

Abstract
During HIV-1 entry, binding of the viral envelope glycoprotein gp120 to the cellular CD4 receptor triggers conformational changes resulting in exposure of new epitopes, the highly conserved CD4-induced (CD4i) epitopes that are essential for subsequent binding to chemokine receptor CCR5 or CXCR4. Due to their functional conservation, CD4i epitopes represent attractive viral targets for HIV-1 entry inhibition. The aim of the present study was to select peptide ligands for CD4i epitopes on native dualtropic (R5X4) HIV-1 envelope (Env) glycoproteins by phage display. Using CD4-activated retroviral particles carrying Env from the R5X4 HIV-1 89.6 strain as the target, we performed screenings of random peptide phage libraries under stringent selection conditions. Selected peptides showed partial identity with amino acids in the extracellular domains of CCR5/CXCR4, including motifs rich in tyrosines and aspartates at the N terminus known to be important for gp120 binding. A synthetic peptide derivative (XD3) corresponding to the most frequently selected phages was optimized for Env binding on peptide arrays. Interestingly, the optimized peptide could bind specifically to gp120 derived from HIV-1 strains with different coreceptor usage, competed with binding of CD4i-specific monoclonal antibody (MAb) 17b, and interfered with entry of both a CCR5 (R5)-tropic and a CXCR4 (X4)-tropic Env pseudotyped virus. This peptide ligand therefore points at unique properties of CD4i epitopes shared by gp120 with different coreceptor usage and could thus serve to provide new insight into the conserved structural details essential for coreceptor binding for further drug development.
HIV-1 entry (reviewed in reference 2) is a multistep process initiated by binding of the outer envelope (Env) glycoprotein gp120 to the CD4 receptor. This results in conformational changes leading to exposure of CD4-induced (CD4i) epitopes for the subsequent obligatory interaction with the chemokine receptor CCR5 or CXCR4. Further conformational changes in gp120 activate the fusion peptide at the N terminus of the transmembrane glycoprotein gp41 and lead to the assembly of a six-helix bundle structure that triggers membrane fusion and internalization of the viral particles. This sequential HIV-1 entry process allows the virus to protect the functionally important and highly conserved Env entry epitopes by limiting their exposure to antibodies which may neutralize the infection (35). The complex HIV-1 entry process offers the opportunity for therapeutic intervention at multiple steps. Thus, the first HIV-1 entry inhibitor approved by the FDA (T20, enfuvirtide [Fuzeon]) is a peptide inhibiting the very last step of entry, i.e., membrane fusion, by interfering with the six-helix bundle formation (39). A second entry inhibitor (maraviroc [Selzentry]), targeting CCR5, was approved by the FDA in 2007 (46). This drug, like others whose development had to be discontinued due to liver toxicities, is a CCR5 antagonist, and its use is restricted to drug-experienced HIV-1-positive patients harboring CCR5 (R5)-tropic HIV-1 strains.
An alternative strategy for HIV-1 entry inhibition is targeting the CD4i epitopes on gp120, which interact with coreceptors. Structural studies have depicted the interactions between the bridging sheet induced in gp120 upon CD4 binding and the extracellular domains of CCR5, in particular, acidic residues and sulfated tyrosines at the N terminus of the receptor (28, 36, 44). Additional interactions occur between the third variable loop (V3) of gp120, which also mediates most of the coreceptor selectivity of infecting HIV-1 isolates, and extracellular domains of CCR5 (13, 15, 29, 41). Targeting viral CD4i epitopes may avoid the toxic effects that accompany most of the current inhibitors against cellular CCR5 and CXCR4 coreceptors and potentially simultaneously inhibit R5- and CXCR4 (X4)-tropic HIV-1 strains, since CD4i epitopes are responsible for binding to both chemokine receptors, whereas the V3 region of gp120 is mainly responsible for coreceptor selectivity (12, 27). Interestingly, to our knowledge no such inhibitors or inhibition approaches are under development, except for the selection of antibody fragments (10, 48, 49) and indirect approaches, such as the induction of neutralizing antibodies against CD4i epitopes (17, 21). Such antibodies are also generated in patients upon natural infection with HIV-1 (7, 16), and their existence proves that CD4i epitopes are a valid target for HIV-1 entry inhibition (20). However, antibodies are not ideally suited to interfere with HIV-1 at this particular step of HIV-1 entry due to their large size. The transient exposure of CD4i epitopes in the entry process seems to be less limiting for inhibition, since smaller single-chain Fv antibody fragments or domain antibodies are able to more potently neutralize at this step than complete parental IgG (37).
On the basis of the arguments presented above, in the present study we selected small-peptide ligands for CD4i epitopes on fusion-competent virus particle-associated gp120 trimers of a dualtropic HIV-1 isolate by phage display. Variants of the most frequently selected peptide sequence that showed identity to the sequences of the domains of the natural N terminus of CCR5 and CXCR4 involved in gp120 binding were synthesized and analyzed functionally in terms of their binding characteristics and their capacity to interfere with HIV-1 entry. Interestingly, a peptide optimized on systematic peptide arrays showed binding to several gp120 proteins, irrespective of CCR5 or CXCR4 coreceptor usage, thus indicating the conserved features of CD4i epitopes essential for infection that may be further exploited for the derivation of entry inhibitors.
MATERIALS AND METHODS
Production of MLV/HIV 89.6 pseudovirions.
Murine leukemia virus (MLV) particles pseudotyped with Env of the dualtropic HIV-1 isolate 89.6 were generated as described previously (45). Briefly, cells of the human stably transfected packaging cell line Fly-syn-GFP, derived from the human fibrosarcoma HT1080 cell line expressing MLV gag and pol, a codon-optimized Rev-independent HIV-1 89.6 env plasmid, and a retroviral vector genome encoding green fluorescent protein (GFP), were cultured to 70% confluence (37°C, 5% CO2) in complete Dulbecco modified Eagle medium (DMEM; Gibco) supplemented with penicillin-streptomycin (1%; Cambrex Biosciences), fetal calf serum (FCS; 10%; Pan Biotech GmbH), glutamine (1%; PAA Laboratories), and puromycin (5 μg/ml; ICN). Cells were washed with prewarmed phosphate-buffered saline (PBS) and cultured in puromycin- and FCS-free DMEM for 2 to 3 days. Pseudovirus-containing culture supernatants were collected, centrifuged (3,000 rpm, 20 min, 4°C), and filtered using a 0.45-μm-pore-size Millipore vacuum-driven filtration device. The supernatants were concentrated using a 100-kDa-molecular mass-cutoff Amicon centrifugal filter device (Millipore) and transferred to Quick-seal polyallomer centrifuge tubes (13.5 ml; Beckman). Pseudovirus particles were pelleted by centrifugation (480,000 × g, 40 min, 4°C) and resuspended in PBS after several washings.
Phage display screenings.
