

Abstract
Autophagy is regulated for IFN-γ-mediated antimicrobial efficacy; however, its molecular effects for IFN-γ signaling are largely unknown. Here, we show that autophagy facilitates IFN-γ-activated Jak2-STAT1. IFN-γ induces autophagy in wild-type but not in autophagy protein 5 (Atg5−/−)-deficient mouse embryonic fibroblasts (MEFs), and, autophagy-dependently, IFN-γ induces IFN regulatory factor 1 and cellular inflammatory responses. Pharmacologically inhibiting autophagy using 3-methyladenine, a known inhibitor of class III phosphatidylinositol 3-kinase, confirms these effects. Either Atg5−/− or Atg7−/− MEFs are, independent of changes in IFN-γ receptor expression, resistant to IFN-γ-activated Jak2-STAT1, which suggests that autophagy is important for IFN-γ signal transduction. Lentivirus-based short hairpin RNA for Atg5 knockdown confirmed the importance of autophagy for IFN-γ-activated STAT1. Without autophagy, reactive oxygen species increase and cause SHP2 (Src homology-2 domain-containing phosphatase 2)-regulated STAT1 inactivation. Inhibiting SHP2 reversed both cellular inflammation and the IFN-γ-induced activation of STAT1 in Atg5−/− MEFs. Our study provides evidence that there is a link between autophagy and both IFN-γ signaling and cellular inflammation and that autophagy, because it inhibits the expression of reactive oxygen species and SHP2, is pivotal for Jak2-STAT1 activation.
Keywords: Autophagy, Inflammation, Interferon, Jak Kinase, Phosphatase, Reactive Oxygen Species (ROS), Signal Transduction, STAT Transcription Factor
Introduction
Autophagy, or autophagocytosis, is required for cellular regulation in response to a variety of stimuli, including starvation, pathogen-associated molecular patterns that are recognized by pattern-recognition receptors such as Toll-like receptors, and cytokines such as tumor necrosis factor (TNF)-α and interferon (IFN)-γ (1, 2). In addition to maintaining cell survival and metabolic homeostasis (3), autophagy provides a cell-autonomous defense system for recognizing viral infections (4, 5) and eliminating intracellular pathogens via the autophagosome-lysosome pathway (6–8).
Proinflammatory cytokine IFN-γ, a type II IFN produced by T cells and natural killer cells, is involved in promoting diverse bioactivities, including antigen processing, intracellular microbial killing, and proinflammation (9). After binding with IFN-γ receptors (IFNGRs),2 IFN-γ typically activates Jak2-STAT1 signaling and then regulates its bioactivities. For Jak2-STAT1 activation, Jak2 is first autophosphorylated at its tyrosine residues (Tyr1007/Tyr1008) and then leads to Jak1 transphosphorylation (Tyr1022/Tyr1023). The activation of Jak1 then phosphorylates IFNGR1 (Tyr440), which induces the recruitment and activation of STAT1 through Jak2-mediated phosphorylation (Tyr701). SOCS1 (suppressor of cytokine signaling-1), SOCS3, and SHP2 (dual-phosphatase Src homology-2 domain-containing phosphatase) provide feedback regulation by suppressing Jak2-STAT1 signaling (9, 10). SOCS1 and SOCS3 interact with IFNGRs, and SHP2 causes the dephosphorylation of Jak2 and STAT1. IFN-γ induces, STAT1-dependently, SOCS1 and SOCS3 expression; however, the mechanisms for SHP2 activation remain undocumented.
IFN-γ uses a process that involves autophagy to increase the eradication of intracellular mycobacteria and chlamydia (6, 11). IFN-inducible immunity-related GTPases (Irgs (immunoreactive glucagons)), such as Irgm1 and Irga6 (6, 11), and IFN-inducible eukaryotic initiation factor (eIF)-2α kinase, protein kinase R (12), are potential autophagic regulators; however, the mechanisms for IFN-γ-induced autophagy are currently undocumented. In addition, the functions of autophagic machinery for IFN-γ-activated Jak2-STAT1 signaling and bioactivities require further investigation. In this study, we examined the role of autophagy and its molecular actions in the IFN-γ-induced Jak2-STAT1 activation and cellular inflammation.
