

Abstract
Epstein-Barr virus (EBV) requires at a minimum membrane-associated glycoproteins gB, gH, and gL for entry into host cells. B-cell entry additionally requires gp42, which binds to gH/gL and triggers viral entry into B cells. The presence of soluble gp42 inhibits membrane fusion with epithelial cells by forming a stable heterotrimer of gH/gL/gp42. The interaction of gp42 with gH/gL has been previously mapped to residues 36 to 81 at the N-terminal region of gp42. In this study, we further mapped this region to identify essential features for binding to gH/gL by use of synthetic peptides. Data from fluorescence polarization, cell-cell fusion, and viral infection assays demonstrated that 33 residues corresponding to 44 to 61 and 67 to 81 of gp42 were indispensable for maintaining low-nanomolar-concentration gH/gL binding affinity and inhibiting B-cell fusion and epithelial cell fusion as well as viral infection. Overall, specific, large hydrophobic side chain residues of gp42 appeared to provide critical interactions, determining the binding strength. Mutations of these residues also diminished the inhibition of B-cell and epithelial cell fusions as well as EBV infection. A linker region (residues 62 to 66) between two gH/gL binding regions served as an important spacer, but individual amino acids were not critical for gH/gL binding. Probing the binding site of gH/gL and gp42 with gp42 peptides is critical for a better understanding of the interaction of gH/gL with gp42 as well as for the design of novel entry inhibitors of EBV and related human herpesviruses.
Epstein-Barr virus (EBV) is a large DNA virus belonging to the family of gammaherpesviruses. The virus is transmitted through saliva, and it can infect epithelial cells, as well as B cells, which provide the host latency reservoir (1, 22). Reactivation of the virus can occur intermittently, allowing virus infection of new hosts (1). Viral reactivation from latency is quickly controlled by the immune system. Primary infection with EBV can lead to the development of infectious mononucleosis. In addition, EBV infection is associated with a variety of human cancers, such as nasopharyngeal carcinoma, Hodgkin's lymphoma, and Burkitt's lymphoma (4, 8, 9, 27, 30). EBV is an enveloped virus which contains a number of membrane glycoproteins required for membrane fusion and viral entry into the host cell. EBV-mediated entry into epithelial cells requires the three viral glycoproteins gB, gH, and gL, which are conserved among herpesviruses, and entry into B cells additionally requires the viral glycoprotein gp42 (7, 16, 17). EBV lacking gp42 can attach to B cells but cannot enter them (29). However, EBV lacking gp42 can still efficiently infect epithelial cells. In fact, gp42 acts as an inhibitor of epithelial cell infection, and recent studies suggest that the level of gp42 in the virion regulates whether EBV preferentially infects epithelial cells or B cells (2). EBV gp42 has been shown to play an essential role in membrane fusion with B cells (7, 16, 17). It binds to human leukocyte antigen class II (HLA class II) proteins expressed on B cells to trigger virus-cell membrane fusion (6, 7, 10, 16, 25).
Interestingly, EBV gp42 occurs in two forms in infected cells, a full-length membrane-bound form and a soluble form generated by proteolytic cleavage that is secreted from infected cells due to loss of the N-terminal transmembrane domain (21). Both the full-length form and the secreted gp42 form bind to gH/gL and HLA class II, and the functional significance of gp42 cleavage is not completely clear. In a virus-free cell-cell fusion assay, enhanced secretion of gp42 promotes fusion with B lymphocytes. Cleavage and secretion of gp42 are necessary for membrane fusion with B lymphocytes (24). However, membrane fusion with epithelial cells is inhibited by the presence of gp42 for both virus infection and cell-cell fusion (14, 29). This is likely due to the formation of a heterotrimeric gH/gL/gp42 complex that is unable to mediate membrane fusion with epithelial cells, possibly due to steric hindrance of gH/gL receptor binding (3, 11).
The interaction of gH/gL and gp42 plays a key role in membrane fusion, but it has not yet been fully understood. The crystal structures of a gH/gL/gp42 complex and gH/gL alone have not been available. Although the crystal structures of gp42 alone and gp42/HLA class II complex have been solved (15, 19), the N-terminal region of gp42 (bound to gH/gL) is not visible in the structures, most likely due to its flexibility. Previous studies have shown that the N-terminal region of gp42 contains multiple functional regions, including a cleavage site that results in the secretion of gp42, a potential homodimerization region, and two segments (15 residues each) required for gH/gL binding (Fig. 1 A) (13, 14). Extensive gp42 N-terminal deletion analysis demonstrated that residues 37 to 56 and 72 to 96 include functional regions of the N terminus of gp42 required to trigger fusion and suggested that some of the residues within residues 67 to 71 are also important. Additional experiments showed that amino acids within segments from residues 47 to 61 and 67 to 81 are critical for binding gH/gL (13). Those two segments are spaced by five residues, which appear to act as a linker. Previous studies also showed that a 46-mer peptide spanning residues 36 to 81, mimicking the full-length gp42, binds to gH/gL and inhibits the formation of gH/gL/gp42 complex, thus blocking membrane fusion with epithelial cells and fusion with B cells (13, 14).
FIG. 1.

Schematic representation of EBV gp42. (A) Representation of wild-type gp42 showing the relative locations of known functional domains. The transmembrane domain is predicted to span residues 9 to 22 and is shown as a gray box. The site of gp42 cleavage is between residues 40 and 42 and is indicated by a black bar. The two gH/gL binding regions, spanning residues 47 to 61 and 67 to 87, are indicted with hatched boxes, flanking the five-residue linker. The C-terminal C-type lectin domain, including the hydrophobic pocket and HLA class II-binding region, is indicated by cross-hatched boxes. The putative dimerization region is indicated by a dotted box. (B) Amino acid sequence of gp42 peptide spanning residues 36 to 81 of the gp42 protein.
In order to obtain a better understanding of the interaction of gp42 with gH/gL, and the role of this interaction in membrane fusion, we probed the gH/gL binding site by using 27 synthetic peptide analogs spanning residues 36 to 81 of gp42. Peptides were tested for binding affinity to soluble gH/gL by using a fluorescence polarization (FP) assay, probed for inhibition of B-cell and epithelial cell fusion in cell-based assays, and finally investigated for their ability to block epithelial cell infection. The data from the FP assay agreed very well with cell-cell fusion data and infection data, providing correlative data for peptide binding affinity and inhibition of cell-cell fusion and infection in an apparent competitive manner. We have defined the minimal length requirements for high-affinity binding to gH/gL and obtained a more detailed map of the key amino acids of the gp42 N terminus that are necessary for optimal gH/gL binding and inhibition of epithelial cell and B-cell membrane fusion.
MATERIALS AND METHODS
Cells.