Virus particles pseudotyped with strain 89.6 Env were coated on 96-well enzyme-linked immunosorbent assay (ELISA) plates (overnight, 4°C; MaxiSorp; Nunc). After three washings (PBS, 2% milk), the plates were blocked (with PBS-5% milk, 2 h at 37°C) and incubated with soluble CD4 (100 nM in PBS-1% milk, 1 h at 37°C). Three random peptide phage libraries (New England Biolabs) were used in parallel to select peptide ligands for CD4i epitopes. Phage libraries (each 5 μl in 100 μl PBS-2% skim milk) were added, and the mixture was incubated overnight with gentle shaking (4°C). After 12 washings (with 400 μl cold PBS-2% milk), bound phages were eluted (250 μl 0.2 M glycine-HCl, pH 2.2, 1 mg/ml bovine serum albumin [BSA], 10 min, 37°C). Eluted phages were transferred to a fresh tube and neutralized (45 μl 1 M Tris-HCl, pH 9.1). Three rounds of positive selections were performed. After each round, the phages were amplified (37°C, 4.5 h) in Escherichia coli and precipitated (with 25% polyethylene glycol [PEG]-2.5 M NaCl by centrifugation at 13,000 rpm for 15 min at 4°C). Phage pellets were resuspended in Tris-buffered saline (200 μl) containing NaN3, vortexed, and stored at 4°C. Each round of positive selection was followed by two consecutive negative selections, one against soluble CD4 (sCD4; 100 ng per well) and one against virus particles pseudotyped with the strain 89.6 Env (coated as described above) in the absence of sCD4. Phages from the positive selection (100 μl plus 100 μl PBS-5% milk/well) were first added to the CD4-coated plates (overnight, 4°C); unbound phages in the supernatants were collected and added to the pseudovirus-coated plate (overnight, 4°C). Finally, phage supernatants were collected for titrations, the next rounds of positive selection, and, after the last biopanning round, analysis of single phages.
Analysis of single-phage clones.
After three rounds of biopanning, single-phage clones were amplified and analyzed for their specificity of binding to CD4i epitopes on gp120 by ELISA using Fly-Env cells with and without sCD4 addition as well as Fly control cells. Fly-syn-GFP cells were cultured as described above in a 96-well plate (5.5 × 104 cells/well, overnight). After gentle washing of the cells with prewarmed medium, the cells were incubated or not with sCD4 (100 nM, 1 h at 37°C, 5% CO2). Each single-phage clone (10 μl) was tested for binding to Fly-Env with sCD4, Fly-Env without sCD4, and Fly cells alone (90 min, 37°C). After bound phages were washed, the phages were detected by a horseradish peroxidase (HRP)-conjugated anti-M13 monoclonal antibody (MAb; New England Biolabs). Single-stranded phage DNA was prepared from the Env binding phages, and the peptide-encoding region was sequenced with primer M13(−96) (CCC TCA TAG TTA GCG TAA CG) on an ABI Prism 310 genetic analyzer.
Peptide synthesis.
Peptides were synthesized by 9-fluorenylmethoxy carbonyl (Fmoc) chemistry on an Applied Biosystems 433A peptide synthesizer. [15N]leucine (Eurisotop GmbH) was protected with the Fmoc group, as described previously (26). [15N]leucine was introduced with a modified Fmoc chemistry protocol. The 15N-labeled peptide was synthesized on NovaSyn TGR resin (Novabiochem), and the unlabeled, sulfated peptide was synthesized on a Rink amide resin (Novabiochem). Biotinylated peptides were synthesized on biotin-PEG NovaTag resin (Novabiochem). For the sulfated peptides, tyrosine without a side chain protecting group was coupled to the resin, and sulfation was done after assembly of the peptide by treatment with dimethylformamide-SO3 complex. The sulfated peptides were cleaved with trifluoroacetic acid (TFA)-H2O-p-cresol (90:5:5; vol/vol/vol) on ice to minimize loss of the sulfate (47). The crude peptides were purified by reverse-phase high-pressure liquid chromatography on a C18 column (Eurospher) and analyzed by electrospray ionization-mass spectrometry (Micromass ZQ 4000; Waters). Sulfated peptides were neutralized with triethylammonium acetate (pH 7) and lyophilized.
Binding of selected peptides to Env. (i) ELISA.
ELISA plates (high binding; Greiner) were coated overnight with culture supernatants (diluted 1:5 in PBS-1% BSA) from CHO cells transfected with pEE14-ADA.C1 or mock-transfected CHO cells, which were obtained from E. Reinherz (Dana Farber Cancer Insititute, Boston, MA). After the plates were blocked for 2 h with PBS-5% BSA, soluble CD4 (40 nM) was added to half of the wells for 1 h at 37°C. After the plates were washed four times with 200 μl PBS-1% BSA, biotinylated peptide XD3 variants with sulfated tyrosine (XD3-ys peptides) were added for 2 h at 37°C and detected with streptavidin-HRP (1:1,500; Dianova).
(ii) Competition ELISA for gp120 binding.
ELISA plates (high binding; Greiner) were coated overnight with 100 ng strain 89.6 gp120 (Immune Technology) per well. After the plates were blocked overnight with 5% (wt/vol) skim milk powder in PBS-Tween 20 (PBS-T), soluble CD4 (40 nM) was added to half of the wells for 1 h at 37°C. Subsequently, the plates were incubated for 2 h with 100 ng per well of biotinylated peptide XD3-ys or 100 ng per well MAb 17b for the positive controls. For competition assays, the wells were first incubated with MAb 17b (2 h, 37°C), followed by incubation with XD3-ys (2 h, 37°C), or vice versa. Peptide XD3-ys was detected with streptavidin-HRP (1:1,500; Dianova) and MAb 17b was detected with HRP-conjugated goat anti-human MAb (Bio-Rad), followed by chemiluminescence imaging. Between all incubations, the plates were washed three times with 300 μl PBS-T.
(iii) NMR.
Nuclear magnetic resonance (NMR) spectra were recorded at 300 K on a 600-MHz Bruker AvanceII spectrometer with a 5 mm CPTXI Z-GRD probe. The spectra were measured with a modified 15N, 1H heteronuclear single quantum correlation spectrocsopy method. Reference spectra were measured with 256 scans and cross spectra with 8,192 scans and a recycling delay of 2.17 s. The mixing time for the cross-correlated relaxation (CCR) evolution was 60 ms. Samples contained 120 μM peptide XD3-ys, 3 μM strain JR-FL gp120 (Immune Technologies), 3 μM sCD4 (R&D Systems), 50 mM NaCl, 50 mM phosphate buffer (pH 6.5), and 7.5% (vol/vol) D2O. CCR rates were obtained by the ratio of the intensity of cross spectra/reference spectra experiment and calculated by equation 1:
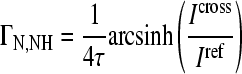 |
(1) |
where ΓN,NH is the CCR rate between the backbone N—H dipole and the chemical shift anisotropy of the backbone nitrogen, τ is mixing time/4, Icross is the cross spectrum, and Iref is the reference spectrum.