EXPERIMENTAL PROCEDURES
Cells and Reagents
Atg5-deficient (Atg5−/−) and Atg7-deficient (Atg7−/−) MEFs were obtained from Dr. N. Mizushima, Department of Medicine, Tokyo Dental and Medical University, Tokyo, Japan, and Dr. M. Komatsu, Laboratory of Frontier Science, Tokyo Metropolitan Institute of Medical Science, Bunkyo-ku, Tokyo, Japan, respectively. Mouse macrophage RAW264.7 cells were obtained from Dr. C. C. Huang, Department of Pediatrics, National Cheng Kung University Hospital, Tainan, Taiwan. Human acute myelogenous leukemia U937 and chronic myelogenous leukemia K562 cells were purchased from the American Type Culture Collection. The human microvascular endothelial cell line-1 (HMEC-1) was passed in culture plates containing endothelial cell growth medium (Cambrex) composed of 2% FBS, 1 μg/ml hydrocortisone, 10 ng/ml epidermal growth factor, and antibiotics. Wild-type (WT), Atg5−/−, or Atg7−/− MEFs, and RAW264.7, U937, and K562 cells were grown in DMEM (Invitrogen) supplemented with 10% heat-inactivated FBS (Invitrogen), 50 units/ml penicillin, and 50 μg/ml streptomycin in a humidified atmosphere with 5% CO2 and 95% air. Recombinant mouse and human cytokine IFN-γ were purchased from PeproTech. Mouse mAb specific for β-actin was purchased from Chemicon International, and anti-mouse LC3 from MBL International. Anti-mouse inducible nitric-oxide synthase was from BD Biosciences. Alexa Fluor 488- and HRP-conjugated goat anti-mouse, goat anti-rabbit, and donkey anti-goat IgG were from Invitrogen.
Antibodies against phospho-Pyk2 (Tyr402), Pyk2, phospho-STAT1α/β (Tyr701), STAT1α/β, phospho-Jak2 (Tyr1007/Tyr1008), Jak2, phospho-Jak1 (Tyr1022/Tyr1023), Jak1, IFN regulatory factor-1 (IRF1), SOCS1, SHP2, and COX IV were purchased from Cell Signaling Technology. Anti-mouse IFNGR1 and IFNGR2 were from Abcam. Tricyclodecan-9-yl-xanthogenate (MG132), 3-methyladenine (3-MA), hydrogen peroxide (H2O2), Hoechst 33342, rapamycin, and LPS were from Sigma-Aldrich. 8-hydroxyls-7-(6-sulfonaphthalen-2-yl)diazenyl-quinoline-5-sulfonic acid (NSC-87877) and caffeic acid phenethyl ester were purchased from Tocris Bioscience. All drug treatments in cells were assessed for their cytotoxic effects using cytotoxicity and viability assays. Doses determined to be harmless were used.
Western Blotting
We harvested the cells and lysed them with a buffer containing 1% Triton X-100, 50 mm Tris (pH 7.5), 10 mm EDTA, 0.02% NaN3, and a protease inhibitor mixture (Roche Applied Science). After they had been freeze-thawed once, the cell lysates were centrifuged at 9,000 × g for 20 min at 4 °C. The supernatants were then collected and boiled in sample buffer for 5 min. After SDS-PAGE, the proteins were transferred to a PVDF membrane (Millipore), blocked in phosphate-buffered saline (PBS)-Tween (PBS plus 0.05% Tween 20) containing 5% skim milk, and probed with primary antibodies overnight at 4 °C. After they had been washed with PBS-T, the blots were incubated with a 1:5,000 dilution of HRP-conjugated secondary antibodies for 1 h at 4 °C. The protein bands were visualized using enhanced chemiluminescence (Pierce).
ELISA
The concentrations of RANTES, IFN-inducible protein-10 (IP-10), and TNF-α in cell-conditioned culture medium were determined using ELISA kits (R&D Systems) according to the manufacturer's instructions.
