

Abstract
The virion host shutoff (VHS) RNase tegument protein released into cells by infecting virus has two effects. Preexisting stable mRNAs (e.g., GAPDH [glyceraldehyde-3-phosphate dehydrogenase]) are rapidly degraded. Stress response RNAs containing AU-rich elements (AREs) in the 3′ untranslated region (3′UTR) are deadenylated and cleaved, but the cleavage products persist for hours, in contrast to the short half-lives of ARE-containing mRNAs in uninfected cells. At late times, the VHS RNase is neutralized by the viral structural proteins VP16 and VP22. A recent study (J. A. Corcoran, W. L. Hsu, and J. R. Smiley, J. Virol. 80:9720-9729, 2006) reported that, at relatively late times after infection, ARE RNAs are rapidly degraded in cells infected with ΔICP27 mutant virus and concluded that ICP27 “stabilizes” ARE mRNAs. We report the following. (i) The rates of degradation of ARE mRNA at early times (3 h) after infection with the wild type or the ΔICP27 mutant virus are virtually identical, and hence ICP27 plays no role in this process. (ii) In noncomplementing cells, VHS RNase or VP22 is not synthesized. Therefore, the only VHS that is active is brought into cells by the ΔICP27 mutant. (ii) The VHS RNase brought into the cells by the ΔICP27 virus is reduced in potency relative to that of wild-type virus. Hence the rapid degradation of ARE mRNAs noted in ΔICP27 mutant-infected cells at late times is similar to that taking place in mock-infected or in ΔVHS RNase mutant-virus-infected cells and does not by itself support the hypothesis that ICP27 stabilizes ARE mRNAs. (iii) Concurrently, we present the first evidence that VHS RNase interacts with ICP27 most likely when bound to cap- and poly(A)-binding proteins, respectively.
The herpes simplex virus type 1 (HSV-1) UL41 gene encoding a protein (virion host shutoff [VHS]) that is carried by the virion into the infected cell and causes the shutoff of host protein synthesis was first reported by Frenkel and coworkers in the late 1980s (24, 25, 26, 43). Although it was long suspected that VHS is an RNase (11, 12, 22, 32, 52), unambiguous evidence that VHS is an endoribonuclease with a substrate specificity of RNase A emerged from studies of a protein purified to homogeneity (46, 47). The VHS RNase is active during the first several hours after infection, and then it is neutralized by proteins made late in infection. Initial reports indicated that the neutralizing protein is VP16 (27, 38). More recent studies from this laboratory showed that an additional late viral protein, VP22, is required for the neutralization of the RNase activity of VHS protein (48). In this report, we show that VHS interacts with still another viral protein, ICP27, and that this protein is required to block degradation of specific mRNAs at times when ICP27 becomes available for this function. Relevant to this report are the following.
Studies on the incorporation of radioactive amino acids into proteins after infection in cells infected with wild-type virus but not with a mutant lacking VHS activity led to the speculation that VHS activity is nonselective (23, 29, 30, 36). Most of the early work was done on model viral mRNAs, and these studies sustained the conclusion that VHS activity was not selective. Even though Soreson and colleagues (41) reported as early as 1991 that VHS mediated the degradation of some mRNAs but spared other mRNAs, most notably, histone mRNAs, the issue was still debated as late as 2004 (reviewed in reference 39).
This laboratory was the first to report that the degradation of mRNA during the early hours after infection by wild-type virus was highly selective (14, 16, 45). Thus, mRNAs that are highly stable in uninfected cells, exemplified by GAPDH (glyceraldehyde-3-phosphate dehydrogenase) and actin mRNAs, were rapidly degraded. In contrast, stress response mRNAs containing AU-rich elements (AREs) in the 3′ untranslated region (3′UTR), exemplified by IEX-1, c-Fos, IkBα, or cox-2, which in uninfected cells share very short half-lives (1, 4, 51), were deadenylated and cleaved at or near the AREs (14). Whereas the 3′ cleavage product rapidly disappeared, the 5′ portion of the cleaved RNA lingered for many hours. Lastly, a subset of mRNAs exemplified by GADD45β and tristetraprolin (TTP) directed the synthesis of corresponding proteins and were not degraded (14). TTP is of particular interest for three reasons: (i) its mRNA contains AREs (3), (ii) TTP physically interacts with VHS protein (15), and (iii) in uninfected cells, TTP binds AREs and sequesters the mRNAs for degradation by exosomes (5, 28).
The VHS protein was also reported to bind cap-binding proteins (9, 17, 18, 19, 31). In addition, this laboratory has shown that VHS is active and degrades mRNA in polyribosomes (49). We proposed a model of degradation of intrinsically stable mRNAs (GAPDH) and of ARE mRNAs on the basis of known structures of translated mRNAs, known pathways of mRNA decay, and the interaction of VHS protein with the cap-binding complex and TTP. In brief, in the course of mRNA translation, it is the poly(A)-binding protein at the 3′ end that physically abuts the cap-binding complex at the 5′ end of the mRNA. In the absence of AREs, VHS is predicted to bind the cap-binding complex and decap the mRNA. This is the normal pathway of degradation of mRNA and would lead to 5′-to-3′ exonucleolytic degradation of the cleaved RNA. In the presence of TTP bound to AREs, VHS is expected to bind TTP and cleave the mRNA at the AREs. The 3′ product of the cleavage would be rapidly degraded 5′ to 3′ in the same manner as the GAPDH mRNA. A dearth or lack of enzymes necessary to degrade the 5′ cleavage product containing an intact cap may account for the lingering of the cleavage product for many hours.
Corcoran and colleagues (6) analyzed the degradation of AU-rich IEX-1 mRNA at relatively late times after infection. The time frame of their studies was selected on the basis of the presence of abundant amounts of ICP27 in the infected cells. They concluded that ICP27 is required to stabilize full-length, polyadenylated IEX mRNA and that VHS is not involved in this process. In the description of their studies, they confused the cleavage events occurring during the early hours after infection when the mRNAs are cleaved and partially degraded in a VHS- but not a ICP27-dependent manner with events taking place at late time intervals. In this report, we have reexamined the degradation of GAPDH and IEX-1 mRNAs at early and late times in infection. Indeed, at late times, the IEX-1 is degraded in ΔICP27 virus-infected cells at a rate similar to that in noninfected or ΔVHS virus-infected cells. Consistent with this finding is the evidence that VHS does not accumulate in ΔICP27 virus-infected cells and that the VHS brought into cells by ICP27 virions is less effective that that brought into cells by wild-type virus. At the same time, we report the first evidence that VHS physically interacts with ICP27 both in lysates of infected cells and in transfected cells.
MATERIALS AND METHODS
Cells and viruses.
