

Abstract
The majority of HIV-1 infections around the world result from non-B clade HIV-1 strains. The CRF01_AE (AE) strain is seen principally in Southeast Asia. AE protease differs by ∼10% in amino acid sequence from clade B protease and carries several naturally occurring polymorphisms that are associated with drug resistance in clade B. AE protease has been observed to develop resistance through a nonactive-site N88S mutation in response to nelfinavir (NFV) therapy, whereas clade B protease develops both the active-site mutation D30N and the nonactive-site mutation N88D. Structural and biochemical studies were carried out with wild-type and NFV-resistant clade B and AE protease variants. The relationship between clade-specific sequence variations and pathways to inhibitor resistance was also assessed. AE protease has a lower catalytic turnover rate than clade B protease, and it also has weaker affinity for both NFV and darunavir (DRV). This weaker affinity may lead to the nonactive-site N88S variant in AE, which exhibits significantly decreased affinity for both NFV and DRV. The D30N/N88D mutations in clade B resulted in a significant loss of affinity for NFV and, to a lesser extent, for DRV. A comparison of crystal structures of AE protease shows significant structural rearrangement in the flap hinge region compared with those of clade B protease and suggests insights into the alternative pathways to NFV resistance. In combination, our studies show that sequence polymorphisms within clades can alter protease activity and inhibitor binding and are capable of altering the pathway to inhibitor resistance.
Human immunodeficiency virus type 1 (HIV-1) is classified into three groups (M, N, and O), of which group M is further classified into nine major clades (A, B, C, D, F, G, H, J, and K) and 43 circulating recombinant forms (CRFs) based on viral genomic diversity (32, 37). The majority of HIV-1 infections across the globe result from non-B clade HIV-1 variants; clade B accounts for only ∼12% of infections (15). However, the development of currently available anti-HIV therapies has been based on the virology of clade B variants. In recent years, several studies have shown that there are clear differences between clades when it comes to viral transmission and the progression to AIDS, an observation which raises questions about the effectiveness of the currently available anti-HIV therapies against the other clades and CRFs (16-18, 39).
HIV-1 protease has been an important drug target in the global effort to curb the progression from HIV infection to AIDS. However, the accumulation of drug-resistant mutations in the protease gene has been a major drawback in using HIV-1 protease inhibitors. The effects of mutations associated with drug resistance in HIV-1 clade B protease have been studied extensively over the years. For the most part, resistance mutation patterns are very similar in HIV-1 clade B and non-B clade proteases (19). However, several alternative resistance pathways have been observed for non-B clade proteases compared with those of clade B protease (1, 12, 13, 26). Limited data are available on how sequence polymorphisms, some of which are associated with drug resistance in clade B protease, might influence the pathway to drug resistance in non-B clade proteases. Furthermore, very little is understood about how sequence polymorphisms in non-B clade proteases affect protease function and inhibitor binding.
HIV-1 CRF01_AE (AE) was the first CRF to be observed in patient populations and is seen principally in Southeast Asia (2, 10, 25). AE protease differs by ∼10% in amino acid sequence from that of clade B protease (Fig. 1A). Interestingly, AE protease develops a different resistance pathway from that of clade B protease to confer resistance to the protease inhibitor nelfinavir (NFV) (1). In patients infected with AE, the protease acquires predominantly the N88S mutation in response to NFV therapy, whereas in patients with clade B infection, the protease acquires the D30N/N88D mutations. The fitness of AE viral strains is thought to be similar to that of HIV-1 group M viral strains (11, 41). However, the effect of AE-specific sequence variations as well as drug resistance substitutions on viral fitness has not been studied extensively.
FIG. 1.

(A) Amino acid sequence alignments of B-WT and AE-WT and NFV-resistant mutants. Residue positions that differ between clade B and AE are indicated in red. NFV resistance mutations are indicated in blue. (B) Ribbon diagram superposition of DRVAE-WT (blue) and DRVAE-N88S (gray). (C) Double-difference plot comparing DRVAE-WT and DRVAE-N88S. (D) Ribbon diagram superposition of clade DRVAE-WT (magenta) and clade DRVB-WT (gray). (E) Double-difference plot comparing DRVAE-WT and DRVB-WT. The color contours in the double-difference plots indicate distance differences of <1.0 Å (black), 1.0 to 0.5 Å (green), 0.5 to 1.0 Å (blue), and >1.0 Å (red).
In the present study, biochemical and biophysical methods were used to determine the effect of sequence polymorphisms in AE protease on enzyme activity and inhibitor binding. Through determination of crystal structures and analysis of changes in hydrogen bonding patterns, a structural rationalization is described for the two different pathways observed for clade B and AE proteases to attain resistance to NFV.
MATERIALS AND METHODS
Protease gene construction.
The clade B wild-type (B-WT) protease gene was generated as previously described (34). The AE wild-type (AE-WT) protease gene was synthesized in fragments (Integrated DNA Technologies, Coralville, IA), with codons optimized for expression in Escherichia coli. The fragments were ligated to form the complete gene, which was then inserted into the pET11a expression vector (Novagen/EMD Chemicals, Gibbstown, NJ). The protease sequence was confirmed by DNA sequencing. The NFV resistance mutations, N88S in AE (AE-N88S) and D30N/N88D in clade B (B-D30N/N88D), were generated by site-directed mutagenesis using a Stratagene QuikChange site-directed mutagenesis kit (Agilent Technologies, La Jolla, CA). Mutagenesis was confirmed by sequencing. The Q7K substitution was introduced to all protease variants to prevent autoproteolysis (38).
Protein expression and purification.
The clade B and AE variants were subcloned into the heat-inducible pXC35 expression vector (American Type Culture Collection [ATCC], Manassas, VA) and transformed into E. coli TAP-106 cells. Protein overexpression, purification, and refolding were carried out as previously described (20). Protein used for crystallographic studies was further purified with a Pharmacia Superdex 75 fast-performance liquid chromatography column (GE Healthcare, Chalfont St. Giles, United Kingdom) equilibrated with refolding buffer (50 mM sodium acetate [pH 5.5], 10% glycerol, 5% ethylene glycol, and 5 mM dithiothreitol).