Mammalian cells were grown in 75-cm2 cell culture flasks (Corning) in medium supplemented with 10% FetalPlex animal serum complex (Gemini Bio-Products) and 1% penicillin-streptomycin (Bio-Whittaker). Mammalian B cells were Daudi B lymphocytes that were EBV positive, express HLA class II and CD21 (American Type Culture Collection), and were modified to stably express T7 RNA polymerase under selection of G418 (900 μg/ml) in complete RPMI 1640 medium (Lonza) (23). Mammalian epithelial cells were human embryonic kidney cells that expressed simian virus 40 large T antigen (293T) (American Type Culture Collection) and were modified to stably express T7 RNA polymerase under selection of 100 μg/ml phleomycin D1 (Zeocin; Invitrogen) in complete Dulbecco's modified Eagle medium (DMEM; Bio-Whittaker). This cell line is also known as line 14, as described previously (20). Chinese hamster ovary K1 (CHO-K1) cells were kindly provided by Nanette Susmarsarski and were grown in complete Ham's F-12 medium (Bio Whittaker). EDTA (1 mM in phosphate-buffered saline [PBS]) or trypsin-EDTA (Bio-Whittaker) was used to detach adherent cells.
Peptides.
Synthetic peptides were obtained commercially through Biobasic, Inc., or Biosynthesis, Inc. The 30-mer peptides and 30-mer mutant peptides (30merMP1 to 30merMP14) were synthesized at desalted purity, while the 30-mer was also synthesized at >95% purity for the purpose of comparison of the effects of purity grades. The longer peptides (33-mer, 35-mers, 41-mer, and 46-mer) were synthesized at >95% purity as determined by high-performance liquid chromatography and mass spectrometry. Peptide FITC30mer is a 30-mer N-terminally labeled with fluorescein isothiocyanate (FITC). All the peptides were used without further purification. Lyophilized peptides were reconstituted in ionized water or Tris buffer (25 mM Tris, pH 7.4, 150 mM NaCl). Peptide concentration was measured by absorbance at 280 nm by using the theoretical 0.1% extinction coefficient values. Peptides were stored at −20°C and stable for several months.
Proteins and antibodies.
Soluble EBV gH/gL protein and gp42 were produced and purified as described elsewhere (14). Briefly, gH/gL and gp42 proteins were obtained using the baculovirus expression system in SF+ and High Five insect cells, respectively. Soluble gH/gL was purified by affinity purification using monoclonal anti-gH/gL antibody E1D1. Expressed six-His-tagged gp42 protein was purified by cobalt metal affinity resin purification. Gel filtration using a Superdex-200 HR 10/30 analytical column (Amersham Biosciences) provided the final purification in running buffers consisting of PBS. Monoclonal antibody E1D1 (anti-gH/gL) was a gift generously provided by L. Hutt-Fletcher (Louisiana State University Health Sciences Center, Shreveport, LA) (26).
FP.
The fluorescence polarization (FP) assay was performed in black, 96-well microplates in triplicates with a total volume of 200 μl in each well. The titration curve of gH/gL was measured by increasing the concentration of gH/gL from 0 to 30 nM while the concentration of FITC30mer was constant at 2.5 nM. For the competition studies, FITC30mer and peptides were mixed together in Tris buffer and then soluble gH/gL was added. This allowed FITC30mer and the competitive peptides to have the same chance of binding to gH/gL. Tris buffer alone and FITC30mer in Tris buffer (free peptide) were included as a blank and control, respectively. For other wells, FITC30mer and gH/gL were added in equal amounts at a final concentration of 2.5 nM in each well. Detergent NP-40 at 0.01% was added to reduce the nonspecific binding. A dilution series of competitive peptides was added until the FP reading became close to the readings of the control (FITC30mer only). Sample mixtures were incubated at room temperature for at least 30 min before the FP value was measured on the Synergy 4 plate reader (Biotek). The data were analyzed using Gen5 (Biotek) and Prism 5.0 (Graphpad Software, San Diego, CA). The 50% inhibitory concentrations (IC50s) as well as the standard errors were determined from the model with nonlinear regression analysis {with an equation of 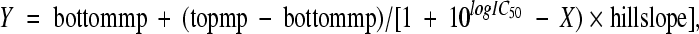 where Y is fluorescence polarization, X is concentration of peptides, and mp is millipolarization} Each measurement was repeated in at least three or four independent experiments, and similar results were obtained.
where Y is fluorescence polarization, X is concentration of peptides, and mp is millipolarization} Each measurement was repeated in at least three or four independent experiments, and similar results were obtained.
Transfection.
CHO-K1 cells were transfected in Opti-MEM I medium (Gibco) by a uniform protocol with Lipofectamine 2000 (Invitrogen), as described previously (23). Cells were plated in a six-well format, and after 24 h, each well received 6 μl of Lipofectamine 2000 and various combinations of expression vectors in the following amounts: 0.5 μg for gH, 0.5 μg for gL, 0.5 μg for gB, 0.8 μg for luciferase, 0.5 μg for green fluorescent protein (GFP), and 1.5 μg for the pCAGGs vector control.
Fusion assay.
CHO-K1 cells were transiently transfected as described above. At 6 h posttransfection, the cells were detached with EDTA and 37,500 cells/well were transferred to a 96-well plate and overlaid with equal numbers of Daudi B target cells or 293T target cells. Fifteen microliters of peptide or Opti-MEM I was added, and the total volume was adjusted to 150 μl with complete RPMI medium. Cells were counted by using a Beckman Coulter Z1 particle counter. Sixteen hours after overlay, cells were washed with PBS and lysed for 10 min with 50 μl passive lysis buffer (Promega) per well. Luciferase activity was measured with a Perkin-Elmer Victor plate reader immediately after the addition of 50 μl/well of luciferase reagent (Promega). For B-cell fusion, 3 μl of soluble gp42 was added to trigger fusion. The added soluble gp42 was the minimum amount of g42 empirically determined to be required for maximum fusion.
Cell-based enzyme-linked immunosorbent assay (CELISA).
To test the effect of peptides on gH/gL surface expression, CHO-K1 cells were transfected with EBV glycoproteins gB, gH, and gL as described above. Twenty-four hours posttransfection, CHO-K1 cells were transferred into a 96-well plate and peptides were added. After 24 h of incubation, the medium was removed, cells were washed with PBS-ABC (PBS with 0.89 g of CaCl2 and 0.89 g of MgCl2-H2O per 8 liters), and primary antibody (anti-gH/gL E1D1) at a 50 nM final concentration was added to PBS-ABC with 3% bovine serum albumin (BSA). Cells were fixed for 10 min in PBS with 2% formaldehyde and 0.2% glutaraldehyde and then blocked with 3% BSA in PBS-ABC. Biotinylated anti-mouse IgG at 1:500 was added as the secondary antibody, followed by streptavidin-horseradish peroxidase at 1:20,000. TMB (3,3′,5,5′-tetramethylbenzidine; Sigma) substrate was added, and quantitative colorimetric measurement was performed at 370 nm by using a Victor plate reader, typically achieving a high signal-over-background measurement by 30 to 60 min.
Cell-free virus infection assay.
GFP-positive EBV was produced by Akata cells (a gift from L. Hutt-Fletcher, Louisiana State University Health Sciences Center, Shreveport, LA) as previously described (12, 18, 28). Briefly, Akata cells were grown in RPMI 1640 medium (Lonza) with 10% FetalPlex animal serum (Gemini Bio-Products), 1% penicillin-streptomycin (BioWhittaker), and 500 μg/ml G418 (Gemini Bio-Products). At a high cell concentration, 4 × 107 cells were removed, 100 μg/ml bacitracin (Sigma Aldrich) was added, and virus was pelleted by centrifugation at 16,000 × g at 4°C for 90 min. Virus was resuspended in 2 ml of DMEM and passed through a 0.8-μm-pore-size filter.