(iv) Peptide arrays.
Peptide arrays were synthesized by Fmoc chemistry at activated PEG spacers on cellulose membranes by automated parallel peptide synthesis on a MultiPep RS instrument (Intavis, Germany), as described previously (5, 32). Replicates of 12-mer peptide variants containing tyrosine sulfate, tyrosine, or phenylalanine at defined positions were generated. In order to preserve the sulfate moiety at the tyrosine, after completion of synthesis, side chains were cleaved by TFA for 8 h on ice. All following washing steps for removal of TFA were performed on ice as well. After rehydration of the membrane, unspecific binding sites were blocked with 5% (wt/vol) skim milk powder in PBS (Lonza) supplemented with 0.05% (vol/vol) PBS-T for 3 h at room temperature. Binding experiments with the gp120 variants (Immune Technology) were performed at a concentration of 50 nM in PBS-T supplemented with 1% (wt/vol) skim milk powder (PBS-TM) for 16 h at 4°C with or without preincubation (1 h at 37°C) of gp120 with 20 nM the purified ectodomain of human CD4 (Immune Technology). The membranes were washed three times for 10 min each time with PBS-T. Bound gp120 was detected with an antihistidine, horseradish peroxidase-conjugated antibody in PBS-T supplemented with 1% (wt/vol) casein, according to the instructions of the manufacturer, followed by chemiluminescence imaging. To confirm the presence of sulfotyrosines in the peptide spots, the membranes were reacted with MAb 1C-A2 (Millipore) diluted 1:1,000 in PBS-TM at 4°C overnight and detected with a goat anti-mouse HRP-conjugated MAb (Dianova) for 30 min at room temperature. HRP was detected by a specific enhanced chemiluminescent substrate detection kit (ECL; Amersham) on X-Omat films (Kodak).
HIV-1 entry inhibition assays.
The antiviral activities of the selected peptides were determined as described previously (19). Briefly, single-round infection assays were performed using luciferase reporter viruses pseudotyped with HIV-1 Env from primary R5-tropic strain D117III and X4-tropic strain NL4-3. The pseudotyped viruses were preincubated with peptide dilutions in DMEM for 1 h at 37°C and then added to U87-CD4-CCR5 and U87-CD4-CXCR4 cells, respectively. After incubation for 48 h at 37°C, the cells were lysed to determine the luciferase activity (firefly luciferase kit; Promega). All assays were performed in triplicate. The negative controls were a scrambled peptide (peptide XD3sc [acetyl-LRWPLYKYHDWQ-NH2]) and a (GS)6× peptide (acetyl-GSGSGSGSGSGS-NH2).
RESULTS
Selection of peptide ligands for CD4i epitopes on dualtropic 89.6 pseudovirions.
Functionally active stable CD4i epitopes, as confirmed by CD4i MAb binding and fusion assays with viral and producer cell-associated gp120 (see supplemental methods, results, and Fig. S1 at http://www.georg-speyer-haus.de/AGdietrich/supplfiles/jvirol/doi10.1128/JVI.00165-10), were used as targets for the selection of peptide ligands from phage-displayed peptide libraries. Three phage libraries expressing random peptide sequences of 7 or 12 amino acids in length were screened in parallel using MLV Gag Pol particles pseudotyped with HIV-1 R5X4 89.6 Env in the presence of sCD4 as the target. In order to enrich for phage binding to CD4i epitopes, we performed two negative selections, one with MLV/89.6 in the absence of sCD4 and one with sCD4 alone (see Fig. S2 at http://www.georg-speyer-haus.de/AGdietrich/supplfiles/jvirol/doi10.1128/JVI.00165-10). Three rounds of biopannings, each consisting of a positive selection followed by two negative selections, were performed. After the last round of biopanning, single amplified phage clones were analyzed by ELISA for binding to producer cell (Fly-syn-GFP)-associated 89.6 Env in the presence or absence of sCD4 (Fig. 1). Stronger binding to Fly-syn-GFP cells expressing 89.6 Env (Fly-Env) than to Fly-syn-GFP cells alone was observed for the selected phages, phages a to e; however, Env binding did not depend on the presence of sCD4 (Fig. 1A). The same was true for phages f to i, which, however, showed much lower reactivities with Fly-Env cells. Sequencing of the peptide-coding regions (Fig. 1B) revealed that phage a was selected 12 times and phage e was selected twice, whereas all others were selected only once. Some of the selected peptide sequences, including that of frequently selected phage a, showed sequence identity to amino acids of the N terminus of CCR5 and CXCR4; in particular, some of them contained the YD motif, known to be implicated in gp120 binding (Fig. 1C) (3). Interestingly, some peptide sequences contained additional amino acid motifs present in extracellular loops 1 and 3 of CCR5, which were previously described to be present in the mimotopes that we selected with two HIV-neutralizing MAbs against CCR5 (MAbs 3A9 and 5C7) and to be involved in HIV-1 entry (33, 34, 42). Due to its high reactivity, frequent selection, and strong sequence similarity to different extracellular regions of coreceptors, phage a was further analyzed. Since sulfation of tyrosines at the N terminus of CCR5 is essential for efficient gp120 binding, the corresponding peptide (XD3), including variants with sulfated tyrosines (XD3-ys), was synthesized.
FIG. 1.

Analyses of phages selected with strain 89.6 Env pseudotyped virus particles preincubated with sCD4. (A) ELISAs showing the reactivity of selected phages (phages a to i) and the wild-type (WT) control phage with 89.6 Env-expressing Fly cells without sCD4 preincubation (black bars) and with sCD4 preincubation (gray bars), as well as with Fly control cells (white bars; no Env expression, sCD4 preincubation). OD, optical density. (B) Sequences of the peptide inserts of the selected phages. Phage a was selected 12 times, and phage e was selected twice. (C) Some peptide inserts (a, b, g, h) show sequence identity with amino acids of the N terminus and motifs in the extracellular domains of the CCR5 receptor marked with arrows. Motifs in boldface type have previously been identified to be implicated in gp120 binding and virus entry. The 3A9/5C7 mimotope corresponds to a composed epitope for 3A9 and 5C7 antibodies against CCR5 identified by us previously to be important for HIV-1 entry (21). The YD-rich motifs are also found at the N terminus of CXCR4.
Synthetic peptide XD3 binds to Env and interferes with MAb 17b binding.