Crystallization and structure determination.
Protease solutions between 1.0 and 2.0 mg ml−1 were equilibrated with a 5-fold molar excess of NFV, darunavir (DRV), and amprenavir (APV) for 1 h on ice. Crystals were grown over a reservoir solution consisting of 126 mM phosphate buffer (pH 6.2), 63 mM sodium citrate, and 18% to 23% ammonium sulfate by the hanging-drop vapor diffusion method. X-ray diffraction data for AE-WT were collected at a BioCARS beamline 14-BM-C at the Advanced Photon Source (Argonne National Laboratory, Argonne, IL) at a wavelength tuned to 0.9 Å with a Quantum 315 CCD X-ray detector (Area Detector Systems Corporation, Poway, CA). Diffraction data for AE-N88S were collected by using beamline X29A at the National Synchrotron Light Source (Brookhaven National Laboratory, Upton, NY) at a wavelength tuned to 1.08 Å with a Quantum 315 charge-coupled-device (CCD) X-ray detector (Area Detector Systems Corporation). Data for the B-D30N/N88D variant was collected in-house with an R-Axis IV imaging plate system (Rigaku Corporation, Tokyo, Japan) mounted on a rotating-anode X-ray source (Rigaku Corporation). All data were collected under cryocooled conditions.
The data were indexed, integrated, and scaled using HKL/HKL-2000 software (HKL Research, Charlottesville, VA) (29). Structure determination and refinement were carried out using the CCP4 program suite (4) as previously described (35). The tensor (T), libration (L), and screw (S) parameter files used in TLS refinement were generated using the TLS motion determination server (30). Model building and real-space refinement were carried out with Coot molecular graphics software (8). Structure comparisons were made by superposing the structures using the Cα atoms of the terminal regions (residues 1 to 9 and 86 to 99) from the two monomers. In the case of the AE complexes, which have multiple orientations for the inhibitor, only the orientation common with the clade B structures was used for analysis. Structures were visualized using PyMol molecular graphics software (6).
Double-difference plots were generated for AE and clade B protease structures to graphically visualize structural differences between the clades, as previously described (35). Briefly, distances between all Cα atoms within the dimer were calculated for each complex. A distance difference matrix was then computed for each atom for a given pair of complexes. The distance difference matrix was then plotted as a contour plot using the gnuplot plotting software (44).
Nomenclature.
The following nomenclature format will be used to refer to each crystal structure: inhibitorprotease variant. Thus, DRV in complex with AE-WT, clade B-WT, AE-N88S, and AE-D30N/N88D protein are designated DRVAE-WT, DRVB-WT, DRVAE-N88S, and DRVB-D30N/N88D, respectively. Prime notation is used to distinguish the two monomers in the protease dimer. For example, residue 30 from the first monomer would be referred to as Asp30, and the same residue from the second monomer would be referred to as Asp30′.
ITC.
Binding affinities and thermodynamic parameters of inhibitor binding to clade B and AE variants were determined by isothermal titration calorimetry (ITC) with a VP isothermal titration calorimeter (MicroCal, LLC, Northampton, MA). The buffer used for all protease and inhibitor solutions consisted of 10 mM sodium acetate (pH 5.0), 2% dimethyl sulfoxide, and 2 mM tris[2-carboxyethyl] phosphine. Binding affinities for all protease variants were obtained by competitive displacement titration using acetyl-pepstatin as the weaker binder. A solution of 30 to 45 μM protease was titrated with 10-μl injections of 200 μM acetyl-pepstatin to saturation. The pepstatin was then displaced by titrating 36 8-μl injections of 200 μM APV or NFV or 41 7-μl injections of 40 μM DRV. Heats of dilution were subtracted from the corresponding heats of reaction to obtain the heat resulting solely from the binding of the ligand to the enzyme. Data were processed and analyzed with the ITC data analysis module (Microcal) for Origin 7 data analysis and graphing software (OriginLab, Northampton, MA). Final results represent the average of at least two measurements.
Measurement of protease activity.
Protease activity was assayed by following each variant's ability to hydrolyze the fluorogenic substrate HiLyte Fluor 488-Lys-Ala-Arg-Val-Leu-Ala-Glu-Ala-Met-Ser-Lys (QXL-520) (AnaSpec, Inc., Fremont, CA) that corresponds to the HIV-1 CA-p2 substrate. The CA-p2 cleavage site was used since it is conserved between HIV-1 clades (7). The assay was carried out in a 96-well plate, and the enzymatic reaction was initiated by adding 20 μl of a solution of 100 to 250 nM protease to 80 μl of substrate solution. The buffer used in all reactions consisted of 10 mM sodium acetate (pH 5.0), 2% dimethyl sulfoxide, and 2 mM tris[2-carboxyethyl]phosphine. Final concentrations in each experiment were 0 to 40 μM substrate and 20 to 50 nM protease. Accurate concentrations of properly folded active protease were determined by carrying out ITC experiments for each variant with acetyl-pepstatin as described in the previous section. Fluorogenic response to protease cleavage was monitored at 23°C using a Victor3 microplate reader (PerkinElmer, Waltham, MA) by exciting the donor molecule at 485 nM and recording emitted light at 535 nM. Data points were acquired every 30 s. The data points in relative fluorescence units (RFU) were converted into concentrations using standard calibration curves generated for HiLyte Fluor 488 at each substrate concentration. In addition to the conversion of RFUs to concentrations, the generation of calibration curves at each substrate concentration allowed us to correct for the inner filter effect (5). Rates of each enzymatic reaction were determined from the linear portion of the data and were fitted against substrate concentrations to determine Km and catalytic turnover rate (kcat) values using VisualEnyzmics enzyme-kinetics software (SoftZymics, Princeton, NJ). Final results for each variant represent the average from at least two experiments.
In order to determine the biochemical fitness of a particular variant in the presence of a given inhibitor, vitality values were calculated using the following equation, based on the vitality function described previously, where Kd is the dissociation constant and kcat/Km is the catalytic efficiency (14, 43).
