

Abstract
Using FrCasE retrovirus-infected newborn mice as a model system, we have shown recently that a long-lasting antiviral immune response essential for healthy survival emerges after a short treatment with a neutralizing (667) IgG2a isotype monoclonal antibody (MAb). This suggested that the mobilization of adaptive immunity by administered MAbs is key for the success in the long term for the MAb-based passive immunotherapy of chronic viral infections. We have addressed here whether the anti-FrCasE protective endogenous immunity is the mere consequence of viral propagation blunting, which would simply give time to the immune system to react, and/or to actual immunomodulation by the MAb during the treatment. To this aim, we have compared viral replication, disease progression, and antiviral immune responses between different groups of infected mice: (i) mice treated with either the 667 MAb, its F(ab′)2 fragment, or an IgM (672) with epitopic specificity similar to that of 667 but displaying different effector functions, and (ii) mice receiving no treatment but infected with a low viral inoculum reproducing the initial viral expansion observed in their infected/667 MAb-treated counterparts. Our data show that the reduction of FrCasE propagation is insufficient on its own to induce protective immunity and support a direct immunomodulatory action of the 667 MAb. Interestingly, they also point to sequential actions of the administered MAb. In a first step, viral propagation is exclusively controlled by 667 neutralizing activity, and in a second one, this action is complemented by FcγR-binding-dependent mechanisms, which most likely combine infected cell cytolysis and the modulation of the antiviral endogenous immune response. Such complementary effects of administered MAbs must be taken into consideration for the improvement of future antiviral MAb-based immunotherapies.
Although monoclonal antibodies (MAbs) principally have been considered for anticancer applications heretofore (62, 64), they now are increasingly being considered to treat severe acute and chronic viral infections (43, 63, 83). The best-studied antiviral MAbs are (i) pavalizumab, a humanized anti-respiratory syncytial virus (RSV) MAb approved by the FDA in 1998 for treating severe lower-respiratory-tract diseases in infants (45); (ii) several anti-human immunodeficiency virus (HIV) MAbs, which have been used in macaque preclinical infection models and in several human trials (4, 5, 19, 27-30, 32, 42, 50, 55, 57, 76-79); and (iii) a few anti-hepatitis C virus (HCV) MAbs, some of which currently are being tested in humans (9, 22, 40). However, other MAbs, some of them of human origin, also have been generated against other human viruses in recent years. Among them are antibodies against Ebola virus (75), West Nile virus (WNV) (48, 53, 54), cytomegalovirus (CMV) (11), avian and human influenza viruses (59, 60, 73, 74), severe acute respiratory syndrome coronavirus (SARS CoV) (81), hepatitis B virus (HBV) (31, 35), Hanta virus (80, 82), and Nipah virus (80, 82). These antiviral MAbs all have been selected on the basis of their neutralizing activity and the possibility that they interfere with the antiviral immune response of treated hosts, because their effector functions have been considered surprisingly little so far. Addressing this question in clinical settings currently is not possible for a variety of reasons that include ethical, technical, and cost concerns. Therefore, we have turned to the neonatal infection of mice by the lethal FrCasE retrovirus as a model system. This model allowed us to show that a very short immunotherapy by a neutralizing MAb of the IgG2a isotype (667 MAb) can permit, in addition to an immediate direct effect on the viral load, the mounting of a long-lasting endogenous antiviral immunity, which is essential for viral control and healthy survival (23-25). Because of the broad therapeutic perspectives opened by this observation, it now is essential to elucidate the molecular and cellular mechanisms underlying this effect.
FrCasE is a simple chimeric mouse retrovirus in which the env gene of the leukemogenic Friend murine leukemia virus (F-MuLV) was replaced by that of the neurodegeneration-inducing CasBr retrovirus (58). When 5 × 104 infectious particles are inoculated into newborn mice under the age of 5 to 6 days, FrCasE can enter the central nervous system (CNS) and induces a neurodegeneration fatal within 1 to 2 months with 100% incidence (15, 23, 41, 58). However, upon infection at a later time, FrCasE can no longer enter the CNS. Instead, it replicates only in the periphery and gives rise to a fatal erythroleukemia preceded by spleen enlargement and a dramatic drop of the hematocrit. Erythroleukemia incidence and incubation period, however, are variable, depending on the inoculum and the date of infection (46).
667 is an IgG2a/κ (44) directed to the main viral receptor-binding site of CasBr Env (16). It displays both in vitro (44) and in vivo (56) neutralizing activities. When rapidly (<2 days) administered for a few days to neonatally FrCasE-infected pups, viral propagation is rapidly blunted, which prevents virus entry in the brain and subsequent neurodegeneration (23). Moreover, all 667-treated mice develop a strong, long-lasting antiviral immune response, which is necessary for them to survive healthy and with no sign of neurodegeneration or of erythroleukemia (23-25) and to resist viral challenges carried out as long as 14 months after first infection (23). Protective antiviral immunity is of a typical TH1 type with humoral and cytotoxic T-cell (CTL) contributions. The anti-FrCasE humoral contribution is high, sustained, and principally of the IgG2a type with both in vitro neutralization- and complement-dependent cytolysis activities (23). Interestingly, it shows typical secondary response characteristics in viral challenge experiments (23), and anti-FrCasE antibodies are transmitted transplacentally and through breastfeeding by mothers to children, where they manifest the same properties as those of 667 in the perinatal infection setting, i.e., they prevent mice from developing neurodegeneration and permit the induction of an endogenous protective antiviral immune response (25). Finally, the CTL response directed to infected cells was shown to be necessary for the protection of FrCasE-infected, 667-treated mice, as the depletion of CD8+ T cells leads to death by retrovirally induced erythroleukemia (24).
At this stage, an important issue is the clarification of whether the anti-FrCasE protective immunity seen in 667 MAb-treated mice is due to actual immunomodulation by the MAb owing to its effector function(s) and/or is the consequence of viral propagation blunting, which would prevent the immune system from being overwhelmed by an excess of antigen and, hence, would give it time to react optimally. To address these two nonexclusive possibilities, we compared here viral propagations, health statuses, and endogenous immune responses in four groups of mice. The first three groups were mice neonatally infected under standard conditions (5 × 104 infectious particles) and treated with either the natural 667 MAb, the antibody effector function-lacking F(ab′)2 fragment of 667, or a neutralizing IgM (672) with effector functions inherently different from those of 667. The last group consisted of mice neonatally infected with a low FrCasE inoculum but not subjected to immunotherapy, which is a condition permitting early viral propagation kinetics similar to those of animals infected and 667 MAb treated under standard conditions. Taken together, our data indicate that the drastic reduction of viral propagation shortly after infection is not sufficient for the induction of protective adaptive immunity and, thereby, point to an immunomodulatory action of 667. Interestingly, they also point to two sequential actions of the administered MAb. In the immediate postinfection period, viral spread is controlled exclusively by 667 neutralizing activity, and later it involves the cytolysis of infected cells owing to FcγR-binding-dependent mechanisms. Finally, our work shows that not all antibody isotypes are equally efficient at protecting infected mice and favoring the mounting of protective immunity, as 672 IgM immunotherapy-treated animals died of erythroleukemia.
MATERIALS AND METHODS
Virus stocks, cell lines, and monoclonal antibody production.
Culture supernatants of Mus dunni embryo fibroblasts transfected with the FrCasE proviral clone (58) were used as viral stocks (56). The anti-murine leukemia virus Env mouse 667 MAb (IgG2a/κ) (44) was purified from hybridoma supernatant and assayed as previously described (16). The anti-CasBr Env mouse IgM/κ 672 MAb (44) was prepared from hybridoma culture supernatant and purified using the IgM purification kit from Pierce. The purified 2.4G2 MAb was obtained from BioXcell.
667 F(ab′)2 fragment preparation.
The 667 MAb and pepsin were mixed at final concentrations of 3 and 1 mg/ml, respectively, in 20 mM sodium acetate buffer (pH 4.0). The mixture was incubated for 7 h at 37°C. The undigested MAb and the Fc fragment were removed by protein A affinity chromatography. The unbound fraction was collected as the F(ab′)2 fragment, and purity was checked by sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis.
Viral titers and in vitro neutralization activity assays.
Viral titers were determined using a focal immunofluorescence assay (FIA) as previously described (23). Dilutions of virus-containing samples were added to 25% confluent Mus dunni cell cultures in the presence of 8 μg/ml of polybrene. The cell-to-cell spread of replication-competent retroviruses was allowed to proceed for 2 days, and focus-forming units (FFU) were visualized by indirect immunofluorescence using the 667 MAb and a fluorescein isothiocyanate (FITC)-conjugated anti-mouse immunoglobulin. The virus neutralization activity of mouse serum samples was assayed as previously described (23). Briefly, 4 × 102 FrCasE FFU were diluted in a 1:1 ratio with serum samples or solutions containing the 667 MAb, the 667 F(ab′)2 fragment, or the 672 MAb and were incubated at 37°C for 1 h. Mixes were used to infect Mus dunni cells cultured in 12-well plates overnight. The infection medium was replaced by fresh culture medium and cells were allowed to reach confluence, at which time FFU were scored as before.
SPOT multiple-peptide synthesis and antibody assay.
A total of 217 overlapping 15-mer amino acid peptides frameshifted by 3 residues and representing the complete CasBr Env sequence were synthesized on a cellulose membrane using the SPOT technique (16, 47). Membrane-bound peptides were probed with 30 ml of 672 MAb-containing cell culture supernatant. MAb binding was revealed by means of an alkaline phosphatase-conjugated rabbit anti-mouse antibody (Sigma).
Viral infection and mice follow-up.
Inbred 129/Sv/Ev mice (H-2Db haplotype) were used in this study. Three-day-old pups were infected intraperitoneally with a viral suspension containing 5 × 104 FFU in 100 μl, as previously described (23, 56). Mice were examined daily for clinical signs of neurodegeneration (reduced weight, tremor, ataxia, and hind-limb paralysis). Erythroleukemia was assayed by measuring the reduction in hematocrit, which is associated with anemia, and spleen swelling by direct abdominal palpation on living animals or by direct examination after euthanasia (24). Mice were bled at the retro-orbital sinus to assay viremia and anti-FrCasE serum immunoglobulin concentrations. After clotting at room temperature for 15 min, blood samples were centrifuged at 6,000 × g for 15 min, and serum aliquots were stored at −20°C until use. For challenge experiments, mice were injected intravenously with a mix containing 25 μl of FrCasE suspension containing 1 × 107 FFU/ml and 1 million infected splenocytes.