Vero, HeLa, HEp-2, and HEK 293T cells obtained from the American Type Culture Collection were propagated in Dulbecco's modified Eagle's minimal essential medium (DMEM) supplemented with 5% newborn calf serum, 5% fetal calf serum (FCS), or 10% FCS. Telomerase-transformed primary human embryonic fibroblasts (HEL), a kind gift of Thomas E. Shenk (Princeton University), were cultured in DMEM supplemented with 10% FCS. HSV-1(F) is the prototype HSV-1 strain used by our laboratory (10). The UL54 mutant virus (vBSΔ27) and complementing cells (Vero 2-2) (40) were a kind gift of Saul Silverstein (Columbia University). The UL41 mutant virus, R2621, was reported elsewhere (33). The d120 mutant lacking both copies of the α4 gene (ΔICP4) (7) was the kind gift Neal A. DeLuca (University of Pittsburgh, Pittsburgh, PA).
Cell infection and treatment.
Cell monolayers were either mock infected or exposed for 1 h to 5 or 10 PFU of the wild-type or a mutant virus per cell at 37°C. Where indicated, at 3 h or 6 h after virus exposure, the cultures were incubated with medium containing actinomycin D (10 μg/ml; Sigma, St. Louis, MO).
Isolation of total and poly(A)+ RNAs and Northern blotting.
Total RNA was extracted at the times indicated in Fig. 1 after actinomycin D addition with the aid of TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. DNase treatment (Applied Biosystems/Ambion, Austin, TX), phenol-chloroform extraction, and ethanol precipitation (Fisher Scientific) were carried out to remove possible DNA contamination. Poly(A)+ mRNAs were isolated from the total RNA samples with the aid of a MicroPoly(A) purist purification kit (Applied Biosystems/Ambion) according to the manufacturer's instructions. Northern blotting was done as previously reported (44). All quantification was performed with a STORM 860 PhosphorImager (Molecular Dynamics, Sunnyvale, CA).
FIG. 1.

Analysis of cellular mRNA decay at an early time (3 h) after infection. (A) Schematic representation of the experiment. (B) Replicate 25-cm2 cultures of HeLa cells were either mock infected (lanes 1 to 4) or infected with 10 PFU per cell of HSV-1(F) (lanes 5 to 8) or the ΔUL41 (lanes 9 to 12) or ΔICP27 (lanes 13 to 16) mutant virus for 1 h. The inoculum was then replaced with fresh complete medium, and the cell cultures were incubated at 37°C for two additional hours. Transcription was then stopped by addition of 10 μg of actinomycin D (Act. D) per ml (time zero). The cells were harvested at the time intervals indicated, and total mRNA was extracted. The accumulation of the GAPDH and IEX-1 transcripts was visualized by Northern blotting. The ethidium bromide staining of 18S rRNA is shown as a loading control. Letters on the right indicate IEX-1 bands A, B, and C. (C) The intensities of the bands at various time points were quantified by phosphorimager analysis in three independent experiments, and the mRNA remaining is plotted as a percentage of that in mock-infected cells (GAPDH mRNA) or relative to time after addition of actinomycin D (IEX-1 mRNA) (averages ± standard errors).
Immunoblotting of electrophoretically separated proteins from cell lysates.
Confluent cell monolayers were either mock infected or exposed to 10 PFU of HSV-1(F) or the ΔICP27 mutant virus per cell and were collected 16 or 24 h after infection. The procedures for harvesting, solubilization, protein quantification, sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transfer to nitrocellulose membranes were done as previously reported (44). The HSV-1 proteins were detected with the mouse monoclonal antibodies for ICP4, ICP0, and ICP8 (Goodwin Institute for Cancer Research, Plantation, FL); the anti-UL38 (50), anti-UL49 (VP22) (2), and anti-UL41 (VHS) (46) rabbit polyclonal antibodies; the anti-US11 monoclonal antibody (35); and the mouse monoclonal antibody against VP16 (LP1), kindly provided by A. Minson, Cambridge University, Cambridge, United Kingdom. The monoclonal anti-FLAG M2 antibody was purchased from Sigma.
Immunoblotting of proteins from purified virions.
Wild-type and recombinant virus virions were purified as described elsewhere (37, 42). Briefly, Vero or Vero 2-2 cells grown in 300-cm2 flasks were exposed to 5 PFU of virus per cell. The cells were harvested 24 h after infection, resuspended, and stored briefly on wet ice in hypotonic buffer and then disrupted in a glass homogenizer with four strokes. Cytoplasmic fractions were individually layered in dextran-10 gradients (1.04 to 1.09 g/cm3) in 1 mM phosphate buffer. The gradients were centrifuged, and virion-containing bands were collected and diluted in 10 mM phosphate buffer. Purified virions were concentrated and resuspended in 100 μl of 10 mM phosphate buffer and stored at −80°C before being processed. The purified virions were resuspended in 4× loading buffer (50 mM Tris-HCl [pH 6.8], 10 mM dithiothreitol, 2% sodium dodecyl sulfate, 0.1% bromophenol blue, 10% glycerol), subjected to electrophoresis on a 10% denaturing polyacrylamide gel, and then transferred to a nitrocellulose membrane. The membrane was reacted with the anti-UL41 (VHS) or anti-UL38 rabbit polyclonal antibody.
Analysis of the shutoff of protein synthesis in infected cells.
Replicate 25-cm2 cultures of HEp-2, Vero, or HeLa cells were either mock infected or infected with 20 PFU of HSV-1(F) or the ΔUL41, ΔICP27, or ΔICP4 mutant virus per cell in the presence of actinomycin D (10 μg/ml) and incubated at 37°C. At 3 h after exposure to virus, the cells were labeled for 1 h with [35S]methionine as previously described (33) and then harvested, solubilized, and resolved in a 12% denaturing polyacrylamide gel. The dried gels were subjected to autoradiography.
Plasmids.
The UL54 (ICP27) coding sequence was amplified from wild-type HSV-1(F) DNA by PCR using the primers ICP27-F (5′-CGGGATCCATGGCGACTGACATTGATATGCTAATTG-3′) and ICP27-R (5′-CGGAATTCCTAAAACAGGGAGTTGCAATAAAAATATTTGCC-3′), which contain a BamHI and an EcoRI restriction site, respectively. For the generation of the N-terminally Flag-tagged version of ICP27, the ICP27-R primer was paired with N-Flag-ICP27 (5′-CGGGATCCATGGATTACAAGGATGACGACGATAAGGCGACTGACATTGATATGCTAATTG-3′), whereas for the generation of the C-terminally Flag-tagged version of ICP27, the ICP27-F primer was paired with C-Flag-ICP27 (5′-CGGAATTCTCACTTATCGTCGTCATCCTTGTAATCCATAAACAGGGAGTTGCAATAAAAA-3′). The PCR fragments were cloned into the pCDNA3.1(+) transfer vector, and the resulting plasmids were named pICP27, pN-Flag-ICP27, and pC-Flag-ICP27, respectively.
HEK 293T cell transient transfection.