Co-immunoprecipitation
For co-immunoprecipitation, 100 μg of cell lysate from WT and Atg5−/− MEFs with or without IFN-γ treatment was incubated together with 5 μg of protein G (Amersham Biosciences) and 2 μg of anti-IFNGR2 IgG overnight at 4 °C. The expression of Jak2 was determined using Western blotting as described above.
EGFP-LC3 Transfection
To monitor autophagosome formation, WT and Atg5−/− MEFs (3 × 104) were transfected with 2 μg of pEGFPC1-LC3. After IFN-γ treatment, the formation of punctate EGFP-LC3 was detected under a confocal fluorescence microscope (Eclipse C1si; Nikon).
Immunostaining followed by Flow Cytometry
To determine IFNGR1, IFNGR2, and STAT1 expression, WT and Atg5−/− MEFs were fixed with 1% formaldehyde in PBS at room temperature for 10 min. After they had been washed twice with PBS, they were stained with rabbit anti-mouse IFNGR1, IFNGR2, and STAT1 at a final concentration of 1 μg/ml for 1 h at room temperature and then incubated with a mixture of Alexa Fluor 488-conjugated goat anti-rabbit IgG. After they had been washed with PBS, cells were analyzed using flow cytometry with excitation set at 488 nm (FACSCalibur; Becton Dickinson). To detect mitochondria in WT and Atg5−/− MEFs, the cells were dyed with 200 nm MitoTracker Green for 30 min at 37 °C, according to the manufacturer's instructions, and analyzed using flow cytometry.
Cell Imaging
For time-lapse fluorescence microscopic observation using a confocal laser scanning microscope (Digital Eclipse C1si-ready; Nikon), EGFP-LC3-transfected WT MEFs were grown on glass-bottom dishes (MatTek Corp.) and then treated with IFN-γ. The formation of autophagosomes with punctate EGFP-LC3 was recorded. For confocal fluorescence microscopic analysis, WT and Atg5−/− MEFs were dyed with 200 nm MitoSOX Red (Invitrogen) for 30 min at 37 °C according to the manufacturer's instructions. After another washing with PBS, the cells were covered with mounting fluid and then visualized.
Luciferase Reporter Assay
For the luciferase reporter assay, the cells were transiently co-transfected, using a reagent (GeneJammer; Stratagene), with IRF1 promoter-driven luciferase reporter (0.2 μg) and 0.01 μg of Renilla luciferase-expressing plasmid (pRL-TK; Promega). Twenty hours after the transfection, the cells were treated with IFN-γ for 1 h, lysed, and then harvested for luciferase and Renilla measurement using a luciferase assay system (Dual-Glo; Promega). For each lysate, the firefly luciferase activity was normalized to the Renilla luciferase activity to assess transfection efficiencies.
Nitrite Assay
NO production was assessed by measuring the accumulated levels of nitrite in the supernatant with the Griess reagent. Briefly, 100 μl of the culture supernatant was reacted with 100 μl of Griess reagent (1% sulfanilamide, 0.1% naphthylethylenediamine dihydrochloride, and 2.5% H3PO4) for 10 min at room temperature. The concentration of nitrite was measured using a microplate reader (Spectra MAX 340PC; Molecular Devices) at 540 nm and calculated using a standard curve of sodium nitrite with ELISA software (Softmax Pro; Molecular Devices).
Cell Proliferation
Seventy-two hours after IFN-γ treatment, the cells were stained with 0.01% trypan blue in a 96-well system, and cell proliferation was measured by counting the number of cells. The assay was independently repeated in three experiments.
Plaque Assay
We propagated a WT herpes simplex virus-1 (HSV-1) strain (KOS) and then titrated it onto Vero cell monolayers. WT and Atg5−/− MEFs that had been treated with IFN-γ for 18 h were infected with the KOS strain at a multiplicity of infection of 0.1 for 48 h. Total cell lysates were transferred to 6-well plates containing monolayers of Vero cells. After 3 days of incubation, the cells were stained with 0.1% crystal violet in 20% methanol and the plaques enumerated using a stereomicroscope.