In order to analyze the individual contributions of particular amino acids, i.e., the tyrosines within XD3, to gp120 binding, peptide arrays with variants containing sulfated tyrosines, unmodified tyrosines, or phenylalanines were synthesized by SPOT synthesis. Since the sulfate moiety at the tyrosines is chemically unstable, we confirmed the integrity of the synthesized peptides by their reactivity with the sulfotyrosine-specific antibody 1C-A12 (Fig. 2A, bottom panel) (30). Binding of R5, X4, or dualtropic Env to the peptide membranes was analyzed both after preincubation with sCD4 and with no preincubation with sCD4 (Fig. 2). Interestingly, all Env proteins analyzed bound to the XD3 peptide variants independently of their coreceptor usage. Moreover, in all cases binding was enhanced after sCD4 preincubation, in contrast to the ELISA data (Fig. 2). The strongest binding was observed for the X4 HXB2 gp120, followed by the dualtropic 89.6 gp120, whereas binding to the R5 gp120 (strains Bal and ADA) was much weaker.
FIG. 2.

Binding of soluble gp120 proteins to peptide variants of XD3 on peptide arrays. (A and B) Peptides are numbered 1 to 16, and their sequences are shown in panel C (sulfated tyrosines are in lowercase and boldface type). Binding of the indicated His-tagged gp120 proteins, both in the presence (+) and the absence (−) of sCD4, was detected with an HRP-conjugated anti-His MAb and visualized by chemiluminescence. The lower panels are controls showing the background reactivity of the anti-His MAb with some peptides in the presence of sCD4 and the strong reactivity of the sulfated peptides with MAb 1C-A12. (B) Longer exposure times (5 times) have been used to visualize binding of R5 Env to the peptides.
The presence of sulfated tyrosines in the peptides generally enhanced the binding of HIV-1 Env proteins compared to the level of binding of the parental peptide. Furthermore, sulfation at the second tyrosine seems to be more critical for binding than sulfation at the first tyrosine, at least for some gp120 proteins (compare the reactivities of peptides 2 and 3 for R5 Bal as well as peptides 12 and 13 for all Env proteins except X4 HXB2 in Fig. 2). Scrambled peptides with sulfated tyrosines also generally showed enhanced binding compared to that of the parental peptide sequences lacking tyrosine sulfation. However, this was not always the case, indicating that the sequence context is also important for the reactivity. To further prove the specificity of binding of the XD3 peptide variants to HIV-1 gp120, we incubated a membrane containing XD3 peptides and control peptides with unrelated viral glycoproteins, influenza virus H5N1 and H1N1 hemagglutinins (see Fig. S3 at http://www.georg-speyer-haus.de/AGdietrich/supplfiles/jvirol/doi10.1128/JVI.00165-10). No binding with the XD3 peptide variants was observed, underlining the specificity for gp120 binding.
Since the presence of sulfated tyrosines in the peptides showed improved binding to Env compared to that of nonsulfated tyrosines, the XD3 parental peptide containing both tyrosines in a sulfated form (XD3-ys) was synthesized. This peptide was further analyzed for binding to Env by CCR by nuclear magnetic resonance spectroscopy and ELISA (Fig. 3). In recent years, CCR, which originates from the interference between two relaxation mechanisms (here, between chemical shift anisotropy and dipolar coupling), has emerged as a valuable method to study protein-ligand interactions (43). For a weakly bound ligand, which is in fast exchange with its free state, the CCR rate is given by the population weighted average of the bound and free state (pL,bound and pL,free, respectively, where L indicates the ligand), according to equation 2:
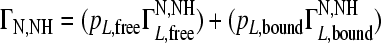 |
(2) |
The apparent CCR ΓN,NH of the peptide increases upon formation of the peptide-protein complex due to its large correlation time, while the CCR rate does not change in case of no complex formation. Thus, binding of peptides can be monitored as CCR (8) by comparing the apparent CCR rate with the CCR rate of the free peptide. Furthermore, from the ratio of the CCR rate of the free peptide and the apparent CCR rate, the molar fractions of the free and complexed peptides can be deduced, giving information about the binding. The CCR rate has previously been used for the study of peptide-protein interactions (1), sugar pucker analysis (25), and the determination of epothilon A binding to tubulin (9).
FIG. 3.

Analysis of binding of the biotinylated and tyrosine sulfated peptide XD3-ys derived from the multiple selected phage a to soluble Env. (A) NMR binding studies of XD3-ys by CCR experiments. The one-dimensional spectra of the 15N-labeled peptide are shown. Traces of the reference spectra (upper panel) and the cross spectra (divided by 2) (lower panel) from free XD3-ys peptide, XD3-ys-gp120, and XD3-ys-gp120-sCD4 are shown. From the ratio of the intensities of the peaks in the reference and cross spectra, the CCR rate can be obtained according to equation 1. Briefly, the increase in peak intensity in the cross spectra of the peptide-protein complex compared to that of the free peptide confirms the complex formation, since the cross-correlated relaxation rate increases with the apparent correlation time. The apparent correlation time is increased upon binding. (B) XD3-ys competes with MAb 17b for binding to CD4i epitopes on soluble 89.6 gp120. Binding of MAb 17b is shown without the XD3-ys peptide (white bars) and after addition of the XD3-ys peptide (black bars), both after preincubation with sCD4 and after no preincubation. Standard deviations are derived from three independent experiments, each in triplicate, with P values (***) being <0.0001. (C) XD3-ys shows concentration-dependent binding to immobilized strain ADA gp140 purified from culture supernatants of CHO cells, both in the presence (black bars) and in the absence (white bars) of sCD4. Background binding to components of culture supernatants from CHO cells that do not express Env is indicated in the presence (bars with diagonal stripes) and in the absence (bars with horizontal stripes) of sCD4. The bars represent the mean values of three replicates, and the corresponding standard deviations are indicated. OD, optical density.
We measured ΓN,NH to study the binding of the XD3-ys peptide to a complex of strain JR-FL gp120-sCD4 (Fig. 3A), using the quantitative CCR experiment, as described previously (8). On the basis of the ratio of the intensities of peptide signals in the cross and the reference spectra, CCR rates were obtained. As expected for a 1.7-kDa peptide, the rate for the free XD3-ys peptide was 0.45 ± 0.08 Hz. In contrast, ΓN,NH was increased to 1.23 ± 0.13 Hz by titration of gp120 onto the free XD3-ys peptide sample, which is explained only by the formation of the XD3-ys peptide-gp120 complex. Additional titration of sCD4 to the XD3-ys peptide-gp120 complex shows no further increase (1.13 ± 0.10 Hz), indicating that sCD4 does not affect the formation of the XD3-ys peptide-gp120 complex. Altogether, the conclusion can be drawn that XD3-ys binds to gp120 but that this binding does not depend on sCD4. From the ratio of the CCR rate of the free peptide to the apparent CCR rate, weak binding of the XD3-ys peptide to gp120 with a Kd (dissociation constant) of ∼300 μM was also concluded on the basis of the overall correlation time of 60 ns for the peptide-gp120 complex.