HEK 293T cells were seeded in 25-cm2 flasks the day before transfection in DMEM containing 10% fetal bovine serum (FBS). The following day, a total of 2 μg of the plasmid DNAs described above was mixed with 12 μl of Plus reagent (Invitrogen) in 400 μl of serum and antibiotic-free DMEM for 15 min. Then, 400 μl of antibiotic-free DMEM containing 8 μl of Lipofectamine (Invitrogen) was added to the plasmid DNA mixture. The mixture was incubated for an additional 15 min and added to the cells in 2 ml of medium. After 3 h, the mixture was removed and fresh DMEM containing 10% FCS was added to the cell cultures. The cells were harvested 48 h after transfection and lysed according to the protocol for the subsequent analyses.
HEp-2 cell infection and 35S labeling.
Replicate 25-cm2 cultures of HEp-2 cell monolayers were either mock infected or exposed to 10 PFU of HSV-1(F) per cell. After 1 h, the inoculum was removed and replaced with methionine-free DMEM containing 100 μCi of [35S]methionine, and the cells were incubated for an additional 18 h before being harvested.
Expression and purification of GST fusion proteins and a GST pulldown assay.
Glutathione S-transferase (GST) fusion proteins containing GST alone or GST fused to full-length VHS were expressed and purified as reported elsewhere (46). For pulldown assays, infected HEp-2 cells or transfected HEK 293T cells were lysed in GST lysis buffer (20 mM Tris [pH] 8.0)-1 mM EDTA-1% Nonidet P-40-200 mM NaCl-0.1 mM sodium orthovanadate-10 mM NaF-2 mM dithiothreitol [DTT]-protease inhibitor mixture [Complete protease mixture; Roche Diagnostics, Indianapolis, IN]) on ice for 1 h. Insoluble material was pelleted by centrifugation at maximum speed in a model 5415C centrifuge (Eppendorf, Boulder, CO) for 10 min at 4°C. The supernatant fluids precleared with 50 μl of a 50% slurry of glutathione beads for 3 h at 4°C were incubated overnight at 4°C with equal amounts of a 50% slurry of glutathione beads bound to GST alone or GST fused to the VHS protein. The beads were pelleted by centrifugation and rinsed five times with GST buffer. The proteins bound to the beads were solubilized in 50 μl of SDS gel loading buffer (2% SDS-5% 2-mercaptoethanol-50 mM Tris [pH 6.8]-2.75% sucrose), heated to 95°C for 5 min, resolved by SDS-PAGE, transferred to a nitrocellulose membrane, and immunoblotted with the mouse monoclonal anti-ICP27 or anti-Flag antibody.
RESULTS
Analysis of cellular mRNA decay in cells infected by the ΔICP27 mutant virus.
We report two series of experiments designed to elucidate the role of ICP27 in the decay of both stable and stress response mRNAs in infected cells.
In the first series, confluent HeLa cell monolayers were either mock infected or exposed to 10 PFU per cell of HSV-1(F) or the ΔUL41 or ΔICP27 mutant virus at 37°C for 1 h. The inoculum was then replaced with fresh medium, and the cells were incubated at 37°C for an additional 2 h (time zero) before transcription was stopped by the addition of 10 μg of actinomycin D per ml (Fig. 1 A). Total RNA was extracted at the time of addition (time zero) and at 1, 2, or 3 h after exposure to the drug. The accumulation of the GAPDH and IEX-1 transcripts was visualized by Northern blotting. The intensities of the bands at various time points were quantified with the aid of a phosphorimager in three independent experiments, and the mRNA remaining is plotted as a percentage of that in mock-infected cells (GAPDH mRNA) or relative to time after addition of actinomycin D (IEX-1 mRNA). The results shown in Fig. 1B and C were as follows. (i) As expected, GAPDH mRNA in cells infected with wild-type virus represented less than 20% of that present in mock-infected cells at the time of addition of actinomycin D (Fig. 1B [bottom], lane 5, and C). In contrast, in ΔUL41 mutant-virus-infected cells, GAPDH mRNA remained relatively unchanged throughout the course of the experiment compared to that in mock-infected cells (Fig. 1B [compare lanes 9 to 12 with lanes 1 to 14] and C). Interestingly, in cells infected by the ΔICP27 mutant virus, the level of GAPDH mRNA at the time of addition of actinomycin D (Fig. 1B, lane 13) was perceptibly higher—40% of the amount of mRNA present in mock-infected cells and 2-fold higher than the amount detected in cells infected with wild-type virus (Fig. 1C). (ii) Infection with HSV-1 activates a stress response and results in the accumulation of a large number of ARE mRNAs. In our studies, IEX-1 mRNA served as the example of a typical ARE mRNA. ARE mRNAs have relatively short half-lives in mock-infected or ΔUL41 mutant-virus-infected cells. In wild-type-virus-infected cells, IEX-1 mRNA was shown to form 3 sets of bands. As marked in Fig. 1B (lanes 5 to 8), the 3 sets of bands consist of intact (∼1.3-kb) poly(A)+ mRNA (band A), a deadenylated (∼1.1-kb) RNA (band B), and a set of truncated (∼0.75-kb) RNAs shown previously to consist of 5′ portions of IEX RNA (44) and designated band C. Visual inspection shows that these 3 sets of bands are present in ΔICP27 mutant-virus-infected cells (Fig. 1B, lanes 13 to 16) and that the bands decay very rapidly in ΔUL41 mutant-virus-infected cells (Fig. 1B, lanes 9 to 12). Quantification of the intensities of the three classes of transcripts together at each given time point in three different experiments and plotting them as a function of time after actinomycin D addition (Fig. 1C) indicated that the total IEX-1 RNA decayed at the same rate in wild-type and ΔICP27 mRNA and at a higher rate in either mock- or ΔUL41 mutant-virus-infected cells. The results presented in Fig. 1 unambiguously indicate that ICP27 plays no role in the degradation of IEX-1 mRNA at early times after infection with wild-type virus.
In the second series of experiments, HeLa cells were infected as reported above, but transcription was arrested by the addition of actinomycin D at 6 h after virus exposure (Fig. 2A). Total and poly(A)+ RNAs were harvested at the time of addition of the drug (time zero) and at various times thereafter and analyzed by Northern blotting for the accumulation of IEX-1 and GAPDH transcripts. The results shown in Fig. 2 were as follows. (i) Fig. 2B shows the electrophoretic patterns of total IEX-1 and GAPDH RNAs. Visual inspection indicates that GAPDH mRNA is abundant in ΔUL41 virus-infected cells (lanes 5 to 8) and largely decayed in wild-type (lanes 1 to 4)- or ΔICP27 mutant-virus (lanes 9 to 12)-infected cells. The amounts of residual GAPDH mRNA did not appear to change throughout the 3-h period, indicating that the degradation process was essentially complete by the time of addition of actinomycin D. (ii) Visual inspection of the RNAs accumulating in ΔUL41 virus-infected cells (Fig. 2B, lanes 5 to 8) indicates that IEX-RNA is largely poly(A)+, that this RNA decays at a barely perceptible rate, and that truncated IEX-1 RNA did not accumulate. In contrast, in cells infected with wild-type virus (lanes 1 to 4) or ΔICP27 mutant virus (lanes 9 to 12), there was significant accumulation of poly(A)− and truncated RNAs. To quantify the decay of the RNAs more accurately, we separated the poly(A)+ and poly(A)− RNAs at each time point as illustrated in Fig. 2C and D. The total and the poly(A)+ RNAs were then quantified and normalized with respect to the amounts detected at time zero (Fig. 2E and F, respectively). The results indicate that in cells infected with the ΔUL41 mutant virus total IEX-1 RNA decayed at a high rate during the first hour after addition of actinomycin D and at a lower rate thereafter. The total IEX-1 RNA in wild-type- or ΔICP27 mutant-virus-infected cells decayed at approximately the same rate. A strikingly different picture emerged from analyses of poly(A)+ RNAs (Fig. 2F). In cells infected with the ΔICP27 mutant virus, the poly(A)+ RNA was degraded at a rate much higher than those of RNAs present at time zero in wild-type-virus- or ΔUL41 mutant-virus-infected cells.
