

Abstract
The binding reaction of the HIV-1 gp120 envelope glycoprotein to the CD4 receptor involves exceptional changes in enthalpy and entropy. Crystal structures of gp120 in unliganded and various ligand-bound states, meanwhile, reveal an inner domain able to fold into diverse conformations, a structurally invariant outer domain, and, in the CD4-bound state, a bridging sheet minidomain. These studies, however, provide only hints as to the flexibility of each state. Here we use amide hydrogen/deuterium exchange coupled to mass spectrometry to provide quantifications of local conformational stability for HIV-1 gp120 in unliganded and CD4-bound states. On average, unliganded core gp120 displayed >10,000-fold slower exchange of backbone-amide hydrogens than a theoretically unstructured protein of the same composition, with binding by CD4 reducing the rate of gp120 amide exchange a further 10-fold. For the structurally constant CD4, alterations in exchange correlated well with alterations in binding surface (P value = 0.0004). For the structurally variable gp120, however, reductions in flexibility extended outside the binding surface, and regions of expected high structural diversity (inner domain/bridging sheet) displayed roughly 20-fold more rapid exchange in the unliganded state than regions of low diversity (outer domain). Thus, despite an extraordinary reduction in entropy, neither unliganded gp120 nor free CD4 was substantially unstructured, suggesting that most of the diverse conformations that make up the gp120 unliganded state are reasonably ordered. The results provide a framework for understanding how local conformational stability influences entropic change, conformational diversity, and structural rearrangements in the gp120-CD4 binding reaction.
Entry by HIV type 1 (HIV-1) into host cells is mediated by the HIV-1 trimeric viral spike, which is composed of three gp120 exterior envelope glycoproteins attached noncovalently to three gp41 transmembrane moieties (reviewed in reference 44). The mechanism of entry is in many ways typical of type 1 viral fusion machinery, with gp120 binding to host receptors on the cell surface and gp41 rearranging into a trimeric coiled-coil, thereby promoting a fusion of viral and host cell membranes (reviewed in reference 8). The HIV-1 envelope glycoproteins, however, utilize a distinct sequence of receptor binding and conformational change involving CD4, substantial CD4-induced structural rearrangements, and coreceptor CCR5 or CXCR4 (9, 15, 43).
The crystal structures of an unliganded gp120 core from the closely related simian immunodeficiency virus (SIV), of various HIV-1 gp120-CD4 complexes, of various HIV-1 gp120-antibody complexes, and of the final postfusion HIV-1 gp41 coiled-coil have been determined (5-7, 23, 29, 30, 38, 42, 49). Structural characterization of the HIV-1 viral spike, meanwhile, is currently available only by modeling or with cryoelectron tomograms (31, 35), the latter for both unliganded, CD4-bound, and antibody b12-bound states of HIV-1. These studies reveal that gp120 is composed of two domains (inner and outer), as well as a bridging sheet minidomain, which forms in the presence of CD4. The gp120 outer domain appears to be structurally constant, whereas the gp120 inner domain (and bridging sheet) displays extensive structural diversity (see Fig. S1 in the supplemental material).
The conformational stability of the HIV-1 gp120 envelope glycoprotein, or its flip side, flexibility, has been the subject of considerable interest and debate. Interest relates to the central role that structural rearrangements and conformational diversity of gp120 play in HIV-1 entry and immune evasion. Debate, meanwhile, has centered on the nature of the unliganded state. Atomic-mobility values (B values) from crystallographic analysis provide hints, as do thermodynamic measurements of CD4- and antibody-induced transitions and glutaraldehyde cross-linking coupled to antigenic analysis (6, 27, 37, 46). Ultimately, however, a spatially resolved description of gp120 conformational stability has been lacking.
To define the local conformational stability of HIV-1 gp120, we used amide hydrogen/deuterium exchange (HDX) coupled to mass spectrometric analysis. Digestion by pepsin allowed us to measure the exchange frequencies for gp120 and CD4 at an average resolution of 16 residues and covering more than 95% of the sequences of core gp120 and two-domain CD4. Fragment-specific exchange rates were processed into free energies of conformational stability and mapped onto published crystal structures. The results provide a spatially resolved view of local conformational stability for HIV-1 gp120 and CD4 in unliganded and complexed states.
MATERIALS AND METHODS
Protein purification.
The HIV-1 gp120 YU2 core construct was prepared as previously described (43). Constructs were expressed in Drosophila Schneider 2 cells under an inducible metallothionein promoter. The two-domain CD4 was produced in Chinese hamster ovary cells (39). Preparations of unliganded gp120, free CD4, and complexed gp120-CD4 were obtained by following procedures previously described (28). Briefly, the supernatant was collected 2 days after transfection and passed through an F105 affinity column. Glycans were removed by digestion with endoglycosidases H and D (Endo H and Endo D) to leave only the protein-proximal N-acetylglucosamine and 1,4-fucose residues. The two-domain CD4 was added to create the complex. Deglycosylated gp120 and the gp120-CD4 complex were passed through a concanavalin A column to remove gp120 with uncleaved glycans. All samples were further purified by gel filtration (Hiload 26/60 Superdex S200 prep grade; Amersham) and concentrated to 1 mg/ml in 30 mM phosphate (pH 7.4)-0.35 M NaCl.
Surface plasmon resonance.
The kinetic constants of gp120 binding to CD4 were measured by a Biacore 3000 surface plasmon resonance spectrometer as described previously (49). Briefly, we immobilized four-domain CD4 onto a CM5 sensor chip to a density of ∼ 500 response units with amine coupling. We passed serial dilutions of deglycosylated or glycosylated gp120 from 400 to 3.125 nM over the chip at 30 μl/min for 5 min; this was followed by a 5-min dissociation phase. The buffer used in the experiments consisted of 10 mM HEPES (pH 7.4), 150 mM NaCl, 3 mM EDTA, and 0.01% surfactant P-20. Sensorgrams were fit globally with BiaEvaluation 4.1 using a 1:1 binding with drifting baseline model so that the χ2 values of the fits were all less than 1% of the maximum number of response units.
Amide hydrogen/deuterium exchange.
The amide hydrogen/deuterium exchange reaction was initiated by mixing 10 μl of gp120 YU2 core, two-domain CD4, or complex solution with 10 μl of deuterated water at 4°C. At five time points between 15 and 1,500 s of exchange, 30 μl of 8 M urea-1 M Tris(2-carboxyethyl) phosphine hydrochloride (TCEP-HCl) was added to quench the exchange reaction and reduce the disulfide bonds in the proteins for 1 min at 1°C. Then, 45 μl of the mixture was injected into the ExSAR system (19), which is kept at 1°C. In the ExSAR system, the sample mixtures were passed through a pepsin column (bed volume, 104 μl; flow rate over the column, 200 μl/min) and the peptic fragments were separated by high-performance liquid chromatography (HPLC; C18, a linear gradient of 12% to 33% acetonitrile at a flow rate of 10 μl/min in 23 min). The eluted peptic fragments were analyzed by a LCQ mass spectrometer (Thermo Fisher).
The deuterium incorporation, D(%), into each peptic fragment for each time point was calculated to take into account the possibility of hydrogen back-exchange during the passage through the ExSAR system (48) by the formula D(%) = (m − m0%)/(m100% − m0%), where m is the centroid mass of a fragment after amide hydrogen/deuterium exchange, m0% is the centroid mass of the fragment from nondeuterated sample, and m100% is the centroid mass of the fragment from the fully deuterated sample. Fully deuterated sample was prepared by mixing 30 μl of protein solution with 30 μl of 100 mM TCEP-HCl in deuterated water, followed by overnight incubation at 60°C. Nondeuterated samples were prepared by mixing 10 μl of protein solution with 10 μl of water.