To further support binding of XD3-ys to CD4i epitopes on gp120, we performed competition experiments with MAb 17b for binding to strain 89.6 gp120 by ELISA. Inhibition of binding of MAb 17b to immobilized gp120 by the XD3-ys peptide was analyzed both with and without previous incubation with sCD4. The experiment was done three independent times, each in triplicate. Figure 3B summarizes these data. MAb 17b binding was inhibited about 30% after sCD4 preincubation and up to 50% in the absence of sCD4. Interestingly, similar results were observed in the reverse experiment, when XD3-ys was bound first and competed by MAb 17b (data not shown). Thus, MAb 17b and XD3-ys share a binding motif within the CD4i epitopes on gp120. After demonstrating binding of XD3-ys to monomeric gp120 (Fig. 3A and B), we further tested binding to the native trimeric form of gp140 (from the R5 ADA strain) by ELISA (Fig. 3C). Interestingly, the data showed that the sulfated peptide bound to ADA gp140, although no binding was detectable in the peptide arrays using monomeric ADA gp120 with this peptide (Fig. 2, peptide 1). Only a weak CD4 dependence of binding was observed at high peptide concentrations.
XD3-ys interferes with HIV-1 Env-dependent transduction.
As the sulfated XD3 peptide was able to bind to CD4i epitopes on viral Env with different coreceptor usage, we next analyzed if this peptide was also able to interfere with HIV-1 entry in single-round infection assays using luciferase reporter viruses pseudotyped with R5-tropic strain D117III and X4-tropic strain NL4-3 Env (Fig. 4). Interestingly, the peptide interfered with entry of both isolates, with the 50% inhibitory concentration values being about 50 μM. Thus, in contrast to peptides derived from the natural CCR5 N terminus, which do not bind to X4 isolates, the sulfated XD3 peptide ligand selected with a dualtropic Env was able to bind to HIV-1 strains independently of their coreceptor usage and to interfere with their entry.
FIG. 4.

Interference with HIV-1 entry of the sulfated XD3 peptide was analyzed in single-round infection assays with R5-tropic (D117III) and X4-tropic (NL4-3) Env pseudotyped luciferase reporter viruses. The infection rate (measured as luciferase activity 48 h after transduction in the cell lysates) decreases with increasing peptide concentrations compared to the rate for virus only (0 μg peptide). The dashed lines correspond to fitted logarithmic curves. Standard deviations refer to triplicate measurements of one representative experiment.
DISCUSSION
In the present study, we used native Env structures associated with pseudoviral particles after CD4 activation in order to select peptides targeting the CD4i coreceptor binding epitopes by phage display. As the target for the biopanning procedure, we used virus particles pseudotyped with the dualtropic strain 89.6 HIV-1 Env after incubation with soluble CD4. The correct exposure of CD4i epitopes and their functionality in Env-mediated membrane fusion under the conditions used were proved by three independent methods: by ELISA with CD4i-specific MAbs; by immunofluorescence microscopy, showing fluorescent dye transfer between labeled Env-expressing cells and unlabeled receptor (CD4 and CCR5)-expressing cells; and by transduction of a marker gene by viral particles pseudotyped with the respective Env (see Fig. S1 at http://www.georg-speyer-haus.de/AGdietrich/supplfiles/jvirol/doi10.1128/JVI.00165-10). The selection conditions applied in the biopanning procedure with viral particles pseudotyped with R5X4 Env were as stringent as possible, with the only difference between positive and negative selections being the presence of soluble CD4 in the former. This stringent setup was chosen to allow enrichment of specific binders to CD4i epitopes and may be the reason why we obtained only a low number (n = 9) of different peptide ligands. Nevertheless, one sequence was selected 12 times (Fig. 1, phage a) and showed interesting features resembling domains of the N terminus and extracellular loops of CCR5 and CXCR4 known to be involved in gp120 binding. In particular, they contained a PxYD motif, which is present at the N terminus of CCR5 and which is essential for binding to the bridging sheet of gp120 (38). Notably, the peptide insert of this phage also contained motifs related to peptide sequences that we previously selected with two MAbs against CCR5 (MAbs 3A9 and 5C7) known to interfere with HIV-1 entry. These peptides contain amino acid motifs at the N terminus and the first and third extracellular domains of CCR5 and mimic the conformational epitopes involved in HIV-1 entry (Fig. 1C, 3A9/5C7 mimotope) (3, 33, 42).
The selected phages strongly bound to native Env of dualtropic strain 89.6 in the ELISA; however, this binding was not CD4 dependent, despite the stringent selection conditions applied during the biopanning procedure (pseudovirions plus CD4 for positive selections versus pseudovirions alone for negative selections). One explanation may be that the filamentous phages with their apically presented peptides are able to bind to CD4i epitopes that are transiently exposed on the flexible structure of gp120 even without a previous interaction with CD4. Similar observations have been made for some antibodies targeting CD4i epitopes that have been reported to also bind in the absence of soluble CD4, although the binding is weaker (reviewed in reference 20; also see Fig. S1B at http://www.georg-speyer-haus.de/AGdietrich/supplfiles/jvirol/doi10.1128/JVI.00165-10). If CD4i epitopes were indeed occasionally exposed on gp120 in the absence of sCD4, this may further explain why the number of phages remaining after the negative selections was low, since part of the CD4i-specific phages will have been eliminated during the second negative selection (see Fig. S2 at http://www.georg-speyer-haus.de/AGdietrich/supplfiles/jvirol/doi10.1128/JVI.00165-10). In addition, NMR binding studies with monomeric strain JR-FL gp120 as well as the ELISA data with soluble strain ADA gp140 showed CD4-independent binding of the sulfated XD3 peptide corresponding to the insert sequence of phage a. However, in this case, CD4 independence may also be attributed to the small size of the peptide compared to the sizes of the phages or antibodies. Nevertheless, we could show that the sulfated XD3 peptide can compete with MAb 17b for binding to the strain 89.6 Env (Fig. 3B), which further proves binding of XD3-ys to CD4i epitopes. Interestingly, in the competition ELISA, MAb 17b displacement by the peptide was lower (30%) after CD4 preincubation than without (50%). Following our arguments made above, this may be due to the small size of the peptide and its better accessibility to the CD4i epitopes, even in the absence of sCD4. In the presence of sCD4, MAb 17b is able to strongly bind to the CD4i epitopes and the peptide is less efficient in competing for binding due to its lower affinity. Notably, an RNA aptamer binding to CD4i epitopes on gp120 also did this in a CD4-independent manner, supporting the findings of the present study (31).
A remarkable feature of the sulfated XD3 peptide is that it binds to both R5 and X4 as well as to dualtropic R5X4 Env on the peptide arrays (Fig. 2). This binding was specific for HIV-1 Env glycoproteins, as no reactivity with influenza virus H5N1 and H1N1 hemagglutinins was observed. The strongest binding was observed for HXB2 gp120, which is in line with a more open structure of Env and a higher neutralization sensitivity of cell line-adapted X4 viruses (40). The dualtropic strain 89.6 gp120 showed intermediate binding signals between the X4 HXB2 and the R5 Env of strains Bal and ADA. In all cases, binding was enhanced after preincubation with soluble CD4. This is in contrast to the findings of the previous binding assays, ELISA and NMR, where binding was mostly CD4 independent. The differences may be attributed to different conditions in the binding assays: whereas for ELISA and NMR the small-peptide ligands are in solution and can easily spontaneously access exposed epitopes in the coreceptor binding region of Env, the peptides are immobilized on the membranes and their access to transiently exposed epitopes on Env is limited. In this case, the soluble large Env proteins have to bind to the immobilized peptides, and this is much easier if epitopes are stably expressed after CD4 induction. This may explain the clear CD4-dependent enhancement of binding seen with the peptide arrays.
