range: 103 to 106 cells (1, 30, 40, 50, 52-54). Previous simulations similar to our present simulations capture observations of HIV-1 diversification in vivo with effective population sizes, C, that span this wide range. In one study, assuming that each cell is infected with M = 3 virions, following observations of infected splenocytes in two patients (28), a result in which C is ∼103 cells captured observations of the time evolution of HIV-1 diversity in patients over extended durations (∼10 years) following seroconversion (63). In another study, assuming multiple infections of cells are rare, as observed in peripheral blood mononuclear cells in four chronically infected patients (26), a much larger value of C (>105) captured experimental observations of the frequency of the least prevalent haplotype in a two-locus/two-allele model (52). We therefore perform simulations with both combinations of values of C and M: low C = 103 to 104 cells and high M = 3 per cell, and high C = 105 and low M, the latter M following a distribution in which 78% of the cells are singly infected, 18% doubly infected, 3% triply infected, and the rest quadruply infected. This latter distribution is consistent with the recently suggested lower limit of 8 to 10% for the frequency of coinfection (51) and is similar to the experimental distribution (26).
Mathematical model of HIV-1 dynamics and evolution.
We constructed a deterministic model of HIV dynamics to predict the frequencies of NVP-resistant genomes when the effective population size is large and the frequency of multiple infections of cells is small. We recognize that genomes of a particular kind are produced by replication from cells they infect and by mutation and recombination of related genomes. Genomes are lost by clearance and by mutation to and recombination with other genomes. The equations below then describe the consequent time evolution of the populations of different genomes:
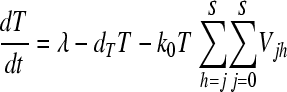 |
(2) |
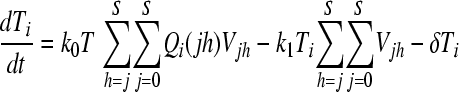 |
(3) |
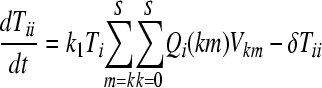 |
(4a) |
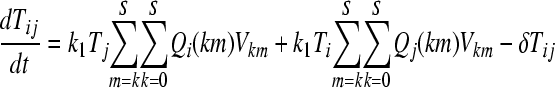 |
(4b) |
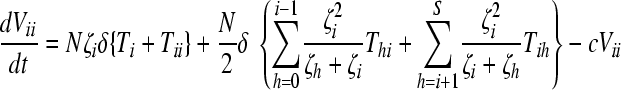 |
(5a) |
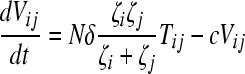 |
(5b) |
Here, we number the different kinds of genomes from 0 to S = 31, with 0 representing the wild type, carrying no NVP resistance mutations, and S representing the genome carrying all five NVP resistance mutations. Vjh is the population of virions containing genomes j and h, where j, h∈{0, 1,…, S} and j ≤ h. Uninfected CD4+ T cells, T, are produced at a constant rate, λ, die at rate dT, and are lost due to infection by free virions, which occur with the rate constant k0 (equation 2). Following infection by a virion Vjh, reverse transcription yields a proviral genome i∈{0, 1,…, S} with a probability Qi(jh). Qi(jh) depends on the mutation rate, μ, the recombination rate, ρ, and the separations between loci, and has been described elsewhere (2). Cells Ti, productively infected with a single provirus i, are produced by infection of T by free virions followed by mutation and recombination events that yield genome i and are lost by further infections, which occur with the rate constant k1, or by death at rate δ (equation 3). k1 < k0 because of virus-induced CD4 downmodulation, which lowers the susceptibility of infected cells to further infections (4, 46). Infection of Ti yields doubly infected cells of two kinds: Tii, carrying two copies of genome i; and Tij, carrying a copy each of distinct genomes i and j (equation 4). Tij cells are produced additionally by infection of Tj. For simplicity, we ignore more than two infections of cells.
Cells Ti and Tii produce homozygous virions Vii (equation 5). We assume that a cell Ti produces Ni = Nζi progeny virions, where 0 ≤ ζi ≤ 1 is the relative fitness of genome i. ζi = 1 if i = 0, the wild type. Furthermore, we assume that a cell Tij produces Nij = (Ni + Nj)/2 progeny virions, comprising homozygous virions Vii and Vjj and heterozygous virions Vij in fractions 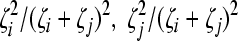 , and
, and 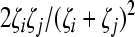 , respectively. The fractions follow from the assumption that in the cell Tij excess genomes of types i and j are produced in proportion to Ni and Nj and are randomly assorted into progeny virions. Note that this description of viral production mimics our simulations above. Virions are cleared at the rate c.
, respectively. The fractions follow from the assumption that in the cell Tij excess genomes of types i and j are produced in proportion to Ni and Nj and are randomly assorted into progeny virions. Note that this description of viral production mimics our simulations above. Virions are cleared at the rate c.
In due course, the different viral populations reach steady states. We obtain the steady-state populations by solving the above equations with the left-hand sides set to zero. The frequency of genome i is then the fraction of genomes of type i at steady state:
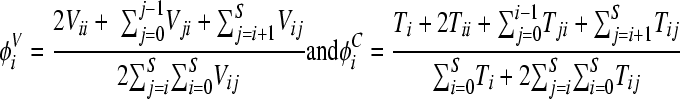 |
(6) |
We employ parameter values representative of HIV-1 infection in vivo (2, 13, 35, 61): λ = 105 cells ml−1 day−1, dT = 0.1 day−1, k0 = 2.4 × 10−8 ml day−1, k1 = 0.7 k0, δ = 1 day−1, c = 23 day−1, and N = 4,792, which yields a basic reproductive ratio of 5, μ = 3 × 10−5 substitutions per site per replication, and ρ = 8.3 × 10−4 crossovers per site per replication. For the NVP resistance mutations we consider, single-nucleotide substitutions yield amino acid changes. We assume that the nucleotide distance between loci is three times the corresponding amino acid distance. The values representing relative fitness of genomes, ζi, are obtained from analysis of growth competition assays (Fig. 2). We solve model equations using a computer program written in C.