Digestion/separation optimization and identification of peptic fragments.
Prior to the submission of unliganded gp120, free CD4, and complexed gp120-CD4 to HDX analysis, the digestion conditions of the proteins and the separation conditions for the peptic fragments were optimized using nondeuterated protein samples (19). In that process, several parameters, including type of protease, denaturation/reduction conditions, and digestion time, were varied to generate ideal sequence coverage and special resolution of the proteins. The peptic fragments were identified by tandem mass spectrometry (MS/MS). Briefly, nondeuterated protein samples were digested and analyzed by MS in the data-dependent MS/MS mode. Nonglycosylated fragments were identified by the SEQUEST search algorithm (45) (Thermo Fisher) using the gp120 YU2 core and two-domain CD4 sequences. Glycosylated peptides were identified manually using the MS/MS fragmentation patterns and cleavage site information. All the results were pooled, and the quality of the peaks was assessed in HDExpress (18).
Data analysis.
The frequency of deuterium incorporation for each peptic fragment of gp120 and CD4 at each time point was measured twice to ensure reproducibility. The averaged frequency of peptic fragments was mapped onto the protein sequence for analysis. Distributions of exchange rates were obtained from these averages using a maximum entropy method (47). The weighted average exchange rate for each peptide was calculated over the distribution of exchange rates (34). Each average rate was used to normalize the average theoretical exchange rate of the same peptide in a completely disordered state. The theoretical exchange rates were determined by calculations that take into account the primary sequence of the peptide fragment (3). This normalized value, or protection factor, is the equilibrium constant for the transition of an ensemble of amide hydrogen-deuterium exchangeable conformations to an ensemble of totally exchange-protected conformations (14, 26, 40). The free energy of conformational stability for each fragment was calculated by substituting protection factor for the equilibrium constant (K) in the equation ΔG = −RTln(K), where ΔG is the free energy of conformational stability, R is the gas constant, and T is temperature.
The free energies of conformational stability for gp120 and CD4 were mapped onto the crystal structures of free CD4, unliganded SIV gp120, and complexed CD4-gp120. Since there were no crystal structures of unliganded HIV-1 gp120, we threaded the sequence of HIV-1 YU2 core into the structure of unliganded SIV gp120 using DeepView/SWISS-MODEL and Swiss-PdbViewer (16). The structure of CD4-bound gp120 was used to fill the disordered residues in the unliganded structure (residues 220 to 228 using SIV gp120 numbering) and the V4 loop. Residues characterized in the SIV gp120 crystal structure but not in HIV-1 gp120 were omitted in the final model. While the unliganded HIV-1 gp120 structure might be better represented by an ensemble of conformations, we used the core SIV structure for clarity and also because it represents at least one crystallographically defined conformation within the ensemble. Also, compared to other gp120 structures, the SIV structure better illustrates the potential for large conformational changes (see Fig. S1 in the supplemental material).
The free energies of conformational stability were compared to the accessible surface area, root mean square deviation, average temperature factors, frequency of residues within secondary structures, and frequency of potential hydrogen bonds to backbone amides for each peptic fragment. These structural statistics were obtained from crystal structures of gp120 and CD4 using the CNS (4), the CCP4 (1), and the HBPLUS Hydrogen Bond Calculator (36) software packages. All comparisons were evaluated through linear regression analysis in GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA) and Microsoft Excel.
Explicit incorporation of complex dissociation into HDX analysis.
With a dissociation constant (Kd) of ∼200 nM for core gp120 and CD4, at equilibrium, approximately 10% of a 1-mg/ml equimolar mixture of gp120 and CD4 separates into uncomplexed, free components. In addition, HDX procedures entail a 2-fold dilution into deuterated solvent, resulting in additional dissociation. A precise HDX analysis of the gp120-CD4 complex therefore requires explicit consideration of complex dissociation.
Prior to dilution, the equilibrium concentration of the gp120-CD4 complex can be calculated from the Kd. After dilution, protein fractions approach equilibrium according to the rate law:
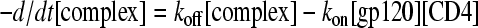 |
(1) |
Substituting [gp120] for [CD4],
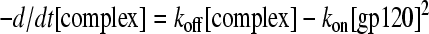 |
(2) |
For initial concentrations, [complex]o and [gp120]o, and conservation of mass,
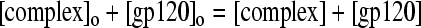 |
(3) |
Rearranging equation 3, we obtain for [gp120]
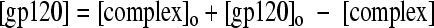 |
(4) |
Substituting equation 4 into equation 2 results in
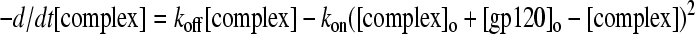 |
(5) |
This can be rearranged for ease of integration:
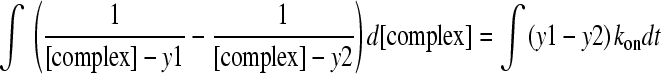 |
(6) |
Where,
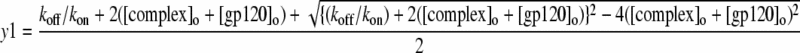 |
(7) |
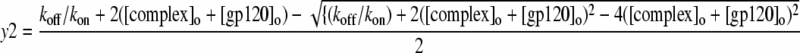 |
(8) |
The solution to this integral is:
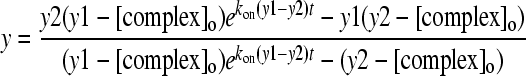 |
(9) |
Thus, by defining kon and koff as well as the concentrations of gp120 and the complex, changes in the fractional concentrations of the complex during exchange reactions can be explicitly determined—as we have done for the complex data for gp120 and CD4.
Statistically valid methods of HDX error determination: use of duplicate measurements and overlapping peptides.
To define the statistically significant differences in local conformational stability, appropriately defined measures of experimental uncertainty were developed. Maximum entropy methods introduced non-Gaussian distributions of rates, which resulted in average extracted rates that did not have meaningful standard deviations. We therefore attempted to develop a scheme that retained measures of uncertainty through a maximum entropy method.
One potential scheme (scheme 1) takes advantage of overlapping fragments in the HDX analysis (see Fig. S2A in the supplemental material). HDX data from all 38 peptic fragments of gp120 were processed into free energies of conformational stability, which were then grouped into the 16 peptic-fragment regions covering gp120, and averages and standard errors of the mean were calculated (see Table S1 in the supplemental material). While this scheme produces statistically valid measurements of uncertainty, only 13 of the 16 peptic-fragment regions contained overlapping fragments. In addition, maximum entropy analysis failed to generate rate distributions with five of the fragments.
A second scheme (scheme 2) relying strictly on the standard deviation generated by the repeat experiment was utilized (see Fig. S2B in the supplemental material). Maximum entropy methods using a common or shared standard deviation for each fragment calculated from averaged data were used to extract rate distributions from nonaveraged exchange curves for 16 peptic fragments that spanned all 16 regions. This resulted in two exchange rates for each peptic fragment. The two rates were then processed into free energies of conformational stability and combined as averages with standard errors of the mean (Table 1). This scheme produced measures of uncertainty for all 16 peptic regions. However, uncertain measurements were generated from a sample size of two. Also, because maximum entropy calculations for both replicates were carried out with the same standard deviation, they were not strictly independent. Comparisons, however, between rates (and errors) calculated by the two error analysis schemes had an R2 of 0.99, indicating that the results from the second scheme could be used with confidence.