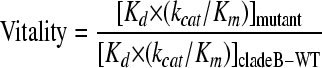 |
The calculated vitality value for B-WT for a particular inhibitor would be 1.0, and vitality values greater than 1.0 would indicate that a given variant had a selective advantage over the same inhibitor, while values lower than 1.0 would indicate that the variant did not have a selective advantage.
RESULTS
Crystal structures.
The AE-WT and NFV-resistant clade B and AE variants were cocrystallized with NFV, DRV, and APV to reveal the structural basis for the altered NFV resistance pathways. In addition, the effects of background polymorphisms in AE-WT on inhibitor binding compared with that of clade B-WT were discerned. Crystals of AE protease in complex with NFV and APV did not diffract to a high resolution; therefore, structural comparisons were carried out for AE and clade B protease in complex with DRV. The structure of DRVB-WT was solved previously in the laboratory and was used for structural comparisons (Protein Data Bank [PDB] code 1T3R). Both DRVAE-WT and DRVAE-N88S crystallized with DRV bound in two orientations in the active site. Crystallographic data and refinement statistics for DRVAE-WT, DRVB-WT, DRVAE-N88S, and DRVB-D30N/N88D are given in Table 1.
TABLE 1.
Crystallographic statistics
| Parametera | Result for indicated variant |
|||
|---|---|---|---|---|
| DRVB-WTb | DRVB-D30N/N88D | DRVAE-WT | DRVAE-N88S | |
| Inhibitor | DRV | DRV | DRV | DRV |
| Resolution (Å) | 1.2 | 2.15 | 1.96 | 1.76 |
| Space group | P 21 21 21 | P 21 21 21 | P 61 | P 61 |
| Z | 4 | 4 | 6 | 6 |
| Cell dimensions (Å) | ||||
| a | 54.9 | 50.9 | 62.2 | 61.9 |
| b | 57.8 | 57.7 | ||
| c | 62.0 | 61.6 | 82.7 | 82.1 |
| Total no. of reflections | 302,022 | 108,838 | 89,284 | 79,445 |
| No. of unique reflections | 55,056 | 10,326 | 12,493 | 17,277 |
| Rsymm (%) | 3.8 | 6.7 | 5.5 | 4.6 |
| Completeness (%) | 95.5 | 99.6 | 93.9 | 97.4 |
| I/σI | 25.0 | 9.6 | 11.2 | 19.6 |
| Rwork (%) | 14.1 | 18.1 | 20.0 | 19.6 |
| Rfree (%) | 17.9 | 23.6 | 25.9 | 23.9 |
| RMSD | ||||
| Bond length (Å) | 0.004 | 0.009 | 0.009 | 0.007 |
| Bond angle | 1.5 | 1.9 | 1.5 | 1.7 |
| PDB code | 1T3R | 3LZV | 3LZS | 3LZU |
Structural comparisons were carried out for AE and clade B DRV complexes by pairwise structural superposition and double-difference plots (Fig. 1B to E). The DRVAE-WT and DRVAE-N88S complexes were structurally similar (Fig. 1B and C). Although the DRVAE-WT and DRVB-WT could be superimposed on each other very well (root mean square deviation [RMSD] of 0.21 Å), there were clear and significant differences between the variants in the main chain at the flap hinge region (residues 33 to 39) and the protease core region (residues 16 to 22) (Fig. 1D and Fig. 2A to D). These differences were further evident by the presence of significant peaks in the double-difference plot (Fig. 1E). The Ile36 side chain in DRVAE-WT packs well against the core region through favorable van der Waals interactions and is shorter than the Met36 in DRVB-WT (Fig. 2C). In addition, the shorter Asp35 in DRVAE-WT further enhances the packing by being flipped inward against the core, while in DRVB-WT, the longer Glu35 is flipped outward into the solvent and forms a salt bridge with Arg57 (Fig. 2D). The packing of the flap hinge and core regions in DRVAE-WT is further stabilized by a hydrogen bond between the carbonyl oxygen of Asp35 and Lys20 NZ atom and is not present in DRVB-WT.
FIG. 2.

(A) Ribbon diagram superposition of DRVAE-WT (blue) and DRVB-WT (gray). The red box indicates the region of the protease molecule highlighted in panels B to D. (B) Structural rearrangement of the flap hinge and core regions between DRVAE-WT (blue) and DRVB-WT (gray). (C) Flap hinge and core regions of DRVAE-WT. (D) Flap hinge and core regions of DRVB-WT protease. Hydrogen bond interactions are indicated by red dashed lines.
The Asp30′ side chain of DRVB-WT does not directly form a hydrogen bond with DRV but indirectly interacts with the N1 atom of DRV through a water molecule-mediated hydrogen bond network (Fig. 3A). In contrast, the Asp30′ side chain of DRVAE-WT forms a direct hydrogen bond with the N1 atom of DRV (Fig. 3B). Residue 30 of both NFV-resistant variants also interacts with the N1 atom of DRV through water molecule-mediated hydrogen bonding (Fig. 3C and D). However, in addition to this interaction, Asn30 of DRVB-D30N/N88D and Asp30 of DRVAE-N88S are oriented away from the active site, enabling them to form hydrogen bonds with Asp88 and Ser88, respectively. In both cases, the NFV resistance mutations stabilize residue 30 away from the active site via hydrogen bonding.
FIG. 3.

Protease inhibitor hydrogen bonding interactions. DRV is shown in orange, and hydrogen bond interactions are indicated by red dashed lines. Since the charged states of the Asp25 carboxyl groups and the position of the O18 hydroxyl hydrogen of DRV are not known, all possible hydrogen bond interactions between the Asp25 carboxyl groups and O18 of the DRV molecules are shown. (A) DRVB-WT (gray). (B) DRVAE-WT (blue). (C) DRVB-D30N/N88D (salmon). (D) DRVAE-N88S (green).
Binding thermodynamics.
To determine the effects of background sequence polymorphisms and NFV resistance mutations on inhibitor binding, the binding thermodynamic parameters of NFV, DRV, and APV binding to WT and resistant AE and clade B variants were determined by isothermal titration calorimetry (Table 2). The AE-WT protease had a 6.9-fold-weaker affinity for NFV and a 2.7-fold-weaker affinity for DRV than the affinities of B-WT protease for NFV and DRV, respectively (Table 2). This result indicates that the AE-WT protease has an inherently weaker affinity for NFV and DRV.