RESULTS
In this study, we considered genomes carrying the common NVP resistance mutations K103N, V106A, Y181C, Y188C, and G190A. Depending on whether each of these loci has a mutation or not, a total of S = 25 − 1 = 31 distinct resistant genomes exist, of which 5 are single mutants, 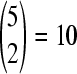 are double mutants, and so on. Our aim was to predict the frequencies of each of these S genomes in a chronically infected individual naïve to antiretroviral therapy. The frequencies depend on the relative fitnesses of the genomes, which remain poorly estimated. Here, we estimated the relative fitness of each of the S genomes by analyzing previously published data from competitive growth assays.
are double mutants, and so on. Our aim was to predict the frequencies of each of these S genomes in a chronically infected individual naïve to antiretroviral therapy. The frequencies depend on the relative fitnesses of the genomes, which remain poorly estimated. Here, we estimated the relative fitness of each of the S genomes by analyzing previously published data from competitive growth assays.
Relative fitness of NVP-resistant genomes.
We considered the experiments of Collins et al. (8), who constructed the 31 NVP-resistant mutants of interest here using site-directed mutagenesis and performed competitive growth assays in which cells were exposed at low MOI simultaneously to the wild type and a mutant and the time evolution of the ratio of the mutant and wild-type viruses was measured using a quantitative real-time RT-PCR-based assay. We employed a model of HIV dynamics in vitro to predict the time evolution of the mutant and wild-type viruses under the conditions of the experiments (see Materials and Methods). We fit model predictions to the experimental data for each of the viable NVP-resistant mutants. Model predictions provide good fits to the data (Fig. 1; see Fig. S1 in the supplemental material) and yield estimates of the relative fitness of the mutant genomes (Fig. 2).
FIG. 1.

Representative fits of our model (equation 1) (lines) to data (symbols) from competitive growth assays (8). The remaining fits are shown in Fig. S1 in the supplemental material.
FIG. 2.

Estimates of the relative fitness of genomes obtained from fits of our model to experimental data (Fig. 1). Error bars represent 95% confidence limits. The relative fitnesses of four mutants found unviable (8) are set to zero (asterisk).
We find that the relative fitness decreases with the number of mutations: the relative fitnesses (mean ± standard deviation [SD]) of the wild type and single, double, triple, quadruple, and quintuple mutants are 1, 0.74 ± 0.14, 0.56 ± 0.19, 0.32 ± 0.19, 0.27 ± 0.26, and 0, respectively. Within a mutant class, the relative fitness of different mutants varies substantially. The relative fitness of single mutants follows K103N > Y181C > G190A > Y188C > V106A, in agreement with Collins et al. (8). Furthermore, we find that some double mutants are fitter than the corresponding single mutants, that some triple mutants are fitter than the corresponding double mutants, and so on. For instance, the V106A/Y181C mutant is fitter than the V106A mutant. Similarly, the K103N/V106A/Y181C mutant is fitter than the K103N/V106A mutant. All single mutants, however, are less fit than the wild type. Thus, whereas individual mutations lower fitness, in combination some mutations increase fitness. The fitness of genomes is thus the result of complex interactions between mutations. We find that our estimates of the relative fitness correlate well with those of Collins et al. (8) (see the Discussion and see Fig. S2 in the supplemental material). We employ our estimates of the relative fitness in our simulations and model to predict the frequencies of NVP-resistant genomes.
Simulations of HIV-1 evolution.
We have previously performed simulations of the genomic diversification of HIV-1 that incorporate the key forces underlying HIV-1 evolution in vivo and that quantitatively capture the time evolution of HIV-1 genomic diversity in patients over extended durations post-seroconversion (63). Here, we apply the simulations to predict the frequencies of NVP-resistant strains. Briefly, we considered synchronous infections of a fixed population of cells. Infection begins with a pool of identical homozygous virions that carry no NVP resistance mutations. Each cell is infected with virions randomly chosen from the viral pool. Following infection, viral RNAs are reverse transcribed into proviral DNA, during which process recombination and mutation introduce genomic diversity. The proviral DNAs produce viral RNAs in proportion to their relative fitness. Viral RNAs are randomly assorted into pairs and released as new virions. Each cell produces progeny virions in proportion to the fitness of the genomes infecting it. Virions are randomly chosen from the resulting viral pool for infection of the next generation of uninfected cells, and the cycle is repeated (see Materials and Methods).
In Fig. 3, we present the evolution of viral diversity and divergence at the five NVP resistance loci we consider. Diversity is a measure of the variation in the viral genomes within an individual at any given time, whereas divergence is a measure of the variation of the viral genomes at any time from the founder genome. Initially, because we consider an individual infected with strains not carrying any NVP resistance mutations, both the diversity and the divergence are zero. With time, mutation and recombination give rise to NVP resistance mutations. Accordingly, the diversity and divergence of genomes increase. NVP resistance mutations, however, carry fitness penalties (Fig. 2). Consequently, selection, along with random genetic drift, curtails viral diversification. Eventually, a balance between these competing forces is established, which here occurs rapidly, and diversity and divergence attain equilibrium.
FIG. 3.

Evolution of the mean viral diversity and divergence at the five NVP resistance loci predicted by our simulations when the cell population is 2,000 and when each cell is infected with 3 virions. The other parameter values are mentioned in Materials and Methods. The inset shows the evolution for the first 150 generations.
In Fig. 4, we show the corresponding evolution of the frequencies of two representative NVP-resistant genomes in two realizations of our simulations. Evidently, the frequencies are subject to large stochastic variations both within and across realizations. We averaged the frequencies for each of the S NVP-resistant genomes over several generations after viral diversity reaches equilibrium and across several realizations of the simulations to obtain the expected frequencies both in the proviral DNA and in the cell-free viral genomic RNA (see Materials and Methods).
FIG. 4.

Evolution of the frequency of the Y188C single mutant (left) and the K103N Y188C double mutant (right) with generations during two realizations (top and bottom) of our simulations. The parameter values employed are the same as in Fig. 3.
Frequencies of NVP-resistant genomes.
