

Abstract
Latent viruses generally defend their host cell against superinfection by nonlatent virulent mutants that could destroy the host cell. Superinfection inhibition thus seems to be a prerequisite for the maintenance of viral latency. Yet viral latency can break down when resistance to superinfection inhibition, known as ultravirulence, occurs. To understand the evolution of viral latency, we have developed a model that analyzes the epidemiology of latent infection in the face of ultravirulence. We show that latency can be maintained when superinfection inhibition and resistance against it coevolve in an arms race, which can result in large fluctuations in virulence. An example is the coevolution of the virulence and superinfection repressor protein of phage λ (cI) and its binding target, the λ oLoR operator. We show that this repressor/operator coevolution is the driving force for the evolution of superinfection immunity groups. Beyond latent phages, we predict analogous dynamics for any latent virus that uses a single repressor for the simultaneous control of virulence and superinfection.
Genomes of cellular organisms are interspaced by a multitude of latent viruses. The human genome, for example, comprises 8% of sequences of retroviral origin (35). The average number of lysogenic phages in bacterial genomes is 2.6, with some genomes containing up to 17 different lysogenic phages (10). In many cases, latent phages equip their hosts with antibiotic resistance or toxicity factors (8). Furthermore, latent infection often poses a treatment problem, since the nonreplicating provirus is unaffected by antiviral drugs (39). Even though latent, nonvirulent viruses are abundant, the evolutionary forces that lead to the evolution of latency and potentially to its breakdown are largely unexplored.
For a virus, latent infection can have genuine advantages. First, limited viral replication can reduce the cost of infection to a level that enables mutualistic interaction with the host (22). Second, the reduced virulence associated with limited viral replication prolongs host survival and increases opportunities for transmission. When transmission is rare, the benefit of increased transmission opportunities can outweigh the cost of reduced viral replication that is associated with latency. In environments with limited horizontal transmission, latent infection that prolongs host survival can therefore evolve spontaneously (3, 4). This is not the case, however, in infected host tissues or bacterial or cell cultures. In such environments, viral particles are readily transmitted between cells and the maintenance of low virulence by limited transmission breaks down. Moreover, high viral densities and abundant between-cell transmission are generally predicted to result in coinfections which select for fast-replicating, virulent mutants (5, 17, 20, 23, 24, 42, 44). Note, however, that defective interfering particles form an important exception (11, 52). Conditions of rapid transmission between cells should therefore select for fast replication and ultimately a loss of latency. When transmission is abundant, maintenance of latency therefore requires additional mechanisms.
An efficient way to maintain latency is superinfection inhibition, the prevention of infection of already-infected cells by other viral mutants of the same viral species. Superinfection inhibition is common and has been demonstrated in viruses of bacteria, plants, and animals (13, 21, 26, 28, 31, 33, 36, 40, 43, 49, 51). Superinfection inhibition can be achieved by interference with the entrance or replication of a superinfecting virus (36). Interestingly, replication interference of the superinfecting virus often occurs by the same mechanisms that grant replication control and latency of the resident virus (13, 25, 36, 38, 43, 45). The simultaneous use of replication repression for the establishment of latency and superinfection inhibition seems to be sufficient for a stable evolution of latency. Yet it is not, since superinfection inhibition by replication repression breaks down as soon as a superinfecting mutant has become resistant to the replication repressor. This case is well described for mutants of phage λ (6).
Phage λ is the best-known example for the control of latent infection, superinfection inhibition, and resistance against it. Phage λ can either integrate into the host genome as a latent prophage and “keep the host cell alive” or alternatively initiate replication and destroy the host cell through lysis. In phage λ, the switch to the lytic cycle is prevented by binding of the virulence repressor cI to the oLoR operator, which overlaps the promoters for the viral lysis and replication genes (29, 45, 46). Likewise, binding of cI to the oLoR operator also prevents replication of a superinfecting phage (Fig. 1A). Resistance to superinfection inhibition occurs by mutations that weaken the affinity of the oLoR operator for the wild-type repressor. Mutants of phage λ that carry mutations in the oLoR region are resistant to superinfection inhibition and can superinfect and destroy cells that carry a prophage (Fig. 1B). For this reason, these operator have been termed ultravirulent (6). Additionally, oLoR operator mutants lose the affinity to their own repressor and enter latency at a low frequency. Nevertheless, site-directed mutagenesis of the repressor and operator has shown that in principle, compensatory mutations in the cI gene can restore repression of a mutated operator and reestablish latency (7, 54, 55). The repressor could therefore potentially coevolve with the operator region and restore lysogeny.
FIG. 1.

Superinfection inhibition and its avoidance by ultravirulence (the example of phage λ). (A) Binding of the virulence repressor (R1) to the replication operator (O1) prevents replication of the resident and the superinfecting virus, and latency is maintained. (B) An ultravirulent operator mutant (O2) avoids repression by the resident wild-type repressor (R1) and will replicate and destroy the latently infected cell.
In spite of the detailed understanding of the repressor-operator interactions, any prediction of coevolution between the λ repressor and operator regions remains complicated since mutations in both elements have pleiotropic effects on virulence and superinfection. Additionally, the relative benefits of ultravirulent and latent mutants depend on the frequency of all other mutants in the population that can be superinfected. The outcome of coevolution is therefore difficult to predict without formal analysis. We have therefore developed a mathematical model for the evolution of virulence and superinfection of a latent virus that is inspired by the biochemical mechanisms of phage λ. This model, however, also applies to other latent viruses that use a replication repression mechanism for the simultaneous control of latency and superinfection.
THEORY
Evolution of latency under superinfection inhibition: epidemiology of a latent virus.