TABLE 2.
Binding thermodynamic parameters for NFV, DRV, and APV binding to AE and clade B variantsa
| Inhibitor and protease variant | Ka (M−1) | Kd (nM) | Kd ratio | ΔH (kcal mol−1) | ΔΔH | −TΔS (kcal mol−1) | Δ(−TΔS) | ΔG (kcal mol−1) | ΔΔG |
|---|---|---|---|---|---|---|---|---|---|
| NFV | |||||||||
| B-WT | (2.6 ± 0.5) × 109 | 0.39 ± 0.07 | 1.0 | 4.4 ± 0.1 | −17.0 | −12.6 ± 0.1 | |||
| B-D30N/N88D | (1.2 ± 0.4) × 108 | 8.1 ± 2.8 | 20.7 | 6.7 ± 0.3 | 2.3 | −17.5 | 0.5 | −10.8 ± 0.2 | 1.8 |
| AE-WT | (3.7 ± 1.0) × 108 | 2.7 ± 0.7 | 6.9 | 5.0 ± 0.3 | 0.6 | −16.6 | 0.9 | −11.5 ± 0.2 | 1.1 |
| AE-N88S | (5.8 ± 1.2) × 107 | 17.2 ± 3.5 | 44.1 | 6.2 ± 0.7 | 1.8 | −16.6 | 0.9 | −10.4 ± 0.1 | 2.2 |
| DRV | |||||||||
| B-WT | (2.2 ± 1.1) × 1011 | 0.004 ± 0.002 | 1.0 | −12.1 ± 0.9 | −3.1 | −15.2 ± 0.3 | |||
| B-D30N/N88D | (3.7 ± 0.7) × 1010 | 0.026 ± 0.005 | 6.5 | −12.5 ± 0.4 | −0.4 | −1.6 | 1.5 | −14.2 ± 0.1 | 1.0 |
| AE-WT | (9.1 ± 0.3) × 1010 | 0.0109 ± 0.0003 | 2.7 | −10.1 ± 0.5 | 2.0 | −4.6 | −1.5 | −14.7 ± 0.02 | 0.5 |
| AE-N88S | (1.1 ± 0.8) × 1010 | 0.087 ± 0.062 | 21.8 | −5.1 ± 3.6 | 7.0 | −8.4 | −5.3 | −13.5 ± 0.4 | 1.7 |
| APV | |||||||||
| B-WT | (2.6 ± 1.3) × 109 | 0.39 ± 0.20 | 1.0 | −7.3 ± 0.9 | −5.3 | −12.6 ± 0.3 | |||
| B-D30N/N88D | (1.2 ± 0.2) × 1010 | 0.08 ± 0.01 | 0.2 | −10.2 ± 1.5 | −2.9 | −3.3 | 2.0 | −13.5 ± 0.09 | −0.9 |
| AE-WT | (3.1 ± 0.2) × 109 | 0.32 ± 0.02 | 0.8 | −5.5 ± 0.3 | −1.8 | −7.3 | −2.0 | −12.70 ± 0.03 | −0.1 |
| AE-N88S | (1.3 ± 0.9) × 1010 | 0.08 ± 0.06 | 0.2 | −5.0 ± 3.6 | 2.3 | −8.6 | −3.3 | −13.6 ± 0.4 | −1.0 |
Ka, association constant; Kd, dissociation constant; H, enthalpy; T, temperature; S, entropy; G, Gibbs free energy.
No significant differences in the enthalpy of NFV binding were observed among any of the variants. Although the binding of DRV to all protease variants was enthalpically favorable, the enthalpic contributions were reduced with the AE variants (−10.1 kcal mol−1 for AE-WT and −5.1 kcal mol−1 for AE-N88S) compared with those for the clade B variants (−12.1 kcal mol−1 for B-WT and −12.5 kcal mol−1 for B-D30N/N88D). As expected, the NFV-resistant variants showed a significant reduction in binding affinity for NFV compared to that of the wild-type variants. With the AE-N88S variant, the affinity for NFV was reduced 44.1-fold (Kd = 17.2 nM) and was far more significant than the D30N/N88D mutations in clade B protease, which reduced the affinity for NFV 20.7-fold (Kd = 8.1 nM). Similarly, the AE-N88S variant had a 21.8-fold-weaker affinity (Kd = 0.087 nM) for DRV compared to a 6.5-fold-weaker affinity (Kd = 0.026 nM) with the B-D30N/N88D variant. Thus, the single N88S substitution in the AE protease has a profound effect on the binding of NFV and DRV.
In contrast to NFV and DRV, clade-specific sequence differences and NFV resistance mutations had only a minimal effect on the affinities for APV of both AE and clade B protease. Despite this, there were some differences in energy parameters. The binding of APV to the clade B variants appeared to be more enthalpically favorable than that to the AE variants. This was compensated for by an increase in the entropic component to the binding energy for the AE proteases.
Protease activity and vitality.
The enzyme-kinetic parameters determined for each clade B and AE variant with the CA-p2 fluorogenic substrate analog are summarized in Table 3. The Km value for B-D30N/N88D protease (35.9 μM) was 2.1-fold greater than that for B-WT protease (16.7 μM). However, the Km values for the AE protease variants (17.5 μM for AE-WT and 19.0 μM for AE-N88S) were similar to that of B-WT protease. The turnover rate for B-D30N/N88D protease (kcat = 0.13 s−1) was significantly lower than that of B-WT protease (kcat = 1.79 s−1). Turnover rates for AE-WT (kcat = 0.7 s−1) and AE-N88S (kcat = 0.2 s−1) were 2.5 and 8.5-fold lower, respectively, than that of clade B-WT. The kcat/Km values, or catalytic efficiency values, for B-D30N/N88D and AE variants were lower than that of B-WT protease. Therefore, the reduction in catalytic efficiency of the B-D30N/N88D protease compared with that of B-WT protease resulted from the combined effects of the Km and kcat values. However, for the AE variants, the lower turnover rates alone were responsible for the reduced catalytic efficiencies. Overall, these results indicate that the polymorphic sequence differences in AE protease can alter the activity profile of the enzyme compared to results with the clade B protease.