Preparation of splenocytes.
Spleens were dissociated by repeated pipetting in RPMI 1640 culture medium (Gibco). Dissociated cells either were used in infectious center assays or were washed twice in phosphate-buffered saline (PBS) containing 2% fetal calf serum (FCS) for flow cytometry analysis. Alternatively, they were incubated in ACK lysis buffer (Biowhittaker) to lyse red blood cells and, after being washed and resuspended in culture medium, were used for different experiments.
SIC assay.
Serial dilutions of spleen cells from simply infected mice or from infected mice treated with either the 667 MAb, the 667 F(ab′)2 fragment, or the 672 MAb were plated onto 25% confluent indicator Mus dunni cells. After 3 days of coculture, spleen infectious centers (SICs) were visualized by immunofluorescence as in FIA (24).
Enzyme-linked immunosorbent assay (ELISA) of anti-FrCasE antibodies.
The 667 MAb and the anti-FrCasE serum antibodies were assayed as described previously (23). Whereas animals of sufficient size were bled at the retro-orbital sinus, the blood of pups was collected following euthanasia. Antibodies used for the standard curve and serum samples were diluted in PBS (0.15 M NaCl, 0.01 M Na phosphate, pH 7) containing 0.1% Tween 20 and 1% bovine serum albumin. The 667 and 672 MAbs were used as standards for anti-FrCasE antibody IgG2a and IgM detection, respectively. Secondary peroxidase-conjugated anti-mouse IgG2a, IgM, and IgG1 rabbit antisera (Serotec) were used as secondary antibodies.
Assay of CD8+ T cells specific for FrCasE-infected cells by flow cytometry.
Splenocytes (5 × 105) were stained with both allophycocyanin-labeled anti-CD8+ T cells (anti-CD8-APC; Becton Dickinson, Heidelberg, Germany) and a phycoerythrin-labeled major histocompatibility complex (MHC) class I H-2Db tetramer (PE-GagL tetramer; Beckman Coulter) specific for the Friend virus GagL epitope (amino acids 85 to 93 of gPr80gag) (Db-GagL tetramers) (68) for 15 min at room temperature in PBA (PBS containing 2% FCS and 0.01% sodium azide). Cells were washed and analyzed by flow cytometry after paraformaldehyde fixation using the FACSCanto II device from Becton Dickinson, and data were processed using the FlowJo program from Three Star.
In vivo cytotoxicity assays.
In the experiments of Fig. 4E to F and 6C, splenocytes prepared from naïve 4-week-old mice were pulsed with either the GagL (12) (specific CTL target cells) or the np396-404 control peptide derived from lymphocytic choriomeningitis virus (LCMV) nucleoprotein (NP) (21) (control cells allowing the measurement of spontaneous cell death) at 37°C for 1 h. Peptides were used at a concentration of 1 mg/ml. After being washed in PBS to eliminate excess peptide, splenocytes loaded with the GagL and the np396-404 peptides were labeled with 5 μM CFSE (CFSEhigh cells) and 0.5 μM CFSE (CFSElow cells), respectively, at 37°C for 10 min in PBS. Cells then were washed again in ice-cold PBS and resuspended in PBS for adoptive transfer. After being counted, they were mixed in a 1:1 ratio. An aliquot was analyzed by flow cytometry to determine the exact CFSElow/CFSEhigh cell ratio. Cells (107 in 0.2 ml of PBS) of the mix were injected intravenously in each recipient 129/Sc/Ev mouse 10 days after the viral challenge performed at week 8 postinfection. Sixteen hours later, mice were euthanized for the flow cytometry quantification of splenic CFSElow and CFSEhigh cells. The CTL activity against GagL-loaded splenocytes was calculated from the ratio of CFSEhigh/CFSElow cells 16 h after adoptive transfer corrected by the CFSEhigh/CFSElow ratio assayed before the grafting of cells to the recipient mice.
Assay of in vivo anti-FrCasE antibody cytolytic activity.
The assay used was adapted from Guyre et al. (26). Noninfected and FrCasE-infected splenocytes were labeled with 0.5 μM (Sp CFSElow) and 5 mM (SpFr CFSEhigh) CSFE, respectively. They then were mixed in a 1:1 ratio and administered intravenously to 2-month-old naive mice or to age-matched mice formerly treated with the 2.4G2 MAb binding and blocking both FcγRII (CD32) and FcγRIII (CD16) (3, 38, 39). Eight μg of 2.4G2 per g of body weight was administered to mice 24, 12, and 1 h before the administration of the splenocyte mix, at which time they received 200 μg of either the 667 MAb, the 667 F(ab′)2 fragment, or the 672 MAb. Five hours later, the animals were euthanized and the ratio of CFSElow/CFSEhigh cells in the spleen was assayed by flow cytometry.
Statistics.
Data are presented as means ± standard errors of the means. Statistical significance between the groups was determined by applying the Mann and Whitney's test to compare two independent groups. A P value of less than 0.05 was considered statistically significant.
Ethics statement.
All procedures for animal handling and experiments were approved by the local animal facility “ComEth” institutional review board under the supervision of the French LR Regional CEEA ethic committee on animal experimentation (Chairman M. Michel, Université Montpellier).
RESULTS
Reduction of viral propagation by passive immunotherapy is not sufficient to explain long-term protection of FrCasE-infected/667 MAb-treated mice.
As mentioned above, a first possibility to explain that a short treatment of neonatally infected mice with the 667 MAb allows long-term healthy survival is that the efficient inhibition of viral propagation simply would give time to the immune system to mount a protective immunity. Should this be true, simply infected mice showing viral propagation kinetics comparable to those observed in FrCasE-infected and 667 MAb-treated mice during the treatment period should survive in the long term and develop a similar protective antiviral immune response. To test this hypothesis, we compared health statuses and antiviral immune responses in mice infected under standard conditions (intraperitoneal injection of a viral suspension containing 5 × 104 FrCasE FFU in 100 μl on day 3 after birth) and treated with the 667 MAb (high-inoculum [HI]-infected/667 MAb treated mice) and age-matched animals infected with a low viral inoculum (LI; intraperitoneal injection of a viral suspension containing 1 to 5 FrCasE FFU in 100 μl on day 3 after birth) but not immunotherapy treated. In these experiments, passive immunotherapy was initiated 1 h after infection, which is a time sufficient to allow infection and efficient viral spread (23-25; also see below), and consisted of three MAb administrations during a period of 5 days (Fig. 1). Notably, previous experiments have shown that protection outcomes of HI-infected mice were identical whether the MAb treatment was initiated 1 h or up to 2 days postinfection (23). Under our experimental conditions, the 667 MAb becomes undetectable in sera of HI-infected/667 MAb-treated mice by day 14 after infection (23). Viral propagation was monitored using three criteria: (i) serum viremia assay, (ii) the flow cytometry quantification of Env-expressing splenocytes, and (iii) the quantification of SICs (see Materials and Methods), i.e., of FrCasE-producing splenocytes (note that the spleen is a major replication organ for MuLVs).
FIG. 1.
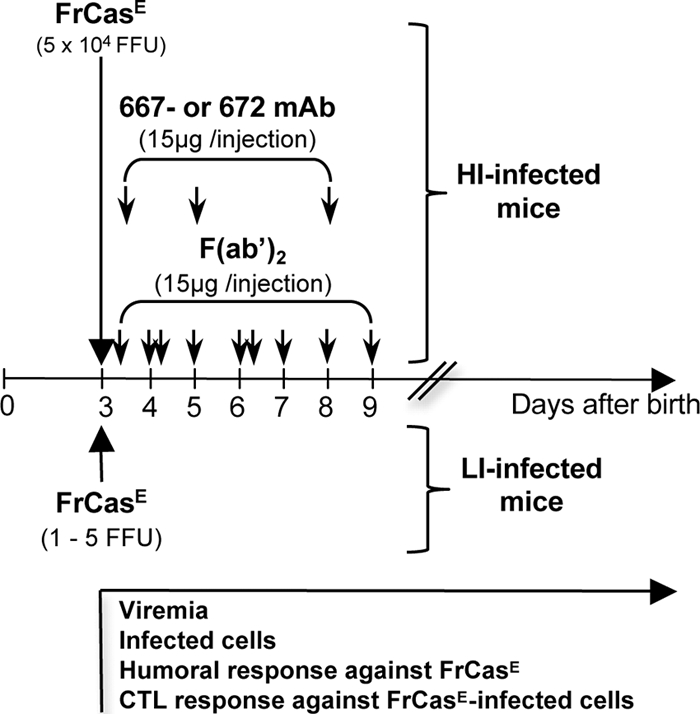
Design of the experiments. 129/Sv/Ev mice were infected on day 3 after birth with either 5 × 104 (HI-infected mice) or 1 to 5 (LI-infected mice) FrCasE FFU. HI-infected, but not LI-infected, mice were subjected to immunotherapy with either the 667 MAb (IgG2a/κ), the 667 F(ab′)2 MAb, or the 672 MAb (IgM/κ) under the conditions indicated in the figure. The 667 F(ab′)2 MAb was administrated every day with double injections on days 4 and 6 after birth, as indicated by double arrows. Mice were monitored from the day of infection onwards as indicated in the text. The analyzed parameters were (i) serum viremia, (ii) Env-expressing splenocytes, (iii) spleen infectious centers (SICs), (iv) endogenous anti-FrCasE IgM and IgG responses, and (v) primary and secondary CTL responses against FrCasE-infected cells. Additional treatments and experiments are indicated in the text.