TABLE 1.
Fragment free energies of conformational stability for unliganded and CD4-bound gp120 with error analysis scheme 2
| Fragment positions | ΔG ± SEM (kcal mol−1) |
ΔΔG ± SEM (kcal mol−1) | Pa | |
|---|---|---|---|---|
| Unliganded gp120 | CD4-bound gp120 | |||
| R81-N92 | −2.6 ± 0.2 | −2.4 ± 0.5 | 0.1 ± 0.5 | 0.8094 |
| N98-E106 | −3.6 ± 0.1 | −5.9 ± 0.0 | −2.3 ± 0.1 | 0.0009 |
| D107-L111 | −2.9 ± 0.2 | −6.4 ± 0.0 | −3.5 ± 0.2 | 0.0023 |
| Q114-F223 | −4.2 ± 0.1 | −5.0 ± 0.0 | −0.7 ± 0.1 | 0.0220 |
| L226-Q258 | −4.1 ± 0.9 | −5.9 ± 0.1 | −1.8 ± 0.9 | 0.1943 |
| L261-L265 | −3.5 ± 0.1 | −5.5 ± 0.0 | −2.0 ± 0.1 | 0.0028 |
| V271-T283 | −3.3 ± 0.1 | −5.0 ± 0.0 | −1.7 ± 0.1 | 0.0029 |
| Q287-L333 | −3.7 ± 0.1 | −3.7 ± 0.1 | 0.1 ± 0.1 | 0.7225 |
| T336-Q344 | −5.6 ± 0.0 | −5.6 ± 0.0 | 0.1 ± 0.0 | 0.2903 |
| L349-F383 | −5.5 ± 0.0 | −6.9 ± 0.2 | −1.4 ± 0.2 | 0.0149 |
| N386-F391 | −3.4 ± 0.2 | −5.2 ± 0.1 | −1.8 ± 0.2 | 0.0078 |
| W393-N413 | −2.8 ± 0.0 | −3.2 ± 0.1 | −0.3 ± 0.1 | 0.0193 |
| L416-M426 | −4.1 ± 0.0 | −5.7 ± 0.1 | −1.6 ± 0.1 | 0.0078 |
| E429-L453 | −4.9 ± 0.3 | −5.9 ± 0.3 | −1.0 ± 0.5 | 0.1543 |
| T455-L483 | −5.1 ± 0.1 | −7.6 ± 0.1 | −2.5 ± 0.1 | 0.0011 |
| Y486-E492 | −4.7 ± 0.1 | −5.3 ± 0.1 | −0.6 ± 0.2 | 0.0676 |
From an unpaired Student t test
NAME.
We sought to process the HDX data for gp120 and CD4 into absolute measures through processes which involved global consideration of experimental time points and the derivation of statistically valid measures of uncertainty. To this end, we derived and utilized a method of normalized assessment of maximum entropy (NAME). NAME entails calculation of free energies of conformational stability from protection factors, which are the exchange rates of an unstructured peptide normalized by the observed exchange rates of the same peptide in the protein structure. The observed rates for peptic fragments are the averages of exchange rate distributions generated by the maximum entropy method from the HDX data (48). The theoretical rates are generated from software for each backbone amide site.
RESULTS
Methodological alterations and overall scheme.
Standard procedures for HDX analysis (19, 22) were followed, although extensive gp120 glycosylation and other gp120-specific considerations necessitated modifications (Fig. 1). First, as rates of exchange between unliganded and CD4-bound states of gp120 were expected to vary dramatically, we sought to account explicitly for the effect of complex dissociation during the exchange reaction. We solved the differential equation describing gp120-CD4 complex dissociation following the 2-fold dilution of the complex into deuterated solvent (see Materials and Methods). Surface plasmon resonance was used to define kon, koff, and Kd of the CD4-gp120 binding reaction, and measurements were made at 5°C to approximate HDX conditions. At 5°C, four-domain CD4 displayed a kon [(2.65 ± 0.09) × 104 M−1s−1], a koff [(1.26 ± 0.05) × 10−2 s−1], and a Kd [(4.8 ± 0.2) × 10−7 M] to deglycosylated YU2 core gp120 that were similar to previously published results with deglycosylated HXBc2 gp120 at 37°C (37). Analogous measurements were also made with glycosylated gp120 (see Table S2 in the supplemental material).
FIG. 1.

Schematic of HDX procedure. Protein expression and preparation were devised to minimize glycan heterogeneity while maintaining biological integrity of the analyzed samples. Nonetheless, heterogeneity was still observed after deglycosylation to the protein-proximal N-acetylglucosamine (GlcNAc), necessitating modification of standard HDX-mass spectrometry analysis to include peptic fragments with no sugar or with GlcNAc or GlcNAc with fucose additions (Fig. 2). The results were processed by normalized assessment of maximum entropy (NAME), a method described in the present study. These alterations have been implemented to allow for calculation of free energies of conformational stabilization from HDX frequencies (http://exon.niaid.nih.gov/HDX_NAME). Asterisks indicate steps applicable to complexed proteins. Error analysis schemes are shown in Fig. S2 in the supplemental material.
Second, to provide valid estimates of error for flexibility measurements, we devised strategies to incorporate information from established maximum entropy methods of data analysis (34) and from overlapping peptides in the mass spectrometry (see Materials and Methods; see also Fig. S2 in the supplemental material). Rates and errors calculated by two different incorporation strategies showed an R2 of 0.99.
Third, we derived and utilized a method of normalized assessment of maximum entropy (NAME), which entailed extraction of rate constants from HDX curves using maximum entropy methods (see Fig. S3 in the supplemental material) followed by normalization with rate constants of theoretically unstructured peptides. The normalized values (protection factors) were then processed into free energies using maximum entropy methods and normalization with intrinsic exchange factors (see Materials and Methods), thereby placing the results on an absolute scale. Since the protection factor is the equilibrium constant for a portion of a protein transitioning from an ensemble of flexible, exchanging states into an ensemble of rigid, nonexchanging states, the free energy associated with it is related to local conformational stability (14, 26). NAME-derived energies per peptic fragment showed correlations (R2) with unprocessed frequencies of 0.36 to 0.68 and 0.75 to 0.77 for unliganded and CD4-bound gp120, respectively (see Table S3 in the supplemental material). NAME has been implemented as a Web application (http://exon.niaid.nih.gov/HDX_NAME); it processes sequence data, amide hydrogen/deuterium exchange, and experimental conditions (temperature and pH) into protection factors and free energies of conformational stability.
HDX sequence coverage and spatial resolution.
We chose to study core gp120 from the YU2 isolate. HIV-1 strain YU2 is a primary CCR5-using isolate which was cloned without cell culture passage (33), and the crystal structure of YU2 core complexed with CD4 had been previously determined (Protein Data Bank accession code 1RZK) (24). Mass spectrometry of YU2 core gp120 produced in Drosophila S2 cells suggested substantial variability in N-linked glycosylation and identified only a small fraction of peptide peaks. Endoglycosidases H and D (Endo H and Endo D) cleave N-linked glycans at the protein-proximal N-acetylglucosamine, reducing the N-linked glycan to a single N-acetylglucosamine and a potential 1,6-fucose. Mass spectrometry of Endo H/D-treated YU2 core after moderate pepsin digestion under optimized reducing conditions resulted in peptide identifications covering 98% of the gp120 sequence.