We consider a culture of susceptible (i.e., uninfected), S, and infected, I, host cells that grow logistically at rates r and ρ, respectively, to a maximal total cell density, K. For simplicity but without a lack of generality, we set this maximal cell density to K = 1 in the formal analysis (note that we relax this assumption in the simulations). An infected cell either transmits the latent provirus to a daughter cell with fidelity δ or, alternatively, the virus is induced and lyses host cells at rate α to produce B viral particles that irreversibly adsorb to host cells at rate bA and subsequently lysogenize with a probability bL (for parameter estimates, see references 34, 50, and 53). Therefore, the infectivity of the virus is measured by the infection rate b = bAbL. Furthermore, we do not distinguish between lysogenic and pseudolysogenic infections (41). All host cells are assumed to die at background rate m. Free viral particles, V, decay at rate mV. The dynamics of susceptible and infected host cells and the number of free viral particles can be described by the following system of equations:
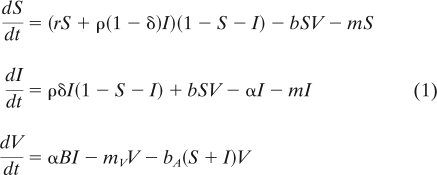 |
A virus can invade a fully susceptible host cell population which is in its equilibrium, Ŝ = 1 − m/r, when the rate of newly established infections through either vertical or horizontal transmission exceeds the rate of host cell death, which is when (see derivation in the Appendix under “Epidemiology of the virus”)
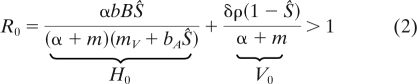 |
where H0 and V0 are the contributions of horizontal and vertical transmission to the basic reproductive ratio, R0, of the parasite (37).
The example of phage λ.
In order to understand the mechanisms that maintain low virulence and latency, we derive the viral life history directly from known biochemical mechanisms of latency and superinfection control of phage λ. The life history of phage λ has several characteristics that lead to obvious choices for model parameters and trade-off functions. First, in the absence of lysis induction by mytomicin C induction or UV irradiation, spontaneous host lysis by phage λ occurs by stochastic induction, which is assumed to have the constant average population induction rate α. In other words, the distribution of the lysis time of the phage is assumed to be exponential, with a mean value of 1/α. We further assume that every lysis event produces a constant number of viral particles, B, irrespective of the timing of the lysis event. A second important aspect of the λ life cycle is its integration into the host genome as a prophage. An integrated prophage has a small effect on host growth rate r (note that the phage nevertheless affects overall population growth through host lysis α). Furthermore, a prophage is efficiently transmitted to daughter cells (45). In the following, we will therefore assume that ρ = r and δ = 1.
When R0 is >1, the virus can invade and reaches a new endemic equilibrium (see “Epidemiology of the virus” in the Appendix). Whether the virus ends up infecting all hosts of the population or coexists with some uninfected hosts at this new equilibrium depends on several parameters and in particular on β = bB, which measures the “transmission efficiency” of the virus. We show in the Appendix that there is a threshold value, βc, of transmission efficiency above which all hosts become infected. Below this threshold, some uninfected hosts remain in the population at the endemic equilibrium.
Evolution of latency.
To study viral evolution, we now consider the competition between two viral strains and extend equation 1 to follow the dynamics of a mutant virus with virulence αM in a population of resident virus with virulence αR. Now the presence of two viral types opens the possibility that free particles of the mutant virus can superinfect cells infected with the resident IR at rate φRM and vice versa (note that the subscripts of φRM indicate the order of infection). In the Appendix (see “Evolution of the virus”), we derive the dynamics of both the resident and mutant strains. We further simplify the evolutionary dynamics by assuming that the mutation rate is relatively low compared to the speed at which the dynamical system driven by the resident virus reaches a new endemic equilibrium. This is a classical assumption in the adaptive dynamics framework (16, 19) that greatly simplifies the analysis of the model because it allows derivation of the condition for the invasion of the mutant at the endemic equilibrium set by the resident virus (see the Appendix for the characterization of this endemic equilibrium):
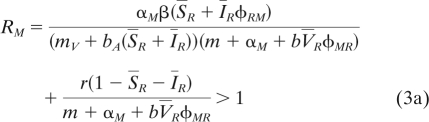 |
This invasion condition can also be written in the following way:
 |
In other words, a mutant virus with a higher virulence than the resident can invade when its additional cost of virulence (on the right side of equation 3b) is compensated by the net rate at which it gains host cells by either infection of new susceptible cells (first term above) or by superinfection of hosts infected by the resident virus. In order to understand the relative role of both the cost and the benefit of modifications of the latency of the virus, we need to examine the biochemical relations between virulence and superinfection in more detail.
Biochemical relations between rate of lysis and superinfection.
In phage λ, the rates of lysis α and superinfection φ are controlled by the binding of the lambda virulence repressor cI to the oLoR operator. Thereby, the probability, P, that a cell remains in the lysogenic state can be expressed by the first-order binding kinetics with repressor concentration c and repressor-operator affinity k as
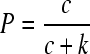 |
(4) |
Note that higher-order kinetics apply for repressor multimers and cooperative binding (29), yet the main results presented below are robust to that. Since the probability of remaining in the lysogenic state is P, the probability of triggering lysis is 1 − P. The rate of cell lysis α of the entire population should be proportional to the probability of cell lysis of individual cells, 1 − P, or
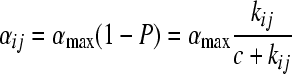 |
(5) |
where kij is the affinity of repressor type i to operator type j. Likewise, the rate of superinfection φij is proportional to the probability that the operator j of a superinfecting phage is not bound in a cell that produces the resident repressor i or
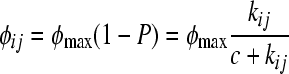 |
(6) |
From equations 5 and 6, we can see that mutations can alter virulence and superinfection either by regulating the concentration of repressor c or by structural changes in the repressor or operator that change their mutual binding affinity, kij. Note that for homologous infections, αij and φij have been shown to be affected by the multiplicity of infection (MOI) and thus by the free virus concentration (34, 54). The role of the MOI during heterologous infection, however, is less known. We therefore do not explore the role of the MOI and assume that the affinity kij is the primary determinant of αij and φij.
