

Abstract
Accurate estimates of virus mutation rates are important to understand the evolution of the viruses and to combat them. However, methods of estimation are varied and often complex. Here, we critically review over 40 original studies and establish criteria to facilitate comparative analyses. The mutation rates of 23 viruses are presented as substitutions per nucleotide per cell infection (s/n/c) and corrected for selection bias where necessary, using a new statistical method. The resulting rates range from 10−8 to10−6 s/n/c for DNA viruses and from 10−6 to 10−4 s/n/c for RNA viruses. Similar to what has been shown previously for DNA viruses, there appears to be a negative correlation between mutation rate and genome size among RNA viruses, but this result requires further experimental testing. Contrary to some suggestions, the mutation rate of retroviruses is not lower than that of other RNA viruses. We also show that nucleotide substitutions are on average four times more common than insertions/deletions (indels). Finally, we provide estimates of the mutation rate per nucleotide per strand copying, which tends to be lower than that per cell infection because some viruses undergo several rounds of copying per cell, particularly double-stranded DNA viruses. A regularly updated virus mutation rate data set will be available at www.uv.es/rsanjuan/virmut.
The mutation rate is a critical parameter for understanding viral evolution and has important practical implications. For instance, the estimate of the mutation rate of HIV-1 demonstrated that any single mutation conferring drug resistance should occur within a single day and that simultaneous treatment with multiple drugs was therefore necessary (72). Also, in theory, viruses with high mutation rates could be combated by the administration of mutagens (1, 5, 21, 44, 53, 83). This strategy, called lethal mutagenesis, has proved effective in cell cultures or animal models against several RNA viruses, including enteroviruses (11, 39, 44), aphtoviruses (83), vesiculoviruses (44), hantaviruses (10), arenaviruses (40), and lentiviruses (15, 53), and appears to at least partly contribute to the effectiveness of the combined ribavirin-interferon treatment against hepatitis C virus (HCV) (13). The viral mutation rate also plays a role in the assessment of possible vaccination strategies (16), and it has been shown to influence the stability of live attenuated polio vaccines (91). Finally, at both the epidemiological and evolutionary levels, the mutation rate is one of the factors that can determine the risk of emergent infectious disease, i.e., pathogens crossing the species barrier (46).
Slight changes of the mutation rate can also determine whether or not some virus infections are cleared by the host immune system and can produce dramatic differences in viral fitness and virulence (75, 90), clearly stressing the need to have accurate estimates. However, our knowledge of viral mutation rates is somewhat incomplete, partly due to the inherent difficulty of measuring a rare and random event but also due to several sources of bias, inaccuracy, and terminological confusion. One goal of our work is to provide an update of published mutation rate estimates, since the last authoritative reviews on viral mutation rates were published more than a decade ago (29, 30). We therefore present a comprehensive review of mutation rate estimates from over 40 original studies and 23 different viruses representing all the main virus types. A second, and perhaps more ambitious, goal of our study is to consolidate the published literature by dealing with what we regard as the two main problems in the field: the use of different units of measurement and the bias caused by selection.
The problem of units is linked to the different modes of replication in viruses. Under “stamping machine” or linear replication, multiple copies are made sequentially from the same template and the resulting progeny strands do not become templates until the progeny virions infect another cell. In contrast, under binary replication, progeny strands immediately become templates and hence the number of molecules doubles in each cycle of strand copying, increasing geometrically. This basic distinction leads to two different definitions of the mutation rate: per strand copying or per cell infection. If replication is stamping machine-like, there is only one cycle of strand copying per infected cell and hence the two units are equivalent. However, binary replication means that the virus completes several cycles of strand copying per cell. The actual replication mode of most viruses is probably intermediate between these two idealized cases, and although it is known to be closer to linear in some viruses (9, 19) and closer to binary in others (26, 55), it is often unknown. This leads to uncertainties in mutation rate estimates. For instance, in the case of poliovirus 1, the estimated rate per strand copying can vary by 10-fold depending on whether stamping machine or binary replication is assumed (27). Typically this difference in the unit of measurement has been overlooked in comparative studies. Here, we express published estimates in the same unit.
The other issue that we address is selection. In general, deleterious mutations tend to be eliminated and hence are less likely to be sampled than neutral ones, introducing a bias in mutation rate estimates. To avoid this problem, selective neutrality is sometimes enforced by the experimenter, such that the number of mutations increases linearly with time (58, 88). The opposite strategy is to focus on lethal mutations, which have necessarily appeared during the last cell infection cycle, thus establishing a direct and time-independent relationship between the observed mutation frequency and the underlying mutation rate (13, 37). In between these two special cases, an explicit correction for selection is needed. Even if the effect of each individual mutation on viral fitness is unknown, the effect of selection can be statistically accounted for as long as the number of mutations sampled for estimating mutation rates is large. We do this here using empirical information about the distribution of mutational fitness effects previously obtained for several viruses (6, 23, 73, 80). Importantly, the basic properties of this distribution appear to be well conserved (78), and hence the proposed method should be applicable to a wide variety of viruses.
Using the resulting mutation rate data, we retest some previously accepted general patterns, suggest new ones, infer the mode of replication of some viruses, and compare the rates of mutation to substitutions with those to insertions/deletions (indels).
METHODS
Definitions and symbols used are summarized in Table 1.
TABLE 1.
Definitions of variables used for estimating mutation rates
| Symbol(s) | Definition |
|---|---|
| μs/n/c | Mutation rate as substitutions per nucleotide per cell infectiona |
| μs/n/r | Mutation rate as substitutions per nucleotide per strand copyinga |
| f, fs | Observed mutation frequency and observed mutation frequency for substitutions only, respectivelya |
| T, Ts | Total no. of mutations observable with a given detection method (mutational target size); subscript s is used to refer to substitutions onlya |
| L | Length (in nucleotides) of the genetic region studied |
| c | No. of cell infection cycles |
| B | No. of viral progeny particles released per infected cell (burst size, or viral yield) |
| N0 | No. of viral particles that start an infection (inoculum size) |
| N1 | No. of viral particles after growth |
| a | Exponential growth rate |
| W | Relative fitness as determined from growth rates and/or burst sizes |
| s | Fitness effect of a mutation, defined as 1 − W |
| E(sv) | Average s value of single random nucleotide substitutions, excluding lethal ones |
| pL | Probability that a single random nucleotide substitution is lethal |
| α | Selection correction factor |
| P0 | Fraction of cultures showing no mutants in the Luria-Delbrück fluctuation test (null class) |
| m | Mutation rate to a given phenotype per strand copying obtained from the Luria-Delbrück fluctuation testa |
| r, rc | No. of cycles of copying and no. of cycles of copying per cell infection, respectively |
| δ | Ratio of indels to total mutations (indel fraction) |
A subscript i is used analogously to refer to indels.
Measuring mutation frequencies and mutational target sizes.
A seemingly simple approach to measuring the mutation frequency (f) is to PCR amplify the nucleic acid of a virus which has been propagated from a genetically homogeneous inoculum for a short time, obtain molecular clones, and sequence them. The set of all mutations that can be sampled using this method (mutational target size) is Ts = 3L for nucleotide substitutions and Ti = L for indels, where L is sequence length. Selection bias needs to be accounted for or corrected as described below. One problem with this method is that some apparent mutations will actually be errors introduced by the PCR/sequencing procedure and this will lead to overestimation of f. The error rate of the method therefore should be calibrated. Alternatively, it is possible to isolate viral clones by picking single plaques, amplify the nucleic acid by PCR, and sequence directly (i.e., without molecular cloning). The way we should then account for selection depends on whether the latter method or the molecular clone sequencing method is used.
Instead of relying solely on sequencing, one can first screen for mutations conferring a specific phenotype. This is often done using a selective agent such as an antiviral, a monoclonal antibody, or a nonpermissive host cell. Nevertheless, mutants still need to be sequenced to determine T. Notice that lethal mutations cannot be scored and do not contribute to T. A potential problem of this approach is that if some mutations are particularly unlikely, T can be underestimated, leading to an overestimation of the mutation rate. Sometimes experiments use neutral reporter genes, typically a transgene, such as, for instance, the lacZ α-complementation sequence. Null mutations in the reporter lead to an observable phenotype. Most indels should produce the null phenotype and hence Ti ≈ L, but only a fraction of nucleotide substitutions will (Ts < 3L). One way to solve this problem is to focus on nonsense substitutions which produce premature stop codons and thus should lead to the null phenotype on most occasions. In this particular case, Ts can be estimated as the total number of possible substitutions leading to a premature stop codon in the reporter gene, i.e., the nonsense mutation target size (13).
Calculation of the mutation rate per cell infection.
Although the details of the calculations can differ slightly depending on the particular study (see Appendix, “Calculation of mutation rates per nucleotide per cell infection”), in general f has to be divided by T and by the number of cell infection cycles, c. For exponentially growing viruses,
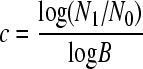 |
(1) |
where N0 and N1 are the initial and final virus titers, respectively, and B is the burst size (or viral yield), which can be determined by routine techniques (7). If all mutations were neutral, they would freely accumulate during the c cycles. However, many mutations will be lost due to selection, and thus a correction factor which accounts for selection bias (α) is needed. The mutation rate to substitutions per nucleotide per cell infection (s/n/c) can be therefore calculated as
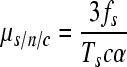 |
(2) |
where the subscript s refers to substitutions. Multiplication by 3 is done because there are three possible substitutions per site. Analogously, for indels, the mutation rate per nucleotide per cell infection (i/n/c) can be obtained as
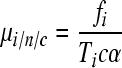 |
(3) |
where the subscript i refers to indels. Finally, we define the indel fraction as
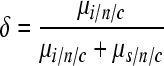 |
(4) |
Correction of selection bias.
In this section we focus on nucleotide substitutions (experiments where indels were scored satisfied neutrality in general). As a first approach, lower- and upper-limit estimates of μs/n/c can be obtained by assuming strict neutrality and lethality, respectively. If all mutations were neutral, the frequency of substitutions, fs, would increase linearly with time, but removal of mutations by selection will make this increase slower than linear. Hence, the assumption of neutrality gives us the following lower-limit estimate:
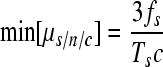 |
(5) |
In contrast, if all mutations were lethal, fs would remain constant through time and thus would not depend on c (it would consist solely of mutants appearing during the last cell infection cycle). However, as long as some mutants produce progeny, fs will increase with c. Hence, the assumption of lethality gives us the following upper-limit estimate:
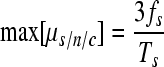 |
(6) |
Notice, however, that the logic of equation 6 holds only if mutation sampling is not affected by selection (nonselective sampling), because otherwise, strongly deleterious and lethal mutations are not observable. This problem occurs, for instance, in plaque sequencing experiments.
By comparing equations 5 and 6 we conclude that, obviously, low c values are preferable because they narrow the estimation interval (i.e., experiments should be carried out over a short time period). To obtain a more accurate estimate of μs/n/c, we have to calculate the selection correction factor α as defined in equation 3, which can be thought of as the reduction in mutation frequency fs due to selection. Equivalently, α = (min[μs/n/c])/μs/n/c, i.e., the ratio between the mutation rate estimate that we would obtain assuming strict neutrality and its actual value. Importantly, α is independent of the magnitude of the mutation rate. Notice that α = 1 for neutral mutations, whereas α = 1/c for lethal mutations, and that these two special cases give us the highest and lowest possible values of α.
We can estimate α from empirical information about the statistical distribution of the fitness effects of random single-nucleotide substitutions, which has been obtained in previous work using site-directed mutagenesis (6, 23, 73, 80). We did so numerically by simulating the effects of mutation and selection. The use of this statistical approach is justified if the mutational target used in the original experiment is representative of mutations occurring elsewhere in the genome, which implies that Ts has to be large. We modeled mutational fitness effects, s, using an exponential distribution (truncated at s = 1) plus a class of lethal mutations occurring with probability pL. This model can be written as
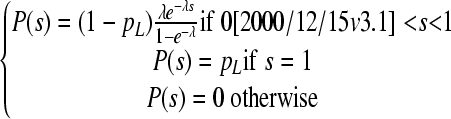 |
(7) |
The exponential distribution has a single parameter λ which equals the reciprocal of its mean. Hence, knowing the average effect of nonlethal mutations E(sv) = 1/λ and the lethal fraction pL, it is possible to predict α as a function of time [notice that since the exponential distribution is truncated at s = 1, E(sv) is indeed different from 1/λ, but this deviation can be ignored for the λ values used here]. Previous work has shown that the exponential plus lethal model allows us to describe with reasonable accuracy the empirical distribution of mutational fitness effects and that realistic parameter values are E(sv) = 0.10 to 0.13 and pL = 0.2 to 0.4 (78). Two-parameter distributions such as the gamma, the beta, or the log normal are generally more accurate than the exponential, but for the purposes of this study, the exponential should be satisfactory.