Spatial resolution (the size of the average HDX-characterized fragment) is experimentally related to the size of the pepsin-generated fragments (17), as well as to the detection limits imposed by the mass spectrometric analysis. Pepsin digestion of unliganded and CD4-bound gp120 resulted in 38 gp120 peptic fragments, which overlapped in 16 independent regions and covered 98% of the gp120 sequence (Fig. 2; see Table S4 in the supplemental material). All 38 fragments were used for analysis in accordance with scheme 1, while only 16 fragments with the highest-quality data covering each of these regions were used for scheme 2 (see Materials and Methods; see Fig. S2 and Table S1 in the supplemental material). The same digestion conditions for free CD4 generated 14 peptic fragments which overlapped in 9 regions and covered 92% of the CD4 sequence (see Table S5 in the supplemental material). Some of the CD4 peptides had poor signal/noise in the peptic digest of the gp120-CD4 complex, presumably related to ion suppression in the more crowded liquid chromatography-elution profiles. Due to these effects, only 9 peptic fragments, which overlapped in 6 regions and covered 57% of the CD4 sequence, were observed with complexed CD4. Only the highest-quality fragments were used for analysis using scheme 2.
FIG. 2.

HDX of HIV-1 gp120 in unliganded and CD4-bound states: peptic fragments, exchange rates, sequences, and secondary structures. The rate of amide deuterium incorporation is related to the stability of backbone-hydrogen bonds, which in turn is reflective of the local conformational stability of the protein. Here the gp120 sequence is shown at the top of each row (labeled “HIV1 YU2”). Residues 252 to 483 form the outer domain of gp120, while the rest of the sequence forms the inner domain. Bars below the sequence correspond to the regions encompassed by 16 peptic fragments (explicitly numbered 1 to 16) selected for the bulk of the HDX analysis (labeled “H/D fragments”). The individual fragments are colored in rainbow fashion from the N to the C terminus in the same way as in Fig. S1 in the supplemental material. Lines under these bars represent overlapping peptic fragments, which were used to estimate experimental error (see Fig. S2 in the supplemental material) (labeled “peptic fragments”). The different styles of the lines indicate differences in glycosylation, as defined by the legend in the lower right. Below these lines, the secondary structure from the unliganded SIV gp120 crystal structure is displayed, followed by horizontal groupings of bars depicting the peptic fragments of unliganded gp120 and their frequency of deuterium incorporation (labeled “unliganded exchange” and depicted for five time points: 15, 50, 150, 500, and 1,500 s, using a gray scale gradient to indicate exchange from ≤90% [white] to ≥10% [black] incorporation, as shown in the lower right key). Under this, the secondary structure from the CD4-bound YU2 core gp120 crystal structure is displayed, followed by horizontal groupings of bars depicting the peptic fragments of CD4-bound gp120 and their frequency of deuterium incorporation (labeled “CD4-bound exchange” and depicted for the same five time points and using the same gray scale gradient as above). In general, the rate of deuterium incorporation was higher at exposed loops (e.g., loop V4) and lower at regions with secondary structure, although when this rate was averaged over the peptic fragments, which were 16 amino acids long on average, this correlation decreased. Overall, unliganded gp120 displayed a significantly lower exchange rate than a theoretically unstructured protein of the same composition, and the rate of deuterium incorporation decreased upon CD4 binding.
Local conformational stability of unliganded gp120.
Having sorted through experimental details related to obtaining HDX measurements on a highly glycosylated sample, we could begin to analyze biologically relevant results, especially with regard to the local conformational stability of unliganded gp120. HDX measurements of unliganded, deglycosylated core gp120 showed good reproducibility, with an average standard deviation of only 4% in deuterium incorporation. Meanwhile, unliganded gp120 HDX measurements across all peptic fragments ranged from 18 to 100% deuterium incorporation, with an average variation between fragments of 31% over the time course of exchange. Variations in HDX measurements were thus ∼8-fold greater than the precision of the measurements.
In light of the observed conformational diversity of core gp120 in various ligand-bound states (see Fig. S1 in the supplemental material), we examined whether this diversity was reflected in heightened exchange, as might be the case if unliganded gp120 was flexibly disordered. We observed an overall protection factor of 2.5 × 104 ± 3.3 × 103 for unliganded gp120 peptic fragments covering 98% of the core. This result indicated that unliganded gp120 exchanges at a rate >10,000-fold lower than an unstructured peptide of the same composition, suggesting that unliganded gp120 is substantially ordered.
In an analysis of the diversity of core gp120, it was clear that most of the structural variation occurs in the gp120 inner domain/bridging sheet region, while the outer domain remained essentially in the same conformation in all crystallographic studies (38). We thus examined domain-specific effects to see if the inner-domain structural diversity was reflected by heightened HDX. We observed that the inner domain showed a 21-fold-more-rapid exchange than the outer domain, a significant difference, but still 7,100-fold slower than that of an unstructured peptide of the same composition (Fig. 3). The results suggest that flexibility might play a role in the conformational diversity of unliganded gp120. However, the results also indicate that a substantial portion of the ensemble of conformations that make up the inner domain/bridging sheet appear to have considerable local conformational stability.
FIG. 3.

Global and domain-specific amide hydrogen/deuterium exchange for gp120 (left) and CD4 (right). The local conformational stability defined by HDX can be analyzed in terms of global and domain-specific effects for both gp120 (outer and inner domains) and CD4 (domain 1, which interacts with gp120, and domain 2, which does not). (A) gp120 domain-specific effects. The frequencies of deuterium incorporation into gp120 peptic fragments when unliganded (red) or CD4 bound (blue) are shown as scatter plots for the outer domain (circles) and the inner domain (triangles). Red and blue horizontal bars indicate averages and standard deviations between peptic fragments, and black horizontal bars indicate standard deviations between experiments. Gray lines connect the same peptic fragments between unliganded and CD4-bound states. Although CD4 interacts primarily with the outer domain, fragments from both the inner and outer domains of gp120 show considerably changes in rate of deuterium incorporation when bound by CD4. (B) CD4 domain-specific effects. The frequencies of deuterium incorporation into CD4 peptic fragments when free (red) or gp120 bound (blue) are shown in a scatter plot, displayed in a manner analogous to that described for panel A. In contrast to the results in panel A, only fragments of CD4 that interact with gp120 show significant changes in rate of deuterium incorporation. (C) Domain-specific protection factors. The average protection factors are shown in bar graphs for gp120 (left) and CD4 (right). Protection factors are the theoretical rates of unstructured peptides divided by the experimentally obtained rates of fragment exchange. Errors were obtained using scheme 2 (see Fig. S2 in the supplemental material).