RESULTS
Can mutations that downregulate repressor concentration invade the population?
We will now consider a case in which only one type of repressor and operator exist which have a mutual affinity, k. Under these circumstances, how will the repressor concentration c evolve? To answer this question, we need to consider the invasion of a mutant virus with the repressor concentration cM, while the resident virus has the repressor concentration cR. The invasion condition of this mutant is obtained after substituting equations 5 and 6 into equation 3. When all the hosts are infected at equilibrium (see “Epidemiology of the virus” in the Appendix for the conditions leading to this scenario), then condition 3b can be readily used to see that a mutant can invade only when αM < αR and therefore when cM > cR. This is because in a fully infected population, a decrease in c will increase the cost of virulence αM and at the same time increase the rate, φMR, at which mutants become superinfected. In a fully infected population, the repressor level should therefore evolve to its physiological attainable maximum.
Note, however, that other assumptions could lead to the evolution of intermediate levels of repressor concentration. First, when susceptible hosts coexist with infected ones at equilibrium, reducing the level of the repressor concentration may increase the number of new infections. This could select for lower repressor levels. Second, the production of the repressor may itself be costly for the infected hosts (e.g., ρ < r), and this would allow selection for intermediate optimal levels of repressor concentration. In the rest of this article, we hence assume this quantity to be constant at c0, and we focus now on the evolution of the affinity between the repressor and the operator.
Can structural mutations in the repressor and the operator invade a wild-type population?
Let us now consider two types of repressor (R1 and R2) and operator (O1 and O2). In order to distinguish viruses by their repressor and operator types, we will refer to the densities of infected hosts, Iij, that carry a virus with repressor type i and operator type j. For simplicity we will assume that matching repressor and operator types (R1O1 and R2O2) have perfect affinity, k11 = k22 = 0, and therefore α11 = α22 = 0. Perfect affinity also implies that the repressor R1 of R1O1 and R1O2 prevents superinfection by viruses with a matching operator O1 and likewise R2 prevents superinfection by R1O2 and R2O2 (i.e., φ11 = φ22 = 0). Assuming perfect affinity, we can now answer whether a structural repressor mutant, R2O1, can invade a wild-type population, R1O1. The situation is similar to mutations that reduce the repressor concentration, mentioned above. In a fully infected host population, a repressor mutant, R2O1, cannot invade a population of R1O1 since it pays the cost of virulence (α21 > α11 = 0), yet the viral particles it produces are unable to superinfect R1O1 cells (see “Invasion of a repressor mutant” in the Appendix).
For an ultravirulent operator mutation (R1O2), the situation is different since this mutation enables superinfection of R1O1 next to its costly increase in virulence (α12 > α11 = 0). An operator mutant, R1O2, can therefore invade when its benefit of superinfection outweighs the cost of virulence. When R1O2 invades, it can either drive the wild-type R1O1 to extinction or coexist with the resident strain (see “Invasion of an ultravirulent mutant” in the Appendix). In both cases, however, this ultravirulent invasion leads to a temporary increase in virulence and free viral particles (see Fig. 3A).
Can a repressor mutant that restores latency invade an ultravirulent population?
In a population that is dominated by the ultravirulent mutant R1O2, a repressor mutation to R2 is highly beneficial, and R2O2 will invade. This is because R2 provides resistance to the ultravirulent R1O2 and at the same time reduces virulence. R2O2 thus has an obvious benefit and will invade and replace the ultravirulent mutant R1O2 (see “Invasion of a compensatory repressor mutation” in the Appendix). This invasion by a nonvirulent, latent strain, R2O2, leads to a temporary decrease in virulence (see Fig. 3A). The new latent strain R2O2 is resistant to its own viral particles and the ultravirulent mutant R1O2 but can superinfect host lysogens of R1O1. Traditionally, viral groups which are resistant to their own particles but able to infect cells infected by other mutants are referred to as superinfection immunity groups (2, 30, 48). The invasion of R2O2 therefore marks the creation of a new superinfection immunity group.
What will happen next?
The coevolution between the repressor and the operator is unlikely to end with two immunity groups. In principle, the mutation to a new operator type, O3, is by no means less likely than the reversion from O2 to O1. A new ultravirulent mutant, if it arises, will invade provided the conditions derived in the Appendix under “Invasion of an ultravirulent mutant” are fulfilled. Dependent on whether operator reversions are more likely than mutations to new operator alleles, two scenarios are possible. In the “reversion scenario,” two immunity groups alternate, driven by the appearance of ultravirulent mutants (Fig. 2 and 3A). These nontransitive interactions enable coexistence as soon as all four mutants are present. This contrasts with the “diversification scenario,” in which new operator alleles invade and are followed by virulence compensation mutations of the repressor (see Fig. 2 and 3B). Whether one scenario is more likely than the other depends on the occurrence of reversion and diversification mutations. In the end, coevolution can be a mixture of both processes. In Fig. 3B, for example, we observed a temporary reversion from immunity group R3O3 to R1O1 before the diversification to immunity group R4O4. Finally, the force that creates immunity groups is the selective advantage of compensatory repressor mutants that provide resistance to both reversion and diversification operator mutants. This process can therefore in principle create as many different immunity groups as there are possible ultravirulent operator alleles. Operator mutations are probably numerous (6, 7). It is only when the system has exhausted the diversity of alleles on the operator that the system switches from the immunity group “invasion stage” to the cyclic “fluctuation stage” (Fig. 3B).
FIG. 2.