We have to pay attention to the way in which fitness is defined. In the above-mentioned studies (6, 23, 73, 78, 80), mutational fitness effects were obtained directly from growth rate ratios as
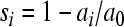 |
(8) |
where a is the exponential growth rate and subscripts i and 0 refer to the mutant and the reference virus, respectively. In order to convert these values to fitness effects per cell infection (our unit of interest here), we need to apply the following transformation:
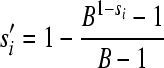 |
(9) |
The reason for this transformation is as follows. By definition, the reference virus increases its numbers by a factor of B after one cell infection cycle. Under exponential growth, the population size equals Nt = N0eat and the time required for the reference virus to complete one cell infection is thus (log B)/a0. After this time, the population size of mutant i will have increased by a factor of 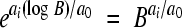 . Therefore, its relative fitness per cell infection is
. Therefore, its relative fitness per cell infection is 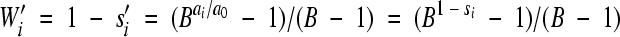 , where the −1 term subtracts the infecting virus.
, where the −1 term subtracts the infecting virus.
After simulating fitness effects using the exponential plus lethal model and converting them to per cell infection units, selection was applied by picking individuals for the next cell infection cycle with probability 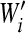 . Generations were assumed to be nonoverlapping, and at each cycle, α was calculated before and after the selection step. The former corresponds to what would be expected if mutation sampling was not affected by selection bias (nonselective sampling), whereas the latter better reflects the situation in which mutation sampling is conditioned by selection (selective sampling). The resulting α values are shown in Fig. 1 for E(sv) = 0.12, pL = 0.3, and a wide range of B values. The use of values of E(sv) ranging from 0.10 to 0.13and of pL ranging from 0.2 to 0.4 had little effect on α in all cases (not shown). To calculate α in the mutation rate estimates appearing on Table 2, specific B values were obtained from the literature, as detailed in Appendix, “Calculation of mutation rates per nucleotide per cell infection.” In all cases, the size of the simulated virus population was N = 104, which is sufficiently high for us to ignore the effects of genetic drift. For simplicity, mutations were assumed to have independent fitness effects (no epistasis), and back mutations were ignored, which seems reasonable in the short term, when forward mutations will greatly outnumber them. Finally, in one study, there were two successive selection regimens (34), and we calculated the overall correction factor as the weighted average of the two α values.
. Generations were assumed to be nonoverlapping, and at each cycle, α was calculated before and after the selection step. The former corresponds to what would be expected if mutation sampling was not affected by selection bias (nonselective sampling), whereas the latter better reflects the situation in which mutation sampling is conditioned by selection (selective sampling). The resulting α values are shown in Fig. 1 for E(sv) = 0.12, pL = 0.3, and a wide range of B values. The use of values of E(sv) ranging from 0.10 to 0.13and of pL ranging from 0.2 to 0.4 had little effect on α in all cases (not shown). To calculate α in the mutation rate estimates appearing on Table 2, specific B values were obtained from the literature, as detailed in Appendix, “Calculation of mutation rates per nucleotide per cell infection.” In all cases, the size of the simulated virus population was N = 104, which is sufficiently high for us to ignore the effects of genetic drift. For simplicity, mutations were assumed to have independent fitness effects (no epistasis), and back mutations were ignored, which seems reasonable in the short term, when forward mutations will greatly outnumber them. Finally, in one study, there were two successive selection regimens (34), and we calculated the overall correction factor as the weighted average of the two α values.
FIG. 1.

Selection-correction factor α as a function of time, measured in cell infection cycles, for different values of the burst size (or viral yield) B. α is mathematically defined in Methods (equation 2 and text). It allows us to quantify the loss in mutation frequency (fs) due to selection and thus to account for selection bias in mutation rate estimates. The expected value of α depends on whether mutation sampling is done in the absence of selection (top), as would be the case for a molecular clone sequencing experiment, or in the presence of selection (bottom), as, for instance, in direct plaque sequencing. The expected α is plotted for eight B values: 3, 10, 30, 100, 300, 103, 3 × 103, and 104. Fitness effects of random single-nucleotide substitutions (defined in equation 8) were assumed to follow an exponential distribution with mean E(sv) = 0.12 plus a class of lethal mutations occurring with probability pL = 0.3 (equation 7).
TABLE 2.
Virus mutation rates expressed as substitutions per nucleotide per cell infection (μs/n/c)
| Group | Virus | Genome size (kb) | Mean μs/n/ca | Individual studies |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Methodb | μs/n/c | Tsc | cc | αc,d | Error source(s)e | δc | Reference(s) | ||||
| ssRNA(+) | Bacteriophage Qβ | 4.22 | (1.1 × 10−3) | T1 | (1.1 × 10−3) | 1 | 1-10 | NDf | T, + | 22 | |
| Tobacco mosaic virus (TMV) | 6.40 | 8.7 × 10−6 | Trans | 8.7 × 10−6 | 723 | 5.7 | 1 | 0.40 | 58 | ||
| Human rhinovirus 14 (HRV-14) | 7.13 | 6.9 × 10−5 | Res | (4.8 × 10−4) | ND | 1/α | 1/c | T, c, α | 93 | ||
| Res | (1.0 × 10−5) | 12 | ND | ND | c, α | 41 | |||||
| Poliovirus 1 (PV-1) | 7.44 | 9.0 × 10−5 | Res | 2.2 × 10−5 | 1 | 2.8-4.6 | ∼1 | T | 17, 30 | ||
| Res | 1.1 × 10−4 | 4 | 2.1 | ND | α | 18 | |||||
| Seq | 3.0 × 10−4 | 8,463 | 3.0 | 0.28 | 90 | ||||||
| Tobacco etch virus (TEV) | 9.49 | 1.2 × 10−5 | Seq | (3.0 × 10−5) | 4,890 | ND | ND | c, α, + | 79 | ||
| Seq | 4.8 × 10−6 | 4,357 | 16-190 | 1 | 0.32 | 88 | |||||
| Hepatitis C virus (HCV) | 9.65 | (1.2 × 10−4) | Seq | (1.2 × 10−4) | 113 | 1/α | 1/c | c, α, + | 13 | ||
| Murine hepatitis virus (MHV) | 31.4 | (3.5 × 10−6) | Seq | (3.5 × 10−6) | 20,163 | 13 | 0.55 | α | 34 | ||
| ssRNA(−) | Vesicular stomatitis virus (VSV) | 11.2 | 3.5 × 10−5 | Res | (6.9 × 10−5) | 2 | 3.0-6.4 | ND | T, α | 27, 43 | |
| Res | 1.8 × 10−5 | 6 | ∼1 | ND | α | 36 | |||||
| Influenza A virus (FLUVA) | 13.6 | 2.3 × 10−5 | Seq | 4.5 × 10−5 | 2,547 | 5 | 0.33 | 70 | |||
| Seq | 7.1 × 10−6 | 2,547 | 7 | 0.28 | 68 | ||||||
| Res | 3.9 × 10−5 | 4 | 17 | ∼1 | c | 84 | |||||
| Influenza B virus (FLUVB) | 14.5 | 1.7 × 10−6 | Seq | 1.7 × 10−6 | 2,547 | 7 | 0.33 | 68 | |||
| dsRNA | Bacteriophage φ6 | 13.4 | 1.6 × 10−6 | Res | 1.6 × 10−6 | ∼5.6 | 1 | ND | T | 9 | |
| Retro | Duck hepatitis B virus (DHBV) | 3.03 | (2.0 × 10−5) | Seq | (2.0 × 10−5) | 1 | ND | ND | T, c, α | 76 | |
| Spleen necrosis virus (SNV) | 7.80 | 3.7 × 10−5 | Trans | 2.4 × 10−5 | 20 | 1 | 1 | 0.25 | 71 | ||
| Trans | 5.8 × 10−5 | 1 | 1 | 1 | T | 24 | |||||
| Murine leukemia virus (MLV) | 8.33 | 3.0 × 10−5 | Trans | 6.0 × 10−6 | 1 | 1 | 1 | T | 89 | ||
| T1 | 4.2 × 10−5 | 4,140 | 1 | 0.48 | 66 | ||||||
| Trans | 1.1 × 10−4 | 76 | 1 | 1 | 0.27 | 29, 69 | |||||
| Bovine leukemia virus (BLV) | 8.42 | 1.7 × 10−5 | Trans | 1.7 × 10−5 | 20 | 1 | 1 | 0.12 | 63 | ||
| Human T-cell leukemia virus type 1 (HTLV-1) | 8.50 | 1.6 × 10−5 | Trans | 1.6 × 10−5 | 20 | 1 | 1 | 0.10 | 60 | ||
| Human immunodeficiency virus | 9.18 | 2.4 × 10−5 | Trans | 4.9 × 10−5 | 20-219 | 1 | 1 | 0.18 | 61, 62, 64 | ||
| type 1 (HIV-1) | Seq | (1.0 × 10−4) | 25,295 | 1 | ND | α, + | 0.35 | 38 | |||
| Trans | 8.7 × 10−5 | 76 | 1 | 1 | + | 0.07 | 47 | ||||
| Trans | 7.3 × 10−7 | 7 | 1 | 1 | 51 | ||||||
| Rous sarcoma virus (RSV) | 9.40 | (1.4 × 10−4) | Other | (1.4 × 10−4) | 3,375 | 1 | ND | c, α | 52 | ||
| ssDNA | Bacteriophage φX174 | 5.39 | 1.1 × 10−6 | Res | 1.3 × 10−6 | 12 | 3.3 | ND | α | 77 | |
| Res | 1.0 × 10−6 | 7 | 1.4 | ND | 12 | ||||||
| Bacteriophage M13 | 6.41 | 7.9 × 10−7 | Trans | 7.9 × 10−7 | 219 | 5.8 | 1 | c | 0.19 | 49 | |
| dsDNA | Bacteriophage λ | 48.5 | 5.4 × 10−7 | Other | 5.4 × 10−7 | 38 | ND | 1 | c | 0.20 | 25, 26 |
| Herpes simplex virus type 1 (HSV-1) | 152 | 5.9 × 10−8 | Res | 5.9 × 10−8 | 76 | 3 | 1 | 0.17 | 31, 54 | ||
| Bacteriophage T2 | 169 | 9.8 × 10−8 | Other | 9.8 × 10−8 | 1,576 | 1 | ∼1 | 0.37 | 26, 55 | ||
Geometric mean calculated when several estimates are available. Less reliable values are shown in parentheses.
Primary method by which mutations were scored. T1, T1 RNase digestion; Trans, phenotypically observable mutations in transgene or a gene complemented in trans; Res, resistance to drugs, antibodies, or restrictive cell types; Seq, direct sequencing of viral RNA/DNA or sequencing of molecular clones (no phenotypic assay). Notice that the T1, Trans, and Res methods are usually followed by sequencing of mutants.
See Table 1 for definition.
α = 1, neutrality was enforced experimentally; α = 1/c, lethal mutations were scored (in this case no c or α values are needed to estimate μs/n/c).
T, short, unknown, or nonrepresentative mutational target; c, unknown number of cell infection cycles; α, selection was present and could not be corrected; +, a large portion of the mutants may be false positives due to sequencing errors, mutations occurring during transfection, or a poor mutation detection method.
ND, not determined.
The simulations described above were performed with Wolfram Mathematica 7.0 and Microsoft Excel 2003. Mathematica notebooks and Excel spreadsheets for obtaining predictions of fs and α as a function of time (c) for any given combination of E(sv), pL, B, and N are available upon request.
Calculation of the mutation rate per strand copying.
In principle, the calculation is the same as for μs/n/c, replacing c by the number of copying cycles, r (also accounting for selection). However, to obtain r from initial and final viral titers, it is necessary to know the replication mode, a condition that is not satisfied in most cases. A specific method for estimating mutation rates per strand copying which avoids this problem is the Luria-Delbrück fluctuation test (55, 56). Its application to viruses has been described elsewhere (12, 36, 55, 81, 85, 86). Briefly, the method consists of seeding from the same source a large number of parallel cultures using a small inoculum, harvesting them, and selecting for a specific phenotype. Although the distribution of the number of mutants per culture depends on the mode of replication and selection, the fraction of cultures showing no selectable mutants (P0) does not. Since mutations are rare and random, their number per culture should follow a Poisson distribution, for which the null class occurs with probability 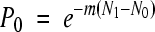 , where m is the rate of mutation to the phenotype per strand copying and N1 − N0 is the absolute amount of growth. Since m is insensitive to selection, no correction of selection bias is needed. However, m can be sensitive to differences in plating efficiency or to phenotypic mixing. The rate of mutation to substitutions per strand copying was calculated as
, where m is the rate of mutation to the phenotype per strand copying and N1 − N0 is the absolute amount of growth. Since m is insensitive to selection, no correction of selection bias is needed. However, m can be sensitive to differences in plating efficiency or to phenotypic mixing. The rate of mutation to substitutions per strand copying was calculated as
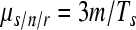 |
(10) |
where m = −(log P0)/(N1 − N0). We obtained no μi/n/r values (i.e., for indels) because all mutations leading to the selectable phenotype were substitutions. An alternative to the null-class method is to use the entire distribution of the number of mutants per culture following the F method described by Drake (26). However, this method requires that mutations are neutral and the replication mode is binary, so we did not use it.
RESULTS AND DISCUSSION
Mutation rates per cell infection.