In terms of still finer spatial resolution, different levels of conformational stability were observed for different regions of unliganded gp120. Here, the free energies were mapped onto the SIV unliganded structure since it better illustrates the possibility of large conformational changes (Fig. 4). Two peptic fragments (the N terminus of the inner domain as well as the V4 loop on the outer domain) (Fig. 4A and Table 1) appeared particularly flexible, exhibiting low ΔG values of conformational stability and complete exchange within 15 s, more than 4 standard deviations above the average exchange rate. Fast exchange for the N terminus (fragment 1, R81-N92) and the V4 loop (fragment 12, W393-N413) was expected, as these regions were mostly unstructured in the unliganded crystal structure (6). Two particularly stable fragments were observed. These encompassed part of the β-sandwich of the inner domain (fragment 16, Y486-E492) and a portion of α2-helix of the outer domain (fragment 9, T336-Q344). The hydrogen-bonding structure inherent in α-helixes may stabilize fragment 9. Fragment 16 is partially buried and intimately associated with the N-terminal β-sandwich, which is the only portion of the inner domain that is conserved between unliganded and CD4-bound states (6, 30), and structural analysis of a gp120 with an intact gp41 interactive region suggests that this β-sandwich is structurally conserved (38). The results suggest that at the extremes of fast and slow exchange, HDX measurements reflect crystallographic expectations. However, domain-specific effects were more subtle, with a far less substantial aggregate difference in HDX between the outer domain (which, crystallographically, appears conformationally stable) and the inner domain/bridging sheet (which, crystallographically, exhibit substantial conformational diversity).
FIG. 4.

HDX-determined conformational stability as mapped onto gp120 and CD4 crystal structures. Combining HDX-determined stability data with atomic-level structural information allows the local conformational stability of gp120 and CD4 to be visualized. (A) Representations of local gp120 conformational stability. Energies of HIV-1 gp120 conformational stability for 16 peptic fragments (Table 1) were mapped onto a homology model of unliganded gp120 (top row) and onto the CD4-bound crystal structure of YU2 core gp120 (second row). Structures are displayed in Cα-worm representation. Peptic fragments are colored and numbered according to their positions in sequence (using the same color scheme described in the legend to Fig. 2 and in Fig. S1 in the supplemental material), with Cα-worm thicknesses corresponding to energies of conformational stability (as shown in the key on the right). Dotted lines indicate regions that were not measured. Although the conformational diversity of unliganded gp120 suggests that it would be represented more accurately as an ensemble of structures, we nonetheless have chosen a single structure, the unliganded SIV crystal structure, to facilitate representation. Alternative representations with gp120 shown in b12-bound and CD4-bound conformations are shown in Fig. S6 in the supplemental material. (B) Molecular surface representations of gp120 and CD4. gp120 and the gp120-binding site on CD4 are colored red, while CD4 and the CD4-binding site on gp120 are colored yellow. The inner and outer domains of gp120 and domains 1 and 2 of CD4 are labeled. (C) Representations of local CD4 conformational stability. Energies of conformational stability for nine peptic fragments of CD4 (see Table S1 in the supplemental material) are mapped onto the free structure of CD4 (left) and the gp120-bound crystal structure from the YU2 core-CD4 complex (right). Structures and colors use a scheme analogous to that defined for panel A. (D) Plot of change in conformational stability of fragments from gp120 (yellow) and CD4 (red) against how much of these fragments fall within the binding interface. CD4 displays change in stability only at the interface, while gp120 displays change outside it. (E) Plot of change in conformational stability of fragments from gp120 and CD4 against the root-mean-square deviations (RMSD) of the same fragments going from unliganded to liganded conformations. There is no correlation between change in stability and crystal structure-implicated motions of the proteins (see Fig. S5 in the supplemental material). Images were generated with PyMOL.
Local conformational stabilities of free and gp120-bound CD4.
The extensive crystallographic structure information available for CD4 (30, 39, 41) as well as the similarity in structure between free and gp120-bound forms suggested that its analysis would serve as a good comparison for the gp120 analysis. HDX measurements of free CD4 were highly reproducible, with an average standard deviation of 3% in deuterium incorporation over all time points, which was 8-fold lower than the average variation between different fragments.
Overall, free CD4 displayed a protection factor of 5.1 × 104 ± 1.3 × 103. This corresponds to an HDX rate 4.5 ± 0.6-fold lower than the average exchange rate for unliganded gp120. The highest level of fragment deuterium incorporation (∼90% at 1,500 s for C16-W28 and F98-T115) was less than that observed for the most flexible portions of unliganded gp120 after 15 s (Table 1; see Table S5 in the supplemental material). Meanwhile, the most stable fragments of free CD4 were comparable in exchange rate to the most stable portions of unliganded gp120.
HDX measurements of gp120-bound CD4 could be made for only six peptic fragments, four fewer than with free CD4, and only five of these fragments contained duplicate observations. Nonetheless, with an average standard deviation of 4.0% in HDX over all time points, these measurements were only slightly less reproducible than those of unliganded gp120, and substantially less (5.9-fold lower) than the average variation in exchange between fragments. When CD4 bound to gp120, its HDX slowed by 7.0 ± 1.7-fold, with a protection factor of 1.5 × 105 ± 3.6 × 104. These overall numbers obscure the fact that a statistically significant reduction in flexibility was observed in only two fragments in domain 1 (P value < 0.001), which is the part of CD4 that contacts gp120 (fragment 3, K29-F43; fragment 4, K46-T81) (Fig. 4B and C; see Table S5 in the supplemental material). Thus, the overall protection factor of domain 1 increased much more than the protection factor of domain 2 (Fig. 3C). Also, this increase in conformational stability correlated significantly with the surface area involved in interface contact (R2 = 0.995, P value of 0.0004 from Pearson's correlation), suggesting that reduction in flexibility was isolated to the site of binding (Fig. 4D).
Local conformational stability for CD4-bound gp120 and comparison with gp120 in an unliganded state.
With HDX measurements for unliganded gp120 as well as for free and gp120-bound CD4 providing a context, we could proceed with an analysis of CD4-bound gp120. HDX measurements, when corrected for complex dissociation, had an average standard deviation of 4.4% deuterium incorporation over all time points. This was 7.5-fold lower than the 33% average variation between peptic fragments and similar in reproducibility to that of unliganded gp120. Globally, a significant reduction in HDX from unliganded gp120 was observed. The overall exchange rate of CD4-bound gp120 was (3.7 ± 0.1) × 10−3 s−1, which was 10 ± 1.8-fold lower than that of unliganded gp120. This translated to a protection factor of 2.46 × 105 ± 9.5 × 103. The inner-domain exchange rate was 11-fold higher than that of the outer domain, compared to 21-fold higher when gp120 was unliganded (Fig. 3C). CD4 binding reduced the exchange of the inner domain by 6-fold and the exchange of the outer domain by 3-fold.
Unlike the case with CD4, the overall reduction in deuterium incorporation was not limited to a small proportion of peptic fragments. We observed reduction in HDX in 15 of 16 peptic fragments. However, reductions for only eight of these fragments achieved statistical significance (Table 1; see Table S1 in the supplemental material). These eight fragments were located primarily on the face of gp120 containing the CD4-binding site. At the binding site, fragments 7 (V271-T283), 10 (L349-F383), 11 (N386-F391), 13 (L416-M426), and 15 (T455-L483) were significantly stabilized. Adjacent to the binding site, α1-helix (fragments 2 [N98-E106] and 3 [D107-L111]) was stabilized; farther from the binding site, on the face of gp120 opposite the site of CD4 binding, fragment 6 (L261-L265) was also stabilized.