Ultravirulent operator mutants drive repressor evolution and create immunity groups. An ultravirulent operator mutant (R1O2) can superinfect and replace the wild type (R1O1). In turn, repressor mutants that compensate virulence (R2O2) are resistant to the ultravirulent-strain (R1O2) and invade. This process creates the two immunity groups R1O1 and R2O2. Now the operator O2 of immunity group R2O2 can either revert to R2O1 (which is ultravirulent with respect to R1O1) or form new operator alleles (e.g., O3). Resistance to the new operator allele O3 can be achieved by mutation to a matching repressor, R3. Origin and compensation of new operator alleles ultimately generate the diversity of immunity groups.
FIG. 3.

Generation and maintenance of immunity groups. (A) Simulation for two immunity groups in the operator reversion scenario. Repeated invasions lead to cycling and coexistence between two immunity groups (R1O1 and R2O2) driven by the appearance of ultravirulent operator mutations (R1O2 and R2O1). Invasions of ultravirulent mutants are marked by a steep increase of free viral particles. (B) New operator mutations drive the repeated repressor compensation and invasion of immunity groups. This invasion phase is followed by a fluctuation phase, in which previous immunity groups reappear. Diversification of immunity groups leads to an overall increase in virulence and free viral particles. We simulated equations in the Appendix (see “Invasion condition”), extended for 2 and 4 immunity groups, using NDSolve in the Mathematica 7.0 software program. Mutations to new operator and repressor types occur at rate 10−9 h−1. Simulation parameters: K = 105 cells; r = ρ = 1.3 h−1; m = 0.1 h−1; mv = 0.01 h−1; αii = 0.01 h−1; αij = 0.6 h−1; φii = 0.01; φij = 0.6; b = 6·10−9 h−1·virus−1; bA = 10−7 h−1·cells−1; B = 100 virus·cells−1; initial condition I11 = 1.
One interesting exception that breaks fluctuation would be the occurrence of a mutant which carries a complete deletion of the operator region known, as λvir. The λvir mutant is insensitive to all repressors and can therefore infect and lyse all immunity groups. At the same time, λvir is a fully lytic phage and cannot form any lysogens. So how can a lysogen persist in the face of operator deletion mutants such as λvir? The invasion condition for λvir is the same as the one derived for an ultravirulent mutant (see “Invasion of an ultravirulent mutant” in the Appendix). The only difference is that a compensatory mutation restoring lysogeny becomes impossible. Our analysis shows that there is a threshold value of transmission efficiency below which the lytic phage will not invade. Above this value, however, the lytic virus will invade and may drive the λ wild type to extinction. Yet because λvir obligately kills its host cells, it may select for host resistance through mutations in the lamB receptor (14). As soon as the host population is resistant at the lamB receptor, λvir is unable to infect and is prone to extinction. On the contrary, the prophage in the genomes of lamB receptor mutant hosts would survive and could “wait for better times” (e.g., an influx of nonresistant lamB hosts). The occurrence of λvir is therefore not likely to eradicate all immunity groups. Interestingly, the escape from host resistance at the lamB receptor could provide an additional benefit of lysogeny, and yet it cannot explain the origin of superinfection immunity groups. In order to explain the origin of immunity groups, we therefore focus on the scenario where latent infection has a low cost and host resistance is primarily caused by superinfection inhibition. In this scenario, the key result of our analysis is that operator mutations which escape repression, as well as repressor mutations that restore repression to new operator types, have very strong fitness advantages and will sequentially invade (Fig. 3A). This process of sequential invasion can potentially explain the generation and coexistence of a very large number of immunity groups (Fig. 3B). It is noteworthy that as the number of immunity groups increases, higher virulence and production of free viral particles will evolve. As the number of immunity groups increases, more and more cells in the population become sensitive to superinfection, and virulence and production of free viral particle may become advantageous. As a consequence, the diversification of immunity groups can lead to levels of higher virulence and can therefore be an important factor for which latency ultimately brakes down.
DISCUSSION
The latent provirus state is generally considered a dormant state that leaves little room for viral evolution. Yet is the viral latent stage really a dormant state? If one considers coevolution between superinfection inhibition and resistance against it (ultravirulence), it becomes apparent that viral evolution plays an essential role in the maintenance of latent infection. Ongoing evolution and diversification of superinfection inhibition types seem to be a hallmark of latency rather than the exception. This view of coevolution between superinfection inhibition mechanisms and avoidance thereof could provide a simple mechanism to explain intrinsic fluctuations in virulence and viral load during latent viral infection and can explain the origin of superinfection immunity groups. In principle, the predictions for these dynamics reach beyond phage λ and are expected for any virus that uses a single repressor for the simultaneous control of latency and superinfection.
The coexistence of superinfection inhibition groups not only is predicted theoretically but also can be observed in natural isolates of lysogenic phage. Lysogenic phages fall into distinct superinfection immunity groups that are resistant to infection by particles from their own group, but susceptible to superinfection by lysogenic phages from other immunity groups (2, 30, 48). Yet the evolutionary process that generates immunity groups has not been described previously. Our model demonstrates that sequential invasion of ultravirulent mutants and compensatory repressor mutations can provide a simple scenario for the evolution of superinfection immunity groups. At the same time, it is apparent from our analysis that the process of diversification into immunity groups prolongs the maintenance of latency in the face of ultravirulent mutants. This prediction should hold for any virus that uses repressor/target matching mechanisms for the simultaneous control of latency and superinfection inhibition.
Control of latency and superinfection by repressor/target matching has been described for several groups of viruses that cause latent or chronic infections, including herpes- and retroviruses. In herpes simplex virus (HSV-1), for example, the latency-associated transcripts (LATs) establish latency and specific superinfection inhibition against other HSV-1 particles (38). Moreover, in foamy virus, the Bet protein blocks the transactivator tas in order to stabilize latency. At the same time, expression of Bet provides superinfection inhibition (43). This suggests that Bet controls the latency and superinfection inhibition by a principle which is analogous to that of the repressor-operator system of lysogenic phages. Similar coevolutionary dynamics could therefore be expected for these groups of herpes- and retroviruses. One hallmark of these coevolutionary dynamics is the repeated resurgence of high viral loads and episodes of high virulence during the invasion of ultravirulent mutants (Fig. 3A and B). One should therefore carefully consider whether the invasion of ultravirulent mutants could explain the fluctuations in viral load that are commonly observed in clinical retrovirus infection (15). Based on our results, we would predict that fluctuations in viral load can occur independently of the immune system and should therefore also be observed in cell culture.