TABLE 3.
Enzyme kinetics parameters for clade B and AE-WT and NFV-resistant variants
| Parameter | Result for indicated variant |
|||
|---|---|---|---|---|
| B-WT | B-D30N/N88D | AE-WT | AE-N88S | |
| Km (μM) | 16.7 ± 6.0 | 35.9 ± 0.1 | 17.5 ± 4.0 | 19.0 ± 0.8 |
| kcat (s−1) | 1.79 ± 0.28 | 0.13 ± 0.09 | 0.70 ± 0.08 | 0.20 ± 0.02 |
| kcat/Km (s−1 μM−1) | 0.11 ± 0.04 | 0.004 ± 0.002 | 0.04 ± 0.01 | 0.010 ± 0.001 |
Vitality values were calculated to determine if the protease variants had a selective advantage over NFV, DRV, and APV. AE-WT and AE-N88S protease had calculated vitality values of 2.52 and 4.01 for NFV, respectively, compared with 0.76 for B-WT (Table 4). However, vitality values for DRV were not significantly different from that of B-WT protease. Vitality values for APV were significantly lower for all variants than for B-WT protease. These results indicate that AE-WT may have a selective advantage over NFV compared to B-WT but that the AE variants may not have a significant selective advantage against DRV or APV relative to B-WT.
TABLE 4.
Vitality values for clade B and AE WT and NFV-resistant variants
| Inhibitor | Result for indicated variant |
||
|---|---|---|---|
| B-D30N/N88D | AE-WT | AE-N88S | |
| NFV | 0.76 | 2.52 | 4.01 |
| DRV | 0.24 | 0.99 | 1.98 |
| APV | 0.01 | 0.30 | 0.02 |
DISCUSSION
Although the majority of HIV-1 patients are infected with non-B forms of the virus, molecular studies have been carried out predominantly with clade B variants. The AE protease has several polymorphisms that are associated with inhibitor resistance in clade B. AE also shows altered patterns of drug resistance to NFV. We have performed detailed studies to determine the effects of sequence polymorphisms on enzyme structure, activity, and inhibitor binding. These analyses led to a structural rationalization for the altered pathways for drug resistance.
AE-WT protease has an inherently weaker affinity for NFV and DRV than that of B-WT, as is evident from the thermodynamic data (Table 2). The weaker affinity observed for NFV is consistent with previously published data for another AE protease variant (3), as well as for clade A protease (42), which is closely related. The inherent weaker affinity for NFV likely allows the AE protease to gain resistance to NFV through a single nonactive-site substitution, N88S. The clade B protease, in contrast, which has a relatively stronger affinity for NFV, requires a combination of an active-site mutation (D30N) and a nonactive-site mutation (N88D) to gain NFV resistance. The ability of the AE-N88S protease to maintain affinity for substrates is evident from our enzyme kinetics data (Table 4), in which the Km value for AE-N88S was comparable to that of AE-WT and B-WT protease. The Km value for clade B-D30N/N88D, on the other hand, was significantly worse than that of the B-WT, likely reflecting the effect of the altered active site.
As an active-site residue, Asp30 plays a key role in substrate recognition by interacting with substrates through side chain-mediated hydrogen bonds with the MA-CA, CA-p2, p1-p6, and p2-NC cleavage sites (36). Therefore, as is evident from our enzyme kinetics data, the D30N/N88D mutations in clade B will likely affect substrate binding and processing. Several studies have observed substrate coevolution in instances in which the protease mutates active-site residues in order to confer inhibitor resistance (22, 23). However, since the AE-N88S protease variant has no active-site mutations, the enzyme retains the ability to effectively recognize substrates while conferring NFV resistance. Therefore, the presence of the N88S substitution in AE protease is unlikely to induce coevolution of the viral substrates in order to maintain effective enzymatic activity.
Despite having Km values that were comparable to that of B-WT protease, both AE-WT and AE-N88S had significantly lower catalytic turnover rates (kcat) than that of the B-WT protease (Table 3). As a result, the catalytic efficiency of the AE variants is lower than that of the B-WT protease. The lower turnover rates of the AE variants could be a direct result of the reduced flexibility of the flap hinge (residues 33 to 39) and core regions (residues 16 to 22) of the protein. Molecular dynamics studies have revealed that hydrophobic sliding of the core region facilitates substrate binding through the opening of the active site (9). The unique hydrogen bonds observed between the flap hinge and the core in the AE variants alter movement of the core, thus impacting the ability of the active site to open up for substrate binding and product release. Based on our enzyme kinetics data, this altered flexibility of the flap hinges in the AE variants has little effect on substrate binding but rather affects the catalytic step of the reaction by slowing down product release.
The higher vitality value observed for AE-WT with NFV provides supporting evidence for the reduction in the efficacy of NFV against the AE protease compared with that of clade B (Table 4). This result is consistent with previous vitality calculations for the clade A protease (42). In addition, these results further highlight the idea that background polymorphic sequence variations in the AE protease can affect the potency of NFV. The suboptimal efficacy of NFV against the AE-WT protease likely permits a nonactive-site variant, AE-N88S, to emerge over variants with active-site mutations to effectively confer resistance to NFV.
The impact on other inhibitors, however, is complex. APV and DRV are chemically very closely related compounds, and similar susceptibility and resistance patterns have been observed for these two inhibitors (31). However, this pattern is not evident for this series of resistant variants. Both the N88S mutation in the AE and the D30N/N88D mutations in the clade B proteases result in hypersusceptibility to APV. Similar results have been observed also for a B-N88S protease variant (24, 45). In contrast, the same substitutions in the protease give rise to even greater resistance to DRV. However, since DRV presents a greater genetic barrier to resistance than APV (33), the in vivo implications of weaker affinity for DRV in the AE variants are likely negligible. Indeed, our calculated vitality values indicate that DRV maintains its potency against the AE variants despite having a weaker affinity for AE-WT and AE-N88S relative to clade B protease.