In Fig. 5, we present the average frequencies of each of the S mutant genomes among the proviral DNA (φiC) and cell-free viral RNA (φiV) predicted by our simulations. We find that mutant frequencies decrease with the number of resistance mutations the genomes carry. Single mutants are present in mean frequencies of ∼10−4 to 10−5, and double mutants are present in mean frequencies of ∼10−8 to 10−9. Triple and higher mutants are absent (not shown). Different mutants within a mutant class occur at different frequencies because of different relative fitness values and frequencies of related mutants. Single mutants, which arise largely from the mutation of the wild type, occur at frequencies that increase with their relative fitness: K103N > Y181C > G190A > Y188C > V106A. Double-mutant frequencies depend additionally on the frequencies of the corresponding single mutants. Thus, for instance, the Y188C/G190A mutant has a higher fitness but lower frequency than the K103N/Y188C mutant because the K103N mutant is fitter and has a higher frequency than the G190A mutant, which results in the higher rate of formation of the K103N/Y188C mutant rather than the Y188C/G190A mutant.
FIG. 5.

The expected frequencies of single (top) and double (bottom) mutants in proviral DNA (left) and in cell-free viral RNA (right) predicted by our simulations. Error bars are standard deviations. The parameter values employed are the same as in Fig. 3.
The standard deviations of the frequencies are large (Fig. 5), indicative of the strong influence of random genetic drift and the resulting stochastic variations. These stochastic variations are observed both across generations within a particular realization as well as across different realizations of the simulations (Fig. 4). Standard deviations of single-mutant frequencies are ∼10−4, often higher than the mean values, and those of double-mutant frequencies are ∼10−6, several orders of magnitude larger than the corresponding mean values, suggesting that large temporal and interpatient variations in the frequencies of resistant mutants are expected due to stochastic effects underlying viral evolution.
Influence of simulation parameters.
We examined here the sensitivity of the above predictions to variations in simulation parameters. Recombination can induce the accumulation (or dissociation) of mutations and alter the frequency of mutant genomes (12). A prerequisite for recombination to induce genomic variation is the infection of individual cells by multiple, distinct virions. We therefore examined the influence of recombination and multiple infections of cells on the frequencies of NVP-resistant genomes. We find that both recombination and multiple infections of cells have no significant effects on the frequencies (Fig. 6). The influence of recombination increases as the separation between resistance loci increases (47, 61). Here, the resistance loci are clustered together (maximum separation of ∼100 amino acids) so that the influence of recombination is expected to be small. An increase in the frequency of multiple infections while keeping all other parameters constant corresponds to an increase in the proviral populations, which do not alter the frequencies of NVP-resistant genomes substantially (see below).
FIG. 6.

The expected frequencies of single (A) and double (B) mutants in proviral DNA predicted by our simulations with a recombination rate of 8.3 × 10−4 crossovers per site per reverse transcription and when each cell is infected with 3 virions (dark gray bars), with no recombination (gray bars), and when each cell is infected with a single virion (light gray bars). Error bars are standard deviations. The remaining parameters are the same as in Fig. 3. The corresponding frequencies in cell-free viral RNA are shown in Fig. S3 in the supplemental material.
The effective population size of HIV-1 may vary across individuals. With the above parameters, our simulations capture the evolution of viral diversity and divergence in patients with effective population sizes in the range ∼103 to 104 cells (63). We therefore examined whether increasing the population size to 104 alters the frequencies of NVP-resistant mutants. We find that with the larger population size, the mean frequencies of both single and double mutants remain largely unchanged, whereas the standard deviations decrease because of the smaller influence of drift (Fig. 7). Triple and higher mutants still do not occur.
FIG. 7.

The expected frequencies of single (A) and double (B) mutants in proviral DNA predicted by our simulations when the populations of cells are 2,000 (dark gray bars) and 10,000 (light gray bars). Error bars are standard deviations. The remaining parameters are the same as in Fig. 3. The corresponding frequencies in cell-free viral RNA are shown in Fig. S4 in the supplemental material.
More generally, current estimates of the effective population size of HIV-1 in vivo display wide variations, ranging from 103 to 106 cells (1, 30, 40, 50, 52-54). In particular, simulations similar to ours but with the assumption that multiple infections of cells are rare, as observed recently in peripheral blood mononuclear cells in four chronically infected patients (26), obtained a much larger value of the effective population size (>105) by comparison with experimental observations of the frequency of the least-prevalent haplotype in a two-locus/two-allele model (52). Here, we therefore performed simulations with a population size of 105 and a low frequency of multiple infections that mimics experimental observations (26) (see Materials and Methods). We find that single and double mutants exist at mean frequencies similar to those predicted above (Fig. 8). Triple and higher mutants are again absent. The larger population size, however, implies a smaller influence of drift. Accordingly, the deviations about the mean values of the mutant frequencies are now smaller than those predicted above. Thus, the standard deviations of the single mutant frequencies are ∼10−4 and those of the double mutants are ∼10−7 (in contrast to ∼10−6 in Fig. 5). Yet, the finding that standard deviations are much larger than the mean values implies that there are significant stochastic variations in the mutant frequencies.
FIG. 8.

The expected frequencies of single (A) and double (B) mutants in proviral DNA predicted by our simulations when the population of cells is 2,000 and each cell is infected with 3 virions (dark gray bars) and when the population of cells is 105 and the frequency of multiple infections of cells follows a distribution in which most cells are singly infected (Materials and Methods) (light gray bars). Error bars are standard deviations. The remaining parameters are the same as in Fig. 3. The corresponding frequencies in cell-free viral RNA are shown in Fig. S5 in the supplemental material.
Frequencies of NVP-resistant genomes from a deterministic model.