FIG. 2.

Analysis of cellular mRNA decay at an early time (6 h) after infection. (A) Schematic representation of the experiment. (B) Replicate 25-cm2 cultures of HeLa cells were infected with 5 PFU per cell of either HSV-1(F) (lanes 1 to 4) or the ΔUL41 (lanes 5 to 8) or ΔICP27 (lanes 9 to 12) mutant virus for 1 h. The inoculum was then replaced with fresh complete medium, and the cell cultures were incubated at 37°C for five additional hours. Transcription was then stopped by addition of 10 μg of actinomycin D (Act. D) per ml (time zero). The cells were harvested at the time intervals indicated, and total RNA was extracted and analyzed as described for Fig. 1B. (C and D) Poly(A)+ mRNA was purified from the total RNA samples as described in Materials and Methods, and the accumulations of IEX-1 (C) and GAPDH (D) transcripts were determined by Northern blotting of total RNA (lanes 1, 4, 7, and 10) and poly(A)− (lanes 2, 5, 8, and 11) and poly(A)+ (lanes 3, 6, 9, and 12) RNA fractions. (E and F) The intensities of IEX-1 bands were quantified from total (E) and poly(A)+ (F) RNAs by phosphorimager analysis and plotted as a function of time after addition of actinomycin D.
The more rapid degradation of poly(A)+ RNAs in cells infected with the ΔICP27 mutant may be due to two non-mutually-exclusive events. The first hypothesis is that VHS RNase was not functioning by 6 h after infection of cells with the ΔICP27 mutant virus. In this instance, the truncated RNAs would continue to persist, whereas the full-length poly(A)+ RNAs would be degraded more rapidly and at a rate compatible with that in mock-infected cells. The second hypothesis is that ICP27 specifically interacts with VHS and blocks the degradation of poly(A)+ RNA. In the sections below, we describe experiments designed to test both hypotheses.
VHS is made in complementing cells infected with the ΔICP27 mutant virus.
ΔICP27 mutant virus is made in Vero cell line 2-2, which carries the gene expressing ICP27 (40). To test whether VHS RNase is made in these cells, confluent cell monolayers of Vero and Vero 2-2 cells were either mock infected or infected with 10 PFU of HSV-1(F) (Vero cells) or the ΔICP27 mutant virus (Vero 2-2 cells) per cell. The cells were harvested 16 and 24 h after infection and processed as described in Materials and Methods. Equal amounts of total protein content were electrophoretically separated on a 10% denaturing polyacrylamide gel, transferred to a nitrocellulose sheet, and reacted with antibodies against ICP4, US3, VHS, VP22, and US11. The results shown in Fig. 3 indicate that both cell lines produced roughly equivalent amounts of viral proteins. Exceptions were decreased amounts of US11 in Vero 2-2 cells infected with the ΔICP27 mutant virus and slower accumulations of VP22 in wild-type-virus-infected Vero cells. The key finding shown in Fig. 3 is that the level of VHS protein accumulating in complementing Vero 2-2 cells infected with the ΔICP27 mutant virus (lanes 5 and 6) was comparable to that observed in Vero cells infected by wild-type virus (lanes 2 and 3).
FIG. 3.

Viral protein synthesis in complementing Vero 2-2 cell lines infected with ΔICP27 mutant virus. Confluent cell monolayers of Vero (lanes 1 to 3) and Vero 2-2 (lanes 4 to 6) cells were either mock infected (lanes 1 and 4, respectively) or infected with 10 PFU per cell of HSV-1(F) (lanes 2 and 3) or the ΔICP27 mutant virus (lanes 5 and 6). The cells were harvested at 16 h (lanes 2 and 5) or 24 h (lanes 3 and 6) after virus exposure and processed as described in Materials and Methods. Equal amounts of proteins were electrophoretically separated on a 10% denaturing polyacrylamide gel, transferred to a nitrocellulose sheet, and reacted with the antibodies made against representative α (ICP4), β (US3), or γ (VHS, VP16, VP22, and US11) proteins.
VHS is packaged in ΔICP27 virions.
The preceding section showed that VHS is made and accumulates in cells infected with the ΔICP27 mutant virus. To determine whether VHS is packaged in virions, HSV-1 virions were purified from lysates of Vero cells infected with the wild-type virus or from lysates of Vero-2-2 cells infected with the ΔICP27 mutant virus according to the procedures of Spear and Roizman (42) and as described in Materials and Methods. Equal amounts of protein from solubilized virions were electrophoretically separated on a denaturing gel, transferred to a nitrocellulose sheet, and probed with polyclonal antibodies directed to the tegument protein VHS and to the capsid protein UL38. The results (Fig. 4) showed that the VHS protein is packaged into ΔICP27 mutant virions (lane 1, upper panel). While the HSV-1(F) virions purified from wild-type-virus-infected Vero cells contain both the 59.5- and 58-kDa forms of VHS (lane 2), only the less phosphorylated form appears to be present in the ΔICP27 virions. The finding of a single VHS band is in agreement with the study by Read et al. (34) reporting that HSV-1(KOS) virions contain only the fast-migrating form of VHS (34).
FIG. 4.
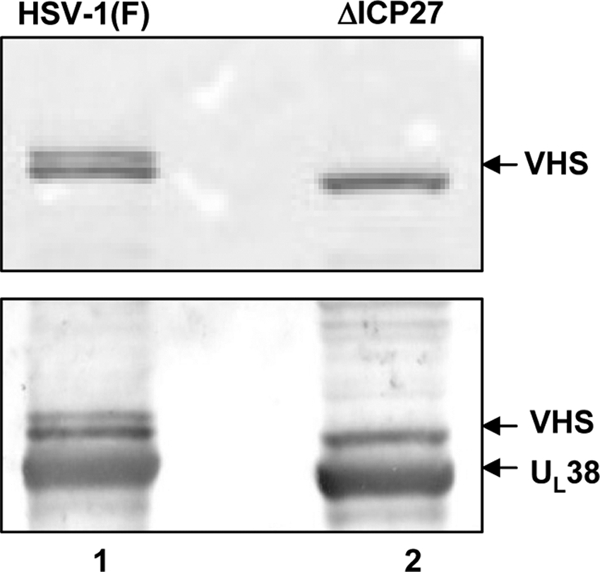
Immunoblot analysis of wild-type HSV-1 and ΔICP27 virions purified from infected Vero or Vero 2-2 cells, respectively. Virions derived from Vero cells infected with wild-type HSV-1(F) (lane 1) or from Vero 2-2 cells infected with the ΔICP27 mutant virus (lane 2) at a multiplicity of infection of 5 PFU/cell and collected 24 h after infection were purified as described in Materials and Methods. Equal amounts of viral proteins were electrophoretically separated on a 10% denaturing polyacrylamide gel, transferred to a nitrocellulose sheet, and probed with rabbit polyclonal antibodies to VHS and UL38.