A close look at the NFVB-WT protease complex reveals an important interaction between the Asp30 residue side chain and the inhibitor bound in the active site. (PDB code 3EKX) (Fig. 4A and B). One of the side chain oxygen atoms of Asp30 forms a direct hydrogen bond with the O38 atom of NFV. Our crystal structures of the NFV-resistant variants show that N88S in AE and N88D in clade B have the ability to interact with residue 30 and orient it away from the active site (Fig. 3B and D) and thereby disrupt the interaction between residue 30 and the inhibitor. These structural observations are similar to interpretations made in previous molecular dynamics studies involving NFV-protease complexes (27, 28). Thus, NFV resistance is likely caused in large part due to the loss of this interaction in the NFV-resistant variants.
FIG. 4.

Hydrogen bond network involving residue 88. (A) Asn88 bridges the terminal helix with Asp30 from the active site and Thr74 from one of the outer beta strands. The red box indicates the region of the protease molecule highlighted in panels B to D. (B) Asn88 in NFVB-WT (PDB code 3EKX). (C) Asp88 in DRVB-D30N/N88D. (D) Ser88 in DRVN88S-WT. Hydrogen bond interactions are indicated by red dashed lines.
Overall, mutations that emerge in response to inhibitor therapy need to have a minimal impact on protease structure and activity to maintain the enzyme's function. The D30N substitution, which is associated with NFV resistance, is one of the few drug-resistant mutations that involve a change in charge. The additional substitution of N88D likely helps preserve the net charge on the protein. In AE, resistance to NFV occurs indirectly with the N88S mutation. Likewise, the sole NFV-resistant alteration, N88S, in the AE protease does not change the overall electrostatics. Thus, in both clade B and AE, NFV resistance is attained with no change to the net charge of the enzyme. In the wild-type variants, Asn88 is one of the few internal hydrogen bonding side chains in the core of the protease monomer. The side chain of Asn88 has a key role in the protease structure bridging the terminal helix, with residues 30 and 31 coming from the active site to the backbone of Thr74 in the center of one of the outer beta strands (Fig. 4A and B). With the substitutions of Asp in clade B and Ser in AE for Asn at position 88 in the NFV-resistant protease variants, the hydrogen bonding network is preserved through the coordination of some key water molecules in the core of the protease monomer (Fig. 4B to D). Thus, mutations confer resistance to NFV through a series of interdependent changes that preserve the structural and electrostatic properties of HIV-1 protease.
In conclusion, protease activity and the response to protease inhibitors can be affected by clade-specific sequence differences. Our findings likely extend beyond HIV-1 protease to other drug targets within HIV and underscore the need to consider clade-specific polymorphisms when developing new drugs and formulating treatment plans. Furthermore, drug resistance pathways observed in the context of clade B viruses cannot be assumed to hold true for other HIV-1 clades.
Acknowledgments
This work was supported by grants from the National Institutes of Health (P01-GM66524) and Tibotec, Inc., to C.A.S. Additionally, this study was supported by a Grant-in-Aid for AIDS research from the Ministry of Health, Labor, and Welfare of Japan (H19-AIDS-007) to W.S.
We thank William Royer, Moses Prabu-Jayabalan, and Madhavi Nalam for helpful discussions and Christina Ng and Brendan Hilbert for assistance with data collection.
We gratefully acknowledge the Mail-In Data Collection Program of the National Synchrotron Light Source, Brookhaven National Laboratory, for collecting X-ray data at the X29A beamline, for which financial support comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the U.S. Department of Energy and from the National Center for Research Resources of the National Institutes of Health. Use of the Advanced Photon Source for X-ray data collection was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract DE-AC02-06CH11357. Use of the BioCARS Sector 14 was supported by the National Institutes of Health, National Center for Research Resources, under grant RR007707.
The protease inhibitors used in this study were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH.
Footnotes
Published ahead of print on 21 July 2010.
REFERENCES
- 1.Ariyoshi, K., M. Matsuda, H. Miura, S. Tateishi, K. Yamada, and W. Sugiura. 2003. Patterns of point mutations associated with antiretroviral drug treatment failure in CRF01_AE (subtype E) infection differ from subtype B infection. J. Acquir. Immune Defic. Syndr. 33:336-342. [DOI] [PubMed] [Google Scholar]
- 2.Carr, J. K., M. O. Salminen, C. Koch, D. Gotte, A. W. Artenstein, P. A. Hegerich, D. St. Louis, D. S. Burke, and F. E. McCutchan. 1996. Full-length sequence and mosaic structure of a human immunodeficiency virus type 1 isolate from Thailand. J. Virol. 70:5935-5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clemente, J. C., R. M. Coman, M. M. Thiaville, L. K. Janka, J. A. Jeung, S. Nukoolkarn, L. Govindasamy, M. Agbandje-McKenna, R. McKenna, W. Leelamanit, M. M. Goodenow, and B. M. Dunn. 2006. Analysis of HIV-1 CRF_01 A/E protease inhibitor resistance: structural determinants for maintaining sensitivity and developing resistance to atazanavir. Biochemistry 45:5468-5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collaborative Computational Project, Number 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760-763. [DOI] [PubMed] [Google Scholar]
- 5.Copeland, R. A. 1996. Enzymes: a practical introduction to structure, mechanism, and data analysis. Wiley-VCH, New York, NY.
- 6.DeLano, W. L. 2002. The PyMol molecular graphics system. DeLano Scientific, San Carlos, CA.