The peptide arrays also revealed improved binding of XD3 peptide variants with sulfated tyrosines independently of their position in the peptide sequences. Furthermore, the presence of sulfated tyrosines was more important for binding than the correct position of neighboring amino acids in the peptide. The importance of sulfated tyrosines for binding to CD4i epitopes on gp120 has previously been described by several groups not only for synthetic peptides derived from the N terminus of CCR5 (6, 14, 15, 23, 24, 28) but also for monoclonal antibodies targeting this region and mimicking the CCR5 receptor by long sulfated CDR3 regions (4, 11, 22, 28). Molecular details of the interactions of sulfated peptides with gp120 have recently been elucidated by NMR and X-ray crystallography and showed the formation of a sulfotyrosine binding pocket at the base of the V3 loop (28). Sulfation is also important for the recognition of syndecans on the surface of cells by gp120, where recognition is mediated via an arginine in the V3 base region (18).
The fact that the sulfated XD3 peptide binds to various Env proteins independently of their tropism underscores the conserved nature of the CD4i epitopes with respect to coreceptor binding. Peptides derived from the CDR3 region of CD4i-specific MAb E51 fused to IgG have also been shown to bind to R5 as well as to X4 Env (22). We could show binding of the CDR3 of MAb 17b, which we used as a positive control on our peptide arrays, to X4 Env (see Fig. S3 at http://www.georg-speyer-haus.de/AGdietrich/supplfiles/jvirol/doi10.1128/JVI.00165-10). In contrast, a sulfated peptide corresponding to the N terminus of CCR5 was reported to bind only to the Env proteins of R5 or dualtropic isolates and not to X4 Env (14). We speculate that the XD3 peptide that was selected with a dualtropic native Env has additional properties reflecting features essential for binding to R5 as well as to X4 Env proteins. Structural analysis could further elucidate the molecular features of CD4i epitopes conserved in HIV-1 strains independently of coreceptor usage. In line with the binding data, the sulfated XD3 peptide interfered with entry of viruses pseudotyped with both R5 (D117III) and X4 (NL4-3) Env proteins in our single-round infection assays. Inhibition of HIV-1 entry was in the high micromolar range and was thus comparable to the inhibition of HIV-1 isolates by sulfated CCR5-derived peptides (14, 24). In view of therapeutic applications, further peptide optimization and increased stability, for example, by multimerization (19) and sulfation by expression from eukaryotic cells, could lead to improved entry inhibitors targeting conserved functional domains of CD4i epitopes on gp120.
Acknowledgments
This work was supported by the Volkswagenstiftung, the Dr. Bodo Sponholz-Stiftung, and the Federal Ministry of Education and Research (BMBF Corus project).
We thank Margot Landersz, Patrizia Schult-Dietrich (GSH), and Lena Lampel (University of Erlangen) for technical assistance. We appreciate the donations of antibodies 445-72 (Susan Zolla-Pazner, New York, NY), 17b (James Robinson, Boston, MA), and CG10 (Jonathan Gershoni, Tel Aviv, Israel) as well as ADA gp140 (Ellis Reinherz). The following reagents were obtained through the NIH AIDS Research and Reference Reagent Program: U87.CD4.CCR5/CXCR4 cells (contributed by H. Deng and D. R. Littman). We thank Meike Vogler and Petra Bernardi for critical reading of the manuscript.
Footnotes
Published ahead of print on 21 July 2010.
REFERENCES
- 1.Bartoschek, S., G. Buurman, B. H. Geierstanger, J. Lapham, and C. Griesinger. 2003. Measurement and ab initio calculation of CSA/dipole-dipole cross-correlated relaxation provide insight into the mechanism of a H2-forming dehydrogenase. J. Am. Chem. Soc. 125:13308-13309. [DOI] [PubMed] [Google Scholar]
- 2.Berger, E. A., P. M. Murphy, and J. M. Farber. 1999. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 17:657-700. [DOI] [PubMed] [Google Scholar]
- 3.Blanpain, C., B. J. Doranz, J. Vakili, J. Rucker, C. Govaerts, S. S. Baik, O. Lorthioir, I. Migeotte, F. Libert, F. Baleux, G. Vassart, R. W. Doms, and M. Parmentier. 1999. Multiple charged and aromatic residues in CCR5 amino-terminal domain are involved in high affinity binding of both chemokines and HIV-1 Env protein. J. Biol. Chem. 274:34719-34727. [DOI] [PubMed] [Google Scholar]
- 4.Bowley, D. R., A. F. Labrijn, M. B. Zwick, and D. R. Burton. 2007. Antigen selection from an HIV-1 immune antibody library displayed on yeast yields many novel antibodies compared to selection from the same library displayed on phage. Protein Eng. Des. Sel. 20:81-90. [DOI] [PubMed] [Google Scholar]
- 5.Brandt, O., U. Dietrich, and J. Koch. 2009. Solid-supported peptide arrays in the investigation of protein-protein and protein-nucleic acid interactions. Curr. Chem. Biol. 3:171-179. [Google Scholar]
- 6.Brower, E. T., A. Schon, J. C. Klein, and E. Freire. 2009. Binding thermodynamics of the N-terminal peptide of the CCR5 coreceptor to HIV-1 envelope glycoprotein gp120. Biochemistry 48:779-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burton, D. R., R. C. Desrosiers, R. W. Doms, W. C. Koff, P. D. Kwong, J. P. Moore, G. J. Nabel, J. Sodroski, I. A. Wilson, and R. T. Wyatt. 2004. HIV vaccine design and the neutralizing antibody problem. Nat. Immunol. 5:233-236. [DOI] [PubMed] [Google Scholar]