Table 2 shows mutation rates, defined as the probability of a nucleotide substitution per nucleotide per cell infection (μs/n/c) obtained from equation 2. For each of 37 studies, we provide the value of μs/n/c and information about the mutational target size (Ts for substitutions), the number of cell infection cycles (c), and the selection correction factor (α) (details of the calculations are in “Calculation of mutation rates per nucleotide per cell infection” in Appendix). The majority of these studies were originally designed to control for selection, such that all mutations were neutral (α = 1) or lethal (cα = 1). In 6 of these 37 studies selection was not controlled for, so we corrected for its effect. The reliability of the mutation rate estimates increases as Ts increases, since mutation sampling becomes more representative. It also increases as c decreases, because there is less time for selection to act, and it increases if α is known. Estimates based on a low Ts, a large c, or an undetermined α or suffering from other problems are shown within parentheses and should be taken with caution. Also, it is clearly desirable that several independent estimates are available for each virus, and so we present average values where possible (since mutation rates vary by orders of magnitude, we used geometric means; i.e., we averaged in log scale). Finally, although the mutation rates in Table 2 refer to nucleotide substitutions, in some cases the mutation rate to indels has been measured and so we show their contribution to the total rate (δ).
Mutation rates per strand copying and inference of replication modes.
Mutation rates defined as the probability of a nucleotide substitution per nucleotide per strand copying (μs/n/r) are shown in Table 3. The availability of these estimates is limited because of the lack of information about the replication modes of many viruses. The values shown were derived using the Luria-Delbrück fluctuation test null-class method (equation 10; details of each calculation can be found in “Calculation of mutation rates per nucleotide per strand copying” in Appendix). Consistently, the mutation rates per strand copying are lower than those per cell infection. For some viruses, the two kinds of estimate are available and we can thus calculate the number of copying cycles per infected cell as rc = μs/n/c/μs/n/r (Table 4). Further, by comparing the observed rc value with its minimum and maximum possible values, we can infer the likely mode of replication. A replication event requires one cycle of strand copying in double-stranded DNA (dsDNA) viruses, and hence min[rc] = 1, which corresponds to the purely stamping machine (linear) replication mode. Single-stranded RNA (ssRNA) viruses produce an intermediate strand of opposite polarity, most dsRNA viruses produce a single positive-sense strand which is later copied to reform dsRNA, and ssDNA viruses are first copied to form dsDNA. Therefore, min[rc] = 2 for all these virus types. This implies that, strictly speaking, fully linear replication is not possible in these viruses. By definition, the number of copying cycles per infected cell is maximal under binary replication and equals max[rc] = log B/log 2. This holds for dsDNA viruses but for ssRNA, dsRNA, and ssDNA viruses we must use max[rc] = log B/log 2 + 1 since there is an additional strand copying from a single strand. Previous work has shown that the mode of replication is close to linear in bacteriophages φX174 (19) and φ6 (9), binary in bacteriophage T2 (55), and probably binary in bacteriophage λ during its lytic phase (26). Comparison of rc with max[rc] and min[rc] (Table 4) confirms these results and also leads us to suggest that replication is close to linear for influenza A virus (FLUVA), intermediate for vesicular stomatitis virus (VSV), and close to binary for poliovirus 1 (PV-1). For φX174 and VSV, we repeated this analysis but used only mutation rates per cell infection and per strand copying obtained from the same study (i.e., not comparing across studies). This gives results consistent with those shown in Table 4 (rc = 1.0 for φX174 and rc = 3.0 for VSV). Finally, in retroviruses, the genomic positive-sense RNA is reverse transcribed to obtain ssDNA, which is copied to form dsDNA, and hence rc = 2 for virus-mediated replication. This is followed by integration into the host chromosome, transcription, and possibly a variable number of host cell replications, implying that rc ≥ 3. However, since proofreading and repair systems are present in these additional host-mediated copying processes (50, 87), they probably contribute little to the overall mutation rate.
TABLE 3.
Virus mutation rates expressed as substitutions per nucleotide per strand copying (μs/n/r)a
| Group | Virus | μs/n/r | Ts | Reference(s) |
|---|---|---|---|---|
| ssRNA(+) | PV-1 | 8.9 × 10−6 | 1 | 82 |
| ssRNA(−) | VSV | 6.0 × 10−6 | 6 | 36 |
| FLUVA | 9.0 × 10−6 | 2-5 | 85, 86 | |
| Measles virus (MV)b | 4.4 × 10−5 | ∼7.5 | 81 | |
| dsRNA | φ6 | 1.4 × 10−6 | ∼5.6 | 9 |
| ssDNA | φX174c | 2.1 × 10−7 | ∼6.4 | 19 |
| 1.0 × 10−6 | 7 | 12 | ||
| dsDNA | λ | 7.9 × 10−8 | 38 | 25, 26 |
| T2 | 2.0 × 10−8 | 1,576 | 26, 55 |
All estimates were obtained using the fluctuation test, except for λ, where the rate per cycle of copying was calculated assuming binary replication (26).
Genome size, 15.9 kb.
Geometric mean, 4.6 × 10−7.
TABLE 4.
Inference of replication modes from the two mutation rate units
| Group | Virus | μs/n/c | μs/n/r | rca | B | Min[rc] | Max[rc]b | Reference(s) |
|
|---|---|---|---|---|---|---|---|---|---|
| μ | B | ||||||||
| ssRNA(+) | PV1 | 9.0 × 10−5 | 8.9 × 10−6 | 10.1 | 1694-6400 | 2.0 | 11.7-13.6 | 17, 18, 30, 82 | 17, 18 |
| ssRNA(−) | VSV | 3.5 × 10−5 | 6.0 × 10−6 | 5.8 | 1250 | 2.0 | 11.3 | 27, 36, 43 | 36 |
| FLUVA | 2.3 × 10−5 | 9.0 × 10−6 | 2.5 | 50 | 2.0 | 6.6 | 68, 70, 84-86 | 68 | |
| dsRNA | φ6 | 1.6 × 10−6 | 1.4 × 10−6 | 1.1 | 76 | 2.0 | 7.2 | 9 | 9 |
| ssDNA | φX174 | 1.1 × 10−6 | 4.6 × 10−7 | 2.4 | 28-180 | 2.0 | 5.8-8.5 | 12, 19, 77 | 19, 20 |
| dsDNA | T2 | 9.8 × 10−8 | 2.0 × 10−8 | 4.9 | 82 | 1.0 | 6.4 | 26, 55 | 26, 55 |
Ratio of μs/n/c to μs/n/r.
Log B/log 2 for dsDNA viruses and log B/log 2 + 1 for all other viruses.
Analysis of general mutation rate patterns.
One of the most general results concerning mutation rates is Drake's rule (26), which states that the mutation rate per genome per strand copying is roughly constant across DNA-based microorganisms, including DNA viruses. There is therefore an inverse relationship between genome size and the mutation rate per nucleotide. However, this rule has not previously been tested using mutation rates expressed per cell infection. This test is necessary because DNA viruses with large genomes show the lowest mutation rates per strand copying but also tend to use binary replication. Binary replication produces more mutations per cell, and this might compensate for the lower rate per strand copying. However, the plot of mutation rates per cell infection against genome size (Fig. 2) indicates that Drake's rule is robust to the choice of units.
FIG. 2.

Relationship between mutation rate and genome size, with major virus groups indicated. Values for viroids and bacteria, the two adjacent levels of biological complexity, are also plotted. The mutation rate is expressed as the number of substitutions per nucleotide per generation, defined as a cell infection in viruses (μs/n/c). We obtained bacterial mutation rates from a previous study (57) and divided them by 1.46 to convert the total rate into a substitution rate, as previously suggested (27). 1, Bacillus; 2, Deinococcus; 3, enterobacteria; 4, Helicobacter; 5, Mycobacterium; 6, Sulfolobus. The star indicates the solitary rate for a viroid (37), a subviral infectious agent constituted by small noncoding RNA. This rate is in substitutions per strand copying, since viroids do not have the equivalent of a generation.
Another general observation is that RNA viruses have a higher mutation rate than DNA viruses (29, 45). However, based on observations that some ssDNA viruses can evolve rapidly, it was recently suggested that they may have mutation rates close to those of RNA viruses (32). We find that the lowest μs/n/c estimate among RNA viruses is 1.6 × 10−6 and the highest among DNA viruses is 1.1 × 10−6. Hence, although our data show no overlap between viral RNA and DNA mutation rates, the separation may be less than is often thought. Indeed, the transition between DNA and RNA viruses appears to be relatively smooth in Fig. 2 and can be partially explained by differences in genome size.
Another question is whether the negative correlation between mutation rate and genome size observed for DNA-based microorganisms is also true for RNA viruses. The main difficulty in testing this is that the range of genome sizes among RNA viruses is only around 1 order of magnitude and the mutation rate estimates have large errors. Despite this limitation, we found a significantly negative correlation between mutation rate and genome size among the combined ssRNA(+), ssRNA(−), and dsRNA viruses (n = 11; Spearman correlation, ρ = −0.618 and P = 0.043; Pearson correlation using log10 scales, r = −0.718 and P = 0.013). However, the mutation rate estimates for the viruses with the smallest and largest genomes (bacteriophage Qβ and murine hepatitis virus [MHV]), which are key for testing this correlation, have problems such as small mutational targets or difficulties in accounting for selection bias (see Appendix, “Calculation of mutation rates per nucleotide per cell infection”). Moreover, the statistical significance of the correlation is lost when the less reliable estimates (shown in parentheses in Table 2) are removed (n = 8; ρ = −0.429, P = 0.289; r = −0.583, P = 0.129). Also, inclusion of retroviruses slightly weakens the correlation (n = 18; ρ = −0.418, P = 0.084; r = −0.550, P = 0.018). Therefore, the current data are consistent with there being a negative relationship between mutation rate and genome size among RNA viruses, but they do not strongly support it.
In previous reviews, it has been proposed that the mutation rates of retroviruses tend to be lower than those of other RNA viruses (29, 59), despite HIV-1 being perhaps the prototypic fast-evolving RNA virus (4, 92). However, there is no evidence for a lower mutation rate in retroviruses (geometric mean μs/n/c = 3.0 × 10−5) than in RNA viruses (μs/n/c = 2.2 × 10−5). Therefore, there is currently no reason to attribute differences between the evolutionary rate of retroviruses and other RNA viruses to differences in mutation rates. It is also worth mentioning that the high rate of evolution of HIV-1 is not extremely different from that of other RNA viruses such as, for instance, FLUVA or foot-and-mouth disease virus (48).
Concerning the fraction of indels compared to total mutations, we observe δ = 0.10 to 0.40 (Table 2) with a mean of 0.24 and a median of 0.20. This is very similar to the estimated δ = 0.21 for a single viroid (37) and is also consistent with the value obtained for several DNA microbes (28). Although it has been suggested that indels are particularly frequent in some RNA viruses (58, 71), our review of the literature confirms that nucleotide substitutions are the most frequent type of spontaneous mutations, being roughly four times more frequent than indels.
Conclusions and outstanding questions.
Whether mutation rates should be expressed per infected cell or per strand copying depends on the questions being addressed and the estimation method. In general, we see several advantages to using cell infection units. First, it is a natural definition of a viral generation, making comparative analyses across different types of virus or between viruses and other organisms more meaningful. Second, most theoretical models in viral population dynamics use this unit. For instance, this figure, together with the rate of infection of new cells, is used to calculate the probability of specific mutations occurring within an infected individual (72) or to predict the outcome of lethal mutagenesis treatments (5). Third, it is more inclusive than the per strand copying rate, since it accounts for other sources of mutation, such as host-mediated editing and copying, or spontaneous damage of the viral nucleic acid. Fourth, it facilitates a clear conceptual separation between the error rate of a viral polymerase and the mutation rate experienced by the virus. On the other hand, although counting cell infection cycles might be easy for animal and bacterial lytic viruses, it is more difficult for persistent viruses, plant viruses, and viruses that integrate in the host genome (e.g., retroviruses and prophages). Also, the complete cell infection cycle includes the extracellular stage, but the duration of this stage can be extremely variable and often indeterminate.
As we have shown, selection can bias mutation rate estimates. Ideally, mutations in the target under study should be strictly neutral or lethal, such that the conversion from mutation frequencies to mutation rates is straightforward (equations 5 and 6). The method based on lethality, for instance, can be implemented by looking at substitutions that produce premature stop codons, provided that no genetic complementation or suppression of stop codons occurs (13), but mutations introduced during PCR amplification need to be taken into account. Another possibility is to use drug dependence, a form of drug resistance in which the ability to grow in the absence of the drug is lost. These mutants can be identified by isolating drug-resistant mutants and assaying them for growth in the absence of the drug. We have also addressed the problem of correcting selection bias when lethality or neutrality is not guaranteed. The selection correction method proposed here can be used in general for converting mutation frequencies into mutation rates, but the calculation of the correction factor α depends on whether the sampling method is selection free (Fig. 1). For instance, in molecular clone sequencing experiments, the efficiency with which mutants are PCR amplified, cloned, and sequenced will not depend on their fitness, and hence mutation sampling is nonselective. In contrast, in experiments where plaques are sequenced directly (i.e., without molecular cloning), highly deleterious or lethal mutations will not be observable, and hence mutation sampling is selective. Our choice of parameters for computing α is based on previous experimental work with a rhabdovirus (80), a potyvirus (6), a levivirus (23), a microvirus (23), and an inovirus (73). Importantly, mutational fitness effects are well conserved across these viruses [E(sv) = 0.10 to 0.13, pL = 0.2 to 0.4] (78), and, given the diversity of this group, we can be relatively confident that the model is realistic for most ssRNA(+), ssRNA(−), and ssDNA viruses infecting animals, plants, or bacteria. In contrast, they are probably not accurate for large ssRNA(+) viruses (e.g., coronaviruses) and dsDNA viruses, and the validity for retroviruses remains to be determined.