Virus aliquots were preincubated with 1 μM peptide for 24 h at 37°C under conditions for testing viral inhibition, during which time human embryonic kidney 293 epithelial cells were plated in a 96-well format nearly at confluence. This infection assay was performed using 50 μl of cell-free virus per well, centrifuged at 2,500 rpm for 1 h at 33°C. Infected cells could be observed producing GFP after 24 h. Fluorescence-activated cell sorting (FACS) with a BD FACSCalibur using a 488-nm laser was used to quantify GFP-expressing cells at 24 h postinfection. The percentage of infected cells was measured by collecting 10,000 total events and gating on the live cell population.
RESULTS
Peptides derived from the N terminus of gp42 bind to gH/gL.
We have previously studied gp42-derived peptides spanning amino acids 36 to 81 by using a fluorescence polarization assay (Fig. 1) (13). In that assay, a FITC-labeled 46-mer (FITC46mer) was used as a peptide binding probe, yielding a 50% effective concentration (EC50) of 5 nM measured using a Beacon 2000 instrument (Invitrogen). In the current study, we have demonstrated that similar binding affinity measurements can be made in a 96-well plate by using a Synergy 4 plate reader (Biotek). The binding of FITC46mer to gH/gL was remeasured, and EC50s similar to the original data measured on the Beacon 2000 (3 to 5 nM) were obtained (Table 1). However, the plate-based FP assay is more efficient and consumes at least 4-fold less peptide probe and protein for each sample.
TABLE 1.
FP and cell fusion data of gp42-derived peptides with different lengths or alanine mutations
| Peptidea | Description | Amino acid sequenceb |
FP IC50 (nM) | IC50 SE (nM) | % B-cell fusion | % epithelial cell fusion |
|---|---|---|---|---|---|---|
| 36 47 61 67 81 | ||||||
| 46-mer | Residues 36-81 | RVAAAAITWVPKPNVEVWPVDPPPPVNFNKTAEQEYGDKEVKLPHW | 3.5 | 0.2 | 8 | 16 |
| 41-mer | Residues 36-81 (62-66 deleted) | RVAAAAITWVPKPNVEVWPVDPPPPV-----AEQEYGDKEVKLPHW | 5.8 | 0.6 | 46 | 14 |
| 35merA1 | Residues 47-81 | KPNVEVWPVDPPPPVNFNKTAEQEYGDKEVKLPHW | 9.6 | 1.4 | 32 | 12 |
| 35merA2 | Residues 42-81 (62-66 deleted) | ITWVPKPNVEVWPVDPPPPV-----AEQEYGDKEVKLPHW | 12.6 | 1.2 | 50 | 14 |
| 33merB1 | Residues 44-81 (62-66 deleted) | WVPKPNVEVWPVDPPPPV-----AEQEYGDKEVKLPHW | 11.7 | 3.2 | 56 | 14 |
| 35merB2 | 5-A substitution at 42-46 | AAAAAKPNVEVWPVDPPPPV-----AEQEYGDKEVKLPHW | 350.0 | 65.0 | 95 | 82 |
| 35merB3 | 2-A substitution at 62-63 | KPNVEVWPVDPPPPVAANKTAEQEYGDKEVKLPHW | 38.6 | 5.3 | 81 | 58 |
| 35merB4 | 2-A substitution at 64-65 | KPNVEVWPVDPPPPVNFAATAEQEYGDKEVKLPHW | 4.2 | 0.3 | 39 | 14 |
| 35merB5 | 2-A substitution at 49-50 | KPAAEVWPVDPPPPVNFNKTAEQEYGDKEVKLPHW | 163.9 | 18.4 | 96 | 84 |
| 35merB6 | 2-A substitution at 52-53 | KPNVEAAPVDPPPPVNFNKTAEQEYGDKEVKLPHW | 711.3 | 215.4 | 100 | 115 |
| 35merB7 | 2-A substitution at 78-79 | KPNVEVWPVDPPPPVNFNKTAEQEYGDKEVKAAHW | 122.2 | 12.8 | 91 | 65 |
| FITC30mer | Residues 47-81 (62-66 deleted) | KPNVEVWPVAAPPPV-----AEQEYGDKEVKLPHW | 1.3 | 0.05 | ND | ND |
| 30-mer (>95%) | Residues 47-81 (62-66 deleted) | KPNVEVWPVAAPPPV-----AEQEYGDKEVKLPHW | 250 | 30 | 102 | 91 |
| 30mer (desalted) | Residues 47-81 (62-66 deleted) | KPNVEVWPVAAPPPV-----AEQEYGDKEVKLPHW | 380 | 40 | ND | ND |
| 30merMP1 | 2-A substitution at 47-48 | AANVEVWPVDPPPPV-----AEQEYGDKEVKLPHW | 410 | 60 | 99 | 85 |
| 30merMP2 | 2-A substitution at 49-50 | KPAAEVWPVDPPPPV-----AEQEYGDKEVKLPHW | 38,400 | 530 | 84 | 72 |
| 30merMP3 | 2-A substitution at 52-53 | KPNVEAAPVDPPPPV-----AEQEYGDKEVKLPHW | >100,000 | NDc | 96 | 98 |
| 30merMP4 | 2-A substitution at 54-55 | KPNVEVWAADPPPPV-----AEQEYGDKEVKLPHW | 15,300 | 5,780 | 95 | 85 |
| 30merMP5 | 2-A substitution at 56-57 | KPNVEVWPVAAPPPV-----AEQEYGDKEVKLPHW | 4,300 | 1,200 | 101 | 107 |
| 30merMP6 | 2-A substitution at 58-59 | KPNVEVWPVDPAAPV-----AEQEYGDKEVKLPHW | 10,000 | 1,900 | 94 | 107 |
| 30merMP7 | 2-A substitution at 60-61 | KPNVEVWPVDPPPAA-----AEQEYGDKEVKLPHW | 2,300 | 890 | 98 | 108 |
| 30merMP8 | 2-A substitution at 68-69 | KPNVEVWPVDPPPPV-----AAAEYGDKEVKLPHW | 1,800 | 550 | 115 | 128 |
| 30merMP9 | 2-A substitution at 70-71 | KPNVEVWPVDPPPPV-----AEQAAGDKEVKLPHW | 19,800 | 4,400 | 95 | 85 |
| 30merMP10 | 2-A substitution at 72-73 | KPNVEVWPVDPPPPV-----AEQEYAAKEVKLPHW | 2,300 | 1,800 | 93 | 106 |
| 30merMP11 | 2-A substitution at 74-75 | KPNVEVWPVDPPPPV-----AEQEYGDAAVKLPHW | 460 | 50 | 97 | 92 |
| 30merMP12 | 2-A substitution at 76-77 | KPNVEVWPVDPPPPV-----AEQEYGDKEAALPHW | 1,400 | 440 | 94 | 99 |
| 30merMP13 | 2-A substitution at 78-79 | KPNVEVWPVDPPPPV-----AEQEYGDKEVKAAHW | 30,300 | 6,790 | 80 | 70 |
| 30merMP14 | 2-A substitution at 80-81 | KPNVEVWPVDPPPPV-----AEQEYGDKEVKLPAA | 10,300 | 1,600 | 98 | 98 |
30-mer was synthesized with >95% purity or desalted purity.