Finally, we employed a deterministic model of HIV-1 dynamics and evolution (see Materials and Methods) to estimate frequencies of the resistant genomes in the limit of an infinitely large effective population size. (Simulations with population sizes larger than ∼105 are computationally prohibitive.) We find that the deterministic model predicts that single- and double-mutant frequencies are similar to the mean values predicted by our simulations presented above (Fig. 9). Thus, the expected higher effective population size of ∼106 cells (52) may also not lead to substantial differences in the frequencies of single and double mutants. The deterministic model predicts, in addition, that triple and higher mutants occur at low frequencies when the population size is sufficiently large. Thus, triple, quadruple, and quintuple mutants occur at frequencies of ∼10−12, ∼10−16, and ∼10−21, respectively. In the cell-free viral population, four unviable mutants (relative fitness = 0) occur at vanishing frequencies (see Fig. S6 in the supplemental material).
FIG. 9.

The expected frequencies of single (A), double (B), triple (C), and quadruple and quintuple (D) mutants in proviral DNA predicted by our simulations (dark gray bars) and our deterministic model (equations 2 to 6) (light gray bars). Error bars represent standard deviations. The parameter values employed are mentioned in Materials and Methods. The corresponding frequencies in cell-free viral RNA are shown in Fig. S6 in the supplemental material.
DISCUSSION
Minority NVP-resistant strains may exist in HIV-1-infected individuals naïve to NVP at frequencies below current assay detection limits and may compromise SDNVP-based efforts to prevent mother-to-child transmission, accelerate the failure of subsequent NNRTI-based antiretroviral therapy, and result in the transmission of drug-resistant genomes. Here, we employ bit-string simulations of viral genomic diversification and a mathematical model of HIV-1 dynamics and evolution to predict the frequencies of minority NVP-resistant strains in chronically infected NVP-naïve individuals. Our simulations incorporate the influence of mutation, multiple infections of cells, recombination, selection, epistatic interactions between multiple loci, and random genetic drift and predict the frequencies of genomes carrying all possible combinations of common NVP resistance mutations. Our model of HIV dynamics predicts frequencies in the limit of an infinitely large effective population size.
We find that the mean frequencies of genomes depend sensitively on the number of mutations the genomes carry. Single mutants occur at mean frequencies of ∼10−4 and double mutants at ∼10−8. These mean frequencies are predicted both by our simulations and the deterministic model and are robust to changes in the recombination rate, the frequency of multiple infections of cells, and the effective population size. We note that the estimates of the mutant frequencies obtained from our simulations are not significantly different from those predicted by our deterministic model (P = 0.41 for double-mutant frequencies based on the one-tailed, paired t test) but are significantly smaller than estimates from earlier approaches (7, 48) (P = 0.04 for double-mutant frequencies based on the one-tailed, paired t test) (see Fig. S7 in the supplemental material). Furthermore, our simulations predict that when the effective population size is small, no triple or higher mutants exist. The deterministic model applies in the limit of an infinitely large effective population size and predicts then that triple, quadruple, and quintuple mutants also exist at frequencies of ∼10−12, ∼10−16, and ∼10−21, respectively.
Chronically infected individuals typically contain ∼107 to 108 infected cells (20). Our predictions thus imply that all single and in some cases double mutants may exist in chronically infected individuals. Triple and higher mutants are unlikely to exist. Given the sensitivity (>0.01%) of current assays, however, several single and all double mutants are unlikely to be detected. The mutation K103N has the highest frequency, ∼0.018%, and is accordingly the most commonly detected (15). Y181C and G190A occur at lower frequencies (∼0.015% and ∼0.012%, respectively) and are thus less readily detected. The frequencies of K103N, Y181C, and G190A are near the detection limits of current assays. Consequently, subtle variations in viral fitness, arising, for instance, from different genetic backgrounds or subtypes (14), may result in frequencies above or below the detection limits in different patients. The present estimates of mutant frequencies thus emphasize the need for the development of more sensitive assays to detect minority resistance mutations in vivo. In addition, our simulations indicate that the standard deviations of the frequencies can be quite large, due to the influence of random genetic drift. Indeed, the standard deviations of the double-mutant frequencies are over 100-fold larger than the corresponding mean frequencies. Thus, large temporal and interpatient variations in the frequencies of NVP-resistant strains may arise due to stochastic effects.
Even when undetected, NVP resistance mutations may exist in patients in substantial numbers (∼103 copies [frequency of ∼10−4]) and may underlie therapy failure and transmission of drug resistance. For instance, although K103N is the predominant mutation in mothers, infants exposed to SDNVP showed predominantly Y181C mutations (37). One explanation is that NVP resistance mutations arise largely de novo in infants, and only a small proportion are transmitted by the mothers (38). Our estimates suggest, alternatively, that Y181C may exist in mothers in undetectable yet comparable frequencies to K103N and may be transmitted preferentially to infants because of its greater fitness in the presence of NVP (42) and/or greater transmissibility.
Long after the administration of SDNVP, NVP resistance mutations fade. The resulting frequencies are expected to be at least as large as those predicted by our simulations for patients not exposed to NVP. We conclude therefore that single-mutant frequencies may not necessarily vanish long after SDNVP. This, together with the relatively small fitness loss of some mutations (relative fitness of ≈0.9 for K103N) may explain the persistence of NVP resistance mutations several years after SDNVP (17, 44). Latently infected cells, which may archive NVP resistance (64), may contribute further to the persistence of NVP resistance mutations. The outcomes of NNRTI-based therapies are thus expected to be similar in NVP-naïve patients and patients in whom therapy is initiated long after SDNVP. Indeed, two studies found no substantial increase in the risk of mother-to-child transmission with repeated usage of SDNVP (18, 36), possibly because the median duration between pregnancies in the latter studies was >20 months, during which period NVP resistance mutations selected following the first SDNVP may have faded to the preexposure levels. Similarly, the risk of failure of NNRTI-based therapy was enhanced when therapy was initiated within 6 months of SDNVP, but no significant enhancement in the risk was observed when therapy was initiated more than 12 months after SDNVP (33, 60).