All control HI-infected/nontreated mice died from neurodegeneration within 1 to 2 months after having shown high viremia (Fig. 2C) and the high productive infection of splenocytes (Fig. 2A and B). In contrast, infection with 1 to 5 FFU led to viral propagation kinetics comparable to that of HI-infected/667 MAb-treated mice during the first 14 days postinfection (peak by day 10 to 12 and return to basal level by day 12 to 14), as assayed at both SIC (Fig. 2D) and Env-expressing splenocyte (Fig. 2E) levels. Consistently with our former studies showing that the 667 MAb treatment does not completely eradicate FrCasE from HI-infected mice (23), the assay of SICs and Env-expressing splenocytes indicated very weak and comparable levels of spleen infection in the two groups of mice. Moreover, serum viremia (limit of detection, 102 FFU/ml) could be detected neither in HI-infected/667 MAb-treated mice, in keeping with our previous data (23), nor in LI-infected/nontreated mice. Of note, a significant increase of both infected and infectious splenocytes was observed by day 15 in LI-infected/nontreated mice but not in HI-infected/667 MAb-treated mice (Fig. 2D and E; also see Discussion). Finally, all of the LI-infected/nontreated mice were protected from the neurodegeneration (presumably because viral propagation was not sufficient to allow FrCasE entry into the brain before day 7 postinfection), as demonstrated by the absence of signal in PCR assays of FrCasE provirus performed in the brain 21 days after infection (see Fig. S1 in the supplemental material). However, all of these mice died from erythroleukemia before week 30 postinfection (Table 1). Erythroleukemia was similar to that developing upon F-MuLV infection in other settings, i.e., it was associated with anemia and dramatic spleen enlargement. This contrasted with the healthy survival of HI-infected/667 MAb-treated mice for more than 30 weeks postinfection (termination of the experiment). Thus, the simple reduction of viral propagation during the first 14 days postinfection, i.e., the time of actual immunotherapy in HI-infected/667 MAb-treated mice, is not sufficient to protect mice from retroviral disease development.
FIG. 2.

Viral propagations in HI-infected/immunotherapy-treated mice versus HI-infected/nontreated animals. A total of 54 newborn mice were used per group of HI-infected/667 MAb-treated and HI-infected/nontreated mice (two animals per time point in three different experiments), and a total of 36 newborn mice were used for each of the other groups (two animals per time point in two different experiments). Viral propagation was monitored using three criteria: spleen infectious centers, Env-expressing splenocytes, and serum viremia. (A, D, and F) SIC assays. Spleen cells from simply infected mice or from infected mice treated with either the 667 MAb, the 667 F(ab′)2 fragment, or the 672 MAb were isolated and plated onto indicator Mus dunni cells. SICs corresponded to the number of infectious centers scored in focal immunofluorescence assays after 3 days of coculture, as described in Materials and Methods. % SICs on the y axis indicates the percentage of infectious splenocytes versus the total number of splenocytes used in the experiment. (B, E, and G) Assay of Env-expressing splenocytes. Splenocytes were purified from the various mice, and Env expression at their surface was quantified by flow cytometry, as described in Materials and Methods. The percentage of Env-expressing splenocytes is indicated on the y axis. (C and H) Serum viremia assay. Viremia was assayed in a focal immunofluorescence assay, as indicated in Materials and Methods. Error bars indicate standard deviations. Statistical significance between the groups was established using the Mann and Whitney test. **, P < 0.01; *, P < 0.05.
TABLE 1.
Pathology development in HI-infected/667 MAb-treated and LI-infected/nontreated mice
| Mouse group | Wk 3 |
Wk 30 |
||
|---|---|---|---|---|
| Survival | Survival | % Hematocrita | Spleen massb (g) | |
| HI infected/667 MAb treated | 30/30c | 30/30c | 50 ± 1 | 0.08 ± 0.04 |
| LI infected/nontreated | 30/30c | 0/30c | 36 ± 2.3 | 1.65 ± 0.3 |
Percent hematocrit was measured as described in Materials and Methods.
Spleens were weighed after sacrifice.
Number of mice in each group.
LI-infected/nontreated mice mount a weaker anti-FrCasE immune response than HI-infected/667 MAb-treated animals.
As a next step, we tested whether erythroleukemia development in LI-infected/nontreated mice correlated with an antiviral immune response weaker than that of HI-infected/667 MAb-treated animals. We first addressed the humoral response against FrCasE by ELISA. From day 10 to 26 postinfection, the anti-FrCasE IgM production profiles were comparable between the groups of mice (Fig. 3A). In contrast, subsequent antiviral IgG production levels were dramatically different, with a weaker response in LI-infected/nontreated mice than in HI-infected/667 MAb-treated animals (>10-fold difference regardless of the time tested until week 30) (Fig. 3B). Consistently, the FrCasE-neutralizing activity of serum taken at week 15 postinfection was higher in HI-infected/667 MAb-treated mice, although the specific neutralizing activities per μg of anti-FrCasE IgG were comparable between the two groups of animals (Fig. 3C). This suggested a quantitative rather than qualitative difference between the humoral responses of LI-infected/nontreated- and HI-infected/667 MAb-treated mice. We then compared the primary and memory CTL responses against infected cells in the two groups of animals. As H-2Db cells infected by most MuLVs present an immunodominant MHC class I-restricted epitope called GagL (52), CD8+ T-cell responses directed against FrCasE-infected cells were quantified by flow cytometry using a fluorescent GagL tetramer during the first 22 days following infection. They showed that LI-infected/nontreated mice mounted a poor, albeit detectable, primary response. This contrasted with the 20-fold stronger response peaking by day 14 postinfection and returning to a nondetectable level by day 20 in HI-infected/667-treated animals (Fig. 4A and B). A weaker memory response also was identified by the GagL tetramer staining of CD8+ T cells in virally challenged LI-infected/nontreated mice 8 weeks after first infection compared to that of HI-infected/667 MAb-treated animals (Fig. 4C and D). Finally, we also verified that differences in CTL numbers actually reflected differences in cytolysis activity in vivo, which was monitored by the in vivo killing of splenocytes grafted to virally challenged mice after prior in vitro labeling with the CFSE vital die and loading with the GagL peptide (Fig. 4E and F). Thus, the simple reduction of viral propagation to a level comparable to that observed in HI-infected/667 MAb-treated animals during the first 14 days postinfection is not sufficient for the mounting of high, long-lasting humoral and cellular immune responses against FrCasE. This strongly suggests an immunomodulatory action of the 667 MAb directing the immune response toward a protective outcome in HI-infected/667 MAb-treated mice.
FIG. 3.

Humoral responses in HI-infected/immunotherapy-treated and LI-infected/nontreated mice. Anti-FrCasE IgM (A) and IgG (B) responses developed in HI-infected/667 MAb-treated- and LI-infected/nontreated mice. Forty neonates per group of animals each were used per experiment as described in the legend to Fig. 1. Two independent experiments were conducted. (C) Anti-FrCasE neutralization activity in sera from HI-infected/immunotherapy-treated and LI-infected/nontreated mice. Neutralization activity was assayed using sera from 15-week-old mice. The presented data are the averages of values obtained from two groups of 15 mice analyzed in independent experiments. In the left panel, the assays of total neutralization activities in the sera from the various mice were conducted using identical volumes of sera collected from these mice. In the right panel, the assays of specific neutralization activities (neutralization activity per weight unit of anti-FrCasE immunoglobulin) were conducted after appropriate dilutions of serum samples so as to have the same amounts of anti-FrCasE immunoglobulins in all samples from HI-infected/immunotherapy-treated mice and from LI-infected/nontreated mice. Anti-FrCasE IgM (D) and total anti-FrCasE IgGs (E) responses developed in HI-infected/667 F(ab′)2-treated and HI-infected/672 MAb-treated mice. Forty neonates per group of animals each were used per experiment as described in the legend to Fig. 1. Two independent experiments were conducted. Error bars indicate standard deviations.
FIG. 4.

CTL response in HI-infected/667 MAb-treated and LI-infected/nontreated mice. Mice were infected as described in Fig. 1 using age-matched noninfected/nontreated mice as controls. (A) Primary CD8+ T-cell response. Typical flow cytometry analyses of Db-GagL+ CD8+ T cells performed on day 14 postinfection are presented. (B) Kinetic analysis of the primary CD8+ T-cell response. The presented data are the averages of values obtained from two mice per time point in each of three independent experiments conducted. (C) Memory CD8+ T-cell response. Typical flow cytometry analysis of Db-GagL+ CD8+ T cells carried out 10 days after a viral challenge performed 8 weeks postinfection. (D) Statistical analysis of the memory CD8+ T-cell response. Eight mice per group were used in three independent experiments conducted as indicated in panel C. (E) Cytotoxic activity against spleen cells loaded with the GagL peptide in mice subjected to a viral challenge. Splenocytes were loaded with GagL or a control peptide and stained in the presence of low and high concentrations of CSFE, respectively. They were administered to mice 10 days after viral challenge in week 8 postinfection. Flow cytometry analyses were carried out on the following day. Typical data are presented in the figure. The left panel indicates the proportion of control- and GagL peptide-loaded splenocytes administered to mice. A reduction in CFSEHigh staining (right peak) in the other two panels is indicative of infected cell lysis. (F) Statistical analysis of cytotoxic activity against spleen cells loaded with the GagL peptide in mice subjected to a viral challenge. Eight mice per group were used in three independent experiments as indicated in panel E. Error bars indicate standard deviations.
The Fc domain of 667 MAb is necessary for healthy survival and efficient mounting of protective immunity in HI-infected/667 MAb-treated mice.