AA mutations are shown in boldface.
ND, not determined.
To define the minimal length requirements for the observed high-affinity binding of the 46-mer, we synthesized two shorter FITC-labeled peptides (Table 1): FITC30mer (residues 47 to 81, lacking the linker from residues 62 to 66) and a FITC-labeled 15-mer (FITC15mer; residues 47 to 61). FP binding experiments showed that FITC15mer did not exhibit binding to gH/gL up to the tested concentration of 100 μM (data not shown), while FITC30mer bound to gH/gL in a manner similar to that of FITC46mer (Fig. 2 A), with an EC50 of 1.3 nM, indicating that peptides as short as 30 residues could bind tightly to gH/gL. Therefore, FITC30mer was used as a probe in place of FITC46mer in the plate-based FP assay, and the 30-mer peptide was used as the starting point to define residues required for the interaction of gp42 with gH/gL. Competition experiments with unlabeled 46-mer peptide and soluble gp42 provided very similar inhibition curves and IC50s at 3 to 5 nM (Fig. 2B).
FIG. 2.
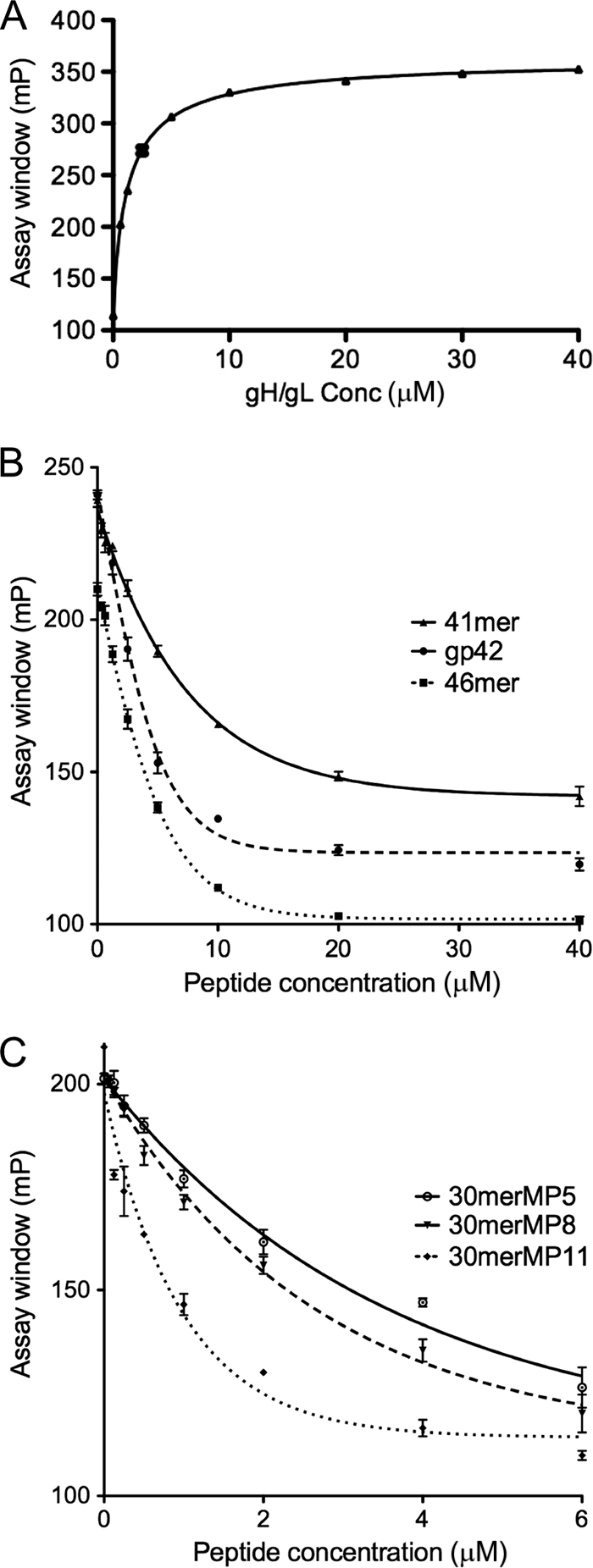
gp42 peptide binding to gH/gL measured by fluorescence polarization. (A) The titration curve of gH/gL protein binding to FITC-labeled gp42 30-mer (FITC30mer; Table 1). The y-axis scale is in millipolarization (mp) units. FITC30mer, at a final concentration of 2.5 nM, was incubated with various concentrations (Conc) of gH/gL in a black, 96-well plate, and fluorescence polarization was measured using a Synergy 4 plate reader (BioTek). Assays were carried out in triplicate, and data were fit using nonlinear regression analysis with Prism 5.0 (Graphpad). The EC50s were calculated based on curve fitting. Representative FP data are shown for unlabeled competitor peptides exhibiting high (B)- and low (C)-affinity binding. FITC30mer was mixed with increasing concentrations of competitor peptides followed by the addition of soluble gH/gL. The IC50s and their standard errors were calculated based on curve fitting with a model of dose-response inhibition (four parameters). (B) Example of competition assay results with high-affinity peptides and the gp42 ectodomain. The gp42 46-mer and 41-mer were selected as representatives for high-affinity-binding peptides that have IC50s in the low-nanomolar-concentration range. The full-length soluble gp42 ectodomain was used as a control and showed an IC50 of 2.5 nM. (C) Example of competition assay results with low-affinity peptides. The 30merMP5, 30merMP8, and 30merMP11 peptides were selected as representatives of low-binding-affinity peptides with IC50s in the low-micromolar-concentration range.
Double alanine scanning of gp42-derived 30-mer.
Because FITC30mer bound to gH/gL with low-nanomolar-concentration affinity, we used double alanine scanning of the 30-mer to identify key residues that contribute to the binding to gH/gL (mutant peptides named 30merMP1 to 30merMP14; Table 1). Two residues (Glu51 and Ala66) were not probed. An E51A point mutation E51A did not show any effect in cell fusion assays and CELISAs in previous studies (13), and we chose not to mutate Ala66 to glycine or other residues. Unexpectedly, the 30-mer lacking the N-terminal FITC label exhibited reduced binding affinity of approximately 190-fold compared to FITC30mer (Table 1). To rule out the possibility that peptide purity might have contributed to the observed difference in binding affinity, the 30-mer was synthesized in both >95% and desalted purity grades. The IC50 of the peptide with 95% purity (250 ± 30 nM) was only slightly better than that of the desalted (380 ± 40 nM) counterpart (Table 1), indicating that the purity of the peptide did not alter binding affinity significantly. The 30-mers with alanine mutations (30merMP1 to 30merMP14) were all synthesized in desalted grade.