Coevolution of replication repression mechanisms not only applies to latent viruses but might also be of importance for viruses which maintain chronic infection by limiting their virulence through a replication repressor. The single-stranded DNA phage M13, for example, limits its damage to the host cell by autorepression of its replication through binding of the viral protein pV to the single-stranded DNA (ssDNA) in order to limit the number of double-stranded DNA (dsDNA) replicative forms (RF). Additionally, pV inhibits translation of the nicking enzyme pII, which is required to relax the supercoiled RF during rolling-circle replication (47). These two repression functions of pV could also prevent replication of superinfecting phage, yet superinfection inhibition in M13 remains to be demonstrated. Nevertheless, superinfection inhibition is known in other filamentous phages which integrate into the host genome (12, 32). Since the mechanisms of replication repression of chronically infecting filamentous phages are analogous to the simultaneous repression of latency and superinfection in latent viruses, similar coevolutionary dynamics between the virulence repressor pV and its binding target (the pII mRNA) should therefore be expected.
The detailed understanding of the molecular mechanisms of superinfection inhibition show that coevolution between the genes that provide superinfection immunity and mutations that avoid it could apply for large groups of latent and chronic viruses. In many other classes of chronic and latent viruses, the invasion of ultravirulent mutants could therefore explain transient increases in viral load and virulence and ultimately could explain the origin of superinfection immunity groups. The existence of immunity groups, however, has rarely been demonstrated outside the context of lysogenic phages. The single exception is the superinfection immunity groups which are habitually used to classify avian leucosis-sarcoma viruses (1). Besides this example, immunity groups in other chronic and latent viruses remain to be discovered. An important empirical and theoretical question that remains to be answered is how many immunity groups can coexist. For the case of phage λ, several mutations that lead to ultravirulence have been described, as well as several repressor mutations that can compensate them (7). In this way, many immunity groups can originate. Yet the pace at which immunity groups appear and disappear remains an important experimental question.
The evolution of virulence and latency is closely linked to the relative benefit of horizontal transmission. Current ideas on the evolution of the latent, nonvirulent infection state that it will evolve when transmission to susceptible hosts is rare (9, 37). In the absence of susceptible hosts, the benefits of vertical transmission to daughter cells can outweigh the benefits of virus production and horizontal transmission and stabilize a nonvirulent alliance between the host and the pathogen (22). Superinfection inhibition acts basically by the same mechanisms, since it immunizes the host population and reduces the proportion of susceptible host cells. In other words, superinfection inhibition renders the production of viral particles useless since these particles are unable to infect immune host cells. Yet the situation is drastically different when the population diverges into several immunity groups. As the number of immunity groups increases, a larger and larger proportion of the population will carry heterologous immunity and can be superinfected. In the presence of many immunity groups, the production of viral particles therefore becomes beneficial, and evolution will act against latency. The appearance of new immunity groups is therefore equivalent to the influx of susceptible cells. Thus, occurrence of immunity groups and influx of susceptible cell are “two sides of the same coin” when they act on the evolution of increased virulence.
Superinfection inhibition and resistance to it are key factors for the dynamics of latency evolution. Any model of latency evolution that does not consider resistance to superinfection inhibition is incomplete, since it misses the possibility of a breakdown of latency due to the diversification of immunity groups. Resistance to superinfection inhibition should therefore be carefully considered to understand the evolutionary dynamics of latent viruses. In particular, in long-term chronic viral infections (e.g., HIV and HSV-1), this mechanism could lead to an intrinsic fluctuation in the viral load and diversification into superinfection immunity groups and might explain the reasons for which viral latency eventually breaks down.
Epidemiology of the virus.
In the absence of the virus, the dynamics of the host is determined by the balance between density-dependent reproduction and mortality of susceptible cells:
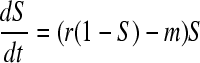 |
(7) |
At the virus-free equilibrium, the host density is thus: Ŝ = 1 − m/r. The ability of a few virus particles to invade the host population can be determined from the dynamics of the parasite when it is rare:
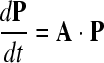 |
(8) |
where P = (I V)T is the vector of parasite densities in two states (in a host or as a free particle), and A is derived from the linearization of equation 1 at the virus-free equilibrium of the host. This matrix can be written as A = F − V, where F refers to fecundity (how many infected host cells are created by infected cells and by free viruses) and V refers to the transition between the two states of the virus:
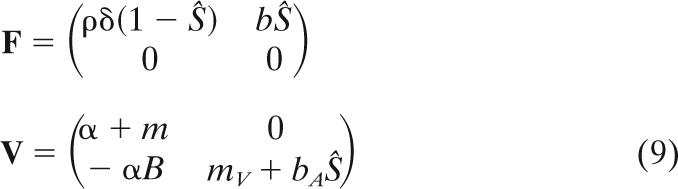 |
The parasite will invade the host virus-free equilibrium when the dominant eigenvalue of A is positive or, equivalently, when the dominant eigenvalue of M = F·V−1 is higher than 1 (18, 27). We focus on the latter criterion because it yields a per-generation measure of the parasite population growth rate. Indeed, the dominant eigenvalue of M is R0, the basic reproductive ratio of the parasite (see equation 2).