IgG2a antibodies are endowed with two important effector functions borne by their Fc fragment (7, 13), which could be involved in the indirect effects of 667 on endogenous antiviral immunity. These effector functions are the binding to the various Fcγ receptors (FcγRs) also recognized by other IgGs and expressed by a number of immune cells and the binding to the C1q component of complement (36). As a first step toward a more formal demonstration of the immunomodulatory action of 667, we compared HI-infected mice treated with either the whole MAb or its F(ab′)2 fragment, which is only capable of virus neutralization. In these experiments, the 667 F(ab′)2 fragment was administered more frequently than intact 667 (Fig. 1) to take into account its 3-fold-shorter in vivo half-life and to maintain its serum concentration at a level comparable to that of 667 during the whole treatment period. Importantly, the neutralizing activity of the 667 F(ab′)2 fragment was identical to that of the 667 MAb in in vitro neutralizing assays (Table 2). In addition to treating mice with similar neutralizing agent concentrations, this constituted a second prerequisite for the validity of our analysis. Viremia in HI-infected/667 F(ab′)2-treated animals was not contained as it was in HI-infected/667 MAb-treated mice, but it was reduced by a 100-fold factor with a viremia onset delayed by 4 days compared to that of HI-infected/nontreated controls (Fig. 2H). Consistently, the number of SICs (Fig. 2F) and Env-positive spleen cells (Fig. 2G) also was delayed and was much higher in HI-infected/667 F(ab′)2-treated mice than in HI-infected/667 MAb-treated ones and lower than in HI-infected/nontreated controls. Finally, all HI-infected/667 F(ab′)2-treated mice died from erythroleukemia within 20 weeks with no neurological signs of any sort (Table 3).
TABLE 2.
In vitro neutralizationa of FrCasE by the 667 MAb, the 667 F(ab′)2 fragment, and 672 MAb
| Antibody | % Inhibition of infection at: |
||||
|---|---|---|---|---|---|
| 0 μg/ml | 10 μg/ml | 5 μg/ml | 2.5 μg/ml | 1.25 μg/ml | |
| 667 MAb | 0 | 91.67 ± 16.67 | 86.11 ± 2.6 | 83.33 ± 2.1 | 72.22 ± 2.78 |
| 672 MAb | 0 | 94.44 ± 13.89 | 75.10 ± 2.78 | 80.56 ± 2.38 | 66.67 ± 5.56 |
| 667 F(ab′)2 | 0 | 92.48 ± 1.91 | 85.99 ± 3.48 | 77.19 ± 6.22 | 61.61 ± 10.13 |
Neutralization is presented as a percentage of the inhibition of infection with FrCasE virus and M. dunni target cells. Values are averages of the results from three independent experiments.
TABLE 3.
Pathology development in HI-infected/immunotherapy-treated mice
| Group | Wk 3 |
Wk 20 |
||
|---|---|---|---|---|
| Survival | Survival | % Hematocrit | Spleen mass (g) | |
| HI infected/667 MAb treated | 20/20a | 20/20a | 50 ± 1.3 | 0.06 ± 0.01 |
| HI infected/672 MAb treated | 11/11a | 0/11a | 38 ± 2.02 | 1.02 ± 0.18 |
| HI infected/667 F(ab′)2 treated | 18/18a | 0/18a | 26 ± 1.7 | 1.8 ± 0.3 |
Number of mice in each group.
We next compared the immune responses in the two groups of animals. Serum levels of antiviral IgMs increased similarly in HI-infected/667 MAb-treated- and HI-infected/667 F(ab′)2-treated mice from day 10 to 26 postinfection (Fig. 3D), but the subsequent total anti-FrCasE IgG response was 40-fold weaker in HI-infected/667 F(ab′)2-treated mice than in HI-infected/667 MAb-treated mice, whatever the time tested (Fig. 3B and E). Notably, the rapid decrease in IgG levels observed after week 15 coincided with the dramatic health deterioration of 667 F(ab)2-treated mice, which were euthanized at week 20 after infection. Not surprisingly, IgG neutralization activity was much higher in HI-infected/667 MAb-treated mice (Fig. 3C), although specific neutralization activities per μg of anti-FrCasE IgGs in the two groups of animals were comparable (Fig. 3C). The primary and memory CTL responses against infected cells in the two groups of animals were much weaker in HI-infected/667 F(ab′)2-treated than in HI-infected/667 MAb-treated mice, whether tested by GagL tetramer staining of CD8+ T cells (Fig. 5A and B) or in in vivo cytolysis activity assays (Fig. 5C). Thus, our data indicate that simple neutralization activity borne by its F(ab′)2 fragment is not sufficient for 667 to ensure not only the efficient reduction of viral propagation during the immunotherapy period but also the development of strong antiviral immunity and the protection of HI-infected mice.
FIG. 5.
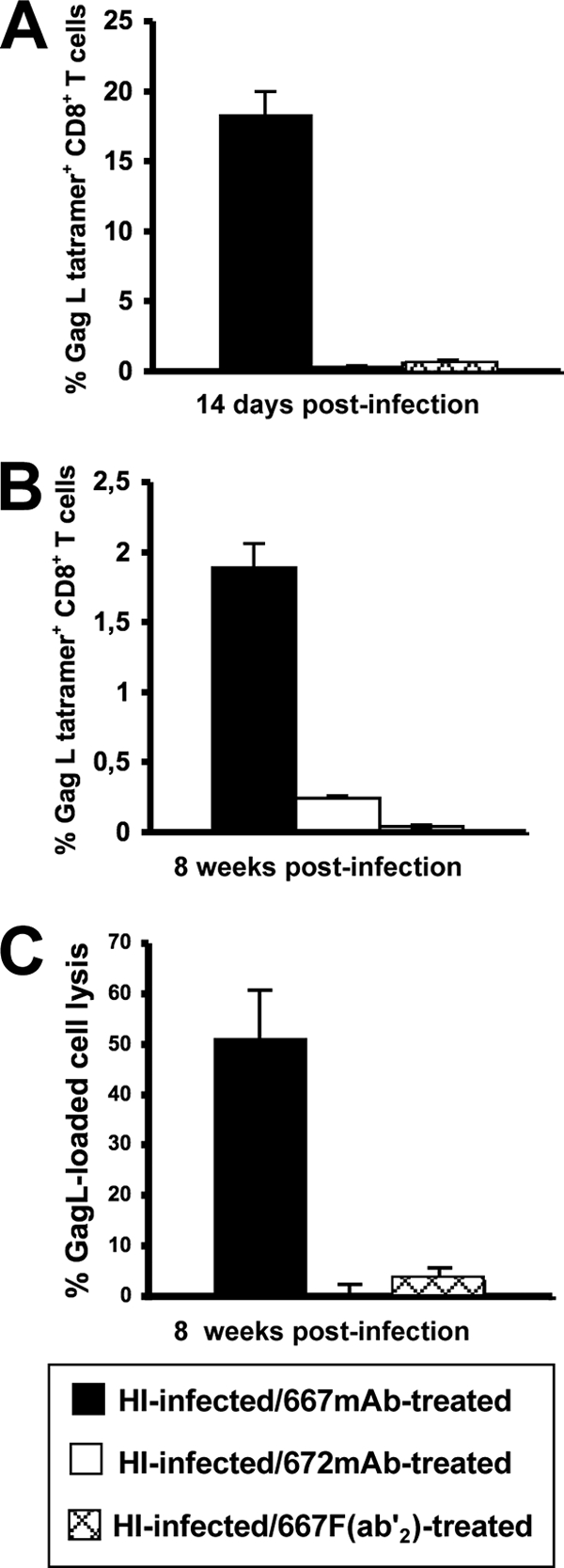
CTL response in HI-infected/667 F(ab′)2-treated and HI-infected/672 MAb-treated mice. Mice were infected as described in the legend to Fig. 1. (A) Primary CD8+ T-cell response. The presented data are the averages of values obtained from two animals analyzed on day 14 postinfection in each of three independent experiments conducted. (B) Memory CD8+ T-cell response. Eight mice per group were used in three independent experiments conducted as indicated in the legend to Fig. 4C. (C) Cytotoxic activity against spleen cells loaded with the GagL peptide in mice subjected to a viral challenge. Experiments were conducted as described for Fig. 4E and F. Eight mice per group were used in three independent experiments as indicated in the legend to Fig. 4E. Error bars indicate standard deviations.
No protective antiviral immunity develops in HI-infected mice treated with a neutralizing IgM.
We searched to identify which of the two effector functions borne by IgG2a is/are instrumental for the development of protective immunity in HI-infected/667 MAb-treated mice. As a first step to this aim, we turned to the immunotherapy of HI-infected animals using a neutralizing IgM, as this isotype is endowed with strong complement-binding activity but lacks the ability to bind to the FcγRs (34, 70). For this, we characterized in further detail the 672 MAb, which is an IgM/κ previously shown to be capable of CasBr neutralization (44). Using the SPOT technique (see Materials and Methods and reference 16), we demonstrated that 672 recognizes the linear epitope THWGLD, which is located in the viral receptor-binding domain of Env (Fig. 6A and B). Interestingly, this motif is contained in that recognized by 667 (16) (Fig. 6B), excluding that the differential in vivo activity between the two MAbs is due to strong epitopic differences. Additionally, we showed that 672 could neutralize FrCasE in vitro similarly to 667 (Table 2). In the immunotherapy treatment of HI-infected mice (Fig. 1), 672 effects were very similar to those of the 667 F(ab′)2 fragment, as all HI-infected/672 MAb-treated mice (i) died of erythroleukemia (Table 3), (ii) showed only partial viral propagation reduction during the second week postinfection (Fig. 2F to H), (iii) showed comparable anti-FrCasE IgM responses but poor IgG ones (Fig. 3D and E), (iv) showed poor neutralization activity of these endogenous IgGs (Fig. 3C), and (v) showed poor CTL activity directed to infected cells (Fig. 5A to C). This strongly suggested that complement activity does not play a major role, if any, in the long-term effect of 667 in HI-infected/667 MAb-treated mice.
FIG. 6.

Characterization of the 672 MAb. (A) Identification of the Env epitope recognized by the 672 MAb. The SPOT analysis was carried out as described in Materials and Methods. Two hundred seventeen overlapping 15-mer amino acid peptides frameshifted by 3 residues and representing the complete CasBr Env were synthesized on a nitrocellulose membrane and probed with 672. Only the region with positive signals is presented. Amino acids are numbered with respect to the first amino acid of the surface protein (pep, peptide; aa, amino acid). (B) Localization of the epitope recognized by 672 in Env. The overall structure of CasBr Env is depicted. The epitope recognized by 672 (gray box) is contained in that recognized by 667 (white box). It is localized within the variable region A (VRA) of the receptor-binding domain (RBD). SS, signal sequence; SU, surface protein; TM, transmembrane protein.
The 667 MAb mediates Fc-associated cell lysis in vivo.