There are several outstanding questions regarding the main evolutionary determinants of virus mutation. For instance, biochemical restrictions might not be sufficient to explain the error-prone nature of RNA virus replication, since fidelity can be increased through single amino acid replacements in the RNA polymerase (74). Also, to investigate the differences between DNA virus and RNA virus mutation rates, more estimates for small DNA viruses are needed, particularly for eukaryotic ssDNA viruses, which are the DNA viruses known to evolve fastest as measured by the number of substitutions that become fixed per year (46). The role played by error-prone host DNA polymerases in determining the mutation rate of DNA viruses is another interesting research avenue. For RNA viruses, it is still unclear whether there is a negative relationship between mutation rate and genome size analogous to Drake's rule. As we have shown, the current data suggest a correlation, but we need more estimates for the largest and smallest RNA viruses to better test this hypothesis. The possibility that the largest RNA viruses, namely, coronaviruses, show low mutation rates is supported by evidence of 3′ exonuclease proofreading activity in their replicases (65). Further, the RNA genome with the highest mutation rate, a hammerhead viroid (37), is 1 order of magnitude smaller than the smallest RNA virus genomes. However, while all viroids have very small genomes, variability studies suggest that they do not all show extremely high mutation rates (33, 35). Finally, it is also unclear whether genome properties other than size, such as genome polarity or structure, can influence the viral mutation rate. dsRNA is less exposed to chemical damage than ssRNA, and ssRNA(−) viruses pack their genetic material densely with nucleoproteins, which might confer protection against mutation.
Finally, we suggest that future mutation rate studies should fulfill the following criteria: the number of cell infection cycles should be as low as possible, the mutational target should be large, and mutations should be neutral or lethal or a correction should be made for selection bias. Adhering to these criteria will help us to get a clearer picture of virus mutation patterns.
Acknowledgments
We are very grateful for the help of John W. Drake and Esteban Domingo. N.C. thanks Alberto Vianelli for his support.
This work was financially supported by grant BFU2008-03978/BMC and the Ramón y Cajal program from the Spanish MICIIN, the Wellcome Trust, the Italian MIUR, the National Institutes of Health, and the Fondazione Cariplo.
APPENDIX
Calculation of mutation rates per nucleotide per cell infection. (i) Bacteriophage Qβ.
An A → G mutant with the mutation at position 40 from the 3′end of the genome was obtained by site-directed mutagenesis, plaque purified, and propagated with a high multiplicity of infection (MOI), such that each passage corresponded to a single infection cycle (22). The fraction of revertants to the wild type was measured after each passage, up to 10 passages, by T1 RNase fingerprinting. A system of linear equations was used to estimate the fraction of revertant phage produced per passage, accounting for the selective disadvantage of the mutant. This gave a most likely value of fs/c = 3.5 × 10−4 revertants per passage (2). Since Ts = 1, equation 2 gives μs/n/c = 3 × 3.5 × 10−4 = 1.1 × 10−3. Transitions are more likely than transversions. Therefore, this might be an overestimation. Also, a mutation rate of 1.1 × 10−3 s/n/c corresponds to more than four mutations per genome, which would probably impose an excessive mutational load for the virus. For this reason, this estimate was used initially by Drake (27) but was discarded later (29, 30).
(ii) TMV.
Tobacco plants constitutively expressing the TMV movement protein (MP) were inoculated with tobacco mosaic virus (TMV) (58). Since the viral MP gene was complemented in trans, selection on this gene was absent or weak (α ≈ 1). Viruses were extracted at 3 days postinoculation, and individual particles were isolated by infecting MP transgenic plants, which form local necrotic lesions. These individual clones were assayed for loss of function of the essential MP gene by inoculating plants not expressing the MP transgene, and null mutants were sequenced. The relevant parameters are f = 0.038, c = 5.7, and L = 804. Twenty-four out of 35 mutations were indels. Assuming that all indels inactivated the gene and using equation 3, μi/n/c = 0.038 × 24/35/5.7/804 = 5.7 × 10−6. For substitutions, the fraction of total mutations that produce the null phenotype was unknown, and nonsense mutations were not observed. The authors used a correction factor of 4.78 derived from DNA-based microbes. According to this and using equation 2, μs/n/c = 0.038 × 4.78 × 11/35/5.7/804 = 1.2 × 10−5. Alternatively, one can use the fraction of lethal substitutions estimated in other viruses, pL = 0.2 to 0.4. Using this to estimate the fraction of substitutions that inactivate the TMV MP protein and taking pL = 0.3, Ts = 3 × 804 × 0.3 = 723, we find that μs/n/c = 3 × 0.038 × 11/35/5.7/723 = 8.7 × 10−6. The two approaches yield similar results (we use the latter). The corresponding indel fraction is δ = 0.40.
(iii) Human rhinovirus 14.
(a) A drug dependence mutation was identified and introduced in a cDNA clone (93). Viruses recovered from this cDNA clone were grown and plated in the presence and absence of the drug to estimate the fraction of revertants to drug sensitivity, f = 1.6 × 10−4, and it was assumed that all revertants were to the wild type (T = Ts = 1) (30). Since the mutant was drug dependent, it had to be grown in the presence of the drug, and thus revertants to the wild type were lethal; i.e., cα = 1. Therefore, μs/n/c = 3 × 1.6 × 10−4 = 4.8 × 10−4. However, reversion to drug sensitivity could be due to mutations other than reversion to the wild type (T > 1), and therefore this probably represents an upper-limit estimate.
(b) A drug resistance frequency of f = 4.0 × 10−5 was obtained, and it was determined that T = Ts = 12, but c and the fitness effects of the mutations were unknown (41). Using equation 6, max[μs/n/c] = 3 × 4.0 × 10−5/12 = 1.0 × 10−5. Notice, however, that despite being an upper limit, this second estimate is much lower than the previous one.
(iv) Poliovirus 1.
(a) A plaque-purified thermosensitive mutant (C5310U) was plated at 33°C and 39°C to obtain the frequency of revertants to the wild type (17). This mutation was approximately neutral at 33°C (α ≈ 1), and only the C-to-U reversion restored growth at 39°C (T = Ts = 1). The average revertant frequency in three isolated plaques was fs = 3.1 × 10−5 and c = 2.8 (30). Hence, μs/n/c = 3 × 3.1 × 10−5/2.8 = 3.3 × 10−5. In a second experiment, the isolated plaques were passaged once in liquid culture, and the observed revertant frequency was fs = 2.3 × 10−5. In the first experiment, the total number of viruses was 1.1 × 109, and therefore, using equation 1 with N0 = 1, we obtain B = (1.1 × 109)1/2.8 = 1,694. In the second experiment, a 10−5 dilution was applied to inoculate the liquid culture, and the average number of viruses after growth was 8.4 × 109. Hence, the amplification factor was (8.4 × 109)/(1.1 × 109) × 105 = 7.6 × 105, which corresponds to log (7.6 × 105)/log 1,694 = 1.8 additional infection cycles. Hence, μs/n/c = 3 × 2.3 × 10−5/(2.8 + 1.8) = 1.5 × 10−5. Taking the geometric mean of the estimates from the two experiments, μs/n/c = 2.2 × 10−5.
(b) The frequency of guanidine-resistant mutants appearing from a guanidine-dependent mutant was measured by plating the virus in the presence and absence of the drug (18). Approximately 2.0 × 106 cells were inoculated with ca. 200 PFU, yielding an average titer of 3.2 × 109 PFU/ml in a total volume of 4 ml after completion of the cytopathic effect (18, 27). Hence, the burst size is B = (3.2 × 109 × 4)/(2.0 × 106) = 6,400, and using equation 1 we obtain c = log (4 × 3.2 × 109/200)/log 6,400 = 2.1. Drake and Holland (30) gave a similar value (c = 2.5). Sequencing showed that the loss of guanidine dependence could be conferred by each of the three possible nucleotide substitutions at position G4804 or an A → G substitution at position 4802 (T = Ts = 4). In two experiments, fs = 1.1 × 10−4 and fs = 5.4 × 10−4. Pooling all data, fs = 3.2 × 10−4. Considering that mutations were probably neutral, i.e., α ≈ 1 (although this was not demonstrated), μs/n/c = 3 × 3.2 × 10−4/2.1/4 = 1.1 × 10−4.
(c) Viruses from transfection of cDNA transcripts were passaged three times at an MOI of 1.0 (c ≈ 3.0), and individual plaques were isolated (90). The 5′ noncoding region and capsid gene (L = 2,821) were sequenced directly from reverse transcription-PCR (RT-PCR) products (i.e., without molecular cloning). Thirteen mutations were observed in 18 plaque-derived viruses. For the wild-type virus, 13 mutations were found after sequencing 50,700 nucleotides in total. Hence, using equation 5, we obtain min[μs/n/c] = 13/50,700/3 = 8.5 × 10−5. No max[μs/n/c] can be obtained since sampling was selective (i.e., the assumption that all mutations are lethal is incompatible with plaque sequencing). The selection correction factor with selective sampling and assuming the same burst size as above (B = 1,694) is α = 0.28 for pL = 0.3 and E(sv) = 0.12. Thus, the corrected estimate is μs/n/c = min[μs/n/c]/α = 8.5 × 10−5/0.28 = 3.0 × 10−4.
(v) Tobacco etch virus (TEV).
(a) Viruses isolated from single necrotic lesions in Chenopodium quinoa were used to infect tobacco plants, and virions were extracted following the appearance of symptoms (79). A region encompassing genome positions 7808 to 9437 was amplified by high-fidelity RT-PCR, and 83 molecular clones were sequenced (Ts = 4,890). Four substitutions were observed. Using equation 6, max[μs/n/c] = 3 × 4/83/4,890 = 3.0 × 10−5. Another reason to consider this estimate as an upper limit is that the observed rate was close to the rate of RT-PCR errors.
(b) Tobacco plants constitutively expressing the TEV polymerase gene NIb were inoculated with TEV (88). Since the viral NIb gene was complemented in trans, selection on this gene was probably absent or weak (α ≈ 1). Samples from 20 plants were taken at different time points ranging from 5 to 60 days postinoculation and used for RT-PCR, cloning, and sequencing. In total, 42 mutations (36 substitutions and 6 indels) were identified in 472 NIb clones (L = 1,536). Since the viral genomic RNA is translated as a polyprotein, indels that modify the reading frame or nonsense mutations in the NIb gene prevent the correct expression of downstream genes (here, the capsid gene). As a first approach, we can focus on these presumably lethal mutations. Of the 36 substitutions, two produced premature stop codons. The number of possible such mutations in the NIb gene is Ts = 251. Hence, μs/n/c = 2/251/472 = 1.7 × 10−5. For indels, μi/n/c = 6/1,536/472 = 8.2 × 10−6, and thus δ = 0.32. Immediately after a stop codon mutant appears in a cell, it can be replicated, transcribed, and packaged normally by the nonmutant proteins present in the cell, but the mutant should be unable to initiate a second infection cycle. Hence, the estimate is in per cell infection units. However, suppression of stop codons or complementation between viruses at a high MOI could allow a subset of mutants to complete several infection cycles, leading to an overestimation of the mutation rate. RT-PCR errors constitute another source of overestimation. As an alternative approach, we can focus on presumably neutral mutations, which are all except nonsense mutations and indels because NIb was trans complemented (Ts = 1,536 × 3 − 251 = 4,357). The viral yield per cell was B = 1,555 as determined in vitro using transfected protoplasts, and it was estimated that c = 3.16 per day; hence, c varied from 16 to 190 (5 to 60 days). According to a regression analysis of the number of mutations on the number of cell infection cycles done in the original publication, μs/n/c = 4.8 × 10−6. The latter value is used. Taking into account that the first approach was expected to produce an overestimation, the two estimates are reasonably consistent.
(vi) Hepatitis C virus.
Over 15,000 molecular clones of the E1-E2 and NS5A regions (L = 472 and L = 743, respectively) obtained from patient serum samples were sequenced. The observed number of nonsense mutations was divided by the number of possible nonsense substitutions in these genes, which was Ts = 113 on average (13), yielding μs/n/c = 1.2 × 10−4. As above, this estimate assumes that all observed nonsense mutations are truly lethal and should be taken as an upper-limit value because we cannot exclude the suppression of stop codons, complementation, or RT-PCR errors.
(vii) Murine hepatitis virus.
Viruses were recovered from a cDNA clone by transfection, seeded into fresh cells, passaged once in standard liquid culture at an MOI of approximately 0.01, plaque purified, and passaged twice plaque to plaque (34). Six plaques were picked, amplified by infecting liquid cultures, and used for direct sequencing (i.e., without molecular cloning). It was estimated that one infection cycle was equivalent to 8 h of growth and, based on this, that the total number of cell infection cycles was c = 13. For the wild-type virus, three mutations were found after sequencing 120,978 nucleotides in total. Hence, using equation 5, we obtain min[μs/n/c] = 3/120,978/13 = 1.9 × 10−6, whereas no max[μs/n/c] can be obtained because mutation sampling was selective. To provide a more accurate estimate, we can use the selection correction method. Plaque-to-plaque passages constituted approximately two-thirds of the total passage time (c1 = 13 × 2/3 = 8.7), although the exact fraction was not provided. Selection is typically relaxed under this passage regimen, and assuming that all mutations except lethal ones accumulated neutrally, we have μs/n/c = min[μs/n/c]/(1 − pL). This defines a correction factor α1 = 1 − pL for this phase. For the standard liquid culture phase (c2 = 4.3 cycles), the correction factor with selective sampling assuming that B = 600 to 700 (42), pL = 0.3, and E(sv) = 0.12 is α2 ≈ 0.26. Using the weighted average to combine α1 and α2, we obtain α = (0.7 × 8.7 + 0.26 × 4.3)/(8.7 + 4.3) = 0.55. Therefore, the corrected estimate of the mutation rate is μs/n/c = 1.9 × 10−6/ 0.55 = 3.5 × 10−6. Notice, however, that our parameterization of the distribution of mutational fitness effects was based on viruses with genome sizes smaller than those of coronaviruses and thus might not be appropriate here. Also, there is some uncertainty in the number of cell infection cycles elapsed. For these reasons, the estimate should be taken with caution.