We recognize several limitations of our approach. First, we assume that the fitness of genomes is determined entirely by their replicative ability, whereas fitness is a combination of infectivity, replicative ability, and survivability. Available data do not allow estimation of differences between genomes in more than one of the latter characteristics with sufficient confidence. For instance, Collins et al. (8) fit their data, which are the same data that we employ—namely, of the ratio of the mutant and wild-type genomes measured at different times, t, in a competitive growth assay—to an exponential, bexp(mt), and assume that the best-fit value of m provides a measure of the relative fitness (fitness coefficient) of the mutant. (b is the initial ratio of the mutant and wild-type genomes.) m thus combines differences in fitness arising from different infectivity, replicative ability, and/or survivability of the virions. Our estimates of the relative fitness correlate well with those of Collins et al. (8) (see Fig. S2 in the supplemental material). An advantage of our analysis is that our estimates of the relative fitness, recognized also as the production rate ratio (65), enable facile determination of viral production as a function of viral fitness and hence are conveniently employed in our simulations and model.
Second, we note that our calculations estimate frequencies of NVP resistance mutations during the chronic infection steady state, where viral diversity at the resistance loci has reached a plateau. If infection is acquired through the transmission of susceptible genomes, then during acute infection, where viral diversity is on the rise, the frequencies of resistant mutants are expected to be lower than current estimates. In contrast, if drug-resistant genomes are acquired through transmission, then the frequencies of resistant mutants may be higher than current estimates. Third, we assume a constant mutation rate at all of the resistance loci. While this is a reasonable approximation given that at all five loci we consider single-nucleotide substitutions yield amino acid changes, the approximation may overestimate the frequencies of the K103N and G190A mutations, which are transversions and may occur at smaller rates compared to the other mutations. The large stochastic variations we predict, however, may subsume changes in the mean frequencies of the K103N and G190A mutations expected from the lower substitution rates. Nonetheless, our estimates of the frequencies of NVP resistance mutations facilitate more accurate assessment of the impact of minority NVP-resistant strains on the outcomes of NVP-based therapies and the transmission of NVP resistance.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grant AI065334. S.G. was supported partly by the Department of Biotechnology, Centre of Excellence for Research on Hepatitis C Virus, India.
Footnotes
Published ahead of print on 28 July 2010.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Achaz, G., S. Palmer, M. Kearney, F. Maldarelli, J. W. Mellors, J. M. Coffin, and J. Wakeley. 2004. A robust measure of HIV-1 population turnover within chronically infected individuals. Mol. Biol. Evol. 21:1902-1912. [DOI] [PubMed] [Google Scholar]
- 2.Arora, P., and N. M. Dixit. 2009. Timing the emergence of resistance to anti-HIV drugs with large genetic barriers. PLoS Comput. Biol. 5:e1000305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonhoeffer, S., C. Chappey, N. T. Parkin, J. M. Whitcomb, and C. J. Petropoulos. 2004. Evidence for positive epistasis in HIV-1. Science 306:1547-1550. [DOI] [PubMed] [Google Scholar]
- 4.Chen, B. K., R. T. Gandhi, and D. Baltimore. 1996. CD4 down-modulation during infection of human T cells with human immunodeficiency virus type 1 involves independent activities of vpu, env, and nef. J. Virol. 70:6044-6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen, H. Y., M. Di Mascio, A. S. Perelson, D. D. Ho, and L. Zhang. 2007. Determination of virus burst size in vivo using a single-cycle SIV in rhesus macaques. Proc. Natl. Acad. Sci. U. S. A. 104:19079-19084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clavel, F., and A. J. Hance. 2004. HIV drug resistance. N. Engl. J. Med. 350:1023-1035. [DOI] [PubMed] [Google Scholar]
- 7.Coffin, J. M. 1995. HIV population-dynamics in-vivo—implications for genetic-variation, pathogenesis, and therapy. Science 267:483-489. [DOI] [PubMed] [Google Scholar]
- 8.Collins, J. A., M. G. Thompson, E. Paintsil, M. Ricketts, J. Gedzior, and L. Alexander. 2004. Competitive fitness of nevirapine-resistant human immunodeficiency virus type 1 mutants. J. Virol. 78:603-611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Cock, K. M., M. G. Fowler, E. Mercier, I. de Vincenzi, J. Saba, E. Hoff, D. J. Alnwick, M. Rogers, and N. Shaffer. 2000. Prevention of mother-to-child HIV transmission in resource-poor countries—translating research into policy and practice. JAMA 283:1175-1182. [DOI] [PubMed] [Google Scholar]
- 10.Dimitrov, D. S., and M. A. Martin. 1995. CD4+ cell turnover. Nature 375:194-195. [DOI] [PubMed] [Google Scholar]