The IC50s of mutant peptides (30merMP1 to 30merMP14) are compared to that of the wild-type 30-mer in Table 1. Mutant peptides exhibited various levels of reduced binding affinity, indicating that some residues are more important than others for binding to gH/gL. Mutations of residue pairs 49-50, 52-53, 54-55, 58-59, 70-71, and 78-79 corresponded to the largest reductions in binding to gH/gL, with IC50s shifted to greater than 10 μM, representing about 30-fold decreases in affinity (Table 1). Mutations of Val52 and Trp53 almost abolished the binding affinity, as the IC50 could not be determined up to the tested concentration of 100 μM. In contrast, the replacement of residue pairs 47-48 and 74-75 with alanines did not show obvious effects on gH/gL binding, while other residues showed an approximately 10-fold reduction in affinity, with IC50s of approximately 1 to 5 μM (Fig. 2C). Based on the IC50s, the residues can be classified into three categories: very important (IC50 of >10 μM), less important (IC50 of 1 to 5 μM), and unimportant (IC50 of <1 μM) for binding to gH/gL. The binding profiles for these mutants, represented as the fold reductions in IC50, are shown in Fig. 3 .
FIG. 3.

Important gp42 residues for binding to gH/gL. The reduction in IC50s of the alanine mutant peptides, relative to the IC50s of the corresponding 30-mer or 30-mer peptides, are plotted against the peptide residues with the corresponding amino acid sequence aligned below. The boxed amino acids (42-46) indicate the 35merB2 peptide with five alanine substitutions relative to 35merA2. The amino acids in brackets (62-63 and 64-65) correspond to 35merB3 and 35merB4 peptides compared to 35merA1 (Table 1). Residues in red are the most important for binding, with mutant peptides exhibiting IC50s greater than 10 μM. The residues in blue are less important, with mutants showing IC50s in the 1 to 5 μM range. Residues in gray are not important, with mutants showing binding similar to that of the wild-type 30-mer, with IC50s less than 1 μM. The error bars were calculated from standard errors of IC50s of related peptides by error propagation. *, the standard error corresponding to residues 52 and 53 cannot be calculated, because the IC50 of 30merMP3 cannot be determined accurately due to very weak binding of 30merMP3 and gH/gL.
Minimal peptide length required for high-affinity binding.
Our previous data showed that shorter peptides spanning residues 36 to 56 (21-mer), 42 to 56 (15-mer), or 67 to 81 (15-mer) bound to gH/gL weakly or did not bind, and they had no effect on either B-cell fusion or epithelial cell fusion (13). To determine the minimal peptide length for high-affinity binding to gH/gL, we further investigated the role of residues of the N terminus of the 30-mer by synthesizing longer peptides, i.e., a 41-mer (starting at Arg36), 35-mer A2 (starting at Ile42), and 33-mer B1 (starting at Trp44). All of these were synthesized without the linker (residues 62 to 66). All three peptides showed low-nanomolar-concentration binding affinities similar to that of the 46-mer (Fig. 4). These results clearly demonstrated that three residues (44WVP) helped to retain the tight binding to gH/gL. Replacing the N-terminal residues with alanines (35merB2) reduced the binding by approximately 30-fold (Fig. 4, compare 35merA2 and 35merB2). The data agreed well with the observation that FITC labeling of the 30-mer increased the binding affinity by 190-fold compared to that of the 30-mer (Table 1). The FITC label (molecular weight [MW], 398) may play a role similar to that of the three bulky, hydrophobic residues (WVP; total MW, 436), potentially anchoring the peptides at the N terminus in binding to gH/gL. However, further extensions beyond Trp44 did not yield better affinity of binding to gH/gL, as shown with the longer 41-mer and 46-mer peptides (Table 1). In addition, FITC labeling of the 46-mer did not enhance its apparent binding affinity (Table 1) or perhaps was beyond the sensitivity of the FP measurement (EC50 of <1 nM). Our data indicate that the 33-mer (residues 44 to 81, without linker residues 62 to 66) represents the minimal length for maintaining nanomolar binding affinity to gH/gL.
FIG. 4.
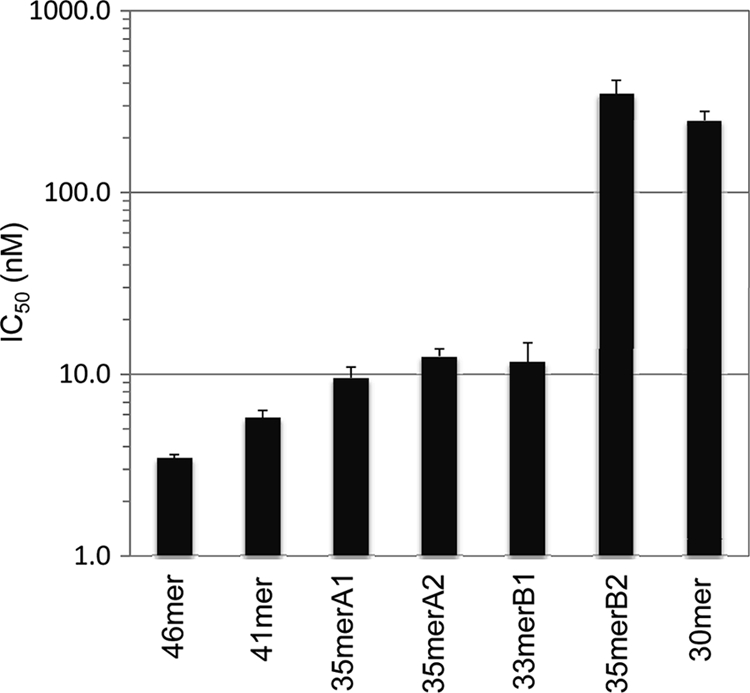
Effects of peptide length and N-terminal sequence on gp42 peptide binding. The IC50s of peptides with different lengths were determined and are shown in bar graph format. The 46-mer, the 41-mer, the 35-mers containing (35merA1) and lacking (35merA2) the linker region, and the 33-mer had similar IC50s of <15 nM. However, the IC50 of the 35-mer with five alanines at the N terminus (35merB2) was 350 nM, similar to that of the 30-mer lacking the linker (250 nM). The error bars were calculated from standard errors of IC50s of related peptides.
Role of the linker (residues 62 to 66) between two gH/gL binding regions.
Our previous deletion mutagenesis data with intact gp42 showed that residues 62 to 66 were not required for cell fusion or gH/gL binding but were flanked by two regions essential for binding. We further examined the role of the five-residue linker (residues 62 to 66) by adding the linker region to the 30-mer peptide (referred to as 35merA1). 35merA1 (with the residue 62-to-66 linker and starting at residue Lys47) and 35merA2 (without the linker but starting with residue Ile42) had similar IC50s of ∼10 nM, compared to 250 nM for the 30-mer (Table 1). These data suggest that the linker might also play a role in gH/gL binding, either by direct side chain interactions or perhaps by providing for a minimal peptide length. The residue specificity of the linker residues was tested by double alanine mutations (35merB3 and 35merB4). The IC50 of 35merB3 (mutation of Asn62 and Phe63) reduced binding 4-fold, which is not a significant change, while that of 35merB4 (mutation of Asn64 and Lys65) is half that of 35merA1. Thr66 was not mutated to alanine, because we expected that it was less likely to cause a large change in affinity. Therefore, the length of the five-residue linker, rather than specific residues, appears more important for maintaining the low-nanomolar-concentration binding affinity, if the peptides were shorter than 35 residues. Individual residues within the linker or the linker itself is not critical for longer peptides to bind with high affinity.