- 7.de Oliveira, T., S. Engelbrecht, E. Janse van Rensburg, M. Gordon, K. Bishop, J. zur Megede, S. W. Barnett, and S. Cassol. 2003. Variability at human immunodeficiency virus type 1 subtype C protease cleavage sites: an indication of viral fitness? J. Virol. 77:9422-9430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emsley, P., and K. Cowtan. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126-2132. [DOI] [PubMed] [Google Scholar]
- 9.Foulkes-Murzycki, J. E., W. R. Scott, and C. A. Schiffer. 2007. Hydrophobic sliding: a possible mechanism for drug resistance in human immunodeficiency virus type 1 protease. Structure 15:225-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao, F., D. L. Robertson, S. G. Morrison, H. Hui, S. Craig, J. Decker, P. N. Fultz, M. Girard, G. M. Shaw, B. H. Hahn, and P. M. Sharp. 1996. The heterosexual human immunodeficiency virus type 1 epidemic in Thailand is caused by an intersubtype (A/E) recombinant of African origin. J. Virol. 70:7013-7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geretti, A. M. 2006. HIV-1 subtypes: epidemiology and significance for HIV management. Curr. Opin. Infect. Dis. 19:1-7. [DOI] [PubMed] [Google Scholar]
- 12.Gomes, P., I. Diogo, M. F. Gonçalves, P. Carvalho, J. Cabanas, M. C. Lobo, and R. Camacho. 2002. Different pathways to nelfinavir genotypic resistance in HIV-1 subtypes B and G. Conference on Retroviruses and Opportunistic Infections, abstract 46. Conference on Retroviruses and Opportunistic Infections, Seattle, WA.
- 13.Grossman, Z., E. E. Paxinos, D. Averbuch, S. Maayan, N. T. Parkin, D. Engelhard, M. Lorber, V. Istomin, Y. Shaked, E. Mendelson, D. Ram, C. J. Petropoulos, and J. M. Schapiro. 2004. Mutation D30N is not preferentially selected by human immunodeficiency virus type 1 subtype C in the development of resistance to nelfinavir. Antimicrob. Agents Chemother. 48:2159-2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gulnik, S. V., L. I. Suvorov, B. Liu, B. Yu, B. Anderson, H. Mitsuya, and J. W. Erickson. 1995. Kinetic characterization and cross-resistance patterns of HIV-1 protease mutants selected under drug pressure. Biochemistry 34:9282-9287. [DOI] [PubMed] [Google Scholar]
- 15.Hemelaar, J., E. Gouws, P. D. Ghys, and S. Osmanov. 2006. Global and regional distribution of HIV-1 genetic subtypes and recombinants in 2004. AIDS 20:W13-23. [DOI] [PubMed] [Google Scholar]
- 16.Hu, D. J., A. Buve, J. Baggs, G. van der Groen, and T. J. Dondero. 1999. What role does HIV-1 subtype play in transmission and pathogenesis?: an epidemiological perspective. AIDS 13:873-881. [DOI] [PubMed] [Google Scholar]
- 17.Kaleebu, P., N. French, C. Mahe, D. Yirrell, C. Watera, F. Lyagoba, J. Nakiyingi, A. Rutebemberwa, D. Morgan, J. Weber, C. Gilks, and J. Whitworth. 2002. Effect of human immunodeficiency virus (HIV) type 1 envelope subtypes A and D on disease progression in a large cohort of HIV-1-positive persons in Uganda. J. Infect. Dis. 185:1244-1250. [DOI] [PubMed] [Google Scholar]
- 18.Kanki, P. J., D. J. Hamel, J. L. Sankale, C. Hsieh, I. Thior, F. Barin, S. A. Woodcock, A. Gueye-Ndiaye, E. Zhang, M. Montano, T. Siby, R. Marlink, I. NDoi, M. E. Essex, and S. MBoup. 1999. Human immunodeficiency virus type 1 subtypes differ in disease progression. J. Infect. Dis. 179:68-73. [DOI] [PubMed] [Google Scholar]
- 19.Kantor, R., D. A. Katzenstein, B. Efron, A. P. Carvalho, B. Wynhoven, P. Cane, J. Clarke, S. Sirivichayakul, M. A. Soares, J. Snoeck, C. Pillay, H. Rudich, R. Rodrigues, A. Holguin, K. Ariyoshi, M. B. Bouzas, P. Cahn, W. Sugiura, V. Soriano, L. F. Brigido, Z. Grossman, L. Morris, A. M. Vandamme, A. Tanuri, P. Phanuphak, J. N. Weber, D. Pillay, P. R. Harrigan, R. Camacho, J. M. Schapiro, and R. W. Shafer. 2005. Impact of HIV-1 subtype and antiretroviral therapy on protease and reverse transcriptase genotype: results of a global collaboration. PLoS Med. 2:e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.King, N. M., L. Melnick, M. Prabu-Jeyabalan, E. A. Nalivaika, S.-S. Yang, Y. Gao, X. Nie, C. Zepp, D. L. Heefner, and C. A. Schiffer. 2002. Lack of synergy for inhibitors targeting a multi-drug-resistant HIV-1 protease. Protein Sci. 11:418-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.King, N. M., M. Prabu-Jeyabalan, P. Wigerinck, M.-P. de Béthune, and C. A. Schiffer. 2004. Structural and thermodynamic basis for the binding of TMC114, a next-generation human immunodeficiency virus type 1 protease inhibitor. J. Virol. 78:12012-12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolli, M., S. Lastere, and C. A. Schiffer. 2006. Co-evolution of nelfinavir-resistant HIV-1 protease and the p1-p6 substrate. Virology 347:405-409. [DOI] [PubMed] [Google Scholar]
- 23.Kolli, M., E. Stawiski, C. Chappey, and C. A. Schiffer. 2009. Human immunodeficiency virus type 1 protease-correlated cleavage site mutations enhance inhibitor resistance. J. Virol. 83:11027-11042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masquelier, B., K. L. Assoumou, D. Descamps, L. Bocket, J. Cottalorda, A. Ruffault, A. G. Marcelin, L. Morand-Joubert, C. Tamalet, C. Charpentier, G. Peytavin, Z. Antoun, F. Brun-Vezinet, and D. Costagliola. 2008. Clinically validated mutation scores for HIV-1 resistance to fosamprenavir/ritonavir. J. Antimicrob. Chemother. 61:1362-1368. [DOI] [PubMed] [Google Scholar]
- 25.Murphy, E., B. Korber, M. C. Georges-Courbot, B. You, A. Pinter, D. Cook, M. P. Kieny, A. Georges, C. Mathiot, F. Barre-Sinoussi, et al. 1993. Diversity of V3 region sequences of human immunodeficiency viruses type 1 from the Central African Republic. AIDS Res. Hum. Retroviruses 9:997-1006. [DOI] [PubMed] [Google Scholar]
- 26.Nunez, M., C. de Mendoza, L. Valer, E. Casas, S. Lopez-Calvo, A. Castro, B. Roson, D. Podzamczer, A. Rubio, J. Berenguer, and V. Soriano. 2002. Resistance mutations in HIV-infected patients experiencing early failure with nelfinavir-containing triple combinations. Med. Sci. Monit 8:CR620-CR623. [PubMed] [Google Scholar]
- 27.Ode, H., S. Matsuyama, M. Hata, T. Hoshino, J. Kakizawa, and W. Sugiura. 2007. Mechanism of drug resistance due to N88S in CRF01_AE HIV-1 protease, analyzed by molecular dynamics simulations. J. Med. Chem. 50:1768-1777. [DOI] [PubMed] [Google Scholar]
- 28.Ode, H., M. Ota, S. Neya, M. Hata, W. Sugiura, and T. Hoshino. 2005. Resistant mechanism against nelfinavir of human immunodeficiency virus type 1 proteases. J. Phys. Chem. B 109:565-574. [DOI] [PubMed] [Google Scholar]
- 29.Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307-326. [DOI] [PubMed] [Google Scholar]
- 30.Painter, J., and E. A. Merritt. 2006. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 62:439-450. [DOI] [PubMed] [Google Scholar]
- 31.Parkin, N., E. Stawiski, C. Chappey, and E. Coakley. 2007. Darunavir/amprenavir cross-resistance in clinical samples submitted for phenotype/genotype combination resistance testing. Conference on Retroviruses and Opportunistic Infections, abstract 607. Conference on Retroviruses and Opportunistic Infections, San Francisco, CA.