- 11.Dimitrov, D. S., R. L. Willey, H. Sato, L. J. Chang, R. Blumenthal, and M. A. Martin. 1993. Quantitation of human immunodeficiency virus type 1 infection kinetics. J. Virol. 67:2182-2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dixit, N. M. 2008. Modelling HIV infection dynamics: the role of recombination in the development of drug resistance. Future HIV Ther. 2:375-388. [Google Scholar]
- 13.Dixit, N. M., M. Markowitz, D. D. Ho, and A. S. Perelson. 2004. Estimates of intracellular delay and average drug efficacy from viral load data of HIV-infected individuals under antiretroviral therapy. Antivir. Ther. 9:237-246. [PubMed] [Google Scholar]
- 14.Eshleman, S. H., D. R. Hoover, S. Chen, S. E. Hudelson, L. A. Guay, A. Mwatha, S. A. Fiscus, F. Mmiro, P. Musoke, J. B. Jackson, N. Kumwenda, and T. Taha. 2005. Nevirapine (NVP) resistance in women with HIV-1 subtype C, compared with subtypes A and D, after the administration of single-dose NVP. J. Infect. Dis. 192:30-36. [DOI] [PubMed] [Google Scholar]
- 15.Eshleman, S. H., M. Mracna, L. A. Guay, M. Deseyve, S. Cunningham, M. Mirochnick, P. Musoke, T. Fleming, M. G. Fowler, L. M. Mofenson, F. Mmiro, and J. B. Jackson. 2001. Selection and fading of resistance mutations in women and infants receiving nevirapine to prevent HIV-1 vertical transmission (HIVNET 012). AIDS 15:1951-1957. [DOI] [PubMed] [Google Scholar]
- 16.Flys, T., D. V. Nissley, C. W. Claasen, D. Jones, C. J. Shi, L. A. Guay, P. Musoke, F. Mmiro, J. N. Strathern, J. B. Jackson, J. R. Eshleman, and S. H. Eshleman. 2005. Sensitive drug-resistance assays reveal long-term persistence of HIV-1 variants with the K103N nevirapine (NVP) resistance mutation in some women and infants after the administration of single-dose NVP: HIVNET 012. J. Infect. Dis. 192:24-29. [DOI] [PubMed] [Google Scholar]
- 17.Flys, T. S., D. Donnell, A. Mwatha, C. Nakabiito, P. Musoke, F. Mmiro, J. B. Jackson, L. A. Guay, and S. H. Eshleman. 2007. Persistence of K103N-containing HIV-1 variants after single-dose nevirapine for prevention of HIV-1 mother-to-child transmission. J. Infect. Dis. 195:711-715. [DOI] [PubMed] [Google Scholar]
- 18.Flys, T. S., M. S. McConnell, F. Matovu, J. D. Church, D. Bagenda, L. Khaki, P. Bakaki, M. C. Thigpen, C. Eure, M. G. Fowler, and S. H. Eshleman. 2008. Nevirapine resistance in women and infants after first versus repeated use of single-dose nevirapine for prevention of HIV-1 vertical transmission. J. Infect. Dis. 198:465-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao, F., and D. Wang. 2007. Minor-drug-resistant HIV populations and treatment failure. Future Virol. 2:293-302. [Google Scholar]
- 20.Haase, A. T., K. Henry, M. Zupancic, G. Sedgewick, R. A. Faust, H. Melroe, W. Cavert, K. Gebhard, K. Staskus, Z. Q. Zhang, P. J. Dailey, H. H. Balfour, A. Erice, and A. S. Perelson. 1996. Quantitative image analysis of HIV-1 infection in lymphoid tissue. Science 274:985-989. [DOI] [PubMed] [Google Scholar]
- 21.Hockett, R. D., J. M. Kilby, C. A. Derdeyn, M. S. Saag, M. Sillers, K. Squires, S. Chiz, M. A. Nowak, G. M. Shaw, and R. P. Bucy. 1999. Constant mean viral copy number per infected cell in tissues regardless of high, low, or undetectable plasma HIV RNA. J. Exp. Med. 189:1545-1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jackson, J. B., P. Musoke, T. Fleming, L. A. Guay, D. Bagenda, M. Allen, C. Nakabiito, J. Sherman, P. Bakaki, M. Owor, C. Ducar, M. Deseyve, A. Mwatha, L. Emel, C. Duefield, M. Mirochnick, M. G. Fowler, L. Mofenson, P. Miotti, M. Gigliotti, D. Bray, and F. Mmiro. 2003. Intrapartum and neonatal single-dose nevirapine compared with zidovudine for prevention of mother-to-child transmission of HIV-1 in Kampala, Uganda: 18-month follow-up of the HIVNET 012 randomised trial. Lancet 362:859-868. [DOI] [PubMed] [Google Scholar]
- 23.Johnson, J. A., J. F. Li, L. Morris, N. Martinson, G. Gray, J. McIntyre, and W. Heneine. 2005. Emergence of drug-resistant HIV-1 after intrapartum administration of single-dose nevirapine is substantially underestimated. J. Infect. Dis. 192:16-23. [DOI] [PubMed] [Google Scholar]
- 24.Johnson, J. A., J. F. Li, X. Wei, J. Lipscomb, D. Irlbeck, C. Craig, A. Smith, D. E. Bennett, M. Monsour, P. Sandstrom, E. R. Lanier, and W. Heneine. 2008. Minority HIV-1 drug resistance mutations are present in antiretroviral treatment-naive populations and associate with reduced treatment efficacy. PLoS Med. 5:1112-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson, V. A., F. Brun-Vezinet, B. Clotet, H. F. Gunthard, D. R. Kuritzkes, D. Pillay, J. M. Schapiro, and D. D. Richman. 2008. Update of the drug resistance mutations in HIV-1: spring 2008. Top. HIV Med. 16:62-68. [DOI] [PubMed] [Google Scholar]
- 26.Josefsson, L., J. Brannstrom, B. Makilato, F. Maldarelli, M. Kearney, W. Shao, J. Mellors, J. Albert, J. Coffin, and S. Palmer. 2010. Analysis of HIV DNA molecules in single infected cells from recently and chronically infected patients, abstr. 289, 17th Conf. Retrovir. Opportunistic Infect., San Francisco, CA.