(viii) Vesicular stomatitis virus.
(a) The frequency of resistance to a monoclonal antibody was measured by plating clonal viral pools or viruses resuspended from plaques in the presence of the antibody (43). Mutations were assumed to be neutral. Virus titers averaged 4.2 × 1011 PFU/ml and 3.7 × 107 PFU/ml for clonal pools and resuspended plaques, respectively (27). Plating 0.1 ml of these stocks yielded f = 1.7 × 10−4 and f = 2.3 × 10−4, respectively. Sequencing showed that there were two possible G → A transitions conferring resistance (T = Ts = 2). Under conditions that restrict viral diffusion, B = 166 for this cell type (14), and B = 1,250 in liquid medium under standard conditions (36). Hence, the formation of a plaque would require approximately log (3.7 × 107 × 0.1)/log 166 = 3.0 cell infection cycles, and an additional log (4.2 × 1011 × 0.1)/log 1,250 = 3.4 cycles would have taken place in clonal pools. Accordingly, μs/n/c = 3 × 2.3 × 10−4/3/2 = 1.2 × 10−4 and μs/n/c = 3 × 1.7 × 10−4/(3 + 3.4)/2 = 4.0 × 10−5 for the resuspended plaques and the clonal pools, respectively, giving a geometric mean of μs/n/c = 6.9 × 10−5. Since the only mutations scored were transitions, which are more likely than transversions, and since neutrality was not guaranteed, this value might be an overestimation.
(b) The average monoclonal antibody resistance frequency obtained from many small cultures undergoing one cell infection cycle or fewer was determined (36), giving f/c = 3.5 × 10−5. It was assumed that T = Ts = 6 from references cited in reference 36. These substitutions were probably close to neutral (α ≈ 1), although this was not directly shown. Under this assumption, μs/n/c = 3 × 3.5 × 10−5/6 = 1.8 × 10−5. The data from this experiment can also be used to estimate the mutation rate per strand copying using the fluctuation test null-class method (see below).
(ix) Influenza virus A.
(a) A single viral plaque was isolated and replated to isolate new plaques (70). The consensus sequence of gene NS (L = 849, Ts = 849 × 3 = 2,547) was obtained for the parental and derived plaques after two amplification passages by direct sequencing of the purified RNA: 3fs/Ts = 7.6 × 10−5, and c = 5. Hence, from equation 5, min[μs/n/c] = 7.6 × 10−5/5 = 1.5 × 10−5, whereas no max[μs/n/c] can be obtained because mutation sampling was selective. The estimated selection correction factor using the exponential plus lethal class model with E(sv) = 0.12, pL = 0.3, c = 5, selective sampling, and B ≈ 50 as estimated in another work (68) is α = 0.33. Thus, μs/n/c = 1.5 × 10−5/ 0.33 = 4.5 × 10−5.
(b) The same method as in the study described above (70) was used, giving 3fs/Ts = 1.4 × 10−5 and c = 7 (48 h postinoculation with a generation time of 7 h, as estimated from one-step growth curves; note that the estimated burst size is B = 50 as shown in Fig. 2 of the original publication) (68). Hence, min[μs/n/c] = 2.0 × 10−6. The estimated selection correction factor using the exponential plus lethal class model with E(sv) = 0.12, pL = 0.3, c = 7, selective sampling, and B = 50 is α = 0.28. Thus, μs/n/c = 2.0 × 10−6/0.28 = 7.1 × 10−6.
(c) Single plaques were isolated after 3 days of growth in cell cultures and used to infect the allantoic cavities of chicken eggs (84). Viruses were harvested after 2 days and plated in the presence and absence of amantadine to score resistant viruses. From 10 independent experiments, the average f values were 4.2 × 10−4 and 1.8 × 10−3 for H1N1 and H2N2 genotypes, respectively. Amantadine resistance was conferred by four different nucleotide substitutions (T = Ts = 4), which were probably neutral (α ≈ 1). In the original publication, the mutation rate per strand copying was estimated by assuming binary replication, but this assumption does not seem to be justified. According to other authors (68) the virus completes a cell infection cycle in ca. 7 h. Thus, after 5 days of growth, c = 17. Thus, μs/n/c = 3 × 4.2 × 10−4/17/4 = 1.9 × 10−5 for H1N1 and μs/n/c = 7.9 × 10−5 for H2N2, with the geometric mean being 3.9 × 10−5.
(x) Influenza virus B.
This estimate for influenza virus B comes from one same study as FLUVA estimate b (68). Here, 3fs/Ts = 4.0 × 10−6 and c = 7. Hence, min[μs/n/c] = 5.7 × 10−7 and max[μs/n/c] = 4.0 × 10−6. Using the exponential plus lethal class model to correct for selection bias as in FLUVA estimate b, we obtain α = 0.33 and μs/n/c = 1.7 × 10−6.
(xi) Bacteriophage φ6.
An amber mutant was grown in amber suppressor cells and plated onto normal (nonpermissive) cells to score revertants (9). The analysis of the number of revertants arising from single bursts (c = 1) yielded 296 mutants in 1,306,864 bursts and B = 76. Thus, f/c = 296/1,306,864/76 = 3.0 × 10−6. However, T was not determined. Discarding indels, there are eight possible single mutations changing an amber stop codon to a non-stop codon. However, some might be lethal and hence not observable. For a fraction of lethal substitutions of pL = 0.2 to 0.4, T = Ts = 4.8 to 6.4. Assuming no selection against viable revertants and taking Ts = 5.6, μs/n/c = 3 × 3.0 × 10−6/5.6 = 1.6 × 10−6. The undetermined Ts value could lead to a maximal underestimation of 5.6-fold and a maximal overestimation of 1.4-fold. Although the E(sv) values of the reversions were unknown, this should not introduce bias here because c = 1. Data from this experiment can also be used to obtain an estimate of the mutation rate per strand copying free of selection bias using the fluctuation test null-class method (see below).
(xii) Duck hepatitis B virus.
The frequency of revertants of a single deleterious nucleotide substitution (G1198A) was studied in viruses obtained from cells transfected with a cDNA clone (76). Ducklings were inoculated with the recovered viruses and the wild type at a 104:1 excess of mutants. At 25 days postinoculation, the ratio of revertants to wild type was obtained by molecular clone sequencing (revertants and the wild type were distinguishable by a neutral molecular marker at another site). This ratio was 0.6, and, correcting for the initial excess of mutants, the revertant frequency was fs = 6.0 × 10−5. This fs value should equal the initial frequency of revertants in the inocula plus the frequency of new revertants that appeared during replication of the mutant before the latter was outcompeted. It was estimated that, at day 25, fs was 4- to 23-fold higher than the initial fs. Hence, the initial fs ranged from 0.3 × 10−5 to 1.5 × 10−5. Multiplying by three to get the mutation frequency to any of the three nucleotides and taking the geometric mean of the interval bounds, c × μs/n/c = 2.0 × 10−5. Since the inoculum came directly from transfected cells, we can assume that the inoculated viruses had undergone approximately one cell infection cycle and thus that μs/n/c = 2.0 × 10−5. The methods used to obtain this estimate are indirect, and thus the value should be taken with caution.
(xiii) Spleen necrosis virus (SNV).
(a) A retroviral vector containing the lacZ α-complementation gene region as a neutral mutational target was used to score null mutations appearing during a single infection cycle (71). Out of 16,867 clones, 37 carried null mutations in the lacZ α-complementation gene region based on the white/blue assay of transformed Escherichia coli colonies. Sequencing showed that 11 were nucleotide substitutions (including two nonsense mutations), 24 were indels (5 frameshifts), and 2 were 15-base G → A hypermutations. The coding region the lacZ region is 258 bases long (280 bases including the promoter region), but the mutational target is smaller because many mutations will not lead to the null phenotype. In a previous study, it was determined that Ts = 219 (3). Hence, for substitutions not caused by host-mediated hypermutation, μs/n/c = 3 × 11/16,867/219 = 8.9 × 10−6. Alternatively, we used the method based on scoring stop codons. Since there are 20 possible nonsense substitutions in the lacZ α-complementation sequence and all should lead to the null phenotype, the mutation rate is μs/n/c = 3 × 2/16,867/20 = 1.8 × 10−5. We used the latter value. Considering that all G → A hypermutations should lead to loss of function and including the promoter in the mutational target (Ts = 280), the G → A mutation rate due to host-mediated hypermutation is 2 × 15/16,867/280 = 6.3 × 10−6. Hence, the total mutation rate to substitutions is μs/n/c = 6.3 × 10−6 + 1.8 × 10−5 = 2.4 × 10−5. For indels, it was determined that Ti = 150 for frameshifts and Ti = 280 for the other indels. Hence, μi/n/c = 5/16,867/150 + 19/16,867/280 = 6.0 × 10−6. The indel ratio is δ = 0.25.
(b) A retroviral vector containing a neo gene with an amber codon (a neutral mutational target) and the hygro gene and was used to score mutations appearing during a single cell infection cycle by selecting clones with G418 resistance (cells containing proviruses revertant to the functional neo gene) and hygromycin resistance (all provirus-containing cells) (24). The amber reversion frequency per cycle was f/c = 2.2 × 10−5. It was shown that 15/17 revertants were to the wild type, whereas the other two were a four-nucleotide insertion and an unidentified mutation. Using reversions to the wild type only, μs/n/c = 3 × 2.2 × 10−5 × 15/17 = 5.8 × 10−5.
(xiv) Murine leukemia virus (MLV).
(a) Using the same method as for SNV estimate b above, the amber reversion frequency per infection cycle was f/c = 4.0 × 10−6 (89). It was shown that 7/14 revertants were to the wild type, whereas the other seven were of an unidentified nature. Using reversions to the wild type only, μs/n/c = 3 × 4.0 × 10−6 × 7/14 = 6.0 × 10−6.
(b) The viral progeny released by a single transformant colony was used to infect fresh cells at a low MOI, which were plated onto solid medium before the release of new viral progeny (66). The resulting infected colonies were analyzed by T1 RNase digestion, covering a target of 1,380 nucleotides (Ts = 3 × 1,380 = 4,140). Three substitutions were detected and confirmed by sequencing after screening of ca. 151,000 nucleotides in total, giving 3fs/c/Ts = 3 × 3/(3 × 151,000) = 2.0 × 10−5. However, selection was not completely absent. Since lethal or strongly deleterious mutations were probably missed, this should be considered a lower-limit estimate. The selection correction factor given by the exponential plus lethal class model using c = 1, pL = 0.3, E(sv) = 0.12, selective sampling, and taking B = 50 from the literature (67) is α = 0.48. Therefore, μs/n/c = 2.0 × 10−5/0.48 = 4.2 × 10−5.
(c) A retroviral vector containing the herpes simplex virus tk gene (a neutral mutational target) and the neo gene was used to score mutations appearing during a single cell infection cycle by selecting tk null mutants with bromouridine and total virus-carrying cells with G418 (69), giving f/c = 0.088. According to Drake et al. (29), of 244 tk− mutants, 114 were gross rearrangements and arose in a mutational target of 2,620 bases. Assuming that all gross rearrangements inactivated the tk gene, the mutation rate to these changes was 0.088 × 114/244/2,620 = 1.6 × 10−5 rearrangements/s/c. The remaining 130 changes were small mutations and arose in a target of 1,128 bases. Among 49 small mutants sequenced, 28 were indels. Hence, μi/n/c = 0.088 × 130/244 × 28/49/1,128 = 2.4 × 10−5. Among nucleotide substitutions, three were nonsense mutations. Given that Ts = 76 for nonsense substitutions in this gene, μs/n/c = 3 × 0.088 × 130/244 × 3/49/76 = 1.1 × 10−4. The indel fraction is thus δ = (1.6 × 10−5 + 2.4 × 10−5)/ (1.1 × 10−4 + 1.6 × 10−5 + 2.4 × 10−5) = 0.27.
(xv) Bovine leukemia virus.
A retroviral vector containing the lacZ α-complementation sequence (neutral mutational target) was used to score mutations appearing during a single cell infection cycle (63). In total, 11/18,009 clones carried null mutations (white E. coli colonies), of which three were nucleotide substitutions (two nonsense mutations) and eight were indels (four frameshifts). Assuming Ts = 219 (see SNV estimate a), μs/n/c = 3 × 3/18,009/219 = 2.3 × 10−6. Using nonsense mutations only, Ts = 20 (see SNV estimate a), and thus μs/n/c = 3 × 2/18,009/20 = 1.7 × 10−5. We used the latter estimate. Ti = 150 for frameshifts and Ti = 280 for other indels (see SNV estimate a), and thus μi/n/c = 4/18,009/150 + 4/18,009/ 280 = 2.3 × 10−6 and δ = 0.12.