VHS is not made in noncomplementing cells infected with the ΔICP27 mutant virus.
In this series of experiments, confluent cell monolayers of HeLa, HEp-2, HEK 293, and HEL cells were either mock infected or infected with 10 PFU of HSV-1(F) or the ΔICP27 mutant virus per cell for either 16 or 24 h. Total cell lysates were prepared as reported in Materials and Methods. Equal amounts of proteins were electrophoretically separated on a 10% denaturing polyacrylamide gel, transferred to a nitrocellulose sheet, and reacted with the antibodies made against representative α (ICP4, ICP0), β (ICP8), or γ (VP16, VHS, VP22, UL38, and US11) proteins. The results (Fig. 5) show that VHS and US11, along with the previously reported VP16 (13, 21), do not accumulate to detectable levels in cells infected with the ΔICP27 mutant virus, consistent with the evidence that cells infected with this mutant virus do not accumulate a subset of late proteins.
FIG. 5.

Viral protein accumulation in ΔICP27 virus-infected human cell lines. Confluent cell monolayers of HeLa (lanes 1 to 5), HEp-2 (lanes 6 to 10), HEK 293T (lanes 11 to 14), and HEL (lanes 15 to 19) cells were either mock infected (lanes 1, 6, and 15) or infected with 10 PFU per cell of HSV-1(F) (lanes 2, 4, 7, 9, 11, 13, 16, and 18) or the ΔICP27 mutant virus (lanes 3, 5, 8, 10, 12, 14, 17, and 19). The cells were harvested 16 h (lanes 2, 3, 7, 8, 11, 12, 16, and 17) or 24 h (lanes 4, 5, 9, 10 13, 14, 18, and 19) after virus exposure and processed as described in Materials and Methods. Equal amounts of proteins were electrophoretically separated on a 10% denaturing polyacrylamide gel, transferred to a nitrocellulose sheet, and reacted with the antibodies made against representative α (ICP4, ICP0), β (ICP8), or γ (VP16, VHS, VP22, UL38 and US11) proteins. Note that ICP4 forms multiple bands, reflecting posttranslational modifications of the protein.
Shutoff of host protein synthesis in cells infected by the ΔICP27 mutant virus.
The purpose of this series of experiments was to determine whether the ΔICP27 mutant virus shuts off host protein synthesis early after infection and in the absence of de novo viral protein synthesis in a manner analogous to that of its wild-type parent. Replicate 25-cm2 flask cultures of HEp-2, Vero, and HeLa cells were mock infected or exposed to 20 PFU of HSV-1(F) or the ΔUL41, ΔICP27, or ΔICP4 mutant virus per cell in the presence of actinomycin D (10 μg/ml). Three hours after exposure to the virus, the cells were labeled for 30 min with [35S]methionine and then harvested, solubilized, resolved on a 12% denaturing polyacrylamide gel and subjected to autoradiography. The intensities of the bands were quantified with the aid of a General Dynamics Storm phosphorimager by assigning the value of 100 to the labeled proteins in the mock-infected lane and a value of 0 to the radioactivity in a blank lane. The results shown in Fig. 6 were as follows. (i) As expected, cellular protein synthesis was dramatically reduced in cells infected with the wild-type (Fig. 6A to C, lane 2) or ΔICP4 mutant (Fig. 6A to C, lane 5) virus. (ii) The observed shutoff of host protein synthesis was dependent on virion-bound VHS activity inasmuch as infection with a virus lacking a functional VHS (Fig. 6A to C, lane 3) did not have impact on global protein synthesis. (iii) Interestingly, the amount of labeled protein accumulating in cells infected with the ΔICP27 mutant virus varied depending on the cell line. Reproducibly, the shutoff of protein synthesis in Vero cells infected with this virus was virtually indistinguishable from that of wild-type HSV-1(F) (Fig. 6B, compare lane 4 to lane 2). In contrast, in HEp-2 and HeLa cells, the shutoff of protein synthesis was only 2-fold lower than that of mock-infected cells (Fig. 6A and C, compare lane 4 with lane 1). These results suggest that the activity of VHS RNase bound to ΔICP27 virions is impaired in HEp-2 and HeLa human cell lines.
FIG. 6.

Shutoff of protein synthesis in HEp-2, Vero, and HeLa cells infected with the ΔICP27 mutant virus. Replicate 25-cm2 cultures of HEp-2 (A), Vero (B), or HeLa (C) cells were either mock infected (lane 1) or infected with 20 PFU per cell of HSV-1(F) (lane 2) or the ΔUL41 (lane 3), ΔICP27 (lane 4), or ΔICP4 (lane 5) mutant virus at 37°C in the presence of 10 μg actinomycin D per ml. After 1 h, the inoculum was replaced with fresh medium containing actinomycin D. Two hours after removal of the inoculum, the cells were labeled for 30 min with [35S]methionine before being harvested. Whole-cell lysates were resolved on a 12% denaturing polyacrylamide gel, dried, and subjected to autoradiography. The intensities of the bands were quantified with the aid of a General Dynamics Storm phosphorimager and are reported as percentages (shown) of the band intensities of mock-infected cells.
VHS interacts with ICP27.
ICP27 has been reported to bind to poly(A)-binding protein (8, 20). One way in which ICP27 may block the degradation of IEX-1 poly(A)+ RNA is to bind to VHS RNase and preclude it from cleaving the RNA. To test the hypothesis that ICP27 and VHS RNase interact, GST or a GST-VHS fusion protein containing full-length VHS was generated in Escherichia coli as previously described (46). The recombinant proteins captured on glutathione-agarose beads were reacted with [35S]methionine-labeled extracts prepared from HEp-2 cells that were infected with 10 PFU of HSV-1(F) per cell for 18 h. After the beads were extensively rinsed, proteins bound to beads were solubilized, subjected to electrophoresis on a denaturing gel, transferred to a nitrocellulose sheet, and exposed to a film. The results (Fig. 7A) were as follows: along with the two predominant bands of 65 and 37 kDa, previously identified as VP16 and VP22, respectively (48), a third band, located just below the one corresponding to VP16, was captured by GST fused to full-length VHS (Fig. 7A, third lane, asterisk). On the basis of apparent molecular mass, we predicted that this new 60- to 65-kDa species may be the α protein ICP27, the product of the UL54 gene. Indeed, as shown in Fig. 7B, that band reacted with an anti-ICP27 monoclonal antibody.