Neither the 667 F(ab′)2 fragment nor the 672 MAb can limit viral propagation in HI-infected mice to a great extent. An obvious possibility to explain viral propagation blunting in 667 MAb immunotherapy-treated animals therefore would be the efficient mediation of the cytolysis of infected cells by 667 owing to its FcγR-binding activity. To test this possibility, we turned to an in vivo cytolysis assay (see Materials and Methods for details) consisting of assaying the survival of FrCasE-infected splenocytes injected intravenously into naïve mice after the administration of either 667 or 672 in the presence or in the absence of the 2.4G2 MAb, as this antibody recognizes two Fcγ receptors (FcγRII or CD32 and FcγRIII or CD16) and thereby blocks antibody-dependent cell cytolysis (ADCC) (3, 38, 39). The 667 F(ab′)2 fragment was used as a negative control in these experiments. The data presented in Fig. 7 indicate a strong FcγR-binding-dependent in vivo cytolysis activity of FrCasE-infected cells for 667, a reduced FcγR-binding-independent cytolysis activity for 672, and none for the 667 F(ab′)2 fragment. Thus, they strengthened the idea that the FcγR-binding effector function of 667 plays a crucial role in the protection of HI-infected mice.
FIG. 7.

In vivo FcγR-dependent cytolytic activities of the 667 MAb, the 667 F(ab′)2 fragment, and the 672 MAb. The assessment of FcγR-dependent cytolytic activities of the 667 MAb, the 667 F(ab′)2 fragment, and the 672 MAb is described in Materials and Methods. Briefly, noninfected and FrCasE-infected splenocytes differentially labeled with high and low concentrations of the vital dye CSFE were administered intravenously to naive mice or to mice formerly treated with the anti-FcγRII and anti-FcγRIII 2.4G2 MAb. At the same time, the animals received either the 667 MAb, the 667 F(ab′)2 fragment, or the 672 MAb. Flow cytometry assay of CFSElow/CFSEhigh ratios from cells recovered from spleens 5 h later to allow the comparison of the in vivo cytolytic activity of the anti-FrCasE MAbs and F(ab′2) in the presence or in the absence of FcγR-blocking MAb. Two mice per group were used in three independent experiments. Error bars indicate standard deviations.
DISCUSSION
We have addressed here whether viral propagation control in the immediate posttreatment period and/or MAb-driven immunomodulation could explain the long-lasting protective antiviral immunity mounted by FrCasE-infected mice treated with the 667 MAb. Four main conclusions can be drawn from our data. First, the drastic reduction of viral propagation during the first week after infection in the period just following infection is sufficient to avoid neurodegeneration. As the permeability of the blood-brain barrier is lost by day 7 after birth in mice (14), it is likely that the limitation of viral spread shortly after infection is sufficient to prevent FrCasE entry into the CNS. Second, virus neutralization by the antigen-binding domain of the MAb, which is essential for viral spread limitation in the immediate postinfection period, is not the exclusive antiviral mechanism at play before the onset of the endogenous antiviral immune response. Rather, the Fc portion of 667, most probably via the FcγRs-mediated lysis of infected cells, plays a crucial role in the direct control of initial viral propagation during the second week after infection. Third, our data rule out that the 667 MAb treatment just gives time to the immune system to react protectively to FrCasE infection but strongly suggest an immunomodulatory action of the administered MAb. Fourth, not all antibody isotypes are equally efficient at protecting mice and helping the mounting of a protective antiviral immunity, as the 672 MAb failed to do so.
Sequential actions of 667 neutralizing and effector functions control viral spread in HI-infected/667 MAb-treated mice.
To address the role of the 667 Fc and to identify the effector function(s) involved in its antiviral activity, we have resorted to immunotherapies of HI-infected mice with both the 667 F(ab′)2 fragment, which lacks IgG2a effector functions, and a neutralizing IgM, 672, which recognizes nearly the same epitope as 667 and is endowed with functions significantly different from IgG2a functions. Interestingly, the outcomes of these experiments were similar, not only because all mice died of erythroleukemia but also with respect to viral propagation and humoral and cellular immune responses. Strikingly, FrCasE spread was controlled in both groups of animals as efficiently as in HI-infected/667 MAb-treated mice for the first 7 days postinfection but less efficiently afterwards (Fig. 2F to H) until the moment the immune reaction became detectable, as assayed at the level of anti-FrCasE IgMs (Fig. 3C to E) and primary CTL responses (Fig. 6). This suggested a two-step action for 667: in the first step, viral replication control would depend exclusively on its neutralizing activity (approximately the first 7 days postinfection), and in a second phase it would depend on the MAb effector function(s) (approximately the following 7 to 10 days) before the endogenous adaptive immunity comes into play. As IgMs, such as 672, are endowed with a much higher complement-dependent cytolysis activity than IgG2a antibodies (33) and 667 displays FcγR-dependent cytolytic activity against FrCasE-infected cells in vivo (Fig. 7), it is most likely that the effector function of 667 involved in virus spread inhibition before the endogenous immune control takes over is binding to FcγR(s) borne by killer cells of the immune system. Taken with the results of others, these data support the notion that effector functions are essential for the efficient direct antiviral activity of MAbs. For example, the viral load reduction of mice infected with influenza virus was much less drastic upon treatment with an F(ab′)2 fragment than with its intact MAb counterpart (49). Additionally, at variance with MAb-treated mice, no protection could be achieved in F(ab′)2-treated animals infected with LCMV (6) or yellow fever virus (69). Finally, mutating the complement-binding site of a neutralizing anti-HIV MAb had no effect on the viral load in simian-human immunodeficiency virus (SHIV)-infected macaques, whereas disabling the FcR-binding site leads to the drastic loss of direct antiviral activity (27, 28). It would be interesting to establish whether MAb neutralizing and effector functions act sequentially in these experiments as we observed in ours.
Possible role of 667 effector function(s) in the mounting of a protective antiviral response in HI-infected/667 MAb-treated mice.
The lack of protective immune responses in HI-infected/667 F(ab′)2-treated and HI-infected/672 MAb-treated mice strongly suggests an immunomodulatory action of 667 in neonatally FrCasE-infected mice, which is an important conclusion of this work. Concerning this effect, the FcγR-binding function is the most interesting effector function to consider. One reason for this is that complement activity is relatively low in mouse neonates, which diminishes the likelihood of a complement-dependent contribution in our experiments. Another, more straightforward, possibility takes into account the biology of both neonatal dendritic cells (DCs) and Fc receptors. Neonatal humoral and cell-mediated immune responses are generally of lower potency and of shorter duration than those generated in adults, especially upon exposure to high doses of antigen (see below) (1, 71). This is largely due not only to the scarcity of neonatal DCs but also to the less effective interaction of these cells with antigens, which results in the weak activation of effector B and T cells (1, 71). Besides this, immune complexes (IC) are more efficient than simple antigens at activating the maturation, antigen presentation, and stimulation of effector lymphocytes by DCs via their interactions with and internalization via their Fc receptors (51). It therefore is reasonable to assume that ICs involving FrCasE and/or infected cells better activate DCs in HI-infected/667 MAb-treated mice than simple viral antigens in HI-infected/nontreated [or even viral antigen/667 F(ab′)2 and viral antigen/672 MAb complexes in HI-infected/667 F(ab′)2- and HI-infected/672 MAb-treated animals, respectively], as recently shown in older HI-infected/667 MAb-treated mice (46). As FcγRs are expressed on other immune cells, it is possible that other mechanisms also come into play in HI-infected/667 MAb-treated mice. This is all the more important because B cells are poorly abundant during early life, and ICs were demonstrated to be more efficient than simple antigens at activating B cells and lengthening plasma cell survival in a variety of situations (51).
A role for viremia blunting in induction of antiviral immunity in neonatally HI-infected/667 MAb-treated mice?
Neonatal immune responses most often are skewed toward tolerance or TH2-type responses upon exposure to high doses of antigen (1, 71). However, decreasing the dose of antigen initially was suggested as a simple way to induce adult-like TH1 responses in neonates (65, 66). Interestingly, one of the two seminal studies was based on mouse infection with the CasBr retrovirus, whose env gene is a constituent of FrCasE (also see below), and this concept received further support in other viral settings. Those included the vaccination of newborn mice with live-attenuated Sendai (72), Herpes simplex (20), and vaccinia (37) viruses capable of very limited propagation. Even though the reduction of viremia early in life is not sufficient on its own to induce long-lasting protective immunity (as shown in this work), it is important to consider that the blunting of viral propagation may collaborate with immunomodulation by 667 to explain the typical TH1 response with IgG2a and CTL contributions developing in neonatally HI-infected/667 MAb-treated mice. This must be considered, because limiting antigen amounts in various experimental models has been shown to favor the expansion of T cells with high-affinity receptors, which are T cells presumably more efficient at eliminating antigens and providing help to B cells (10, 61, 67). One possibility is that the fitness of the CD8+ T-cell response would (at least partially) depend on limited bystander inflammation at the time of inoculation, as shown in infections of adult mice with various inocula of polyomavirus (2).
We would, however, emphasize that the contribution of viral propagation blunting to long-term antiviral immunity might be both time and virus dependent. The idea of restriction to a narrow window of time is supported by the observation that the infection of 2-week-old mouse infants with a low LCMV inoculum led to only limited expansion of CD8+ effector T cells and the absence of memory CD8+ T cells (8). The idea of virus type dependency stems from the different outcomes of infections of neonatal mice with low inocula of two related retroviruses, CasBr (17, 18, 66) and FrCasE (this work), despite apparently very similar experimental conditions. In the former case, mice are protected from the retroviral disease and develop a strong TH1 memory response (18, 66) that can be reactivated easily during adulthood (17), whereas in the latter case, mice die after having developed a poor and ineffective primary CD8+ T-cell response lacking memory.
Evolution of the humoral anti-FrCasE response in HI-infected/667 MAb-treated mice.