(xvi) Human T-cell leukemia virus type 1.
A retroviral vector containing the lacZ α-complementation sequence (neutral mutational target) was used to score mutations appearing during a single cell infection cycle (60). Of 36,561 clones analyzed, 33 carried null mutations in the target (white E. coli colonies), of which 19 were single-nucleotide substitutions (four nonsense mutations), 1 was a double mutation (referred to as a hypermutation in the original study), and 13 were indels (including seven frameshifts). Assuming Ts = 219 (see SNV estimate a), μs/n/c = 3 × 19/36,561/219 = 7.1 × 10−6. Using nonsense mutations only, Ts = 20 (see SNV estimate a, and thus μs/n/c = 3 × 4/36,561/20 = 1.6 × 10−5; we use the latter). The rate of hypermutation appears to be low, and we did not attempt to calculate it since it was based on a single observation and corresponded to a double mutant, for which Ts was undetermined (the assumption that all double mutants inactivate the target cannot be made). Ti = 150 for frameshifts and Ti = 280 for other indels (see SNV estimate a), and thus μi/n/c = 7/36,561/150 + 6/36,561/280 = 1.9 × 10−6 and δ = 0.10.
(xvii) Human immunodeficiency virus type 1.
(a) A retroviral vector containing the lacZ α-complementation sequence (neutral mutational target) was used to score mutations appearing during a single cell infection cycle in several related studies (61, 62, 64). In the first study (64), f/c = 70/15,424 (66 mutant clones, with 4 of them carrying two mutations). The mutational spectrum was constituted by 46 nucleotide substitutions (six nonsense mutations [29]) and 24 indels (17 frameshifts). Given that Ts = 219 (see SNV estimate a), μs/n/c = 3 × 46/15,424/219 = 4.1 × 10−5. Using nonsense mutations only (see SNV estimate a), Ts = 20 and μs/n/c = 3 × 6/15,424/20 = 5.8 × 10−5 (the latter value is used). For indels, using the Ti values given in SNV estimate a, μi/n/c = 17/15,424/150 + 7/15,424/280 = 9.0 × 10−6 and δ = 0.13. In a second study (62), the same method was used to score mutations in vpr null mutants and in vpr null mutants complemented in trans by virus producer cells. This showed that vpr reduces the viral mutation rate by approximately 3-fold. In the presence of a functional vpr protein provided in trans, f/c = 0.006. The mutational spectrum was unknown, but assuming that it was similar to the one reported in the previous study (64), nucleotide substitutions should constitute approximately two-thirds of all the observed mutations. Given that Ts = 219, μs/n/c s = 3 × 0.006 × 2/3/219 = 5.5 × 10−5. In a third study (61), the same method was used to score mutations in the absence or presence of the antiretroviral drugs zidovudine (AZT) and lamivudine (3TC), as well as in viruses encoding reverse transcriptase variants resistant to these drugs. The average mutation frequency per cycle from three independent experiments for the wild-type virus was f/c = 0.005 (0.004, 0,005, and 0.006) in the absence of drugs. Sequencing of 40 mutant clones showed that 22 carried nucleotide substitutions (there were three additional G → A hypermutants, but these are not counted here because the numbers of substitutions carried by each hypermutant were not provided), 6 carried frameshifts, and 2 carried other indels. Taking Ts = 219 for substitutions, Ti = 150 for frameshifts, and Ti = 280 for other indels, μs/n/c = 3 × 0.005 × 22/40/219 = 3.7 × 10−5, μi/n/c = 0.005 × 6/20/150 + 0.005 × 2/20/280 = 1.2 × 10−5, and δ = 0.22. The geometric mean of the three μs/n/c values is 4.9 × 10−5. The average of the two indel fractions is δ = 0.18.
(b) To score mutations appearing during a single cell infection cycle, pseudotyped viruses were obtained by cotransfecting 293T cells with a viral vector defective for the env gene and a helper plasmid (38). HeLa cells were infected with these viruses, selected for antibiotic resistance, cloned, and used for DNA amplification and subcloning using a phage λ library. Sequencing of six nearly full-length viral genomes (9,072 nucleotides on average, Ts = 27,216) yielded four nucleotide substitutions and no indels. Hence, μs/n/c = 3 × 4/6/27,216 = 7.3 × 10−5. In another assay, lymphocytes were infected with the pseudotyped viruses, sorted by fluorescence using flow cytometry, cloned, and used for DNA amplification by long-range PCR and direct sequencing of PCR products. Sequencing of seven large portions of viral genomes (7,791 nucleotides on average, Ts = 23,373) yielded eight nucleotide substitutions and three indels. Hence, μs/n/c = 3 × 8/7/23,373 = 1.5 × 10−4. Taking the geometric mean of the two estimates, μs/n/c = 1.0 × 10−4. The average Ts is 25,295. For indels, μi/n/c = 3/7/7,791 = 5.5 × 10−5, and δ = 0.35. The fraction of stop codon mutations was unusually high (4/12), and no synonymous substitutions were observed, suggesting that cell clones receiving inactive viruses were favored, and selection acting on the virus during the cell infection cycle was not controlled for. Also, the fraction of mutations resulting from transfection was unknown. Finally, the pseudotyped viruses lacked vpr, which may have an effect on the mutation rate (62). These factors could have led to a high number of false positives, and thus this estimate should be taken with caution.
(c) A retroviral vector that contained all cis elements required for replication, regulatory and accessory virus genes, and reporter genes tk and hygro but which lacked the gag, pol, and env genes was constructed and used to score mutations appearing during one cell infection cycle by cotransfecting the vector with helper plasmids carrying the missing genes (47). The hygro gene confers resistance to hygromycin, whereas the tk gene confers sensitivity to bromouridine. Hygromycin was used for selecting cells carrying the vector and bromouridine to score null mutations in the tk gene (996 nucleotides). In total, 349/15,930 clones were mutant, giving f/c = 0.022. Sequencing of 43 mutants indicated that 13/43 mutations were indels. Hence μi/n/c = 0.022 × 13/43/996 = 9.7 × 10−6. The fraction of nucleotide substitutions that produced the tk null phenotype was unknown, and it was not indicated which mutations produced stop codons. The number of possible mutations to stop codons in this gene is 76, and, using information from previous studies (29, 69), it is expected that approximately 1/7 of the observed nucleotide substitutions produced such mutations (see MLV estimate c). Hence μs/n/c = 3 × 0.022 × 30/43/7/ 76 = 8.7 × 10−5, and δ = 0.072. Mutations arising during transfection were not controlled, potentially introducing false positives.
(d) A retroviral vector containing the gag and pol genes and two reporter genes, bsd and eYFP, but defective for env, regulatory, and accessory genes was used to score mutations appearing during one cell infection cycle (51). The eYFP gene encodes the yellow fluorescent protein and was used to count the total number of cells carrying the vector. 293T cells stably expressing the vector were transfected with a helper plasmid to yield pseudotyped viruses, which were used to transduce fresh cells. The bsd gene encoded resistance to basticidin but had a premature ochre stop codon such that only cells receiving a revertant virus would be resistant to blasticidin. This gave f/c = (2.0 to 4.0) × 10−6, and sequencing of 16 revertants showed nine single-nucleotide substitutions (to the wild type or three other codons), five apparent G → A hypermutations, and two deletions. Here, T is unknown for indels and hypermutations, but for single nucleotide substitutions, Ts = 7. Using the latter and taking f/c = 3.0 × 10−6, μs/n/c = 3 × 3.0 × 10−6 × 9/16/7 = 7.3 × 10−7. Additional assays were carried out with HIV-1 and other retroviruses by directly cotransfecting cells with the vector and the helper plasmid instead of using stable eYFP producers, but it was shown that transfection was a significant source of mutation and hence these data did not provide a reliable estimate of the mutation rate.
(xviii) Rous sarcoma virus.
A single transformant colony was isolated and viruses released from it were used to infect fresh cells, which were plated onto soft agar before new viruses could be released (52). The resulting colonies were amplified, and the purified RNA was assayed for mutations by denaturing gradient gel analysis. Several domains of known size were analyzed using RNA from 58 colonies, which was the equivalent of 65,250 nucleotides (Ts = 3 × 65,250/58 = 3,375). Nine mutations were found, giving f = 9/65,250 = 1.4 × 10−4. Only one mutation was confirmed by sequencing. Assuming no superinfection, these mutations appeared during a single cell infection cycle. However, selection was probably present. Additionally, the number of provirus copies per cell was unknown, and hence this estimate has to be taken with caution.
(xix) Bacteriophage φX174.
(a) Approximately 340 independent wells were infected with an average of 2 PFU each and incubated overnight (77). Lysates were plated onto the selective strain E. coli gro89, a defective mutant with a mutation of the rep gene, which encodes a DNA helicase required for particle maturation, to score for phages with the ability to infect this strain. From 12 wells, it was determined that f = 1.7 × 10−5. The average final number of PFU per well was 6.6 × 107, and B = 180 as estimated in another study (20). Thus, using equation 1, c = log (6.6 × 107/2)/log 180 = 3.3. Sequencing of 156 independent mutants showed that T = Ts = 12. Since Ts was determined, there is no selection bias due to lethal mutations. Some bias could exist due to a nonlethal effect. However, since c was small, this should not produce a large deviation in the estimate (probably less than 2-fold). Neglecting this effect, μs/n/c = 3 × 1.7 × 10−5/3.3/12 = 1.3 × 10−6.
(b) A plaque-purified virus was used to infect 216 independent cultures with an average of 231 PFU each (12). Cultures were incubated until an average of 3.3 × 105 PFU per culture was produced and then plated onto E. coli gro87, a rep gene mutant similar to the one used in phage φX174 estimate a. A total of 239 mutants were scored, and thus f = 239/3.3 × 10−5/216 = 3.4 × 10−6. Sequencing of 47 clones showed that T = Ts = 7. Taking B = 180 (20), c = log (3.3 × 105/231)/log 180 = 1.4. The fact that Ts was determined implies that there was no selection bias due to lethal mutations. Also, the bias due to nonlethal effects should be small, because c was close to 1.0. Thus, μs/n/c = 3 × 3.4 × 10−6/7/1.4 = 1.0 × 10−6. Data from this experiment can also be used obtain an estimate of the mutation rate per strand copying free of selection bias (see below).
(xx) Bacteriophage M13.
A single plaque of a recombinant virus carrying the lacZ α-complementation sequence (258 bases) as a neutral mutational target (α = 1) was used to inoculate a large E. coli culture and incubated overnight (49). Viral DNA was extracted and transfected to score null mutations in the lacZ sequence (based on the blue/white colony assay). After discarding 11 false positives, f = 117/199,655, with 67 plaques containing single-nucleotide substitutions and 50 containing indels (11 frameshifts and 39 deletions or rearrangements). In a previous study (3), it was determined that Ts = 219. Thus, c × μs/n/c = 3 × 67/199,655/219 = 4.6 × 10−6. For indels, assuming Ti = 150 for frameshifts and Ti = 280 for other indels (see SNV estimate a), c × μi/n/c = 11/150/199,655 + 39/280/199,655 = 1.1 × 10−6. At the very least, c = 3 (two cycles during the formation of the plaque and another one during the infection of the liquid culture). According to Drake (26), the initial and final viral population sizes were 1 and ca. 1.0 × 1015, respectively. Our own unpublished data suggest that under relatively optimal conditions, the exponential growth rate of the virus is ca 4.0 h−1 and the duration of the cell infection cycle is ca. 1 h. According to this and assuming exponential growth, an increase in population size by a factor of 1.0 × 1015 would require approximately c = 8.6. Taking the average of 3 and 8.6, c = 5.8. This gives μs/n/c = 4.6 × 10−6/ 5.8 = 7.9 × 10−7, μi/n/c = 1.1 × 10−6/5.8 = 1.9 × 10−7, and δ = 0.19. The most evident source of error in this estimate is the undetermined c value. This could lead to a maximal underestimation of 1.9-fold and a maximal overestimation which, although not determined, probably does not exceed 1.5-fold.
(xxi) Bacteriophage λ.
Estimates of the mutation rate per nucleotide per strand copying were obtained under lytic growth conditions, where replication is thought to be mainly binary, yielding μs/n/r = 7.9 × 10−8 and μi/n/r = 2.0 × 10−8 (26) (see estimate per strand copying for this phage, below). Using B = 115 (20), there should be rc = log 115/log 2 = 6.8 cycles of copying per cell infection. Neutrality was satisfied (α = 1), and thus μs/n/c = 6.8 × 7.9 × 10−8 = 5.4 × 10−7, μi/n/c = 6.8 × 2.0 × 10−8 = 1.4 × 10−7, and δ = 0.20.
(xxii) Herpes simplex virus.
Ganciclovir was used to detect null mutations in the tk gene in cell culture (54), giving f = 6.0 × 10−5 (31). Sequencing revealed 45 indels, 17 missense mutations, and 5 nonsense mutations (67 mutations in total). There are Ts = 76 possible nonsense substitutions in this gene, c = 3 (31), L = 1,128, and tk null mutations are selectively neutral in cell culture in the absence of the drug (α = 1) (8). Hence, μs/n/c = 3 × 6.0 × 10−5 × 5/67/3/76 = 5.9 × 10−8, μi/n/c = 6.0 × 10−5 × 45/67/3/1128 = 1.2 × 10−8, and δ = 0.17.
(xxiii) Bacteriophage T2.