FIG. 7.

VHS pulls down ICP27 from lysates of infected or transfected cells. (A and B) One hour after mock infection or exposure to 10 PFU of HSV-1(F) per cell, confluent HEp-2 cell monolayers were incubated in methionine-free DMEM containing [35S]methionine. The cells were harvested 18 h after infection and lysed in GST lysis buffer (see Materials and Methods). Precleared cell lysates were reacted with GST (lane 2) or GST fused to full-length VHS (VHS-FL) (lane 3) for pulldown assays, resolved by SDS-PAGE, and transferred to a nitrocellulose membrane. Lane 1 contains 1/10 of the whole-cell lysate volume. (A) Autoradiographic image of proteins pulled down by GST-VHS. (B) Immunoblotting with anti-ICP27 mouse monoclonal antibody. The arrows indicate the location of ICP27. (C) HEK 293T cells were transiently transfected with plasmids encoding the empty pcDNA 3.1(+) vector (pcDNA) (lanes 1 and 5) or the vector expressing full-length ICP27 (pICP27) (lanes 2 and 6) or expressing the two Flag-tagged versions of ICP27 (pN-Flag-ICP27 [lanes 3 and 7] and pC-Flag-ICP27 [lanes 4 and 8]), which differ in location of Flag. Soluble cell extracts were prepared 48 h after transfection, reacted with glutathione-agarose beads bound to GST fused to full-length VHS (lanes 5 to 8). The proteins bound to the beads were subjected to electrophoresis on a denaturing polyacrylamide gel, transferred to a nitrocellulose membrane, and reacted with antibodies against Flag (upper blot) or ICP27 (lower blot). The immunoreactivity of Flag and ICP27 from a 1/10 volume of whole-cell lysates is also shown.
In a series of additional studies, pcDNA 3.1(+) encoding full-length UL54 (pICP27) or two Flag-tagged versions, one N-terminally tagged (pN-Flag-ICP27) and the other C-terminally tagged (pC-Flag-ICP27), were transiently transfected into HEK 293T cells. Forty-eight hours after transfection, soluble cell lysates were reacted with glutathione-agarose beads bound to the full-length GST-VHS fusion protein and processed as described in Materials and Methods. The proteins eluted from the beads were subjected to electrophoresis on a denaturing polyacrylamide gel, transferred to a nitrocellulose membrane, and reacted with antibodies against either ICP27 or Flag. The results shown in Fig. 7C verified the conclusion that VHS interacts with ICP27 in the absence of other viral proteins. However, we cannot exclude at the moment the possibility that this interaction might be mediated by a cellular protein. Furthermore, we observed the presence of ICP27 cleavage products in the total cell lysates (Fig. 7B, lanes 2 to 4, lower panel) which are not pulled down by VHS (lanes 6 to 8). The cleavage pattern observed with the Flag antibody (Fig. 7B, lane 4, upper panel) clearly indicates that the cleavage occurs in proximity to the C terminus of the protein. It is not the purpose of this study to investigate the significance of the observed ICP27 cleavage products.
DISCUSSION
Infection of cells by HSV-1 results in a very strong cellular stress response. This takes place in at least two rounds, at the time of exposure of the cell to the virus and after activation of protein kinase R (PKR). A large but as-yet-undefined fraction of the stress response mRNAs contain AREs in their 3′UTR (51). In uninfected cells, stress response mRNAs have relatively short half-lives. In HSV-1-infected cells, the half-lives of these RNAs are much longer. Along with retaining intact mRNA, the cells retain deadenylated mRNA as well as the 5′ portion of the RNAs cleaved 3′ of the AREs. The results show that in the absence of VHS RNase introduced into the cells by an infecting virus, the half-lives of ARE RNAs are short and similar to those of uninfected cells. At least at early times after infection, the evidence that VHS RNase is responsible for the endonucleolytic cleavage of the ARE RNAs is unambiguous, as reported previously (15, 44), and reinforced by the results presented in Fig. 1. A key player in this process is tristetraprolin: a protein induced after infection that binds to AREs and, in uninfected cells, recruits the mRNAs into exosomes, where they are degraded. VHS RNase interacts with tristetraprolin (15) to cleave the mRNAs in a manner that leads to their persistence.
A key component of the stress response degradation machinery is the VHS RNase. This enzyme is brought into the cell by the infecting virus and is active during early times after infection. The duration of this activity of the VHS RNase after infection is not known. The only data available are that it or both the enzyme introduced during infection and the newly synthesized mRNA are neutralized by at least two viral proteins: VP16 and VP22 (27, 38, 45). VHS RNase also degrades stable mRNAs (e.g., GAPDH mRNA) that lack AREs. The degradation is extremely rapid; by 3 h after infection (the initiation time of the experiment whose results are shown in Fig. 1), only traces of GAPDH mRNA remain. Finally, some mRNAs appear to be resistant to VHS RNase nucleolytic activity (14).
The central issue examined here is the report by Corcoran et al. (6), who noted that at late times after infection, the degradation of ARE mRNA IEX-1 in ΔICP27 mutant-virus-infected cells is more rapid than in wild-type-virus-infected cells. The conclusion drawn from this result is that the ΔICP27 virus stabilizes IEX-1 mRNA. Although the results were based on analyses of mRNA stability at relatively late times, the conclusion was stated to apply to IEX-1 RNA stability at all times after infection. Those authors did not consider the possibilities that ΔICP27 mutant-virus-infected cells are partially deficient in VHS RNase and that, hence, the rapid degradation of the IEX-1 mRNA in ΔICP27 virus-infected cells is due to a natural process of recruitment and rapid degradation of the mRNAs in exosomes unhindered by VHS RNase, as occurs in uninfected or in ΔUL41 virus-infected cells. Neither did they consider the possibility that, if ICP27 indeed spares ARE mRNAs late in infection, the VHS may interact in some fashion with ICP27. In this report. we examined both issues.
In this article, we report three findings. First, VHS RNase is made in complementing cells in which ΔΙCP27 mutant virus stock is made and is packaged into the progeny virions, as shown in Fig. 4. Second, VHS RNase along with other γ proteins does not accumulate in noncomplementing cells infected with the ΔICP27 mutant virus. A key issue raised by this observation is whether the endonucleolytic activity is due solely to the VHS RNase introduced by the infecting virus or whether newly made VHS RNase in the infected cells contributes to the degradation of the mRNA. Third, the data show that the ΔICP27 virus effectively shuts off protein synthesis in Vero cells in the presence of actinomycin D at the time of exposure to the virus but that the shutoff is only partial in HEp-2 and HeLa human cell lines. The implication of this finding is that the VHS RNase activity of the ΔICP27 mutant virus is at least partially defective.