In the following, we focus on the simplified life cycle of lambda where ρ = r and δ = 1. We also define the new compound parameter β = bB, which refers to the “transmission efficiency” of the virus. When R0 is >1, the parasite invades the host virus-free equilibrium. This requires the transmission efficiency of the virus to be sufficiently high:
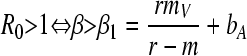 |
(10) |
If the above condition is fulfilled, the system reaches a new endemic equilibrium. Whether or not the virus coexists with uninfected cells at this endemic equilibrium also depends on the transmission efficiency of the virus. When β1 < β < β2 with
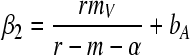 |
(11) |
then uninfected and infected hosts coexist at the endemic equilibrium:
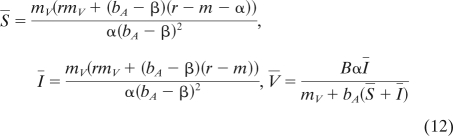 |
When β > βc = β2, however, the population reaches a fully infected state where
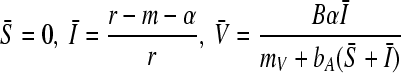 |
(13) |
Evolution of the virus. (i) Invasion condition.
Let us now assume that there are two different types of virus in the population. To determine which strain will outcompete the other, we have to follow the dynamics of the new system with a resident virus and a mutant virus (the subscripts R and M, below, refer to the resident and the mutant, respectively):
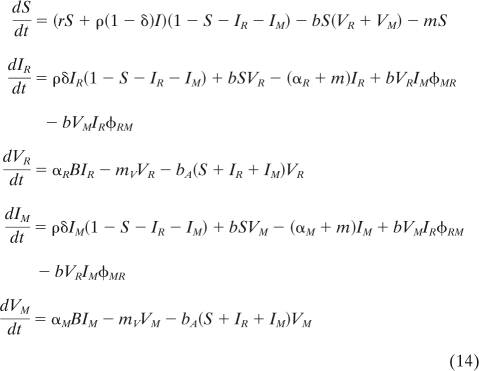 |
Let us now assume that the system has reached an endemic equilibrium (see above section) and that a new mutant virus appears (i.e., its frequency is initially rare). Will this mutant invade the virus population? Because we assume that the resident system has reached an endemic equilibrium and that the mutant is initially rare, the invasion condition can be derived from the linearization of the above system near the endemic equilibrium of the resident. We use again the next-generation method to derive the invasion condition of the mutant (18, 27). The mutant will invade when the dominant eigenvalue RM of MM = FM·VM−1 is higher than one, where
 |
This yields the following invasion condition:
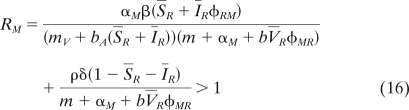 |
In the main text we give the invasion condition under the simplifying assumption that ρ = r and δ = 1.
(ii) Invasion of a repressor mutant.
Let us assume a population of R1O1 virus at its endemic equilibrium. Can a repressor mutant, R2O1, invade the resident population? For the sake of simplicity, we can assume perfect repression of matching alleles or α11 = 0, φ11 = 0, while α21 > 0 and φ21 > 0. In this case, uninfected hosts never coexist with the infected hosts at the endemic equilibrium (see “Epidemiology of the virus,” above), and the condition (equation 3b) reduces to α21 < 0. This condition is never fulfilled, and consequently, a repressor mutant cannot invade.
(iii) Invasion of an ultravirulent mutant.
Let us assume again a population of R1O1 virus at its endemic equilibrium. Can an ultravirulent mutant, R1O2 (with a mutation in the operator that weakens the binding affinity with the repressor), invade the resident population? For the sake of simplicity, we can first assume perfect repression of matching alleles: α11 = 0, φ11 = 0, while α12 > 0 and φ12 > 0. In this case, uninfected hosts never coexist with the infected hosts at the endemic equilibrium (see “Epidemiology of the virus,” above), and the condition (equation 3b) reduces to β > β1/φ12. There is thus a threshold value of β above which the ultravirulent mutant can invade.
When the ultravirulent mutant can invade, it can either reach fixation or coexist with the avirulent resident strain. The assumption that perfect matching of alleles leads to perfect repression of lysis simplifies this scenario. Indeed, in this case the resident avirulent strain R1O1 behaves like a susceptible host, while the mutant R1O2 behaves like the invading virus in the system analyzed in “Epidemiology of the virus,” above. Consequently, the two strains can coexist when β1 < φ12β < β2, with α = α12 in the above equations for β2 (see “Epidemiology of the virus”).
(iv) Invasion of a compensatory repressor mutation.
When R1O2 has invaded the resident R1O1 population, it can either go to fixation or coexist with R1O1. This new equilibrium can be invaded by a compensatory repressor mutant, R2O2, that restores lysogeny. Indeed, using again the simplifying assumption of perfect repression of matching alleles, the condition for invasion of R2O2 (equation 3b) is α12 > 0. This condition is always fulfilled and leads to the invasion of R2O2. This new immunity group may either go to fixation or coexist neutrally with the strain R1O1, belonging to the first immunity group.
Acknowledgments
We thank Michael Hochberg and Olivier Tenaillon and two anonymous reviewers for stimulating comments on a previous version and Jean-Baptiste Ferdy for inspiring discussions.
S.G. acknowledges financial support from CNRS, ANR 07 JCIC 0128 “EPICE” and ERC starting grant 203054 “EVOLEPID”. T.W.B. was partly funded by the Ubbo Emmius scholarship of the University of Groningen.
Footnotes
Published ahead of print on 21 July 2010.