Mutations at the rII locus (L = 3,136) producing rapid plaque growth (phenotype r) were scored in single bursts (55). After discarding cases in which the mutant was probably present in the inoculum, 420 mutants were scored in 22,615 bursts (c = 1), and it was determined that B = 82. This gives f/c = 420/22,615/82 = 2.3 × 10−4. Mutations were probably close to neutral (α ≈ 1), and deviations from neutrality should not produce a strong bias since c = 1. The mutational spectrum of this gene was analyzed for the closely related bacteriophage T4 (26). Fifteen nonsense mutations (all of which should produce the phenotype) and 21 missense mutations were identified in a 435-base region of the locus, and nonsense mutations were expected to represent ca. 0.073 of all random substitutions. Hence, the expected number of substitutions (including those that did not produce the r phenotype) is 15/0.073 = 206, indicating that 21/206 = 0.102 of missense mutations produced the r phenotype. In another assay, it was shown that among 121 observed rII mutants, 27 were single-nucleotide substitutions and 94 were indels. Ts is not simply 3L because many mutations were not observable. Since nonsense mutations represented approximately 0.073 of all mutations and 0.102 of missense mutations were observable, Ts = 3L × [0.073 + 0.102 × (1 − 0.073)] = 1,576. Thus, μs/n/c = 3 × 2.3 × 10−4 × 27/121/1,576 = 9.8 × 10−8, μi/n/c = 2.3 × 10−4 × 94/121/3,136 = 5.7 × 10−8, and δ = 0.37.
Calculation of mutation rates per nucleotide per strand copying. (i) Poliovirus 1.
The reversion frequency of an amber mutant was measured by plating the virus in permissive and selective cells (82). Only one substitution (UAG to UCG) was viable (T = Ts = 1), and selection against the amber mutant was strong. Despite selection, it is still possible to use the null-class method to estimate the mutation rate. Out of 27 independent cultures assayed, 23 showed no revertants (P0 = 23/27). The average number of PFU per culture was 1.8 × 104, but the plating efficiency of the amber mutant was approximately three times lower than that of the revertant, so the corrected number is 5.4 × 104 PFU. Hence, using equation 10, μs/n/r = −3 × log (23/27)/(5.4 × 104) = 8.9 × 10−6.
(ii) Vesicular stomatitis virus.
The monoclonal antibody resistance frequency was determined from many small cultures undergoing one cell infection cycle or fewer, and the mutation rate to the phenotype using the null-class method was m = 1.2 × 10−5 s/r (36). Ts = 6 from references cited in reference 36, and thus μs/n/r = 3 × 1.2 × 10−5/6 = 6.0 × 10−6.
(iii) Influenza virus A.
Single plaques were isolated and replated in the presence of antihemagglutinin monoclonal antibody (85). The mutation rate was estimated using the null-class method. Sequencing of a large number of clones (86) indicated that Ts = 2 and Ts = 5 for each of the two antibodies used. The rates of mutation to the resistant phenotype were converted into mutation rates per nucleotide accordingly, yielding μs/n/r = 3.0 × 10−5 and μs/n/r = 2.7 × 10−6, with geometric mean μs/n/r = 9.0 × 10−6.
(iv) Measles virus.
A fluctuation test was performed by growing a total of 227 cultures, each inoculated with 200 PFU, and testing them for the presence of antihemagglutinin monoclonal antibody-resistant mutants (81). Mutation rates were estimated using the null-class method. Cultures were divided in three experimental blocks, yielding m = 1.4 × 10−4 s/r, m = 6.0 × 10−5 s/r, and m = 1.7 × 10−4 s/r (geometric mean, 1.1 × 10−4 s/r). Four different nucleotide substitutions were identified after sequencing five clones. Given the low number of clones sequenced, it is likely that Ts > 4. Drake and Holland (30) argued that the most likely value is Ts = 7.5, assuming a Poisson distribution of mutation counts across sites. However, if some mutations are more likely than others, this assumption may not be satisfied. Independent evidence comes from a previous work (see reference 81), in which three additional resistance mutations were found, yielding Ts = 7 in total. Hence, it seems that the value Ts = 7.5 given by Drake and Holland is robust. According to this, μs/n/r = 3 × 1.1 × 10−4/7.5 = 4.4 × 10−5.
(v) Bacteriophage φ6.
An amber mutant was grown in cells encoding an amber suppressor (permissive cells) and plated in normal (nonpermissive) cells after a single burst to score the appearance of amber revertants (9). An estimate of ms = 2.7 × 10−6 s/r was obtained using the null-class method. There are eight possible ways of reverting an amber mutation through single-nucleotide substitutions. However, some might be lethal and hence not observable. For pL = 0.2 to 0.4, Ts = 4.8 to 6.4. Taking Ts = 5.6, μs/n/r = 3 × 2.7 × 10−6/5.6 = 1.4 × 10−6. Error in this estimate comes mainly from the undetermined T value, which could produce a maximal underestimation of 5.6-fold and a maximal overestimation of 1.4-fold.
(vi) Bacteriophage φX174.
(a) A fluctuation test was carried out using the reversion of an amber mutation as the selectable phenotype (19). In each individual culture, the number of initial PFU was high but the virus underwent a single cell infection cycle. For each of three amber mutants, P0 = 646/740, P0 = 679/778, and P0 = 510/602. The corresponding burst sizes were B = 167, B = 28, and B = 51, and the final numbers of PFU were 9.0 × 107, 5.5 × 107, and 2.6 × 109, respectively. Hence, N1 − N0 = 9.0 × 107/740 − 9.0 × 107/740/167 = 1.2 × 105 for the first mutant, and analogously, N1 − N0 = 6.8 × 104 and 4.2 × 106 for the second and third mutants, respectively. Using the null-class method, m = 1.1 × 10−6 s/r, m = 2.0 × 10−6 s/r, and m = 3.9 × 10−8 s/r, respectively, with geometric mean ms = 4.5 × 10−7 s/r. If all amber revertants were to the wild type, μs/n/r = 3 × 4.5 × 10−7 = 1.4 × 10−6. However, there are eight possible single-nucleotide revertants, and if all were viable, the mutation rate would be μs/n/r = 3 × 4.5 × 10−7/8 = 1.7 × 10−7. For this phage, the estimated lethal fraction is P = 0.2 (23), and hence the expected number of viable revertants is 8 × 0.8 = 6.4, which gives μs/n/r = 2.1 × 10−7.
(b) A plaque-purified virus was used to infect 216 independent cultures with an average of 231 PFU each (12). The mutation rate was calculated using the null-class method. m = 2.3 × 10−6 s/r, and sequencing of 47 clones showed that Ts = 7. Hence, μs/n/r = 3 × 2.3 × 10−6/7 = 1.0 × 10−6.
(vii) Bacteriophage λ.
Drake (26) reported a mutation rate to loss of function of gene cI of 2.0 × 10−5 per strand copying, based on previous work (25). Screening of over 300 cI null mutants showed that ca. 5% of the mutations were to amber stop codons (26). The number of possible substitutions leading to amber stop codons in the cI gene is Ts = 38. Therefore, μs/n/r = 3 × 2 × 10−5 × 0.05/38 = 7.9 × 10−8.
(viii) Bacteriophage T2.
The appearance of the r phenotype was scored using single-burst experiments (55). Given that replication is close to binary (55) and assuming that r mutants are neutral, the F method described by Drake can be used and yields a rate of mutation to the phenotype of 4.8 × 10−5 (26). However, it is also possible to use the null-class method: from 22,615 bursts, 85 gave one or more mutants (after correcting for the presence of mutants in the inoculum), and B = 82. Hence, m = −log [(22,615 − 85)/22,615]/82 = 4.6 × 10−5, which is consistent with the estimate obtained with the F method. Ts = 1,576, and 27/121 mutations were nucleotide substitutions (see calculation of the mutation rate per cell infection cycle for this same phage). Hence, μs/n/r = 3 × 4.6 × 10−5 × 27/121/1,576 = 2.0 × 10−8.
Footnotes
Published ahead of print on 21 July 2010.
REFERENCES
- 1.Anderson, J. P., R. Daifuku, and L. A. Loeb. 2004. Viral error catastrophe by mutagenic nucleosides. Annu. Rev. Microbiol. 58:183-205. [DOI] [PubMed] [Google Scholar]
- 2.Batschelet, E., E. Domingo, and C. Weissmann. 1976. The proportion of revertant and mutant phage in a growing population, as a function of mutation and growth rate. Gene 1:27-32. [DOI] [PubMed] [Google Scholar]
- 3.Bebenek, K., J. Abbotts, J. D. Roberts, S. H. Wilson, and T. A. Kunkel. 1989. Specificity and mechanism of error-prone replication by human immunodeficiency virus-1 reverse transcriptase. J. Biol. Chem. 264:16948-16956. [PubMed] [Google Scholar]
- 4.Belshaw, R., A. Gardner, A. Rambaut, and O. G. Pybus. 2008. Pacing a small cage: mutation and RNA viruses. Trends Ecol. Evol. 23:188-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bull, J. J., R. Sanjuán, and C. O. Wilke. 2007. Theory of lethal mutagenesis for viruses. J. Virol. 81:2930-2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carrasco, P., F. de la Iglesia, and S. F. Elena. 2007. Distribution of fitness and virulence effects caused by single-nucleotide substitutions in tobacco etch virus. J. Virol. 81:12979-12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carter, J., and V. Saunders. 2007. Virology: principles and applications. Wiley, New York, NY.
- 8.Challberg, M. D., and T. J. Kelly. 1989. Animal virus DNA replication. Annu. Rev. Biochem. 58:671-717. [DOI] [PubMed] [Google Scholar]
- 9.Chao, L., C. U. Rang, and L. E. Wong. 2002. Distribution of spontaneous mutants and inferences about the replication mode of the RNA bacteriophage φ6. J. Virol. 76:3276-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung, D. H., Y. Sun, W. B. Parker, J. B. Arterburn, A. Bartolucci, and C. B. Jonsson. 2007. Ribavirin reveals a lethal threshold of allowable mutation frequency for Hantaan virus. J. Virol. 81:11722-11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crotty, S., C. E. Cameron, and R. Andino. 2001. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc. Natl. Acad. Sci. U. S. A. 98:6895-6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cuevas, J. M., S. Duffy, and R. Sanjuán. 2009. Point mutation rate of bacteriophage ΦX174. Genetics 183:747-749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cuevas, J. M., F. González-Candelas, A. Moya, and R. Sanjuán. 2009. The effect of ribavirin on the mutation rate and spectrum of hepatitis C virus in vivo. J. Virol. 83:5760-5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cuevas, J. M., A. Moya, and R. Sanjuán. 2005. Following the very initial growth of biological RNA viral clones. J. Gen. Virol. 86:435-443. [DOI] [PubMed] [Google Scholar]
- 15.Dapp, M. J., C. L. Clouser, S. Patterson, and L. M. Mansky. 2009. 5-Azacytidine can induce lethal mutagenesis in human immunodeficiency virus type 1. J. Virol. 83:11950-11958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davenport, M. P., L. Loh, J. Petravic, and S. J. Kent. 2008. Rates of HIV immune escape and reversion: implications for vaccination. Trends Microbiol. 16:561-566. [DOI] [PubMed] [Google Scholar]
- 17.de la Torre, J. C., C. Giachetti, B. L. Semler, and J. J. Holland. 1992. High frequency of single-base transitions and extreme frequency of precise multiple-base reversion mutations in poliovirus. Proc. Natl. Acad. Sci. U. S. A. 89:2531-2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de la Torre, J. C., E. Wimmer, and J. J. Holland. 1990. Very high frequency of reversion to guanidine resistance in clonal pools of guanidine-dependent type 1 poliovirus. J. Virol. 64:664-671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Denhardt, D. T., and R. B. Silver. 1966. An analysis of the clone size distribution of ΦX174 mutants and recombinants. Virology 30:10-19. [DOI] [PubMed] [Google Scholar]
- 20.De Paepe, M., and F. Taddei. 2006. Viruses' life history: towards a mechanistic basis of a trade-off between survival and reproduction among phages. PLoS Biol. 4:e193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Domingo, E. 2006. Quasispecies: concept and implications for virology. Springer, New York, NY.
- 22.Domingo, E., R. A. Flavell, and C. Weissmann. 1976. In vitro site-directed mutagenesis: generation and properties of an infectious extracistronic mutant of bacteriophage Qβ. Gene 1:3-25. [DOI] [PubMed] [Google Scholar]
- 23.Domingo-Calap, P., J. M. Cuevas, and R. Sanjuán. 2009. The fitness effects of random mutations in single-stranded DNA and RNA bacteriophages. PLoS Genet. 5:e1000742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dougherty, J. P., and H. M. Temin. 1988. Determination of the rate of base-pair substitution and insertion mutations in retrovirus replication. J. Virol. 62:2817-2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dove, W. F. 1968. The genetics of the lambdoid phages. Annu. Rev. Genet. 2:305-340. [Google Scholar]
- 26.Drake, J. W. 1991. A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. U. S. A. 88:7160-7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drake, J. W. 1993. Rates of spontaneous mutation among RNA viruses. Proc. Natl. Acad. Sci. U. S. A. 90:4171-4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Drake, J. W. 2009. Avoiding dangerous missense: thermophiles display especially low mutation rates. PLoS Genet. 5:e1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drake, J. W., B. Charlesworth, D. Charlesworth, and J. F. Crow. 1998. Rates of spontaneous mutation. Genetics 148:1667-1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drake, J. W., and J. J. Holland. 1999. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. U. S. A. 96:13910-13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drake, J. W., and C. B. Hwang. 2005. On the mutation rate of herpes simplex virus type 1. Genetics 170:969-970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duffy, S., L. A. Shackelton, and E. C. Holmes. 2008. Rates of evolutionary change in viruses: patterns and determinants. Nat. Rev. Genet. 9:267-276. [DOI] [PubMed] [Google Scholar]
- 33.Durán-Vila, R., S. F. Elena, J. A. Daròs, and R. Flores. 2008. Structure and evolution of viroids, p. 43-64. In E. Domingo, C. R. Parrish, and J. J. Holland (ed.), Origin and evolution of viruses. Elsevier, New York, NY.