Comparison of key binding residue interactions in the 30-mer and 35-mer peptides.
We selected three pairs of residues that showed the largest contributions to gH/gL binding in the 30-mers used in the alanine scanning experiments to be incorporated into the higher-affinity 35-mer peptide containing the linker. The binding data obtained demonstrate that the relative magnitude of the affinity changes in the alanine-mutated 35-mers agrees well with that of the 30-mers, in direct comparison to the corresponding wild-type peptides (Fig. 5). Mutations of residues Val52 and Trp53 (35merB6 and 30merMP3) dramatically reduced the binding by 74-fold for 35merB6 and >260-fold for 30merMP3, while the peptides containing the dual Asn49 and Val50 mutations, 35merB5 and 30merMP2, were reduced by 17-fold and 100-fold, respectively. Leu78 and Pro79 mutations (35merB7 and 30merMP13) reduced binding by 12-fold for 35merB7 and 80-fold for 30merMP13. Overall, the relative patterns of changes were the same in the 30-mers and the 35-mers. Mutations of Val52 and Trp53 affected the binding affinity the most in both peptides, followed by the mutations of Asn49 and Val50, with the mutations of Leu78 and Pro79 having the least effect, though it was still significant. Generally, the affinity reduction caused by double alanine mutations in the 30-mers was about four to seven times that observed in the 35-mers (Fig. 5).
FIG. 5.

Comparison of effects of alanine mutations in the 30-mer and 35-mer peptides. The IC50s of the 30-mer and 35-mer peptides with the same double alanine mutations were compared to those of the corresponding peptides with the wild-type residues. The relative fold reductions in IC50s of the 30-mers and 35-mers were similar. Overall, the affinity reduction caused by mutations in the 30-mers was about four to seven times that caused by mutations in the 35-mers. The error bars were calculated from standard errors of IC50s of related peptides by error propagation. *, the standard error corresponding to residues 52 and 53 cannot be calculated for 30merMP3, because its IC50 cannot be determined accurately due to very loose binding of 30merMP3 and gH/gL.
We also investigated whether the gp42-derived peptides can form secondary structures in solution by using circular dichroism (CD) spectroscopy. The 46-mer, 41-mer, 33-mer, and 30-mer peptides were tested at concentrations of 25 μM, 50 μM, and 100 μM at both 20°C and 4°C. The results clearly showed that these four peptides adopt a typical random coil configuration on their own (data not shown), indicating that these short peptides do not form a well-defined secondary structure in solution.
Cell fusion inhibition with gp42-derived peptides.
To correlate the biochemical data on peptide binding affinity obtained in the FP assay with functional inhibition in membrane fusion, a cell-cell fusion assay was used. We have previously shown that a 46-mer peptide spanning residues 36 to 81 (Fig. 1B and Table 1) binds soluble gH/gL with high affinity, similar to secreted gp42 (13). The peptide effectively interferes with epithelial cell fusion, potentially by occupying and blocking an epithelial cell receptor binding surface that overlaps the gp42 binding site contained within gH/gL. Since the peptide does not contain the HLA class II binding domain and associated hydrophobic pocket, which we have previously shown to be essential for the induction of B-cell membrane fusion, it can also inhibit B-cell fusion by blocking secreted gp42 from binding to gH/gL.
As expected from our previous studies, B-cell and epithelial cell fusions were efficiently inhibited by the 46-mer and the 41-mer in a dose-dependent manner (Fig. 6 ). In contrast, the 30-mer peptide, spanning the two previously identified core gH/gL binding motifs (residues 47 to 81, with the linker region deleted), was not able to inhibit membrane fusion. The reduced affinity of the shorter peptide results in an inability to inhibit membrane fusion in epithelial cells (Fig. 6C and D) and an inability to compete with soluble gp42 for gH/gL binding within B cells (Fig. 6A and B). As expected from the in vitro binding data, the alanine mutants (30merMP1 to 30merMP14; Table 1) also did not inhibit fusion in B cells (Fig. 6A) and epithelial cells (Fig. 6C).
FIG. 6.


Inhibition of B-cell fusion and epithelial cell fusion by gp42-derived peptides. Inhibition of fusion by the various peptides was determined using a cell-based fusion assay as detailed in Materials and Methods. (A and B) For B-cell fusion, CHO-K1 cells were transfected with gB, gH, gL, and a T7 luciferase reporter construct. Six hours posttransfection, soluble gp42 (sgp42) was added upon overlay with Daudi B cells expressing T7 luciferase. (C and D) For epithelial cell fusion, CHO-K1 cells were also transfected with gB, gH, gL, and a T7 luciferase reporter. Six hours posttransfection, 293T cells expressing T7 polymerase were overlaid. Sixteen hours after overlay for both fusion assays, luciferase activity was read and normalized to the level corresponding to no peptide addition. This control was set to 100%. Error bars represent standard deviations for the normalized values representing the average of at least three independent experiments. Panels A and C show a comparison of all peptides screened for gH/gL binding. Panels B and D show dose-dependent inhibition of all peptides showing an IC50 of <10 nM.
The truncated peptides that retained higher affinity in the FP binding assay, including 35merA1 (residues 47 to 61 and 67 to 81), 35merA2 (residues 42 to 81), and 33merB1 (residues 44 to 81), were all able to inhibit membrane fusion (Fig. 6). Mutation of key residues (49-50, 52-53, and 78-79) that are important for high-affinity binding to gH/gL (in 35merB5, 35merB6, and 35merB7, respectively) correspondingly resulted in a loss of the ability to inhibit cell-cell fusion (Fig. 6). Similarly, the replacement of the important N-terminal amino acids 42 to 46 with alanines (35merB2) resulted in a loss of fusion inhibition activity in both cell types (Fig. 6). Alanine substitutions in the linker region, at residue pairs 62-63 (35merB3) and 64-65 (35merB4), which had modest effects on peptide binding, also resulted in good fusion inhibition (Fig. 6). Overall, the ability to inhibit cell-cell fusion correlates very well with the corresponding IC50s measured in the FP assay.
To verify that the addition of the gp42 peptides did not influence fusion by reducing the expression of gH/gL in transfected cells, we confirmed that gH/gL surface expression was not altered by the soluble peptides (Fig. 7). For these experiments, CHO-K1 cells were transfected with EBV glycoproteins gB, gH, and gL as done for the cell-cell fusion assay, peptides were added without the addition of target cells, and gH/gL expression was monitored by CELISA 16 h after peptide addition.
FIG. 7.

Surface expression of gH/gL in the presence of gp42 peptides. Expression was measured by CELISA using monoclonal E1D1 antibody, secondary biotinylated anti-mouse IgG antibody, tertiary streptavidin-horseradish peroxidase, and TMB substrate. Color development was measured by absorbance at 370 nm. The data shown are averages of at least three independent experiments. gH/gL expression was normalized to the level corresponding to no peptide addition, which was set to 100%. Error bars represent standard deviations for the normalized values.
Inhibition of epithelial cell infection by gp42-derived peptides.