Despite a seeming visual correlation between site of CD4 binding and reduction in gp120 HDX (Fig. 4A), an R2 value of only 0.01 was observed between changes in free energies of conformational stability and surface area involved in the binding site on gp120 (Fig. 4D). In part, this may relate to the size of some of the peptic fragments, which may obscure reductions in flexibility. For example, fragment 14 (E429-L453) is 35 residues in length but makes contact only at residues 429 and 430. A more biologically relevant effect may relate to the large structural rearrangement induced in the gp120 inner domain by CD4 binding. Notably, the largest reduction in HDX occurs at the α-helix spanned by fragment 3 (D107-L111), which does not contact CD4. This corroborates the observation by Chen et al. (6) that charged residues are buried during the conformational change between unliganded and CD4-bound states of gp120 at this α-helix. Ultimately, however, a comprehensive molecular explanation is difficult, as unliganded gp120 is likely best represented by a diverse ensemble of structures.
DISCUSSION
Conformational diversity of gp120 is a central feature of its biological function in entry and immune evasion. In terms of immune evasion, part of the conformational diversity relates to the large rearrangements induced by CD4, allowing for highly conserved surfaces that make up the coreceptor-binding surface to be hidden from the immune system prior to CD4 binding at the cell surface (27, 32). Part also relates to the decoy strategies involving the elicitation of nonneutralizing antibodies (7). The bottom line is that gp120 in many ways resembles the conformational machines involved in fusion, rather than the generally more rigid receptor-binding components (38). While the unusual nature of gp120 has been explored with methods ranging from X-ray crystallography to titration calorimetry and glutaraldehyde fixation of antigenic populations, none of these allow for a spatially resolved portrait of local conformational stability. Here we use HDX to establish such a portrait of HIV-1 gp120, in unliganded and CD4-bound states.
The usefulness of HDX as a probe of local conformational stability is well established (2, 10-12, 14, 19-21, 25, 26), and a number of different HDX methodologies have been developed. Virtually all of these use mass spectroscopy to assess the degree of deuterium incorporation in dismantled fragments, frozen after amide hydrogen/deuterium exchange by low pH and then reassembled into a spatially resolved portrait of the intact protein. We modified established methods to measure local conformational stability (Fig. 1). Many of these modifications were previously anticipated (e.g., maximum entropy methods, which extract the maximally unbiased probability distributions of exchange rates from the frequencies of exchange and their standard deviations) (34, 47). We used previously described software from S. W. Englander and colleagues to calculate the rate constants of the peptic fragments investigated by HDX as theoretically unstructured peptides (see reference 3). These rate constants were normalized by the HDX data to obtain protection factors, which allow our observations to be placed on an absolute scale (14, 26). The free energies calculated from protection factors are measures of conformational stability (13). We observed that NAME-derived energies showed significant correlation with unprocessed exchange frequencies. The average R2 values between NAME energies and unprocessed frequencies were 0.50 for unliganded gp120 and 0.76 for CD4-bound gp120 (see Table S3 in the supplemental material). When mapped onto the crystal structures of unliganded and CD4-bound gp120, HDX frequencies at 50 s generated a picture of conformational stability (see Fig. S4 in the supplemental material), extremely similar to the one produced by the free energies (Fig. 4).
We analyzed the correlation between our HDX-determined free energies of local conformational stability and crystal structure-determined parameters of B value, accessible surface area, and frequencies of hydrogen bonding and β-sheet/α-helix (see Fig. S5 in the supplemental material). Interestingly, the only statistically significant correlations were between CD4-bound gp120 and average accessible surface areas, between CD4-bound gp120 and frequencies of β-sheets/α-helices, or between gp120-bound CD4 and B value. Our results show that the conformational stabilization of gp120 by CD4 is complex, involving the fixation of regions both distal and proximal to the binding site. This is in contrast to the conformational stabilization of CD4 by gp120, which is localized to the site on CD4 that binds gp120. These observations corroborate crystal structures showing large, global conformational changes in the inner domain of gp120 and small, localized ones in CD4. However, our HDX data suggest that local conformational stability does not predispose a particular region in either gp120 or CD4 to conformational changes but does correspond to larger refolding reactions of domains. For example, crystal structures of SIV unliganded gp120 and HIV-1 CD4-bound gp120 show substantial structural rearrangement in the inner domain but little alteration of the outer domain (6, 30). Such asymmetry was observed with HDX for both HIV-1 unliganded and CD4-bound gp120: the outer domain was more than an order of magnitude more stable than the inner domain (Fig. 3C). However, no statistically significant correlation was observed between local changes in structure and changes in conformational stability over the peptic fragments (Fig. 4E).
Our results indicate that unliganded gp120 is not unstable. How then to explain the extraordinary reduction in entropy in the gp120-CD4 binding reaction if both unliganded gp120 and free CD4 are not unstructured? Theory indicates that reductions in entropy can come from two factors: protein folding or solvent release. Crystallographic studies suggest that at least part of the entropic reduction relates to the release of ordered solvent from the surface of a highly hydrated unliganded gp120 (6, 30). However, while changes in surface burial correlated well with changes in conformational stability for CD4 (R2 = 0.99), no correlation was observed with gp120, suggesting that solvent exclusion plays a general but complicated role in conformational stability. Likely a combination of solvent effects and the sampling of fewer, more stable, structural conformations together explain observed entropic changes (Fig. 5).
FIG. 5.

Model of the transition from unliganded to CD4-bound states of gp120. HDX results combined with numerous crystal structures suggest that the unliganded state of HIV-1 gp120 consists of a diverse ensemble of relatively stable conformations, with slow interconversion between different, highly solvated conformations. The left side of the figure shows Cα-ribbon representations for polypeptides from different gp120 crystal structures, colored from the N terminus (red) to the C terminus (purple) as in Fig. 2 and 4 and Fig. S1 in the supplemental material. Blue spheres represent solvating waters. HDX of unliganded and CD4-bound gp120 confirm the conformational stabilization of gp120 by CD4, particularly around the surface of gp120 involved in contact with CD4. However, much of the stabilization is not within the interface and therefore does not correlate with solvent exclusion. Also, regions in the inner domain (with no CD4 contact) appear to have been more stabilized than those in the outer domain (which actually contact CD4). This is in line with evidence from crystal structures of gp120 bound to different ligands that show greater conformational differences in the inner domain. The right side of the figure illustrates the changes in gp120 upon CD4 complexation. Two features are highlighted, the change in gp120 conformational stability and the loss of solvent at the interface of gp120 with CD4. Overall, this is consistent with a model of CD4 inducing allosteric changes that fix the conformation of one side of gp120. Surfaces on gp120 facing away from the site of CD4 interaction are only slightly affected.
Recently, analysis of HIV-1 gp120 with intact N and C termini (38) indicated that a “layered” gp120 architecture, with considerable conformational diversity in the inner domain, may account for a portion of the high entropy content of the unliganded state. The gp120 layers are not unstructured in the manner of a flexible loop but exist as a diverse ensemble of conformations, each significantly different from the other and each with significant conformational stability, all anchored to a conformationally invariant β-sandwich. Binding by CD4 reduces this diverse and highly entropic, though not unstructured, ensemble to a “single” conformation. In line with this, our results show that gp120 fragments spanning the β-sandwich (fragments 1 [R81-N92], 5 [Q114-F223], and 16 [Y486-E492]) display much less change in conformational stability upon CD4 binding than fragments spanning the layers (fragments 2 [N98-E106], 3 [D107-L111], and 4 [Q114-F223]) (Table 1). In particular, our data suggest that stabilization of the α-helix spanned by fragment 3 (D107-L111) is important for the conformational convergence in the inner domain. The present HDX analysis of HIV-1 gp120 thus provides not only definition of local conformational flexibility but also insight into the thermodynamic and structural parameters governing the CD4-gp120 binding reaction.