In essence, one explanation for the rapid degradation of poly(A)+ mRNA at relatively late times after infection with the ΔICP27 virus is that the VHS RNase activity is essentially exhausted; no new VHS RNase enzyme is made in the noncomplementing cells, and hence, at late times, the IEX-1 mRNA is degraded at a rate similar to that observed in ΔUL41 virus-infected and in noninfected cells.
We have also probed a key element in the non-mutually-exclusive hypothesis that ICP27 protects certain mRNAs from degradation. In this series of experiments, we demonstrated that VHS RNase interacts with ICP27 both in transfected, uninfected cells and in infected cells. This is a novel, previously unreported finding. The rationale underlying the experiment is that VHS RNase has been reported to bind to components of the translational complex bound to the cap structure (9, 17, 18, 19, 31), whereas ICP27 was reported to bind to the poly(A)-binding protein (8, 20). In the course of the translation process, the cap structure and the proteins bound to it are in juxtaposition to the poly(A)-binding protein. One model of the ensuing events is that ICP27, responsible for the shuttling of certain mRNAs late in infection, binds the VHS RNase in the cap structure and prevents it from cleaving the transported mRNAs. This is most likely the mechanism that spares mRNAs late in infection from nonneutralized VHS RNase.
To impute a similar function for the stabilization of IEX-1 mRNA, it would have to be shown that the mRNA encoding IEX-1 binds ICP27 at its poly(A)-binding site. The fact of the matter is that IEX-1 protein cannot be detected after the first hour after infection in the lysates of wild-type-virus-infected cells. At least with respect to the rapid degradation of IEX-1 mRNA in cells late after infection with the ΔICP27 mutant virus, the data favor the hypothesis that, in these cells, VHS RNase activity is deficient. The degradation of GAPDH does not support the presence of active VHS RNase since it is virtually all degraded before 3 h after infection, the time zero point of the experiment illustrated in Fig. 1.
Acknowledgments
These studies were aided by National Cancer Institute grant CA115662.
Footnotes
Published ahead of print on 14 July 2010.
REFERENCES
- 1.Bakheet, T., B. R. Williams, and K. S. Khabar. 2003. ARED 2.0: an update of AU-rich element mRNA database. Nucleic Acids Res. 31:421-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blaho, J. A., C. Mitchell, and B. Roizman. 1994. An amino acid sequence shared by the herpes simplex virus 1 alpha regulatory proteins 0, 4, 22, and 27 predicts the nucleotidylylation of the UL21, UL31, UL47, and UL49 gene products. J. Biol. Chem. 269:17401-17410. [PubMed] [Google Scholar]
- 3.Brooks, S. A., J. E. Connolly, and W. F. Rigby. 2004. The role of mRNA turnover in the regulation of tristetraprolin expression: evidence for an extracellular signal-regulated kinase-specific, AU-rich element-dependent, autoregulatory pathway. J. Immunol. 172:7263-7271. [DOI] [PubMed] [Google Scholar]
- 4.Chen, C. Y., and A. B. Shyu. 1995. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem. Sci. 20:465-470. [DOI] [PubMed] [Google Scholar]
- 5.Chen, C. Y., R. Gherzi, S. E. Ong, E. L. Chan, R. Raijmakers, G. J. Pruijn, G. Stoecklin, C. Moroni, M. Mann, and M. Karin. 2001. AU-binding proteins recruit the exosome to degrade ARE-containing mRNAs. Cell 107:451-464. [DOI] [PubMed] [Google Scholar]
- 6.Corcoran, J. A., W. L. Hsu, and J. R. Smiley. 2006. Herpes simplex virus ICP27 is required for virus-induced stabilization of the ARE-containing IEX-1 mRNA encoded by the human IER3 gene. J. Virol. 80:9720-9729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeLuca, N. A., A. McCarthy, and P. A. Schaffer. 1985. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J. Virol. 56:558-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dobrikova, E., M. Shveygert, R. Walters, and M. Gromeier. 2010. Herpes simplex virus proteins ICP27 and UL47 associate with polyadenylate-binding protein and control its subcellular distribution. J. Virol. 84:270-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doepker, R. C., W. L. Hsu, H. A. Saffran, and J. R. Smiley. 2004. Herpes simplex virus virion host shutoff protein is stimulated by translation initiation factors eIF4B and eIF4H. J. Virol. 78:4684-4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ejercito, P. M., E. D. Kieff, and B. Roizman. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J. Gen. Virol. 2:357-364. [DOI] [PubMed] [Google Scholar]
- 11.Elgadi, M. M., C. E. Hayes, and J. R. Smiley. 1999. The herpes simplex virus vhs protein induces endoribonucleolytic cleavage of target RNAs in cell extracts. J. Virol. 73:7153-7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elgadi, M. M., and J. R. Smiley. 1999. Picornavirus internal ribosome entry site elements target RNA cleavage events induced by the herpes simplex virus virion host shutoff protein. J. Virol. 73:9222-9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellison, K. S., R. A. Maranchuck, K. L. Mottet, and J. R. Smiley. 2005. Control of VP16 translation by the herpes simplex virus type 1 immediate-early protein ICP27. J. Virol. 79:4120-4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Esclatine, A., B. Taddeo, L. Evans, and B. Roizman. 2004. The herpes simplex virus 1 UL41 gene-dependent destabilization of cellular RNAs is selective and may be sequence-specific. Proc. Natl. Acad. Sci. U. S. A. 101:3603-3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esclatine, A., B. Taddeo, and B. Roizman. 2004. Herpes simplex virus 1 induces cytoplasmic accumulation of TIA-1/TIAR and both synthesis and cytoplasmic accumulation of tristetraprolin, two cellular proteins that bind and destabilize AU-rich RNAs. J. Virol. 78:8582-8592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esclatine, A., B. Taddeo, and B. Roizman. 2004. The UL41 protein of herpes simplex virus mediates selective stabilization or degradation of cellular mRNAs. Proc. Natl. Acad. Sci. U. S. A. 101:18165-18170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Everly, D. N., Jr., P. Feng, I. S. Mian, and G. S. Read. 2002. mRNA degradation by degradation by the virion host shutoff protein (UL41) of herpes simplex virus: genetic and biochemical evidence that UL41 is a nuclease. J. Virol. 76:8560-8571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng, P., D. N. Everly, Jr., and G. S. Read. 2001. mRNA decay during herpesvirus infections: interaction between a putative viral nuclease and a cellular translation factor. J. Virol. 75:10272-10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feng, P., D. N. Everly, Jr., and G. S. Read. 2005. mRNA decay during herpes simplex virus (HSV) infections: protein-protein interactions involving the HSV virion host shutoff protein and translation factors eIF4H and eIF4A. J. Virol. 79:9651-9664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fontaine-Rodriguez, E. C., T. J. Taylor, M. Olesky, and D. M. Knipe. 2004. Proteomics of herpes simplex virus infected cell protein 27: association with translation initiation factors. Virology 330:487-492. [DOI] [PubMed] [Google Scholar]