Two points deserve comment concerning the anti-FrCasE humoral immunity in the four groups of animals of this study. The first point concerns the apparent similarity of IgM responses whatever the infection and treatment conditions (Fig. 3 and 6), which suggests the absence of an immunomodulatory action of FrCasE/667 ICs at that level. However, we would like to underline that IgM responses were assessed only at the quantitative level. At this stage of investigation, we cannot formally exclude a possible immunomodulatory effect of the immunotherapy on the humoral response via the selection of distinct idiotype repertoires in HI-infected/667 MAb-treated and HI-infected/667 F(ab′)2, HI-infected/672 MAb, or LI-infected/nontreated mice (also see below). Future work will address this point. The second point concerns the dramatic differences between anti-FrCasE IgG responses in HI-infected/667 MAb-treated mice on the one hand and LI-infected/nontreated, HI-infected/667 F(ab′)2, and HI-infected/672 MAb-treated animals on the other (Fig. 3 and 6). Two periods were clearly distinguishable in our experiments. A first early period extended from the immunotherapy to week 10 postinfection and already was characterized by more abundant anti-FrCasE IgGs in the former group of animals compared to those of the other two. A second late period began by week 10, at which time the IgG response increased not only dramatically but also synchronously in all HI-infected/667 MAb-treated mice, whereas it remained very low in the other two groups. It is plausible that 667/FrCasE ICs played a role at the beginning of the early period to induce a stronger antiviral IgG response, possibly by favoring the IgM-to-IgG class switch and/or by boosting the endogenous IgG response in HI-infected/667 MAb-treated mice. However, such a mechanism cannot be considered at later time points, as 667 no longer is detectable in 667 MAb-treated mice 2 weeks after its first administration (23). Further investigations are necessary to elucidate the mechanisms programming this humoral response enhancement.
In conclusion, we have shown in this study that the anti-FrCasE protective immunity developing in neonatally HI-infected/667 MAb-treated mice is not the sole consequence of the blunting of viral propagation by 667, which would have given sufficient time to the immune system of infected pups to react efficiently. Rather, it results from a genuine immunomodulatory effect of the administered antibody dependent on Fc fragment-associated function(s). As the effector properties of the various antibody isotypes are significantly different, our data strongly suggest that the choice of the neutralizing MAb isotype will be critical not only for immediate antiviral effects, as shown in the immunotherapies of SHIV-infected macaques cited above (27, 28), but also for inducing endogenous protective antiviral immunity.
Supplementary Material
Acknowledgments
M.P.'s laboratory is designated an Equipe Labelisée funded by the Ligue Nationale contre le Cancer. This work also was supported by the Programme Blanc of the Agence Nationale pour la Recherche (contract NT09_558711) and by an ARC grant. R.N. was supported by fellowships from the French Ministry of Research (MRT) and the Fondation pour la Recherche Médicale.
We are grateful to Eric Kremer for the careful reading of the manuscript and helpful comments.
Footnotes
Published ahead of print on 7 July 2010.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Adkins, B., C. Leclerc, and S. Marshall-Clarke. 2004. Neonatal adaptive immunity comes of age. Nat. Rev. Immunol. 4:553-564. [DOI] [PubMed] [Google Scholar]
- 2.Andrews, N. P., C. D. Pack, V. Vezys, G. N. Barber, and A. E. Lukacher. 2007. Early virus-associated bystander events affect the fitness of the CD8 T cell response to persistent virus infection. J. Immunol. 178:7267-7275. [DOI] [PubMed] [Google Scholar]
- 3.Araujo-Jorge, T., M. T. Rivera, A. el Bouhdidi, M. Daeron, and Y. Carlier. 1993. An Fc gamma RII-, Fc gamma RIII-specific monoclonal antibody (2.4G2) decreases acute Trypanosoma cruzi infection in mice. Infect. Immun. 61:4925-4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armbruster, C., G. M. Stiegler, B. A. Vcelar, W. Jager, N. L. Michael, N. Vetter, and H. W. Katinger. 2002. A phase I trial with two human monoclonal antibodies (hMAb 2F5, 2G12) against HIV-1. AIDS 16:227-233. [DOI] [PubMed] [Google Scholar]
- 5.Baba, T. W., V. Liska, R. Hofmann-Lehmann, J. Vlasak, W. Xu, S. Ayehunie, L. A. Cavacini, M. R. Posner, H. Katinger, G. Stiegler, B. J. Bernacky, T. A. Rizvi, R. Schmidt, L. R. Hill, M. E. Keeling, Y. Lu, J. E. Wright, T. C. Chou, and R. M. Ruprecht. 2000. Human neutralizing monoclonal antibodies of the IgG1 subtype protect against mucosal simian-human immunodeficiency virus infection. Nat. Med. 6:200-206. [DOI] [PubMed] [Google Scholar]
- 6.Baldridge, J. R., and M. J. Buchmeier. 1992. Mechanisms of antibody-mediated protection against lymphocytic choriomeningitis virus infection: mother-to-baby transfer of humoral protection. J. Virol. 66:4252-4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baudino, L., F. Nimmerjahn, S. Azeredo da Silveira, E. Martinez-Soria, T. Saito, M. Carroll, J. V. Ravetch, J. S. Verbeek, and S. Izui. 2008. Differential contribution of three activating IgG Fc receptors (FcgammaRI, FcgammaRIII, and FcgammaRIV) to IgG2a- and IgG2b-induced autoimmune hemolytic anemia in mice. J. Immunol. 180:1948-1953. [DOI] [PubMed] [Google Scholar]
- 8.Belnoue, E., P. Fontannaz-Bozzotti, S. Grillet, P. H. Lambert, and C. A. Siegrist. 2007. Protracted course of lymphocytic choriomeningitis virus WE infection in early life: induction but limited expansion of CD8+ effector T cells and absence of memory CD8+ T cells. J. Virol. 81:7338-7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burioni, R., M. Perotti, N. Mancini, and M. Clementi. 2008. Perspectives for the utilization of neutralizing human monoclonal antibodies as anti-HCV drugs. J. Hepatol. 49:299-300. [DOI] [PubMed] [Google Scholar]
- 10.Busch, D. H., and E. G. Pamer. 1999. T cell affinity maturation by selective expansion during infection. J. Exp. Med. 189:701-710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cekinovic, D., M. Golemac, E. P. Pugel, J. Tomac, L. Cicin-Sain, I. Slavuljica, R. Bradford, S. Misch, T. H. Winkler, M. Mach, W. J. Britt, and S. Jonjic. 2008. Passive immunization reduces murine cytomegalovirus-induced brain pathology in newborn mice. J. Virol. 82:12172-12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen, W., H. Qin, B. Chesebro, and M. A. Cheever. 1996. Identification of a gag-encoded cytotoxic T-lymphocyte epitope from FBL-3 leukemia shared by Friend, Moloney, and Rauscher murine leukemia virus-induced tumors. J. Virol. 70:7773-7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Congy-Jolivet, N., A. Probst, H. Watier, and G. Thibault. 2007. Recombinant therapeutic monoclonal antibodies: mechanisms of action in relation to structural and functional duality. Crit. Rev. Oncol. Hematol. 64:226-233. [DOI] [PubMed] [Google Scholar]
- 14.Czub, M., S. Czub, F. J. McAtee, and J. L. Portis. 1991. Age-dependent resistance to murine retrovirus-induced spongiform neurodegeneration results from central nervous system-specific restriction of virus replication. J. Virol. 65:2539-2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Czub, S., W. P. Lynch, M. Czub, and J. L. Portis. 1994. Kinetic analysis of spongiform neurodegenerative disease induced by a highly virulent murine retrovirus. Lab. Investig. 70:711-723. [PubMed] [Google Scholar]
- 16.Dreja, H., L. Gros, S. Villard, E. Bachrach, A. Oates, C. Granier, T. Chardes, J. C. Mani, M. Piechaczyk, and M. Pelegrin. 2003. Monoclonal antibody 667 recognizes the variable region A motif of the ecotropic retrovirus CasBrE envelope glycoprotein and inhibits Env binding to the viral receptor. J. Virol. 77:10984-10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fadel, S. A., L. G. Cowell, S. Cao, D. A. Ozaki, T. B. Kepler, D. A. Steeber, and M. Sarzotti. 2006. Neonate-primed CD8+ memory cells rival adult-primed memory cells in antigen-driven expansion and anti-viral protection. Int. Immunol. 18:249-257. [DOI] [PubMed] [Google Scholar]
- 18.Fadel, S. A., D. A. Ozaki, and M. Sarzotti. 2002. Enhanced type 1 immunity after secondary viral challenge in mice primed as neonates. J. Immunol. 169:3293-3300. [DOI] [PubMed] [Google Scholar]
- 19.Ferrantelli, F., R. A. Rasmussen, K. A. Buckley, P. L. Li, T. Wang, D. C. Montefiori, H. Katinger, G. Stiegler, D. C. Anderson, H. M. McClure, and R. M. Ruprecht. 2004. Complete protection of neonatal rhesus macaques against oral exposure to pathogenic simian-human immunodeficiency virus by human anti-HIV monoclonal antibodies. J. Infect. Dis. 189:2167-2173. [DOI] [PubMed] [Google Scholar]