- 8.Carlomagno, T., I. C. Felli, M. Czech, R. Fischer, M. Sprinzl, and C. Griesinger. 1999. Transferred cross-correlated relaxation: application to the determination of sugar pucker in an aminoacylated tRNA-mimetic weakly bound to EF-Tu. J. Am. Chem. Soc. 121:1945-1948. [Google Scholar]
- 9.Carlomagno, T., V. M. Sanchez, M. J. Blommers, and C. Griesinger. 2003. Derivation of dihedral angles from CH-CH dipolar-dipolar cross-correlated relaxation rates: a C-C torsion involving a quaternary carbon atom in epothilone A bound to tubulin. Angew. Chem. Int. ed. Engl. 42:2515-2517. [DOI] [PubMed] [Google Scholar]
- 10.Chen, W., Z. Zhu, Y. Feng, and D. S. Dimitrov. 2008. Human domain antibodies to conserved sterically restricted regions on gp120 as exceptionally potent cross-reactive HIV-1 neutralizers. Proc. Natl. Acad. Sci. U. S. A. 105:17121-17126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choe, H., W. Li, P. L. Wright, N. Vasilieva, M. Venturi, C. C. Huang, C. Grundner, T. Dorfman, M. B. Zwick, L. Wang, E. S. Rosenberg, P. D. Kwong, D. R. Burton, J. E. Robinson, J. G. Sodroski, and M. Farzan. 2003. Tyrosine sulfation of human antibodies contributes to recognition of the CCR5 binding region of HIV-1 gp120. Cell 114:161-170. [DOI] [PubMed] [Google Scholar]
- 12.Cocchi, F., A. L. DeVico, A. Garzino-Demo, A. Cara, R. C. Gallo, and P. Lusso. 1996. The V3 domain of the HIV-1 gp120 envelope glycoprotein is critical for chemokine-mediated blockade of infection. Nat. Med. 2:1244-1247. [DOI] [PubMed] [Google Scholar]
- 13.Cormier, E. G., and T. Dragic. 2002. The crown and stem of the V3 loop play distinct roles in human immunodeficiency virus type 1 envelope glycoprotein interactions with the CCR5 coreceptor. J. Virol. 76:8953-8957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cormier, E. G., M. Persuh, D. A. Thompson, S. W. Lin, T. P. Sakmar, W. C. Olson, and T. Dragic. 2000. Specific interaction of CCR5 amino-terminal domain peptides containing sulfotyrosines with HIV-1 envelope glycoprotein gp120. Proc. Natl. Acad. Sci. U. S. A. 97:5762-5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cormier, E. G., D. N. Tran, L. Yukhayeva, W. C. Olson, and T. Dragic. 2001. Mapping the determinants of the CCR5 amino-terminal sulfopeptide interaction with soluble human immunodeficiency virus type 1 gp120-CD4 complexes. J. Virol. 75:5541-5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Decker, J. M., F. Bibollet-Ruche, X. Wei, S. Wang, D. N. Levy, W. Wang, E. Delaporte, M. Peeters, C. A. Derdeyn, S. Allen, E. Hunter, M. S. Saag, J. A. Hoxie, B. H. Hahn, P. D. Kwong, J. E. Robinson, and G. M. Shaw. 2005. Antigenic conservation and immunogenicity of the HIV coreceptor binding site. J. Exp. Med. 201:1407-1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Denisova, G., B. Stern, D. Raviv, J. Zwickel, N. I. Smorodinsky, and J. M. Gershoni. 1996. Humoral immune response to immunocomplexed HIV envelope glycoprotein 120. AIDS Res. Hum. Retroviruses 12:901-909. [DOI] [PubMed] [Google Scholar]
- 18.de Parseval, A., M. D. Bobardt, A. Chatterji, U. Chatterji, J. H. Elder, G. David, S. Zolla-Pazner, M. Farzan, T. H. Lee, and P. A. Gallay. 2005. A highly conserved arginine in gp120 governs HIV-1 binding to both syndecans and CCR5 via sulfated motifs. J. Biol. Chem. 280:39493-39504. [DOI] [PubMed] [Google Scholar]
- 19.Dervillez, X., A. Huther, J. Schuhmacher, C. Griesinger, J. H. Cohen, D. von Laer, and U. Dietrich. 2006. Stable expression of soluble therapeutic peptides in eukaryotic cells by multimerisation: application to the HIV-1 fusion inhibitory peptide C46. Chem. Med. Chem. 1:330-339. [DOI] [PubMed] [Google Scholar]
- 20.DeVico, A. L. 2007. CD4-induced epitopes in the HIV envelope glycoprotein, gp120. Curr. HIV Res. 5:561-571. [DOI] [PubMed] [Google Scholar]
- 21.DeVico, A. L., R. Rahman, J. Welch, R. Crowley, P. Lusso, M. G. Sarngadharan, and R. Pal. 1995. Monoclonal antibodies raised against covalently crosslinked complexes of human immunodeficiency virus type 1 gp120 and CD4 receptor identify a novel complex-dependent epitope on gp 120. Virology 211:583-588. [DOI] [PubMed] [Google Scholar]
- 22.Dorfman, T., M. J. Moore, A. C. Guth, H. Choe, and M. Farzan. 2006. A tyrosine-sulfated peptide derived from the heavy-chain CDR3 region of an HIV-1-neutralizing antibody binds gp120 and inhibits HIV-1 infection. J. Biol. Chem. 281:28529-28535. [DOI] [PubMed] [Google Scholar]
- 23.Farzan, M., S. Chung, W. Li, N. Vasilieva, P. L. Wright, C. E. Schnitzler, R. J. Marchione, C. Gerard, N. P. Gerard, J. Sodroski, and H. Choe. 2002. Tyrosine-sulfated peptides functionally reconstitute a CCR5 variant lacking a critical amino-terminal region. J. Biol. Chem. 277:40397-40402. [DOI] [PubMed] [Google Scholar]
- 24.Farzan, M., N. Vasilieva, C. E. Schnitzler, S. Chung, J. Robinson, N. P. Gerard, C. Gerard, H. Choe, and J. Sodroski. 2000. A tyrosine-sulfated peptide based on the N terminus of CCR5 interacts with a CD4-enhanced epitope of the HIV-1 gp120 envelope glycoprotein and inhibits HIV-1 entry. J. Biol. Chem. 275:33516-33521. [DOI] [PubMed] [Google Scholar]
- 25.Felli, I. C., C. Richter, C. Griesinger, and H. Schwalbe. 1999. Determination of RNA sugar pucker mode from cross-correlated relaxation in solution NMR spectroscopy. J. Am. Chem. Soc. 121:1956-1957. [DOI] [PubMed] [Google Scholar]
- 26.Goodman, M. 2004. Houben-Weyl synthesis of peptides and peptidomimetics, vol. E22. Georg Tieme Verlag, Stuttgart, Germany.