In order to test the effects of the different gp42-derived peptides on viral entry, epithelial cells were infected with GFP-expressing EBV produced by Akata cells. Virus was preincubated for 24 h with 1 μM gp42-derived peptide or anti-gH/gL antibody E1D1 as a control. The effect of gH/gL antibody or peptide on the infection of epithelial cells was measured by FACS analysis of GFP expression. The gH/gL antibody and peptides that were shown to inhibit cell-cell fusion (46-mer, 41-mer, 35merA1, 35merA2, 33merB1, and 35merB4) showed a significant reduction of infection to ∼25% of the level for control infected cells. In contrast, cells infected with virus preincubated with peptides that did not block cell-cell fusion showed only a modest reduction (30-mer) or no reduction (30merMP3) of infection (Fig. 8). Our data confirm that all of the gp42 peptides with tight binding to gH/gL, mimicking the intact gp42 protein, inhibited EBV infection of epithelial cells.
FIG. 8.

Inhibition of epithelial cell infection by gp42-derived peptides. The graph shows the inhibitory effect of gp42-derived peptides on EBV infection of epithelial cells. Virus was incubated with gp42-derived peptide or anti-gH/gL antibody E1D1 (gH/gL monoclonal antibody [mAb]) as a control at 1 μM. FACS analysis was used to measure the GFP-expressing cells, and data were normalized by setting the EBV-only control to 100% infection so that each experimental condition demonstrates a fraction of uninhibited 100% infection. The data shown represent the averages of three independent experiments.
DISCUSSION
In the current study, we identified essential residues of gp42 for gH/gL binding and membrane fusion by using gp42-derived peptides spanning residues 36 to 81. The results from FP binding, cell fusion, and viral infection assays confirm that two previously defined gH/gL binding regions (residues 47 to 61 and 67 to 81) are necessary, but these 30 residues are not sufficient to retain high-level binding affinity for gH/gL. The minimal 30-mer peptide containing these regions exhibits a 70-fold-reduced binding affinity for gH/gL compared to that of the 46-mer (residues 36 to 81) or the full-length gp42 protein. This 30-mer is also unable to inhibit B-cell or epithelial cell fusion. Interestingly, the high-affinity binding of the 30-mer could be restored by adding three amino acids to the N terminus, by FITC labeling at the N terminus, or by inserting the linker residues in the middle of the peptide. Mutation of the added amino acids at the N terminus to alanines abolished this increased affinity, indicating that these residues form important side chain interactions with gH/gL. In contrast, while the insertion of residues 62 to 66 restored binding, these linker residues could be mutated without large effects on the affinity. Together, the results demonstrate that a 33-mer peptide from the gp42 N terminus retains high-affinity binding to gH/gL, that the N-terminal residues (43 to 46) interact specifically with gH/gL, and that the linker from residues 62 to 66 may contribute to peptide affinity by providing proper spacing of two separable peptide binding regions.
Alanine scanning mutants highlighted residues that are important for binding to gH/gL, with the most dramatic effects on affinity being localized to the N-terminal residue 44-to-61 segment. The most important residues for binding appear to include hydrophobic residues with large side chains, such as WVP (44 to 46) and VW (52-53), which may act to anchor the peptides in specific gH/gL binding pockets. While the most important residues for binding appear to lie in the N-terminal region of the gp42 sequence, alanine substitutions throughout the peptide have major effects on binding, suggesting that both the N- and C-terminal regions interact with gH/gL by using specific side chain interactions. Mutation of the gp42 charged residues appeared to have only limited effects on the binding affinity for gH/gL, indicating that these do not play a major role in the interaction.
As described above, we determined that the linker region (residues 62 to 66) plays an important role when peptides are shorter than 35 residues but is an unnecessary spacer when the peptides are longer. Without the structure of the complexes of these peptides bound to gH/gL, it is difficult to understand what role the linker may play in the shorter peptides. Alanine mutations incorporated into the 30-mer and 35-mer sequences, lacking and including the linker region, respectively, exhibit the same pattern of affinity changes, indicating that the mutated residues interact similarly with gH/gL in the shorter and longer peptides. It is possible that the deletion of the linker generates a binding penalty for all of the peptides, short and long, but that this is more evident in the shorter peptides as a large decrease in affinity. It is clear from our current studies that the length of the linker, not the sequence, is most important for this effect.
Our cell-cell fusion data with the series of peptide analogs correlate extremely well with binding affinity data obtained in the FP assay. These observations support the conclusion that fusion inhibition for both B cells and epithelial cells occurs by a simple competitive mechanism that relies on occupancy of the gH/gL binding site for gp42. Binding of the gp42-derived peptide to gH/gL competes with wild-type gp42 for binding to the gH/gL complex, thereby inhibiting gH/gL/gp42 complex formation in B-cell fusion assays, although competition for endogenously expressed gp42 is difficult to achieve with externally added peptide. The peptides also appear to compete for binding of the gH/gL complex to a specific receptor in epithelial cells, leading to an inhibition of fusion. Recent studies have shown that soluble forms of human integrins αvβ6 and αvβ8 triggered EBV gB- and gH/gL-mediated epithelial cell fusion, and this observation led to the suggestion that these may be epithelial receptors (5).
We tested all gp42-derived peptides, which were able to inhibit cell-cell fusion, as well as the 30-mer and 30merMP3 peptides, which were not able to inhibit fusion, for their ability to inhibit virus infection. Our data with epithelial cell targets showed a good correlation between peptide binding affinity, cell-cell fusion data, and viral infection data. All peptides that bound with high affinity were able to inhibit membrane fusion and also reduced infection to nearly one-quarter of what was observed in uninhibited controls, whereas the two control peptides did not reduce infection significantly. In contrast, when peptides were examined in a B-cell infection assay, no inhibition was observed (data not shown). This was likely due to the coexpressed viral gp42 forming tight membrane-bound gH/gL/gp42 complexes, resulting in gp42 that could not be displaced by the peptide. This finding is consistent with the results observed for peptide inhibition of B-cell membrane fusion in which even high concentrations of peptide can achieve only a modest decrease in cell-cell fusion when transfected gp42 is present.
In summary, the FP binding data, cell-cell fusion data, and viral infection data demonstrate that the affinity of binding of gp42-derived peptides to gH/gL is tightly correlated with the peptide inhibitory activity. We have identified a minimal 33-mer peptide that retains high-affinity binding and inhibitory activity and mapped key regions of the gp42 sequence that likely interact with gH/gL side chain-specific pockets. These studies should help guide the design of other peptides or peptidomimetics as inhibitors of EBV entry and perhaps provide useful tools for the further study of the gH/gL epithelial cell receptor.
Acknowledgments
We thank Lindsey Hutt-Fletcher for her generous gift of the E1D1 antibody cell line. We thank the members of the Longnecker, Jardetzky, and Spear laboratories for help and support.
This research was supported by grants AI076183 (to R.L. and T.S.J.) and AI067048 (to R.L.) from the National Institute of Allergy and Infectious Diseases and CA117794 from the National Cancer Institute (to R.L. and T.S.J.). This work was also supported in part by a predoctoral fellowship from Northwestern University's Biotechnology Training Program from the NIH (to A.N.K.).