- 32.Plantier, J. C., M. Leoz, J. E. Dickerson, F. De Oliveira, F. Cordonnier, V. Lemee, F. Damond, D. L. Robertson, and F. Simon. 2009. A new human immunodeficiency virus derived from gorillas. Nat. Med. 15:871-872. [DOI] [PubMed] [Google Scholar]
- 33.Poveda, E., C. de Mendoza, L. Martin-Carbonero, A. Corral, V. Briz, J. Gonzalez-Lahoz, and V. Soriano. 2007. Prevalence of darunavir resistance mutations in HIV-1-infected patients failing other protease inhibitors. J. Antimicrob. Chemother. 60:885-888. [DOI] [PubMed] [Google Scholar]
- 34.Prabu-Jeyabalan, M., E. Nalivaika, N. M. King, and C. A. Schiffer. 2004. Structural basis for coevolution of the human immunodeficiency virus type 1 nucleocapsid-p1 cleavage site with a V82A drug-resistant mutation in viral protease. J. Virol. 78:12446-12454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prabu-Jeyabalan, M., E. A. Nalivaika, K. Romano, and C. A. Schiffer. 2006. Mechanism of substrate recognition by drug-resistant human immunodeficiency virus type 1 protease variants revealed by a novel structural intermediate. J. Virol. 80:3607-3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prabu-Jeyabalan, M., E. A. Nalivaika, and C. A. Schiffer. 2002. Substrate shape determines specificity of recognition for HIV-1 protease: analysis of crystal structures of six substrate complexes. Structure 10:369-381. [DOI] [PubMed] [Google Scholar]
- 37.Robertson, D. L., J. P. Anderson, J. A. Bradac, J. K. Carr, B. Foley, R. K. Funkhouser, F. Gao, B. H. Hahn, M. L. Kalish, C. Kuiken, G. H. Learn, T. Leitner, F. McCutchan, S. Osmanov, M. Peeters, D. Pieniazek, M. Salminen, P. M. Sharp, S. Wolinsky, and B. Korber. 2000. HIV-1 nomenclature proposal. Science 288:55-56. [DOI] [PubMed] [Google Scholar]
- 38.Rose, J. R., R. Salto, and C. S. Craik. 1993. Regulation of autoproteolysis of the HIV-1 and HIV-2 proteases with engineered amino acid substitutions. J. Biol. Chem. 268:11939-11945. [PubMed] [Google Scholar]
- 39.Spira, S., M. A. Wainberg, H. Loemba, D. Turner, and B. G. Brenner. 2003. Impact of clade diversity on HIV-1 virulence, antiretroviral drug sensitivity and drug resistance. J. Antimicrob. Chemother. 51:229-240. [DOI] [PubMed] [Google Scholar]
- 40.Surleraux, D. L., A. Tahri, W. G. Verschueren, G. M. Pille, H. A. de Kock, T. H. Jonckers, A. Peeters, S. De Meyer, H. Azijn, R. Pauwels, M. P. de Bethune, N. M. King, M. Prabu-Jeyabalan, C. A. Schiffer, and P. B. Wigerinck. 2005. Discovery and selection of TMC114, a next generation HIV-1 protease inhibitor. J. Med. Chem. 48:1813-1822. [DOI] [PubMed] [Google Scholar]
- 41.Tebit, D. M., I. Nankya, E. J. Arts, and Y. Gao. 2007. HIV diversity, recombination and disease progression: how does fitness “fit” into the puzzle? AIDS Rev. 9:75-87. [PubMed] [Google Scholar]
- 42.Velazquez-Campoy, A., M. J. Todd, S. Vega, and E. Freire. 2001. Catalytic efficiency and vitality of HIV-1 proteases from African viral subtypes. Proc. Natl. Acad. Sci. U. S. A. 98:6062-6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Velazquez-Campoy, A., S. Vega, E. Fleming, U. Bacha, Y. Sayed, H. W. Dirr, and E. Freire. 2003. Protease inhibition in African subtypes of HIV-1. AIDS Rev. 5:165-171. [PubMed] [Google Scholar]
- 44.Williams, T., and C. Kelley. Accessed 28 January 2009. gnuplot, version 4.2. http://www.gnuplot.info/.
- 45.Ziermann, R., K. Limoli, K. Das, E. Arnold, C. J. Petropoulos, and N. T. Parkin. 2000. A mutation in human immunodeficiency virus type 1 protease, N88S, that causes in vitro hypersensitivity to amprenavir. J. Virol. 74:4414-4419. [DOI] [PMC free article] [PubMed] [Google Scholar]