Supplementary Material
Acknowledgments
We thank S. W. Englander and Z. Zhang for Excel macros and LAPLACE software used in data analysis, E. Freire, Q. Sattentau, A. Schön, L. Shapiro, and members of the Structural Biology Section, Vaccine Research Center, for discussions or comments on the manuscript, V. Gopalan and Y. Huyen for implementing a Web-based version of the HDX analysis, M. Posner for antibody F105, J. Stuckey for assistance with figures, and the NIH AIDS Research and Reference Reagent Program for CD4.
Support for this work was provided by the NIH Intramural Research Program, a grant from the Bill and Melinda Gates Foundation Grand Challenges in Global Heath Initiative, and the NIH/Oxford/Cambridge Graduate Partnerships Program.
Footnotes
Published ahead of print on 21 July 2010.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Acta Crystallogr. D. Biol. Crystallogr. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760-763. [DOI] [PubMed] [Google Scholar]
- 2.Baerga-Ortiz, A., C. A. Hughes, J. G. Mandell, and E. A. Komives. 2002. Epitope mapping of a monoclonal antibody against human thrombin by H/D-exchange mass spectrometry reveals selection of a diverse sequence in a highly conserved protein. Protein Sci. 11:1300-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bai, Y., J. S. Milne, L. Mayne, and S. W. Englander. 1993. Primary structure effects on peptide group hydrogen exchange. Proteins 17:75-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brunger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54:905-921. [DOI] [PubMed] [Google Scholar]
- 5.Chan, D. C., D. Fass, J. M. Berger, and P. S. Kim. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263-273. [DOI] [PubMed] [Google Scholar]
- 6.Chen, B., E. M. Vogan, H. Gong, J. J. Skehel, D. C. Wiley, and S. C. Harrison. 2005. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature 433:834-841. [DOI] [PubMed] [Google Scholar]
- 7.Chen, L., Y. D. Kwon, T. Zhou, X. Wu, S. O'Dell, L. Cavacini, A. J. Hessell, M. Pancera, M. Tang, L. Xu, Z. Y. Yang, M. Y. Zhang, J. Arthos, D. R. Burton, D. S. Dimitrov, G. J. Nabel, M. R. Posner, J. Sodroski, R. Wyatt, J. R. Mascola, and P. D. Kwong. 2009. Structural basis of immune evasion at the site of CD4 attachment on HIV-1 gp120. Science 326:1123-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colman, P. M., and M. C. Lawrence. 2003. The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol. 4:309-319. [DOI] [PubMed] [Google Scholar]
- 9.Dalgleish, A. G., P. C. Beverley, P. R. Clapham, D. H. Crawford, M. F. Greaves, and R. A. Weiss. 1984. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 312:763-767. [DOI] [PubMed] [Google Scholar]
- 10.Davidson, W., L. Frego, G. W. Peet, R. R. Kroe, M. E. Labadia, S. M. Lukas, R. J. Snow, S. Jakes, C. A. Grygon, C. Pargellis, and B. G. Werneburg. 2004. Discovery and characterization of a substrate selective p38alpha inhibitor. Biochemistry 43:11658-11671. [DOI] [PubMed] [Google Scholar]
- 11.Ehring, H. 1999. Hydrogen exchange/electrospray ionization mass spectrometry studies of structural features of proteins and protein/protein interactions. Anal. Biochem. 267:252-259. [DOI] [PubMed] [Google Scholar]
- 12.Engen, J. R., and D. L. Smith. 2001. Investigating protein structure and dynamics by hydrogen exchange MS. Anal. Chem. 73:256A-265A. [DOI] [PubMed] [Google Scholar]
- 13.Englander, S. W., J. J. Englander, R. E. McKinnie, G. K. Ackers, G. J. Turner, J. A. Westrick, and S. J. Gill. 1992. Hydrogen exchange measurement of the free energy of structural and allosteric change in hemoglobin. Science 256:1684-1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Englander, S. W., and N. R. Kallenbach. 1983. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. Biophys. 16:521-655. [DOI] [PubMed] [Google Scholar]
- 15.Feng, Y., C. C. Broder, P. E. Kennedy, and E. A. Berger. 1996. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272:872-877. [DOI] [PubMed] [Google Scholar]
- 16.Guex, N., and M. C. Peitsch. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714-2723. [DOI] [PubMed] [Google Scholar]
- 17.Hamuro, Y., S. J. Coales, K. S. Molnar, S. J. Tuske, and J. A. Morrow. 2008. Specificity of immobilized porcine pepsin in H/D exchange compatible conditions. Rapid Commun. Mass Spectrom. 22:1041-1046. [DOI] [PubMed] [Google Scholar]
- 18.Hamuro, Y., S. J. Coales, J. A. Morrow, K. S. Molnar, S. J. Tuske, M. R. Southern, and P. R. Griffin. 2006. Hydrogen/deuterium-exchange (H/D-Ex) of PPARgamma LBD in the presence of various modulators. Protein Sci. 15:1883-1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamuro, Y., S. J. Coales, M. R. Southern, J. F. Nemeth-Cawley, D. D. Stranz, and P. R. Griffin. 2003. Rapid analysis of protein structure and dynamics by hydrogen/deuterium exchange mass spectrometry. J. Biomol. Tech. 14:171-182. [PMC free article] [PubMed] [Google Scholar]
- 20.Hamuro, Y., L. Wong, J. Shaffer, J. S. Kim, D. D. Stranz, P. A. Jennings, V. L. Woods, Jr., and J. A. Adams. 2002. Phosphorylation driven motions in the COOH-terminal Src kinase, CSK, revealed through enhanced hydrogen-deuterium exchange and mass spectrometry (DXMS). J. Mol. Biol. 323:871-881. [DOI] [PubMed] [Google Scholar]
- 21.Hoofnagle, A. N., K. A. Resing, E. J. Goldsmith, and N. G. Ahn. 2001. Changes in protein conformational mobility upon activation of extracellular regulated protein kinase-2 as detected by hydrogen exchange. Proc. Natl. Acad. Sci. U. S. A. 98:956-961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horn, J. R., B. Kraybill, E. J. Petro, S. J. Coales, J. A. Morrow, Y. Hamuro, and A. A. Kossiakoff. 2006. The role of protein dynamics in increasing binding affinity for an engineered protein-protein interaction established by H/D exchange mass spectrometry. Biochemistry 45:8488-8498. [DOI] [PubMed] [Google Scholar]
- 23.Huang, C. C., F. Stricher, L. Martin, J. M. Decker, S. Majeed, P. Barthe, W. A. Hendrickson, J. Robinson, C. Roumestand, J. Sodroski, R. Wyatt, G. M. Shaw, C. Vita, and P. D. Kwong. 2005. Scorpion-toxin mimics of CD4 in complex with human immunodeficiency virus gp120 crystal structures, molecular mimicry, and neutralization breadth. Structure 13:755-768. [DOI] [PubMed] [Google Scholar]