- 27.Hartley, O., P. J. Klasse, Q. J. Sattentau, and J. P. Moore. 2005. V3: HIV's switch-hitter. AIDS Res. Hum. Retroviruses 21:171-189. [DOI] [PubMed] [Google Scholar]
- 28.Huang, C. C., S. N. Lam, P. Acharya, M. Tang, S. H. Xiang, S. S. Hussan, R. L. Stanfield, J. Robinson, J. Sodroski, I. A. Wilson, R. Wyatt, C. A. Bewley, and P. D. Kwong. 2007. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science 317:1930-1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang, C. C., M. Tang, M. Y. Zhang, S. Majeed, E. Montabana, R. L. Stanfield, D. S. Dimitrov, B. Korber, J. Sodroski, I. A. Wilson, R. Wyatt, and P. D. Kwong. 2005. Structure of a V3-containing HIV-1 gp120 core. Science 310:1025-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kehoe, J. W., N. Velappan, M. Walbolt, J. Rasmussen, D. King, J. Lou, K. Knopp, P. Pavlik, J. D. Marks, C. R. Bertozzi, and A. R. Bradbury. 2006. Using phage display to select antibodies recognizing post-translational modifications independently of sequence context. Mol. Cell. Proteomics 5:2350-2363. [DOI] [PubMed] [Google Scholar]
- 31.Khati, M., M. Schuman, J. Ibrahim, Q. Sattentau, S. Gordon, and W. James. 2003. Neutralization of infectivity of diverse R5 clinical isolates of human immunodeficiency virus type 1 by gp120-binding 2′F-RNA aptamers. J. Virol. 77:12692-12698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koch, J., and M. Mahler. 2002. Peptide arrays on membrane supports. Springer, Heidelberg, Germany.
- 33.Konigs, C., A. Pustowka, J. Irving, C. Kessel, K. Klich, V. Wegner, M. J. Rowley, I. R. Mackay, W. Kreuz, C. Griesinger, and U. Dietrich. 2007. Peptide mimotopes selected with HIV-1-blocking monoclonal antibodies against CCR5 represent motifs specific for HIV-1 entry. Immunol. Cell Biol. 85:511-517. [DOI] [PubMed] [Google Scholar]
- 34.Konigs, C., M. J. Rowley, P. Thompson, M. A. Myers, M. Scealy, J. M. Davies, L. Wu, U. Dietrich, C. R. Mackay, and I. R. Mackay. 2000. Monoclonal antibody screening of a phage-displayed random peptide library reveals mimotopes of chemokine receptor CCR5: implications for the tertiary structure of the receptor and for an N-terminal binding site for HIV-1 gp120. Eur. J. Immunol. 30:1162-1171. [DOI] [PubMed] [Google Scholar]
- 35.Kwong, P. D., M. L. Doyle, D. J. Casper, C. Cicala, S. A. Leavitt, S. Majeed, T. D. Steenbeke, M. Venturi, I. Chaiken, M. Fung, H. Katinger, P. W. Parren, J. Robinson, D. Van Ryk, L. Wang, D. R. Burton, E. Freire, R. Wyatt, J. Sodroski, W. A. Hendrickson, and J. Arthos. 2002. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 420:678-682. [DOI] [PubMed] [Google Scholar]
- 36.Kwong, P. D., R. Wyatt, J. Robinson, R. W. Sweet, J. Sodroski, and W. A. Hendrickson. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Labrijn, A. F., P. Poignard, A. Raja, M. B. Zwick, K. Delgado, M. Franti, J. Binley, V. Vivona, C. Grundner, C. C. Huang, M. Venturi, C. J. Petropoulos, T. Wrin, D. S. Dimitrov, J. Robinson, P. D. Kwong, R. T. Wyatt, J. Sodroski, and D. R. Burton. 2003. Access of antibody molecules to the conserved coreceptor binding site on glycoprotein gp120 is sterically restricted on primary human immunodeficiency virus type 1. J. Virol. 77:10557-10565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu, S., S. Fan, and Z. Sun. 2003. Structural and functional characterization of the human CCR5 receptor in complex with HIV gp120 envelope glycoprotein and CD4 receptor by molecular modeling studies. J. Mol. Model. 9:329-336. [DOI] [PubMed] [Google Scholar]
- 39.Liu, S., W. Jing, B. Cheung, H. Lu, J. Sun, X. Yan, J. Niu, J. Farmar, S. Wu, and S. Jiang. 2007. HIV gp41 C-terminal heptad repeat contains multifunctional domains. Relation to mechanisms of action of anti-HIV peptides. J. Biol. Chem. 282:9612-9620. [DOI] [PubMed] [Google Scholar]
- 40.Moore, J. P., and D. D. Ho. 1995. HIV-1 neutralization: the consequences of viral adaptation to growth on transformed T cells. AIDS 9(Suppl. A):S117-S136. [PubMed] [Google Scholar]
- 41.Nolan, K. M., A. P. Jordan, and J. A. Hoxie. 2008. Effects of partial deletions within the human immunodeficiency virus type 1 V3 loop on coreceptor tropism and sensitivity to entry inhibitors. J. Virol. 82:664-673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O'Connor, K. H., C. Konigs, M. J. Rowley, J. A. Irving, L. C. Wijeyewickrema, A. Pustowka, U. Dietrich, and I. R. Mackay. 2005. Requirement of multiple phage displayed peptide libraries for optimal mapping of a conformational antibody epitope on CCR5. J. Immunol. Methods 299:21-35. [DOI] [PubMed] [Google Scholar]
- 43.Reif, B., M. Hennig, and C. Griesinger. 1997. Direct measurement of angles between bond vectors in high-resolution NMR. Science 276:1230-1233. [DOI] [PubMed] [Google Scholar]
- 44.Rizzuto, C. D., R. Wyatt, N. Hernandez-Ramos, Y. Sun, P. D. Kwong, W. A. Hendrickson, and J. Sodroski. 1998. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science 280:1949-1953. [DOI] [PubMed] [Google Scholar]
- 45.Siegert, S., S. Thaler, R. Wagner, and B. S. Schnierle. 2005. Assessment of HIV-1 entry inhibitors by MLV/HIV-1 pseudotyped vectors. AIDS Res. Ther. 2:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vandekerckhove, L., C. Verhofstede, and D. Vogelaers. 2008. Maraviroc: integration of a new antiretroviral drug class into clinical practice. J. Antimicrob. Chemother. 61:1187-1190. [DOI] [PubMed] [Google Scholar]
- 47.Yagami, T., S. Shiwa, S. Futaki, and K. Kitagawa. 1993. Evaluation of the final deprotection system for the solid-phase synthesis of Tyr(SO3H)-containing peptides with 9-fluorenylmethyloxycarbonyl (Fmoc)-strategy and its application to the synthesis of cholecystokinin (CCK)-12. Chem. Pharm. Bull. (Tokyo) 41:376-380. [DOI] [PubMed] [Google Scholar]
- 48.Zhang, M. Y., Y. Shu, D. Rudolph, P. Prabakaran, A. F. Labrijn, M. B. Zwick, R. B. Lal, and D. S. Dimitrov. 2004. Improved breadth and potency of an HIV-1-neutralizing human single-chain antibody by random mutagenesis and sequential antigen panning. J. Mol. Biol. 335:209-219. [DOI] [PubMed] [Google Scholar]
- 49.Zhang, M. Y., Y. Shu, I. Sidorov, and D. S. Dimitrov. 2004. Identification of a novel CD4i human monoclonal antibody Fab that neutralizes HIV-1 primary isolates from different clades. Antiviral Res. 61:161-164. [DOI] [PubMed] [Google Scholar]