REFERENCES
- 1.Adkins, H. B., S. C. Blacklow, and J. A. Young. 2001. Two functionally distinct forms of a retroviral receptor explain the nonreciprocal receptor interference among subgroups B, D, and E avian leukosis viruses. J. Virol. 75:3520-3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allison, H. E., M. J. Sergeant, C. E. James, J. R. Saunders, D. L. Smith, R. J. Sharp, T. S. Marks, and A. J. McCarthy. 2003. Immunity profiles of wild-type and recombinant shiga-like toxin-encoding bacteriophages and characterization of novel double lysogens. Infect. Immun. 71:3409-3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson, R., and R. May. 1982. Coevolution of hosts and parasites. Parasitology 85:411-426. [DOI] [PubMed] [Google Scholar]
- 4.van Baalen, M. V., and M. W. Sabelis. 1995. The milker-killer dilemma in spatially structured predator-prey interactions. Oikos 74:391-400. [Google Scholar]
- 5.van Baalen, M. V., and M. W. Sabelis. 1995. The dynamics of multiple infection and the evolution of virulence. Am. Nat. 146:881-910. [DOI] [PubMed] [Google Scholar]
- 6.Bailone, A., and R. Devoret. 1978. Isolation of ultra-virulent mutants of phage lambda. Virology 84:547-550. [DOI] [PubMed] [Google Scholar]
- 7.Benson, N., C. Adams, and P. Youderian. 1992. Mutant λ repressors with increased operator affinities reveal new, specific protein-DNA contacts. Genetics 130:17-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brabban, A. D., E. Hite, and T. R. Callaway. 2005. Evolution of foodborne pathogens via temperate bacteriophage-mediated gene transfer. Foodborne Pathog. Dis. 2:287-303. [DOI] [PubMed] [Google Scholar]
- 9.Bull. J. J., I. J. Molineux, and W. R. Rice. 1991. Selection of benevolence in a host-parasite system. Evolution 45:875-882. [DOI] [PubMed] [Google Scholar]
- 10.Casjens, S. 2003. Prophages and bacterial genomics: what have we learned so far? Mol. Microbiol. 49:277-300. [DOI] [PubMed] [Google Scholar]
- 11.Chao, L., K. Hanley, C. Burch, C. Dahlberg, and P. Turner. 2000. Kin selection and parasite evolution: higher and lower virulence with hard and soft selection. Q. Rev. Biol. 75:261-275. [DOI] [PubMed] [Google Scholar]
- 12.Cheng, C., H. Wang, H. Bau, and T. Kuo. 1999. The primary immunity determinant in modulating the lysogenic immunity of the filamentous bacteriophage cf. J. Mol. Biol. 287:867-876. [DOI] [PubMed] [Google Scholar]
- 13.Christen, L., J. Seto, and E. Niles. 1990. Superinfection exclusion of vaccinia virus in virus-infected cell-cultures. Virology 174:35-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clément, J. M., E. Lepouce, C. Marchal, and M. Hofnung. 1983. Genetic study of a membrane protein: DNA sequence alterations due to 17 lamB point mutations affecting adsorption of phage lambda. EMBO J. 2:77-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daar, E. S., T. Moudgil, R. d. Meyer, and D. D. Ho. 1991. Transient high levels of viremia in patients with primary human immunodeficiency virus type 1 infection. N. Engl. J. Med. 324:961-964. [DOI] [PubMed] [Google Scholar]
- 16.Day, T., and S. Gandon. 2007. Applying population-genetic models in theoretical evolutionary epidemiology. Ecol. Lett. 10:876-888. [DOI] [PubMed] [Google Scholar]
- 17.de Roode, J. C., R. Pansini, S. J. Cheesman, M. E. H. Helinski, S. Huijben, A. R. Wargo, A. S. Bell, B. H. K. Chan, D. Walliker, and A. F. Read. 2005. Virulence and competitive ability in genetically diverse malaria infections. Proc. Natl. Acad. Sci. U. S. A. 102:7624-7628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diekmann, O., J. A. Heesterbeek, and J. A. Metz. 1990. On the definition and the computation of the basic reproduction ratio R0 in models for infectious diseases in heterogeneous populations. J. Math Biol. 28:365-382. [DOI] [PubMed] [Google Scholar]
- 19.Dieckmann, U., J. A. J. Metz, M. W. Sabelis, and K. Sigmund. 2002. Adaptive dynamics of infectious diseases: in pursuit of virulence management. Cambridge University Press, Cambridge, United Kingdom.
- 20.Ebert, D., and J. J. Bull. 2003. Challenging the trade-off model for the evolution of virulence: is virulence management feasible? Trends Microbiol. 11:15-20. [DOI] [PubMed] [Google Scholar]
- 21.Ellenberg, P., M. Edreira, and L. Scolaro. 2004. Resistance to superinfection of Vero cells persistently infected with Junin virus. Arch. Virol. 149:507-522. [DOI] [PubMed] [Google Scholar]
- 22.Ferdy, J., and B. Godelle. 2005. Diversification of transmission modes and the evolution of mutualism. Am. Nat. 166:613-627. [DOI] [PubMed] [Google Scholar]
- 23.Frank, S. A. 1996. Models of parasite virulence. Q. Rev. Biol. 71:37-78. [DOI] [PubMed] [Google Scholar]
- 24.Gandon, S., V. A. Jansen, and M. van Baalen. 2001. Host life history and the evolution of parasite virulence. Evolution 55:1056-1062. [DOI] [PubMed] [Google Scholar]
- 25.Heinrich, J., M. Velleman, and H. Schuster. 1995. The tripartite immunity system of phages P1 and P7. FEMS Microbiol. Rev. 17:121-126. [DOI] [PubMed] [Google Scholar]
- 26.Huang, I., W. Li, J. Sui, W. Marasco, H. Choe, and M. Farzan. 2008. Influenza A virus neuraminidase limits viral superinfection. J. Virol. 82:4834-4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hurford, A., D. Cownden, and T. Day. 2010. Next-generation tools for evolutionary invasion analyses. J. R. Soc. Interface 7:561-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hutchison, C. A., and R. L. Sinsheimer. 1971. Requirement of protein synthesis for bacteriophage phi X174 superinfection exclusion. J. Virol. 8:121-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson, A., A. Poteete, G. Lauer, R. Sauer, G. Ackers, and M. Ptashne. 1981. Lambda-repressor and cro—components of an efficient molecular switch. Nature 294:217-223. [DOI] [PubMed] [Google Scholar]