- 24.Huang, C. C., M. Venturi, S. Majeed, M. J. Moore, S. Phogat, M. Y. Zhang, D. S. Dimitrov, W. A. Hendrickson, J. Robinson, J. Sodroski, R. Wyatt, H. Choe, M. Farzan, and P. D. Kwong. 2004. Structural basis of tyrosine sulfation and VH-gene usage in antibodies that recognize the HIV type 1 coreceptor-binding site on gp120. Proc. Natl. Acad. Sci. U. S. A. 101:2706-2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hvidt, A., and K. Linderstrom-Lang. 1954. Exchange of hydrogen atoms in insulin with deuterium atoms in aqueous solutions. Biochim. Biophys. Acta 14:574-575. [DOI] [PubMed] [Google Scholar]
- 26.Hvidt, A., and S. O. Nielsen. 1966. Hydrogen exchange in proteins. Adv. Protein Chem. 21:287-386. [DOI] [PubMed] [Google Scholar]
- 27.Kwong, P. D., M. L. Doyle, D. J. Casper, C. Cicala, S. A. Leavitt, S. Majeed, T. D. Steenbeke, M. Venturi, I. Chaiken, M. Fung, H. Katinger, P. W. Parren, J. Robinson, D. Van Ryk, L. Wang, D. R. Burton, E. Freire, R. Wyatt, J. Sodroski, W. A. Hendrickson, and J. Arthos. 2002. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 420:678-682. [DOI] [PubMed] [Google Scholar]
- 28.Kwong, P. D., R. Wyatt, E. Desjardins, J. Robinson, J. S. Culp, B. D. Hellmig, R. W. Sweet, J. Sodroski, and W. A. Hendrickson. 1999. Probability analysis of variational crystallization and its application to gp120, the exterior envelope glycoprotein of type 1 human immunodeficiency virus (HIV-1). J. Biol. Chem. 274:4115-4123. [DOI] [PubMed] [Google Scholar]
- 29.Kwong, P. D., R. Wyatt, S. Majeed, J. Robinson, R. W. Sweet, J. Sodroski, and W. A. Hendrickson. 2000. Structures of HIV-1 gp120 envelope glycoproteins from laboratory-adapted and primary isolates. Structure 8:1329-1339. [DOI] [PubMed] [Google Scholar]
- 30.Kwong, P. D., R. Wyatt, J. Robinson, R. W. Sweet, J. Sodroski, and W. A. Hendrickson. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwong, P. D., R. Wyatt, Q. J. Sattentau, J. Sodroski, and W. A. Hendrickson. 2000. Oligomeric modeling and electrostatic analysis of the gp120 envelope glycoprotein of human immunodeficiency virus. J. Virol. 74:1961-1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Labrijn, A. F., P. Poignard, A. Raja, M. B. Zwick, K. Delgado, M. Franti, J. Binley, V. Vivona, C. Grundner, C. C. Huang, M. Venturi, C. J. Petropoulos, T. Wrin, D. S. Dimitrov, J. Robinson, P. D. Kwong, R. T. Wyatt, J. Sodroski, and D. R. Burton. 2003. Access of antibody molecules to the conserved coreceptor binding site on glycoprotein gp120 is sterically restricted on primary human immunodeficiency virus type 1. J. Virol. 77:10557-10565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li, Y., J. C. Kappes, J. A. Conway, R. W. Price, G. M. Shaw, and B. H. Hahn. 1991. Molecular characterization of human immunodeficiency virus type 1 cloned directly from uncultured human brain tissue: identification of replication-competent and -defective viral genomes. J. Virol. 65:3973-3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lisal, J., T. T. Lam, D. E. Kainov, M. R. Emmett, A. G. Marshall, and R. Tuma. 2005. Functional visualization of viral molecular motor by hydrogen-deuterium exchange reveals transient states. Nat. Struct. Mol. Biol. 12:460-466. [DOI] [PubMed] [Google Scholar]
- 35.Liu, J., A. Bartesaghi, M. J. Borgnia, G. Sapiro, and S. Subramaniam. 2008. Molecular architecture of native HIV-1 gp120 trimers. Nature 455:109-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McDonald, I. K., and J. M. Thornton. 1994. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238:777-793. [DOI] [PubMed] [Google Scholar]
- 37.Myszka, D. G., R. W. Sweet, P. Hensley, M. Brigham-Burke, P. D. Kwong, W. A. Hendrickson, R. Wyatt, J. Sodroski, and M. L. Doyle. 2000. Energetics of the HIV gp120-CD4 binding reaction. Proc. Natl. Acad. Sci. U. S. A. 97:9026-9031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pancera, M., S. Majeed, Y. A. Ban, L. Chen, C. C. Huang, L. Kong, Y. D. Kwon, J. Stuckey, T. Zhou, J. E. Robinson, W. R. Schief, J. Sodroski, R. Wyatt, and P. D. Kwong. 2010. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc. Natl. Acad. Sci. U. S. A. 107:1166-1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ryu, S. E., P. D. Kwong, A. Truneh, T. G. Porter, J. Arthos, M. Rosenberg, X. P. Dai, N. H. Xuong, R. Axel, R. W. Sweet, et al. 1990. Crystal structure of an HIV-binding recombinant fragment of human CD4. Nature 348:419-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Truhlar, S. M., C. H. Croy, J. W. Torpey, J. R. Koeppe, and E. A. Komives. 2006. Solvent accessibility of protein surfaces by amide H/2H exchange MALDI-TOF mass spectrometry. J. Am. Soc. Mass Spectrom. 17:1490-1497. [DOI] [PubMed] [Google Scholar]
- 41.Wang, J. H., Y. W. Yan, T. P. Garrett, J. H. Liu, D. W. Rodgers, R. L. Garlick, G. E. Tarr, Y. Husain, E. L. Reinherz, and S. C. Harrison. 1990. Atomic structure of a fragment of human CD4 containing two immunoglobulin-like domains. Nature 348:411-418. [DOI] [PubMed] [Google Scholar]
- 42.Weissenhorn, W., A. Dessen, S. C. Harrison, J. J. Skehel, and D. C. Wiley. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426-430. [DOI] [PubMed] [Google Scholar]
- 43.Wu, L., N. P. Gerard, R. Wyatt, H. Choe, C. Parolin, N. Ruffing, A. Borsetti, A. A. Cardoso, E. Desjardin, W. Newman, C. Gerard, and J. Sodroski. 1996. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature 384:179-183. [DOI] [PubMed] [Google Scholar]
- 44.Wyatt, R., and J. Sodroski. 1998. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 280:1884-1888. [DOI] [PubMed] [Google Scholar]
- 45.Yates, J. R., III, J. K. Eng., A. L. McCormack, and D. Schieltz. 1995. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 67:1426-1436. [DOI] [PubMed] [Google Scholar]
- 46.Yuan, W., J. Bazick, and J. Sodroski. 2006. Characterization of the multiple conformational states of free monomeric and trimeric human immunodeficiency virus envelope glycoproteins after fixation by cross-linker. J. Virol. 80:6725-6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang, Z., W. Li, T. M. Logan, M. Li, and A. G. Marshall. 1997. Human recombinant [C22A] FK506-binding protein amide hydrogen exchange rates from mass spectrometry match and extend those from NMR. Protein Sci. 6:2203-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang, Z., and D. L. Smith. 1993. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 2:522-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou, T., L. Xu, B. Dey, A. J. Hessell, D. Van Ryk, S. H. Xiang, X. Yang, M. Y. Zhang, M. B. Zwick, J. Arthos, D. R. Burton, D. S. Dimitrov, J. Sodroski, R. Wyatt, G. J. Nabel, and P. D. Kwong. 2007. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature 445:732-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.