- 34.Eckerle, L. D., X. Lu, S. M. Sperry, L. Choi, and M. R. Denison. 2007. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J. Virol. 81:12135-12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flores, R., C. Hernández, A. E. Martínez de Alba, J. A. Daròs, and F. Di Serio. 2005. Viroids and viroid-host interactions. Annu. Rev. Phytopathol. 43:117-139. [DOI] [PubMed] [Google Scholar]
- 36.Furió, V., A. Moya, and R. Sanjuán. 2005. The cost of replication fidelity in an RNA virus. Proc. Natl. Acad. Sci. U. S. A. 102:10233-10237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gago, S., S. F. Elena, R. Flores, and R. Sanjuán. 2009. Extremely high mutation rate of a hammerhead viroid. Science 323:1308. [DOI] [PubMed] [Google Scholar]
- 38.Gao, F., Y. Chen, D. N. Levy, J. A. Conway, T. B. Kepler, and H. Hui. 2004. Unselected mutations in the human immunodeficiency virus type 1 genome are mostly nonsynonymous and often deleterious. J. Virol. 78:2426-2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Graci, J. D., D. A. Harki, V. S. Korneeva, J. P. Edathil, K. Too, D. Franco, E. D. Smidansky, A. V. Paul, B. R. Peterson, D. M. Brown, D. Loakes, and C. E. Cameron. 2007. Lethal mutagenesis of poliovirus mediated by a mutagenic pyrimidine analogue. J. Virol. 81:11256-11266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grande-Pérez, A., E. Lázaro, P. Lowenstein, E. Domingo, and S. C. Manrubia. 2005. Suppression of viral infectivity through lethal defection. Proc. Natl. Acad. Sci. U. S. A. 102:4448-4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heinz, B. A., R. R. Rueckert, D. A. Shepard, F. J. Dutko, M. A. McKinlay, M. Fancher, M. G. Rossmann, J. Badger, and T. J. Smith. 1989. Genetic and molecular analyses of spontaneous mutants of human rhinovirus 14 that are resistant to an antiviral compound. J. Virol. 63:2476-2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hirano, N., K. Fujiwara, and M. Matumoto. 1976. Mouse hepatitis virus (MHV-2). Plaque assay and propagation in mouse cell line DBT cells. Jpn. J. Microbiol. 20:219-225. [PubMed] [Google Scholar]
- 43.Holland, J. J., J. C. de la Torre, D. A. Steinhauer, D. K. Clarke, E. A. Duarte, and E. Domingo. 1989. Virus mutation frequencies can be greatly underestimated by monoclonal antibody neutralization of virions. J. Virol. 63:5030-5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holland, J. J., E. Domingo, J. C. de la Torre, and D. A. Steinhauer. 1990. Mutation frequencies at defined single codon sites in vesicular stomatitis virus and poliovirus can be increased only slightly by chemical mutagenesis. J. Virol. 64:3960-3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holland, J. J., K. Spindler, F. Horodyski, E. Grabau, S. Nichol, and S. VandePol. 1982. Rapid evolution of RNA genomes. Science 215:1577-1585. [DOI] [PubMed] [Google Scholar]
- 46.Holmes, E. C. 2009. The evolution and emergence of RNA viruses. Oxford University Press, Oxford, United Kingdom.
- 47.Huang, K. J., and D. P. Wooley. 2005. A new cell-based assay for measuring the forward mutation rate of HIV-1. J. Virol. Methods 124:95-104. [DOI] [PubMed] [Google Scholar]
- 48.Jenkins, G. M., A. Rambaut, O. G. Pybus, and E. C. Holmes. 2002. Rates of molecular evolution in RNA viruses: a quantitative phylogenetic analysis. J. Mol. Evol. 54:156-165. [DOI] [PubMed] [Google Scholar]
- 49.Kunkel, T. A. 1985. The mutational specificity of DNA polymerase-beta during in vitro DNA synthesis. Production of frameshift, base substitution, and deletion mutations. J. Biol. Chem. 260:5787-5796. [PubMed] [Google Scholar]
- 50.Kunkel, T. A. 2004. DNA replication fidelity. J. Biol. Chem. 279:16895-16898. [DOI] [PubMed] [Google Scholar]
- 51.Laakso, M. M., and R. E. Sutton. 2006. Replicative fidelity of lentiviral vectors produced by transient transfection. Virology 348:406-417. [DOI] [PubMed] [Google Scholar]
- 52.Leider, J. M., P. Palese, and F. I. Smith. 1988. Determination of the mutation rate of a retrovirus. J. Virol. 62:3084-3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Loeb, L. A., J. M. Essigmann, F. Kazazi, J. Zhang, K. D. Rose, and J. I. Mullins. 1999. Lethal mutagenesis of HIV with mutagenic nucleoside analogs. Proc. Natl. Acad. Sci. U. S. A. 96:1492-1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu, Q., Y. T. Hwang, and C. B. Hwang. 2002. Mutation spectra of herpes simplex virus type 1 thymidine kinase mutants. J. Virol. 76:5822-5828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luria, S. E. 1951. The frequency distribution of spontaneous bacteriophage mutants as evidence for the exponential rate of phage reproduction. Cold Spring Harbor Symp. Quant. Biol. 16:463-470. [DOI] [PubMed] [Google Scholar]
- 56.Luria, S. E., and M. Delbrück. 1943. Mutations of bacteria from virus sensitivity to virus resistance. Genetics 28:491-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lynch, M. 2006. The origins of eukaryotic gene structure. Mol. Biol. Evol. 23:450-468. [DOI] [PubMed] [Google Scholar]
- 58.Malpica, J. M., A. Fraile, I. Moreno, C. I. Obies, J. W. Drake, and F. García-Arenal. 2002. The rate and character of spontaneous mutation in an RNA virus. Genetics 162:1505-1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mansky, L. M. 1998. Retrovirus mutation rates and their role in genetic variation. J. Gen. Virol. 79:1337-1345. [DOI] [PubMed] [Google Scholar]
- 60.Mansky, L. M. 2000. In vivo analysis of human T-cell leukemia virus type 1 reverse transcription accuracy. J. Virol. 74:9525-9531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mansky, L. M., and L. C. Bernard. 2000. 3′-Azido-3′-deoxythymidine (AZT) and AZT-resistant reverse transcriptase can increase the in vivo mutation rate of human immunodeficiency virus type 1. J. Virol. 74:9532-9539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mansky, L. M., S. Preveral, L. Selig, R. Benarous, and S. Benichou. 2000. The interaction of vpr with uracil DNA glycosylase modulates the human immunodeficiency virus type 1 in vivo mutation rate. J. Virol. 74:7039-7047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mansky, L. M., and H. M. Temin. 1994. Lower mutation rate of bovine leukemia virus relative to that of spleen necrosis virus. J. Virol. 68:494-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mansky, L. M., and H. M. Temin. 1995. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 69:5087-5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Minskaia, E., T. Hertzig, A. E. Gorbalenya, V. Campanacci, C. Cambillau, B. Canard, and J. Ziebuhr. 2006. Discovery of an RNA virus 3′→5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl. Acad. Sci. U. S. A. 103:5108-5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Monk, R. J., F. G. Malik, D. Stokesberry, and L. H. Evans. 1992. Direct determination of the point mutation rate of a murine retrovirus. J. Virol. 66:3683-3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Niwa, O., A. Decleve, M. Liberman, and H. S. Kaplan. 1973. Adaptation of plaque assay methods to the in vitro quantitation of the radiation leukemia virus. J. Virol. 12:68-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nobusawa, E., and K. Sato. 2006. Comparison of the mutation rates of human influenza A and B viruses. J. Virol. 80:3675-3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parthasarathi, S., A. Varela-Echavarría, Y. Ron, B. D. Preston, and J. P. Dougherty. 1995. Genetic rearrangements occurring during a single cycle of murine leukemia virus vector replication: characterization and implications. J. Virol. 69:7991-8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Parvin, J. D., A. Moscona, W. T. Pan, J. M. Leider, and P. Palese. 1986. Measurement of the mutation rates of animal viruses: influenza A virus and poliovirus type 1. J. Virol. 59:377-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pathak, V. K., and H. M. Temin. 1990. Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: substitutions, frameshifts, and hypermutations. Proc. Natl. Acad. Sci. U. S. A. 87:6019-6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Perelson, A. S. 2002. Modelling viral and immune system dynamics. Nat. Rev. Immunol. 2:28-36. [DOI] [PubMed] [Google Scholar]
- 73.Peris, J. B., P. Davis, J. M. Cuevas, M. R. Nebot, and R. Sanjuán. 2010. Distribution of fitness effects caused by single-nucleotide substitutions in bacteriophage f1. Genetics 185:603-609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pfeiffer, J. K., and K. Kirkegaard. 2003. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. U. S. A. 100:7289-7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pfeiffer, J. K., and K. Kirkegaard. 2005. Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS Pathog. 1:e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pult, I., N. Abbott, Y. Y. Zhang, and J. Summers. 2001. Frequency of spontaneous mutations in an avian hepadnavirus infection. J. Virol. 75:9623-9632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Raney, J. L., R. R. Delongchamp, and C. R. Valentine. 2004. Spontaneous mutant frequency and mutation spectrum for gene A of ΦX174 grown in E. coli. Environ. Mol. Mutagen. 44:119-127. [DOI] [PubMed] [Google Scholar]
- 78.Sanjuán, R. 2010. Mutational fitness effects in RNA and ssDNA viruses: common patterns revealed by site-directed mutagenesis studies. Philos. Trans. R. Soc. Lond. 365:1975-1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sanjuán, R., P. Agudelo-Romero, and S. F. Elena. 2009. Upper-limit mutation rate estimation for a plant RNA virus. Biol. Lett. 5:394-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sanjuán, R., A. Moya, and S. F. Elena. 2004. The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus. Proc. Natl. Acad. Sci. U. S. A. 101:8396-8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schrag, S. J., P. A. Rota, and W. J. Bellini. 1999. Spontaneous mutation rate of measles virus: direct estimation based on mutations conferring monoclonal antibody resistance. J. Virol. 73:51-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sedivy, J. M., J. P. Capone, U. L. RajBhandary, and P. A. Sharp. 1987. An inducible mammalian amber suppressor: propagation of a poliovirus mutant. Cell 50:379-389. [DOI] [PubMed] [Google Scholar]
- 83.Sierra, S., M. Dávila, P. R. Lowenstein, and E. Domingo. 2000. Response of foot-and-mouth disease virus to increased mutagenesis: influence of viral load and fitness in loss of infectivity. J. Virol. 74:8316-8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stech, J., X. Xiong, C. Scholtissek, and R. G. Webster. 1999. Independence of evolutionary and mutational rates after transmission of avian influenza viruses to swine. J. Virol. 73:1878-1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Suárez, P., J. Valcárcel, and J. Ortín. 1992. Heterogeneity of the mutation rates of influenza A viruses: isolation of mutator mutants. J. Virol. 66:2491-2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Suárez-López, P., and J. Ortín. 1994. An estimation of the nucleotide substitution rate at defined positions in the influenza virus haemagglutinin gene. J. Gen. Virol. 75:389-393. [DOI] [PubMed] [Google Scholar]
- 87.Thomas, M. J., A. A. Platas, and D. K. Hawley. 1998. Transcriptional fidelity and proofreading by RNA polymerase II. Cell 93:627-637. [DOI] [PubMed] [Google Scholar]
- 88.Tromas, N., and S. F. Elena. 2010. The rate and spectrum of spontaneous mutations in a plant RNA virus. Genetics 185:983-989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Varela-Echavarría, A., N. Garvey, B. D. Preston, and J. P. Dougherty. 1992. Comparison of Moloney murine leukemia virus mutation rate with the fidelity of its reverse transcriptase in vitro. J. Biol. Chem. 267:24681-24688. [PubMed] [Google Scholar]
- 90.Vignuzzi, M., J. K. Stone, J. J. Arnold, C. E. Cameron, and R. Andino. 2006. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439:344-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vignuzzi, M., E. Wendt, and R. Andino. 2008. Engineering attenuated virus vaccines by controlling replication fidelity. Nat. Med. 14:154-161. [DOI] [PubMed] [Google Scholar]
- 92.Wain-Hobson, S. 1993. The fastest genome evolution ever described: HIV variation in situ. Curr. Opin. Genet. Dev. 3:878-883. [DOI] [PubMed] [Google Scholar]
- 93.Wang, W., W. M. Lee, A. G. Mosser, and R. R. Rueckert. 1998. WIN 52035-dependent human rhinovirus 16: assembly deficiency caused by mutations near the canyon surface. J. Virol. 72:1210-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]