- 30.Kameyama, L., L. Fernandez, J. Calderon, A. Ortiz-Rojas, and T. Patterson. 1999. Characterization of wild lambdoid bacteriophages: detection of a wide distribution of phage immunity groups and identification of a Nus-dependent, nonlambdoid phage group. Virology 263:100-111. [DOI] [PubMed] [Google Scholar]
- 31.Karpf, A., E. Lenches, E. Strauss, J. Strauss, and D. Brown. 1997. Superinfection exclusion of alphaviruses in three mosquito cell lines persistently infected with Sindbis virus. J. Virol. 71:7119-7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kimsey, H., and M. Waldor. 2004. The CTX phi repressor RstR binds DNA cooperatively to form tetrameric repressor-operator complexes. J. Biol. Chem. 279:2640-2647. [DOI] [PubMed] [Google Scholar]
- 33.Kliem, M., and B. Dreiseikelmann. 1989. The superimmunity gene sim of bacteriophage P1 causes superinfection exclusion. Virology 171:350-355. [DOI] [PubMed] [Google Scholar]
- 34.Kourilsky, P., and A. Knapp. 1974. Lysogenization by bacteriophage lambda. III. Multiplicity dependent phenomena occurring upon infection by lambda. Biochimie 56:1517-1523. [PubMed] [Google Scholar]
- 35.Lander, E., A. Williams, Y. Wolf, K. Wolfe, S. Yang, R. Yeh, F. Collins, M. Guyer, J. Peterson, A. Felsenfeld, K. Wetterstrand, A. Patrinos, and M. Morgan. 2001. Initial sequencing and analysis of the human genome. Nature 409:860-921. [DOI] [PubMed] [Google Scholar]
- 36.Lee, Y., D. Tscherne, S. Yun, I. Frolov, and C. Rice. 2005. Dual mechanisms of pestiviral superinfection exclusion at entry and RNA replication. J. Virol. 79:3231-3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lipsitch, M., S. Siller, and M. A. Nowak. 1996. The evolution of virulence in pathogens with vertical and horizontal transmission. Evolution 50:1729-1741. [DOI] [PubMed] [Google Scholar]
- 38.Mador, N., A. Panet, and I. Steiner. 2002. The latency-associated gene of herpes simplex virus type 1 (HSV-1) interferes with superinfection by HSV-1. J. Neurovirol. 8:97-102. [DOI] [PubMed] [Google Scholar]
- 39.Marcello, A. 2006. Latency: the hidden HIV-1 challenge. Retrovirology 3:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McAllister, W. T., and C. L. Barrett. 1977. Superinfection exclusion by bacteriophage T7. J. Virol. 24:709-711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller, R. V., and M. J. Day. 2008. Contribution of lysogeny, pseudolysogeny and starvation to phage ecology. In S. T. Abedon (ed.), Bacteriophage ecology. Cambridge University Press, Cambridge, United Kingdom.
- 42.Mosquera, J., and F. R. Adler. 1998. Evolution of virulence: a unified framework for coinfection and superinfection. J. Theor. Biol. 195:293-313. [DOI] [PubMed] [Google Scholar]
- 43.Nethe, M., B. Berkhout, and A. van der Kuyl. 2005. Retroviral superinfection resistance. Retrovirology 2:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nowak, M., and R. May. 1994. Superinfection and the evolution of parasite virulence. Proc. R. Soc. Lond. Ser. Biol. Sci. 255:81-89. [DOI] [PubMed] [Google Scholar]
- 45.Oppenheim, A. B., O. Kobiler, J. Stavans, D. L. Court, and S. Adhya. 2005. Switches in bacteriophage lambda development. Annu. Rev. Genet. 39:409-429. [DOI] [PubMed] [Google Scholar]
- 46.Ptashne, M. 1992. A genetic switch: phage lambda and higher organisms. Blackwell Publishers, Cambridge, MA.
- 47.Russel, M., and P. Model. 2006. Filamentous phage. In S. T. Abedon and R. Calendar (ed.), The bacteriophages. Oxford University Press, Bethesda, MD.
- 48.Serra-Moreno, R., J. Jofre, and M. Muniesa. 2008. The cI repressors of Shiga toxin-converting prophages are involved in coinfection of Escherichia coli strains, which causes a down regulation in the production of Shiga toxin 2. J. Bacteriol. 190:4722-4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simon, K., J. Cardamone, P. Whitakerdowling, J. Youngner, and C. Widnell. 1990. Cellular mechanisms in the superinfection exclusion of vesicular stomatitis-virus. Virology 177:375-379. [DOI] [PubMed] [Google Scholar]
- 50.Stopar, D., and S. T. Abedon. 2008. Modeling bacteriophage population growth. In S. T. Abedon (ed.), Bacteriophage ecology. Cambridge University Press, Cambridge, United Kingdom.
- 51.Susskind, M., D. Botstein, and A. Wright. 1974. Superinfection exclusion by P22 prophage in lysogens of Salmonella-Typhimurium. 3. Failure of superinfecting phage DNA to enter siea+ lysogens. Virology 62:350-366. [DOI] [PubMed] [Google Scholar]
- 52.Turner, P. E., and L. Chao. 1999. Prisoner's dilemma in an RNA virus. Nature 398:441-443. [DOI] [PubMed] [Google Scholar]
- 53.Weitz, J., Y. Mileyko, R. Joh, and E. Voit. 2008. Collective decision making in bacterial viruses. Biophys. J. 95:2673-2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wharton, R. P., and M. Ptashne. 1985. Changing the binding specificity of a repressor by redesigning an alpha-helix. Nature 316:601-605. [DOI] [PubMed] [Google Scholar]
- 55.Youderian, P., A. Vershon, S. Bouvier, R. T. Sauer, and M. M. Susskind. 1983. Changing the DNA-binding specificity of a repressor. Cell 35:777-783. [DOI] [PubMed] [Google Scholar]