- 27.Jourdain, G., N. Ngo-Giang-Huong, S. Le Coeur, C. Bowonwatanuwong, P. Kantipong, P. Leechanachai, S. Ariyadej, P. Leenasirimakul, S. Hammer, and M. Lallemant. 2004. Intrapartum exposure to nevirapine and subsequent maternal responses to nevirapine-based antiretroviral therapy. N. Engl. J. Med. 351:229-240. [DOI] [PubMed] [Google Scholar]
- 28.Jung, A., R. Maier, J. P. Vartanian, G. Bocharov, V. Jung, U. Fischer, E. Meese, S. Wain-Hobson, and A. Meyerhans. 2002. Multiply infected spleen cells in HIV patients. Nature 418:144. [DOI] [PubMed] [Google Scholar]
- 29.Keele, B. F., E. E. Giorgi, J. F. Salazar-Gonzalez, J. M. Decker, K. T. Pham, M. G. Salazar, C. X. Sun, T. Grayson, S. Y. Wang, H. Li, X. P. Wei, C. L. Jiang, J. L. Kirchherr, F. Gao, J. A. Anderson, L. H. Ping, R. Swanstrom, G. D. Tomaras, W. A. Blattner, P. A. Goepfert, J. M. Kilby, M. S. Saag, E. L. Delwart, M. P. Busch, M. S. Cohen, D. C. Montefiori, B. F. Haynes, B. Gaschen, G. S. Athreya, H. Y. Lee, N. Wood, C. Seoighe, A. S. Perelson, T. Bhattacharya, B. T. Korber, B. H. Hahn, and G. M. Shaw. 2008. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 105:7552-7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kouyos, R. D., C. L. Althaus, and S. Bonhoeffer. 2006. Stochastic or deterministic: what is the effective population size of HIV-1? Trends Microbiol. 14:507-511. [DOI] [PubMed] [Google Scholar]
- 31.Lapadula, G., I. Izzo, F. Gargiulo, G. Paraninfo, F. Castelnuovo, E. Quiros-Roldan, G. Cologni, F. Ceresoli, N. Manca, G. Carosi, and C. Torti. 2008. Updated prevalence of genotypic resistance among HIV-1 positive patients naive to antiretroviral therapy: a single center analysis. J. Med. Virol. 80:747-753. [DOI] [PubMed] [Google Scholar]
- 32.Levy, D. N., G. M. Aldrovandi, O. Kutsch, and G. M. Shaw. 2004. Dynamics of HIV-1 recombination in its natural target cells. Proc. Natl. Acad. Sci. U. S. A. 101:4204-4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lockman, S., R. L. Shapiro, L. M. Smeaton, C. Wester, I. Thior, L. Stevens, F. Chand, J. Makhema, C. Moffat, A. Asmelash, P. Ndase, P. Arimi, E. van Widenfelt, L. Mazhani, V. Novitsky, S. Lagakos, and M. Essex. 2007. Response to antiretroviral therapy after a single, peripartum dose of nevirapine. N. Engl. J. Med. 356:135-147. [DOI] [PubMed] [Google Scholar]
- 34.Loubser, S., P. Balfe, G. Sherman, S. Hammer, L. Kuhn, and L. Morris. 2006. Decay of K103N mutants in cellular DNA and plasma RNA after single-dose nevirapine to reduce mother-to-child HIV transmission. AIDS 20:995-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mansky, L. M., and H. M. Temin. 1995. Lower in vivo mutation rate of human-immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 69:5087-5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinson, N. A., D. K. Ekouevi, F. Dabis, L. Morris, P. Lupodwana, B. Tonwe-Gold, P. Dhlamini, R. Becquet, J. G. Steyn, V. Leroy, I. Viho, G. E. Gray, and J. A. McIntyre. 2007. Transmission rates in consecutive pregnancies exposed to single-dose nevirapine in Soweto, South Africa and Abidjan, Cote d'Ivoire. J. Acquir. Immun. Defic. Syndr. 45:206-209. [DOI] [PubMed] [Google Scholar]
- 37.Martinson, N. A., L. Morris, G. Gray, D. Moodley, V. Pillay, S. Cohen, P. Dhlamini, A. Puren, S. Bhayroo, J. Steyn, and J. A. McIntyre. 2007. Selection and persistence of viral resistance in HIV-infected children after exposure to single-dose nevirapine. J. Acquir. Immun. Defic. Syndr. 44:148-153. [DOI] [PubMed] [Google Scholar]
- 38.McIntyre, J., and N. Martinson. 2007. Prophylactic regimens to prevent mother-to-child transmission of HIV and their effect on future therapy. Therapy 4:797-805. [Google Scholar]
- 39.Metzner, K. J., S. G. Giulieri, S. A. Knoepfel, P. Rauch, P. Burgisser, S. Yerly, H. F. Gunthard, and M. Cavassini. 2009. Minority quasispecies of drug-resistant HIV-1 that lead to early therapy failure in treatment-naive and -adherent patients. Clin. Infect. Dis. 48:239-247. [DOI] [PubMed] [Google Scholar]
- 40.Nijhuis, M., C. A. B. Boucher, P. Schipper, T. Leitner, R. Schuurman, and J. Albert. 1998. Stochastic processes strongly influence HIV-1 evolution during suboptimal protease-inhibitor therapy. Proc. Natl. Acad. Sci. U. S. A. 95:14441-14446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Novak, R. M., L. Chen, R. D. MacArthur, J. D. Baxter, K. H. Hullsiek, G. Peng, Y. Xiang, C. Henely, B. Schmetter, J. Uy, M. van den Berg-Wolf, and M. Kozal. 2005. Prevalence of antiretroviral drug resistance mutations in chronically HIV-infected, treatment-naive patients: implications for routine resistance screening before initiation of antiretroviral therapy. Clin. Infect. Dis. 40:468-474. [DOI] [PubMed] [Google Scholar]
- 42.Paintsil, E., A. Margolis, J. A. Collins, and L. Alexander. 2006. The contribution of HIV fitness to the evolution pattern of reverse transcriptase inhibitor resistance. J. Med. Virol. 78:425-430. [DOI] [PubMed] [Google Scholar]
- 43.Palmer, S., V. Boltz, F. Maldarelli, M. Kearney, E. K. Halvas, D. Rock, J. Falloon, R. T. Davey, Jr., R. L. Dewar, J. A. Metcalf, J. W. Mellors, and J. M. Coffin. 2006. Selection and persistence of non-nucleoside reverse transcriptase inhibitor-resistant HIV-1 in patients starting and stopping non-nucleoside therapy. AIDS 20:701-710. [DOI] [PubMed] [Google Scholar]
- 44.Palmer, S., V. Boltz, N. Martinson, F. Maldarelli, G. Gray, J. McIntyre, J. Mellors, L. Morris, and J. Coffin. 2006. Persistence of nevirapine-resistant HIV-1 in women after single-dose nevirapine therapy for prevention of maternal-to-fetal HIV-1 transmission. Proc. Natl. Acad. Sci. U. S. A. 103:7094-7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Piatak, M., M. S. Saag, L. C. Yang, S. J. Clark, J. C. Kappes, K. C. Luk, B. H. Hahn, G. M. Shaw, and J. D. Lifson. 1993. High-levels of HIV-1 in plasma during all stages of infection determined by competitive PCR. Science 259:1749-1754. [DOI] [PubMed] [Google Scholar]
- 46.Piguet, V., F. Gu, M. Foti, N. Demaurex, J. Gruenberg, J. L. Carpentier, and D. Trono. 1999. Nef-induced CD4 degradation: a diacidic-based motif in Nef functions as a lysosomal targeting signal through the binding of beta-COP in endosomes. Cell 97:63-73. [DOI] [PubMed] [Google Scholar]
- 47.Rhodes, T., H. Wargo, and W.-S. Hu. 2003. High rates of human immunodeficiency virus type 1 recombination: near-random segregation of markers one kilobase apart in one round of viral replication. J. Virol. 77:11193-11200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ribeiro, R. M., S. Bonhoeffer, and M. A. Nowak. 1998. The frequency of resistant mutant virus before antiviral therapy. AIDS 12:461-465. [DOI] [PubMed] [Google Scholar]
- 49.Ribeiro, R. M., L. Qin, L. L. Chavez, D. Li, S. G. Self, and A. S. Perelson. 2010. Estimation of the initial viral growth rate and basic reproductive number during acute HIV-1 infection. J. Virol. 84:6096-6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rodrigo, A. G., E. G. Shpaer, E. L. Delwart, A. K. N. Iversen, M. V. Gallo, J. Brojatsch, M. S. Hirsch, B. D. Walker, and J. I. Mullins. 1999. Coalescent estimates of HIV-1 generation time in vivo. Proc. Natl. Acad. Sci. U. S. A. 96:2187-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rouzine, I., E. Gubankova, M. Kearney, S. Palmer, F. Maldarelli, and J. Coffin. 2008. Estimate of the frequency of HIV-1 co-infection in vivo, abstr. 345. 15th Conf. Retrovir. Opportunistic Infect., Boston, MA.