Footnotes
Published ahead of print on 28 July 2010.
REFERENCES
- 1.Amon, W., and P. J. Farrell. 2005. Reactivation of Epstein-Barr virus from latency. Rev. Med. Virol. 15:149-156. [DOI] [PubMed] [Google Scholar]
- 2.Borza, C. M., and L. M. Hutt-Fletcher. 2002. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat. Med. 8:594-599. [DOI] [PubMed] [Google Scholar]
- 3.Borza, C. M., A. J. Morgan, S. M. Turk, and L. M. Hutt-Fletcher. 2004. Use of gHgL for attachment of Epstein-Barr virus to epithelial cells compromises infection. J. Virol. 78:5007-5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burkitt, D. 1962. A tumour syndrome affecting children in tropical Africa. Postgrad. Med. J. 38:71-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chesnokova, L. S., S. L. Nishimura, and L. M. Hutt-Fletcher. 2009. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8. Proc. Natl. Acad. Sci. U. S. A. 106:20464-20469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haan, K. M., W. W. Kwok, R. Longnecker, and P. Speck. 2000. Epstein-Barr virus entry utilizing HLA-DP or HLA-DQ as a coreceptor. J. Virol. 74:2451-2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haan, K. M., S. K. Lee, and R. Longnecker. 2001. Different functional domains in the cytoplasmic tail of glycoprotein B are involved in Epstein-Barr virus-induced membrane fusion. Virology 290:106-114. [DOI] [PubMed] [Google Scholar]
- 8.Henle, W., and G. Henle. 1970. Evidence for a relation of Epstein-Barr virus to Burkitt's lymphoma and nasopharyngeal carcinoma. Bibl. Haematol. 1970:706-713. [DOI] [PubMed] [Google Scholar]
- 9.Henle, W., and G. Henle. 1969. The relation between the Epstein-Barr virus and infectious mononucleosis, Burkitt's lymphoma and cancer of the postnasal space. East Afr. Med. J. 46:402-406. [PubMed] [Google Scholar]
- 10.Hutt-Fletcher, L. M. 2007. Epstein-Barr virus entry. J. Virol. 81:7825-7832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hutt-Fletcher, L. M., and C. M. Lake. 2001. Two Epstein-Barr virus glycoprotein complexes. Curr. Top. Microbiol. Immunol. 258:51-64. [DOI] [PubMed] [Google Scholar]
- 12.Hutt-Fletcher, L. M., and S. M. Turk. 2001. Virus isolation. Methods Mol. Biol. 174:119-123. [DOI] [PubMed] [Google Scholar]
- 13.Kirschner, A. N., A. S. Lowrey, R. Longnecker, and T. S. Jardetzky. 2007. Binding-site interactions between Epstein-Barr virus fusion proteins gp42 and gH/gL reveal a peptide that inhibits both epithelial and B-cell membrane fusion. J. Virol. 81:9216-9229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kirschner, A. N., J. Omerovic, B. Popov, R. Longnecker, and T. S. Jardetzky. 2006. Soluble Epstein-Barr virus glycoproteins gH, gL, and gp42 form a 1:1:1 stable complex that acts like soluble gp42 in B-cell fusion but not in epithelial cell fusion. J. Virol. 80:9444-9454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirschner, A. N., J. Sorem, R. Longnecker, and T. S. Jardetzky. 2009. Structure of Epstein-Barr virus glycoprotein 42 suggests a mechanism for triggering receptor-activated virus entry. Structure 17:223-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li, Q., C. Buranathai, C. Grose, and L. M. Hutt-Fletcher. 1997. Chaperone functions common to nonhomologous Epstein-Barr virus gL and varicella-zoster virus gL proteins. J. Virol. 71:1667-1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McShane, M. P., and R. Longnecker. 2004. Cell-surface expression of a mutated Epstein-Barr virus glycoprotein B allows fusion independent of other viral proteins. Proc. Natl. Acad. Sci. U. S. A. 101:17474-17479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Molesworth, S. J., C. M. Lake, C. M. Borza, S. M. Turk, and L. M. Hutt-Fletcher. 2000. Epstein-Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. J. Virol. 74:6324-6332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mullen, M. M., K. M. Haan, R. Longnecker, and T. S. Jardetzky. 2002. Structure of the Epstein-Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Mol. Cell 9:375-385. [DOI] [PubMed] [Google Scholar]
- 20.Omerović, J., L. Lev, and R. Longnecker. 2005. The amino terminus of Epstein-Barr virus glycoprotein gH is important for fusion with epithelial and B cells. J. Virol. 79:12408-12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ressing, M. E., D. van Leeuwen, F. A. Verreck, S. Keating, R. Gomez, K. L. Franken, T. H. Ottenhoff, M. Spriggs, T. N. Schumacher, L. M. Hutt-Fletcher, M. Rowe, and E. J. Wiertz. 2005. Epstein-Barr virus gp42 is posttranslationally modified to produce soluble gp42 that mediates HLA class II immune evasion. J. Virol. 79:841-852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rickinson, A. B., and E. Kieff. 2007. Epstein-Barr virus, p. 2655-2700. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 5th ed. Lippincott-Williams & Wilkins, Philadelphia, PA.
- 23.Silva, A. L., J. Omerovic, T. S. Jardetzky, and R. Longnecker. 2004. Mutational analyses of Epstein-Barr virus glycoprotein 42 reveal functional domains not involved in receptor binding but required for membrane fusion. J. Virol. 78:5946-5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sorem, J., T. S. Jardetzky, and R. Longnecker. 2009. Cleavage and secretion of Epstein-Barr virus glycoprotein 42 promote membrane fusion with B lymphocytes. J. Virol. 83:6664-6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spriggs, M. K., R. J. Armitage, M. R. Comeau, L. Strockbine, T. Farrah, B. Macduff, D. Ulrich, M. R. Alderson, J. Mullberg, and J. I. Cohen. 1996. The extracellular domain of the Epstein-Barr virus BZLF2 protein binds the HLA-DR beta chain and inhibits antigen presentation. J. Virol. 70:5557-5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strnad, B. C., T. Schuster, R. Klein, R. F. Hopkins III, T. Witmer, R. H. Neubauer, and H. Rabin. 1982. Production and characterization of monoclonal antibodies against the Epstein-Barr virus membrane antigen. J. Virol. 41:258-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takada, K. 2001. Role of Epstein-Barr virus in Burkitt's lymphoma. Curr. Top. Microbiol. Immunol. 258:141-151. [DOI] [PubMed] [Google Scholar]
- 28.Takada, K., K. Horinouchi, Y. Ono, T. Aya, T. Osato, M. Takahashi, and S. Hayasaka. 1991. An Epstein-Barr virus-producer line Akata: establishment of the cell line and analysis of viral DNA. Virus Genes 5:147-156. [DOI] [PubMed] [Google Scholar]
- 29.Wang, X., and L. M. Hutt-Fletcher. 1998. Epstein-Barr virus lacking glycoprotein gp42 can bind to B cells but is not able to infect. J. Virol. 72:158-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wei, W. I., and J. S. Sham. 2005. Nasopharyngeal carcinoma. Lancet 365:2041-2054. [DOI] [PubMed] [Google Scholar]