- 21.Fontaine-Rodriguez, E. C., and D. M. Knipe. 2008. Herpes simplex virus ICP27 increases translation of a subset of viral late mRNAs. J. Virol. 82:3538-3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones, F. E., C. A. Smibert, and J. R. Smiley. 1995. Mutational analysis of the herpes simplex virus virion host shutoff protein: evidence that vhs functions in the absence of other viral proteins. J. Virol. 69:4863-4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krikorian, C. R., and G. S. Read. 1991. In vitro mRNA degradation system to study the virion host shutoff function of herpes simplex virus. J. Virol. 65:112-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwong, A. D., and N. Frenkel. 1987. Herpes simplex virus-infected cells contain a function(s) that destabilizes both host and viral mRNAs. Proc. Natl. Acad. Sci. U. S. A. 84:1926-1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwong, A. D., J. A. Kruper, and N. Frenkel. 1988. Herpes simplex virus virion host shutoff function. J. Virol. 62:912-921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwong, A. D., and N. Frenkel. 1989. The herpes simplex virus virion host shutoff function. J. Virol. 63:4834-4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lam, Q., C. A. Smibert, K. E. Koop, C. Lavery, J. P. Capone, S. P. Weinheimer, and J. R. Smiley. 1996. Herpes simplex virus VP16 rescues viral mRNA from destruction by the virion host shutoff function. EMBO J. 15:2575-2581. [PMC free article] [PubMed] [Google Scholar]
- 28.Lykke-Andersen, J., and E. Wagner. 2005. Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes Dev. 19:351-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oroskar, A. A., and G. S. Read. 1987. A mutant of herpes simplex virus type 1 exhibits increased stability of immediate-early (alpha) mRNAs. J. Virol. 61:604-606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oroskar, A. A., and G. S. Read. 1989. Control of mRNA stability by the virion host shutoff function of herpes simplex virus. J. Virol. 63:1897-1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Page, H. G., and G. S. Read. 28 April 2010. The virion host shutoff endonuclease (UL41) of herpes simplex virus interacts with the cellular cap-binding complex eIF4F. J. Virol. doi: 10.1128/JVI.00166-10. [DOI] [PMC free article] [PubMed]
- 32.Pak, A. S., D. N. Everly, K. Knight, and G. S. Read. 1995. The virion host shutoff protein of herpes simplex virus inhibits reporter gene expression in the absence of other viral gene products. Virology 211:491-506. [DOI] [PubMed] [Google Scholar]
- 33.Poon, A. P. W., and B. Roizman. 1997. Differentiation of the shutoff of protein synthesis by virion host shutoff and mutant γ134.5 genes of herpes simplex virus 1. Virology 229:98-105. [DOI] [PubMed] [Google Scholar]
- 34.Read, G. S., B. M. Karr, and K. Knight. 1993. Isolation of a herpes simplex virus type 1 mutant with a deletion in the virion host shutoff gene and identification of multiple forms of the vhs (UL41) polypeptide. J. Virol. 67:7149-7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roller, R. J., and B. Roizman. 1992. The herpes simplex virus 1 RNA binding protein US11 is a virion component and associates with ribosomal 60S subunits. J. Virol. 66:3624-3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schek, N., and S. L. Bachenheimer. 1985. Degradation of cellular mRNAs induced by a virion-associated factor during herpes simplex virus infection of Vero cells. J. Virol. 55:601-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sciortino, M. T., M. Suzuki, B. Taddeo, and B. Roizman. 2001. RNAs extracted from herpes simplex virus 1 virions: apparent selectivity of viral but not cellular RNAs packaged in virions. J. Virol. 75:8105-8116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smibert, C. A., B. Popova, P. Xiao, J. P. Capone, and J. R. Smiley. 1994. Herpes simplex virus VP16 forms a complex with the virion host shutoff protein vhs. J. Virol. 68:2339-2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smiley, J. R. 2004. Herpes simplex virus virion host shutoff protein: immune evasion mediated by a viral RNase? J. Virol. 78:1063-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soliman, T. M., R. M. Sandri-Goldin, and S. J. Silverstein. 1997. Shuttling of the herpes simplex virus type 1 regulatory protein ICP27 between the nucleus and the cytoplasm mediates the expression of late proteins. J. Virol. 71:9188-9197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soreson, C. M., P. A. Hart, and J. Ross. 1991. Analysis of herpes simplex virus-induced mRNA destabilizing activity using an in vitro mRNA decay system. Nucleic Acid Res. 19:4459-4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spear, P. G., and B. Roizman. 1972. Proteins specified by herpes simplex virus. V. Purification and structural proteins of the herpes virion. J. Virol. 9:143-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strom, T., and N. Frenkel. 1987. Effects of herpes simplex virus on mRNA stability. J. Virol. 61:2198-2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taddeo, B., A. Esclatine, W. Zhang, and B. Roizman. 2003. The stress-inducible immediate-early responsive gene IEX-1 is activated in cells infected with herpes simplex virus 1, but several viral mechanisms, including 3′ degradation of its RNA, preclude expression of the gene. J. Virol. 77:6178-6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taddeo, B., A. Esclatine, and B. Roizman. 2004. Post-transcriptional processing of cellular RNAs in herpes simplex virus-infected cells. Biochem. Soc Trans. 32:697-701. [DOI] [PubMed] [Google Scholar]
- 46.Taddeo, B., W. Zhang, and B. Roizman. 2006. The UL41 protein of herpes simplex virus 1 degrades RNA by endonucleolytic cleavage in absence of other cellular or viral proteins. Proc. Natl. Acad. Sci. U. S. A. 103:2827-2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taddeo, B., and B. Roizman. 2006. The virion host shutoff protein (UL41) of herpes simplex virus 1 is an endoribonuclease with a substrate specificity similar to that of RNase A. J. Virol. 80:9341-9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taddeo, B., M. T. Sciortino, W. Zhang, and B. Roizman. 2007. Interaction of herpes simplex virus RNase with VP16 and VP22 is required for the accumulation of the protein but not for accumulation of mRNA. Proc. Natl. Acad. Sci. U. S. A. 104:12163-12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taddeo, B., W. Zhang, and B. Roizman. 2009. The virion-packaged endoribonuclease of herpes simplex virus 1 cleaves mRNA in polyribosomes. Proc. Natl. Acad. Sci. U. S. A. 106:12139-12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ward, P. L., W. O. Ogle, and B. Roizman. 1996. Assemblons: nuclear structure defined by aggregation of immature capsids and some tegument proteins of herpes simplex virus 1. J. Virol. 70:4623-4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilusz, C. J., M. Wormington, and S. W. Peltz. 2001. The cap-to-tail guide to mRNA turnover. Nat. Rev. Mol. Cell Biol. 2:237-246. [DOI] [PubMed] [Google Scholar]
- 52.Zelus, B. D., R. S. Stewart, and J. Ross. 1996. The virion host shutoff protein of herpes simplex virus type 1: messenger ribonucleolytic activity in vitro. J. Virol. 70:2411-2419. [DOI] [PMC free article] [PubMed] [Google Scholar]