- 52.Rouzine, I. M., and J. M. Coffin. 1999. Linkage disequilibrium test implies a large effective population number for HIV in vivo. Proc. Natl. Acad. Sci. U. S. A. 96:10758-10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seo, T. K., J. L. Thorne, M. Hasegawa, and H. Kishino. 2002. Estimation of effective population size of HIV-1 within a host: a pseudomaximum-likelihood approach. Genetics 160:1283-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shriner, D., R. Shankarappa, M. A. Jensen, D. C. Nickle, J. E. Mittler, J. B. Margolick, and J. I. Mullins. 2004. Influence of random genetic drift on human immunodeficiency virus type I env evolution during chronic infection. Genetics 166:1155-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Siliciano, J. D., J. Kajdas, D. Finzi, T. C. Quinn, K. Chadwick, J. B. Margolick, C. Kovacs, S. J. Gange, and R. F. Siliciano. 2003. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4(+) T cells. Nat. Med. 9:727-728. [DOI] [PubMed] [Google Scholar]
- 56.Simen, B. B., J. F. Simons, K. H. Hullsiek, R. M. Novak, R. D. MacArthur, J. D. Baxter, C. L. Huang, C. Lubeski, G. S. Turenchalk, M. S. Braverman, B. Desany, J. M. Rothberg, M. Egholm, and M. J. Kozal. 2009. Low-abundance drug-resistant viral variants in chronically HIV-infected, antiretroviral treatment-naive patients significantly impact treatment outcomes. J. Infect. Dis. 199:693-701. [DOI] [PubMed] [Google Scholar]
- 57.Simon, V., D. D. Ho, and Q. A. Karim. 2006. HIV/AIDS epidemiology, pathogenesis, prevention, and treatment. Lancet 368:489-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Speirs, C., E. van Nimwegen, D. Bolton, M. Zavolan, M. Duvall, S. Angleman, R. Siegel, A. S. Perelson, and M. J. Lenardo. 2005. Analysis of human immunodeficiency virus cytopathicity by using a new method for quantitating viral dynamics in cell culture. J. Virol. 79:4025-4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stafford, M. A., L. Corey, Y. Z. Cao, E. S. Daar, D. D. Ho, and A. S. Perelson. 2000. Modeling plasma virus concentration during primary HIV infection. J. Theor. Biol. 203:285-301. [DOI] [PubMed] [Google Scholar]
- 60.Stringer, J. S. A., M. S. McConnell, J. Kiarie, O. Bolu, T. Anekthananon, T. Jariyasethpong, D. Potter, W. Mutsotso, C. B. Borkowf, D. Mbori-Ngacha, P. Muiruri, J. O. Ong'ech, I. Zulu, L. Njobvu, B. Jetsawang, S. Pathak, M. Bulterys, N. Shaffer, and P. J. Weidle. 2010. Effectiveness of non-nucleoside reverse-transcriptase inhibitor-based antiretroviral therapy in women previously exposed to a single intrapartum dose of nevirapine: a multi-country, prospective cohort study. PLoS Med. 7:e1000233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suryavanshi, G. W., and N. M. Dixit. 2007. Emergence of recombinant forms of HIV: dynamics and scaling. PLoS Comput. Biol. 3:2003-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.UNAIDS. 2009. AIDS epidemic update: December 2009. UNAIDS, Geneva, Switzerland. http://data.unaids.org/pub/Report/2009/JC1700_Epi_Update_en.pdf.
- 63.Vijay, N. N. V., Vasantika, R. Ajmani, A. S. Perelson, and N. M. Dixit. 2008. Recombination increases human immunodeficiency virus fitness, but not necessarily diversity. J. Gen. Virol. 89:1467-1477. [DOI] [PubMed] [Google Scholar]
- 64.Wind-Rotolo, M., C. Durand, L. Cranmer, A. Reid, N. Martinson, M. Doherty, B. L. Jilek, J. Kagaayi, A. Kizza, V. Pillay, O. Laeyendecker, S. J. Reynolds, S. H. Eshleman, B. Lau, S. C. Ray, J. D. Siliciano, T. C. Quinn, and R. F. Siliciano. 2009. Identification of nevirapine-resistant HIV-1 in the latent reservoir after single-dose nevirapine to prevent mother-to-child transmission of HIV-1. J. Infect. Dis. 199:1301-1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu, H., Y. Huang, C. Dykes, D. Liu, J. Ma, A. S. Perelson, and L. M. Demeter. 2006. Modeling and estimation of replication fitness of human immunodeficiency virus type 1 in vitro experiments by using a growth competition assay. J. Virol. 80:2380-2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.