- 20.Franchini, M., C. Abril, C. Schwerdel, C. Ruedl, M. Ackermann, and M. Suter. 2001. Protective T-cell-based immunity induced in neonatal mice by a single replicative cycle of herpes simplex virus. J. Virol. 75:83-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gairin, J. E., H. Mazarguil, D. Hudrisier, and M. B. Oldstone. 1995. Optimal lymphocytic choriomeningitis virus sequences restricted by H-2Db major histocompatibility complex class I molecules and presented to cytotoxic T lymphocytes. J. Virol. 69:2297-2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galun, E., N. A. Terrault, R. Eren, A. Zauberman, O. Nussbaum, D. Terkieltaub, M. Zohar, R. Buchnik, Z. Ackerman, R. Safadi, Y. Ashur, S. Misrachi, Y. Liberman, L. Rivkin, and S. Dagan. 2007. Clinical evaluation (phase I) of a human monoclonal antibody against hepatitis C virus: safety and antiviral activity. J. Hepatol. 46:37-44. [DOI] [PubMed] [Google Scholar]
- 23.Gros, L., H. Dreja, A. L. Fiser, M. Plays, M. Pelegrin, and M. Piechaczyk. 2005. Induction of long-term protective antiviral endogenous immune response by short neutralizing monoclonal antibody treatment. J. Virol. 79:6272-6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gros, L., M. Pelegrin, H. A. Michaud, S. Bianco, J. Hernandez, C. Jacquet, and M. Piechaczyk. 2008. Endogenous cytotoxic T-cell response contributes to the long-term antiretroviral protection induced by a short period of antibody-based immunotherapy of neonatally infected mice. J. Virol. 82:1339-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gros, L., M. Pelegrin, M. Plays, and M. Piechaczyk. 2006. Efficient mother-to-child transfer of antiretroviral immunity in the context of preclinical monoclonal antibody-based immunotherapy. J. Virol. 80:10191-10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guyre, C. A., D. Gomes, K. A. Smith, J. M. Kaplan, and M. A. Perricone. 2008. Development of an in vivo antibody-mediated killing (IVAK) model, a flow cytometric method to rapidly evaluate therapeutic antibodies. J. Immunol. Methods 333:51-60. [DOI] [PubMed] [Google Scholar]
- 27.Hessell, A. J., L. Hangartner, M. Hunter, C. E. Havenith, F. J. Beurskens, J. M. Bakker, C. M. Lanigan, G. Landucci, D. N. Forthal, P. W. Parren, P. A. Marx, and D. R. Burton. 2007. Fc receptor but not complement binding is important in antibody protection against HIV. Nature 449:101-104. [DOI] [PubMed] [Google Scholar]
- 28.Hessell, A. J., P. Poignard, M. Hunter, L. Hangartner, D. M. Tehrani, W. K. Bleeker, P. W. Parren, P. A. Marx, and D. R. Burton. 2009. Effective, low-titer antibody protection against low-dose repeated mucosal SHIV challenge in macaques. Nat. Med. 15:951-954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hessell, A. J., E. G. Rakasz, P. Poignard, L. Hangartner, G. Landucci, D. N. Forthal, W. C. Koff, D. I. Watkins, and D. R. Burton. 2009. Broadly neutralizing human anti-HIV antibody 2G12 is effective in protection against mucosal SHIV challenge even at low serum neutralizing titers. PLoS Pathog. 5:e1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hofmann-Lehmann, R., J. Vlasak, R. A. Rasmussen, S. Jiang, P. L. Li, T. W. Baba, D. C. Montefiori, B. J. Bernacky, T. A. Rizvi, R. Schmidt, L. R. Hill, M. E. Keeling, H. Katinger, G. Stiegler, L. A. Cavacini, M. R. Posner, and R. M. Ruprecht. 2002. Postnatal pre- and postexposure passive immunization strategies: protection of neonatal macaques against oral simian-human immunodeficiency virus challenge. J. Med. Primatol. 31:109-119. [DOI] [PubMed] [Google Scholar]
- 31.Hong, H. J., C. J. Ryu, H. Hur, S. Kim, H. K. Oh, M. S. Oh, and S. Y. Park. 2004. In vivo neutralization of hepatitis B virus infection by an anti-preS1 humanized antibody in chimpanzees. Virology 318:134-141. [DOI] [PubMed] [Google Scholar]
- 32.Huber, M., V. von Wyl, C. G. Ammann, H. Kuster, G. Stiegler, H. Katinger, R. Weber, M. Fischer, H. Stoiber, H. F. Gunthard, and A. Trkola. 2008. Potent human immunodeficiency virus-neutralizing and complement lysis activities of antibodies are not obligatorily linked. J. Virol. 82:3834-3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imai, M., C. Landen, R. Ohta, N. K. Cheung, and S. Tomlinson. 2005. Complement-mediated mechanisms in anti-GD2 monoclonal antibody therapy of murine metastatic cancer. Cancer Res. 65:10562-10568. [DOI] [PubMed] [Google Scholar]
- 34.Kaetzel, C. S. 2005. The polymeric immunoglobulin receptor: bridging innate and adaptive immune responses at mucosal surfaces. Immunol. Rev. 206:83-99. [DOI] [PubMed] [Google Scholar]
- 35.Kim, S. H., Y. W. Shin, K. W. Hong, K. H. Chang, K. H. Ryoo, S. H. Paik, J. M. Kim, B. Brotman, W. Pfahler, and A. M. Prince. 2008. Neutralization of hepatitis B virus (HBV) by human monoclonal antibody against HBV surface antigen (HBsAg) in chimpanzees. Antivir. Res. 79:188-191. [DOI] [PubMed] [Google Scholar]
- 36.Klaus, G. G., M. B. Pepys, K. Kitajima, and B. A. Askonas. 1979. Activation of mouse complement by different classes of mouse antibody. Immunology 38:687-695. [PMC free article] [PubMed] [Google Scholar]
- 37.Kovarik, J., M. Gaillard, X. Martinez, P. Bozzotti, P. H. Lambert, T. F. Wild, and C. A. Siegrist. 2001. Induction of adult-like antibody, Th1, and CTL responses to measles hemagglutinin by early life murine immunization with an attenuated vaccinia-derived NYVAC(K1L) viral vector. Virology 285:12-20. [DOI] [PubMed] [Google Scholar]
- 38.Kummer, U., U. Zengerle, J. Pischel, B. Trautmann, R. Mailhammer, and N. Sidell. 2001. Increased in vivo mitogenicity of anti-TCR/CD3 monoclonal antibody through reduced interaction with Fcgamma receptors. Immunol. Lett. 75:153-158. [DOI] [PubMed] [Google Scholar]
- 39.Kurlander, R. J., D. M. Ellison, and J. Hall. 1984. The blockade of Fc receptor-mediated clearance of immune complexes in vivo by a monoclonal antibody (2.4G2) directed against Fc receptors on murine leukocytes. J. Immunol. 133:855-862. [PubMed] [Google Scholar]
- 40.Law, M., T. Maruyama, J. Lewis, E. Giang, A. W. Tarr, Z. Stamataki, P. Gastaminza, F. V. Chisari, I. M. Jones, R. I. Fox, J. K. Ball, J. A. McKeating, N. M. Kneteman, and D. R. Burton. 2008. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 14:25-27. [DOI] [PubMed] [Google Scholar]
- 41.Lynch, W. P., S. J. Robertson, and J. L. Portis. 1995. Induction of focal spongiform neurodegeneration in developmentally resistant mice by implantation of murine retrovirus-infected microglia. J. Virol. 69:1408-1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manrique, A., P. Rusert, B. Joos, M. Fischer, H. Kuster, C. Leemann, B. Niederost, R. Weber, G. Stiegler, H. Katinger, H. F. Gunthard, and A. Trkola. 2007. In vivo and in vitro escape from neutralizing antibodies 2G12, 2F5, and 4E10. J. Virol. 81:8793-8808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marasco, W. A., and J. Sui. 2007. The growth and potential of human antiviral monoclonal antibody therapeutics. Nat. Biotechnol. 25:1421-1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McAtee, F. J., and J. L. Portis. 1985. Monoclonal antibodies specific for wild mouse neurotropic retrovirus: detection of comparable levels of virus replication in mouse strains susceptible and resistant to paralytic disease. J. Virol. 56:1018-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mejías, A., and O. Ramilo. 2008. Review of palivizumab in the prophylaxis of respiratory syncytial virus (RSV) in high-risk infants. Biologics 2:433-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Michaud, H. Gomard, T. Gros, L. Thiolon, K. Nasser, R. Jacquet, C. Hernandez, J. Piechaczyck, M., and M. Pelgrin. 2010. A crucial role for infected-cell/antibody immune complexes in the enhancement of endogenous antiviral immunity by short passive immunotherapy. PLoS Pathog. 6:e1000948. [DOI] [PMC free article] [PubMed]
- 47.Molina, F., D. Laune, C. Gougat, B. Pau, and C. Granier. 1996. Improved performances of spot multiple peptide synthesis. Pept. Res. 9:151-155. [PubMed] [Google Scholar]
- 48.Morrey, J. D., V. Siddharthan, H. Wang, J. O. Hall, R. T. Skirpstunas, A. L. Olsen, J. L. Nordstrom, S. Koenig, S. Johnson, and M. S. Diamond. 2008. West Nile virus-induced acute flaccid paralysis is prevented by monoclonal antibody treatment when administered after infection of spinal cord neurons. J. Neurovirol. 14:152-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mozdzanowska, K., J. Feng, and W. Gerhard. 2003. Virus-neutralizing activity mediated by the Fab fragment of a hemagglutinin-specific antibody is sufficient for the resolution of influenza virus infection in SCID mice. J. Virol. 77:8322-8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakowitsch, S., H. Quendler, H. Fekete, R. Kunert, H. Katinger, and G. Stiegler. 2005. HIV-1 mutants escaping neutralization by the human antibodies 2F5, 2G12, and 4E10: in vitro experiments versus clinical studies. AIDS 19:1957-1966. [DOI] [PubMed] [Google Scholar]
- 51.Nimmerjahn, F., and J. V. Ravetch. 2008. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 8:34-47. [DOI] [PubMed] [Google Scholar]
- 52.Ohlén, C., M. Kalos, L. E. Cheng, A. C. Shur, D. J. Hong, B. D. Carson, N. C. Kokot, C. G. Lerner, B. D. Sather, E. S. Huseby, and P. D. Greenberg. 2002. CD8(+) T cell tolerance to a tumor-associated antigen is maintained at the level of expansion rather than effector function. J. Exp. Med. 195:1407-1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oliphant, T., M. Engle, G. E. Nybakken, C. Doane, S. Johnson, L. Huang, S. Gorlatov, E. Mehlhop, A. Marri, K. M. Chung, G. D. Ebel, L. D. Kramer, D. H. Fremont, and M. S. Diamond. 2005. Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat. Med. 11:522-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oliphant, T., G. E. Nybakken, M. Engle, Q. Xu, C. A. Nelson, S. Sukupolvi-Petty, A. Marri, B. E. Lachmi, U. Olshevsky, D. H. Fremont, T. C. Pierson, and M. S. Diamond. 2006. Antibody recognition and neutralization determinants on domains I and II of West Nile Virus envelope protein. J. Virol. 80:12149-12159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parren, P. W., P. A. Marx, A. J. Hessell, A. Luckay, J. Harouse, C. Cheng-Mayer, J. P. Moore, and D. R. Burton. 2001. Antibody protects macaques against vaginal challenge with a pathogenic R5 simian/human immunodeficiency virus at serum levels giving complete neutralization in vitro. J. Virol. 75:8340-8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pelegrin, M., M. Marin, A. Oates, D. Noel, R. Saller, B. Salmons, and M. Piechaczyk. 2000. Immunotherapy of a viral disease by in vivo production of therapeutic monoclonal antibodies. Hum. Gene Ther. 11:1407-1415. [DOI] [PubMed] [Google Scholar]
- 57.Poignard, P., R. Sabbe, G. R. Picchio, M. Wang, R. J. Gulizia, H. Katinger, P. W. Parren, D. E. Mosier, and D. R. Burton. 1999. Neutralizing antibodies have limited effects on the control of established HIV-1 infection in vivo. Immunity 10:431-438. [DOI] [PubMed] [Google Scholar]
- 58.Portis, J. L., S. Czub, C. F. Garon, and F. J. McAtee. 1990. Neurodegenerative disease induced by the wild mouse ecotropic retrovirus is markedly accelerated by long terminal repeat and gag-pol sequences from nondefective Friend murine leukemia virus. J. Virol. 64:1648-1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Prabakaran, M., N. Prabhu, F. He, Q. Hongliang, H. T. Ho, J. Qiang, T. Meng, M. Goutama, and J. Kwang. 2009. Combination therapy using chimeric monoclonal antibodies protects mice from lethal H5N1 infection and prevents formation of escape mutants. PLoS One 4:e5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prabhu, N., M. Prabakaran, H. T. Ho, S. Velumani, J. Qiang, M. Goutama, and J. Kwang. 2009. Monoclonal antibodies against the fusion peptide of hemagglutinin protect mice from lethal influenza A virus H5N1 infection. J. Virol. 83:2553-2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rees, W., J. Bender, T. K. Teague, R. M. Kedl, F. Crawford, P. Marrack, and J. Kappler. 1999. An inverse relationship between T cell receptor affinity and antigen dose during CD4(+) T cell responses in vivo and in vitro. Proc. Natl. Acad. Sci. U. S. A. 96:9781-9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reichert, J. M. 2008. Monoclonal antibodies as innovative therapeutics. Curr. Pharm. Biotechnol. 9:423-430. [DOI] [PubMed] [Google Scholar]
- 63.Reichert, J. M. 2007. Trends in the development and approval of monoclonal antibodies for viral infections. BioDrugs 21:1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reichert, J. M., C. J. Rosensweig, L. B. Faden, and M. C. Dewitz. 2005. Monoclonal antibody successes in the clinic. Nat. Biotechnol. 23:1073-1078. [DOI] [PubMed] [Google Scholar]
- 65.Ridge, J. P., E. J. Fuchs, and P. Matzinger. 1996. Neonatal tolerance revisited: turning on newborn T cells with dendritic cells. Science 271:1723-1726. [DOI] [PubMed] [Google Scholar]
- 66.Sarzotti, M., D. S. Robbins, and P. M. Hoffman. 1996. Induction of protective CTL responses in newborn mice by a murine retrovirus. Science 271:1726-1728. [DOI] [PubMed] [Google Scholar]
- 67.Savage, P. A., J. J. Boniface, and M. M. Davis. 1999. A kinetic basis for T cell receptor repertoire selection during an immune response. Immunity 10:485-492. [DOI] [PubMed] [Google Scholar]
- 68.Schepers, K., M. Toebes, G. Sotthewes, F. A. Vyth-Dreese, T. A. Dellemijn, C. J. Melief, F. Ossendorp, and T. N. Schumacher. 2002. Differential kinetics of antigen-specific CD4+ and CD8+ T cell responses in the regression of retrovirus-induced sarcomas. J. Immunol. 169:3191-3199. [DOI] [PubMed] [Google Scholar]
- 69.Schlesinger, J. J., and S. Chapman. 1995. Neutralizing F(ab′)2 fragments of protective monoclonal antibodies to yellow fever virus (YF) envelope protein fail to protect mice against lethal YF encephalitis. J. Gen. Virol. 76:217-220. [DOI] [PubMed] [Google Scholar]
- 70.Shibuya, A., N. Sakamoto, Y. Shimizu, K. Shibuya, M. Osawa, T. Hiroyama, H. J. Eyre, G. R. Sutherland, Y. Endo, T. Fujita, T. Miyabayashi, S. Sakano, T. Tsuji, E. Nakayama, J. H. Phillips, L. L. Lanier, and H. Nakauchi. 2000. Fc alpha/mu receptor mediates endocytosis of IgM-coated microbes. Nat. Immunol. 1:441-446. [DOI] [PubMed] [Google Scholar]
- 71.Siegrist, C. A., and R. Aspinall. 2009. B-cell responses to vaccination at the extremes of age. Nat. Rev. Immunol. 9:185-194. [DOI] [PubMed] [Google Scholar]
- 72.Siegrist, C. A., F. Saddallah, C. Tougne, X. Martinez, J. Kovarik, and P. H. Lambert. 1998. Induction of neonatal TH1 and CTL responses by live viral vaccines: a role for replication patterns within antigen presenting cells? Vaccine 16:1473-1478. [DOI] [PubMed] [Google Scholar]
- 73.Simmons, C. P., N. L. Bernasconi, A. L. Suguitan, K. Mills, J. M. Ward, N. V. Chau, T. T. Hien, F. Sallusto, Q. Ha do, J. Farrar, M. D. de Jong, A. Lanzavecchia, and K. Subbarao. 2007. Prophylactic and therapeutic efficacy of human monoclonal antibodies against H5N1 influenza. PLoS Med. 4:e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sui, J., W. C. Hwang, S. Perez, G. Wei, D. Aird, L. M. Chen, E. Santelli, B. Stec, G. Cadwell, M. Ali, H. Wan, A. Murakami, A. Yammanuru, T. Han, N. J. Cox, L. A. Bankston, R. O. Donis, R. C. Liddington, and W. A. Marasco. 2009. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 16:265-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Takada, A., H. Ebihara, S. Jones, H. Feldmann, and Y. Kawaoka. 2007. Protective efficacy of neutralizing antibodies against Ebola virus infection. Vaccine 25:993-999. [DOI] [PubMed] [Google Scholar]
- 76.Trkola, A., H. Kuster, P. Rusert, B. Joos, M. Fischer, C. Leemann, A. Manrique, M. Huber, M. Rehr, A. Oxenius, R. Weber, G. Stiegler, B. Vcelar, H. Katinger, L. Aceto, and H. F. Gunthard. 2005. Delay of HIV-1 rebound after cessation of antiretroviral therapy through passive transfer of human neutralizing antibodies. Nat. Med. 11:615-622. [DOI] [PubMed] [Google Scholar]
- 77.Trkola, A., H. Kuster, P. Rusert, V. von Wyl, C. Leemann, R. Weber, G. Stiegler, H. Katinger, B. Joos, and H. F. Gunthard. 2008. In vivo efficacy of human immunodeficiency virus neutralizing antibodies: estimates for protective titers. J. Virol. 82:1591-1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Veazey, R. S., R. J. Shattock, M. Pope, J. C. Kirijan, J. Jones, Q. Hu, T. Ketas, P. A. Marx, P. J. Klasse, D. R. Burton, and J. P. Moore. 2003. Prevention of virus transmission to macaque monkeys by a vaginally applied monoclonal antibody to HIV-1 gp120. Nat. Med. 9:343-346. [DOI] [PubMed] [Google Scholar]
- 79.Walker, L. M., S. K. Phogat, P. Y. Chan-Hui, D. Wagner, P. Phung, J. L. Goss, T. Wrin, M. D. Simek, S. Fling, J. L. Mitcham, J. K. Lehrman, F. H. Priddy, O. A. Olsen, S. M. Frey, P. W. Hammond, G. Miiro, J. Serwanga, A. Pozniak, D. McPhee, O. Manigart, L. Mwananyanda, E. Karita, A. Inwoley, W. Jaoko, J. Dehovitz, L. G. Bekker, P. Pitisuttithum, R. Paris, S. Allen, S. Kaminsky, T. Zamb, M. Moyle, W. C. Koff, P. Poignard, and D. R. Burton. 2009. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326:285-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhu, Z., K. N. Bossart, K. A. Bishop, G. Crameri, A. S. Dimitrov, J. A. McEachern, Y. Feng, D. Middleton, L. F. Wang, C. C. Broder, and D. S. Dimitrov. 2008. Exceptionally potent cross-reactive neutralization of Nipah and Hendra viruses by a human monoclonal antibody. J. Infect. Dis. 197:846-853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhu, Z., S. Chakraborti, Y. He, A. Roberts, T. Sheahan, X. Xiao, L. E. Hensley, P. Prabakaran, B. Rockx, I. A. Sidorov, D. Corti, L. Vogel, Y. Feng, J. O. Kim, L. F. Wang, R. Baric, A. Lanzavecchia, K. M. Curtis, G. J. Nabel, K. Subbarao, S. Jiang, and D. S. Dimitrov. 2007. Potent cross-reactive neutralization of SARS coronavirus isolates by human monoclonal antibodies. Proc. Natl. Acad. Sci. U. S. A. 104:12123-12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhu, Z., A. S. Dimitrov, K. N. Bossart, G. Crameri, K. A. Bishop, V. Choudhry, B. A. Mungall, Y. R. Feng, A. Choudhary, M. Y. Zhang, Y. Feng, L. F. Wang, X. Xiao, B. T. Eaton, C. C. Broder, and D. S. Dimitrov. 2006. Potent neutralization of Hendra and Nipah viruses by human monoclonal antibodies. J. Virol. 80:891-899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhu, Z., A. S. Dimitrov, S. Chakraborti, D. Dimitrova, X. Xiao, C. C. Broder, and D. S. Dimitrov. 2006. Development of human monoclonal antibodies against diseases caused by emerging and biodefense-related viruses. Expert Rev. Anti Infect. Ther. 4:57-66. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.