Detecting ROS
ROS generation was determined using CM-H2DCFDA (Invitrogen). Cells (106/sample) were incubated with 200 nm CM-H2DCFDA for 30 min according to the manufacturer's instructions, washed with PBS, and then immediately measured using a microplate reader (Fluoroskan Ascent; Thermo Electron Corp.) with excitation at 488 nm and emission at 520 nm.
Lentivirus-based RNAi Transfection
SHP2 and Atg5 knockdown in Atg5−/− MEFs, HMEC-1, and K562 was done using lentiviral transduction to stably express short hairpin RNA (shRNA) that targeted mouse and human SHP2, respectively. Mouse and human shSHP2 clones were obtained from the National RNAi Core Facility (Institute of Molecular Biology/Genomic Research Center, Academia Sinica, Taiwan). The mouse and human libraries were referred to as TRC-Mm 1.0 and TRC-Hs 1.0, respectively. The constructs that were used in the MEFs (shRNA target sequence TRCN0000029877 5′-CGTGTTAGGAACGTCAAAGAA-3′ for mouse SHP2–1; TRCN0000029878 5′-GACATCCTTATTGACATCATT-3′ for mouse SHP2–2), the K562 cells (shRNA target sequence TRCN0000005003 5′-CGCTAAGAGAACTTAAACTTT-3′ for human SHP2), and the HMEC-1 cells (shRNA target sequence TRCN0000151963 5′-CCTGAACAGAATCATCCTTAA-3′ for human Atg5), and control shLuc (shRNA target sequence TRCN0000072247 5′-GAATCGTCGTATGCAGTGAAA-3′ for luciferase) were used to generate recombinant lentiviral particles. Human TE671 cells were co-transfected with pCMVdeltaR8.91 and pMD.G (two helper plasmids; gifts from Dr. H. K. Sytwu, Graduate Institute of Life Sciences, National Defense Medical Center, Taiwan) plus pLKO.1-puro-shRNA, using a transfection reagent (GeneJammer; Stratagene). The transfected cells were incubated at 37 °C in an atmosphere of 5% CO2 for 24 h, and then the medium was replaced with fresh medium. Cell supernatants containing the viral particles were harvested at 36, 48, 60, and 72 h after transfection. The supernatants were filtered using a 0.45-μm low-protein-binding filter and concentrated by centrifugation at 20,000 × g at 4 °C for 3 h using a JA25.50 (Beckman) rotor. The virus pellets were resuspended with fresh medium and stored at −80 °C. Atg5−/− MEFs, HMEC-1, and K562 cells were transduced with lentivirus at an appropriate multiplicity of infection in complete growth medium supplemented with 8 μg/ml Polybrene. After 24 h of transduction, protein expression was monitored using Western blotting.
PCR
Probes for mitochondrial DNA-encoded cytochrome c oxidase subunit I (COX I) and nucleus-encoded GAPDH genes were generated by PCR amplification with a Porter Thermal Cycler (Infinigen Biotechnology). Primers for the COX I and GAPDH probes corresponded to nucleotides ACTATACTACTACTAACAGACCG (forward) and GGTTCTTTTTTTCCGGAGTA (reverse; PCR product of 177 bp) and GGGAAGCCCATCACCATCT (forward) and GCCTCACCCCATTTGATGTT (reverse; PCR product of 58 bp), respectively. Total DNA was extracted using the Genomic DNA Mini kit (Qiagen). PCR conditions were an initial denaturation at 95 °C for 5 min followed by 30 rounds of cycling at 95 °C for 1 min, 60 °C for 50 s, and then 72 °C for 20 s. The band intensities were quantified directly from the stained agarose gels using video imaging and a densitometry software system (GelDoc-It Imaging System; UVP, Upland, CA).
Statistical Analysis
Data are means ± S.D. from three independent experiments and were analyzed using one-way analysis of variance and then a two-tailed, paired t test for experiments involving two paired groups. Statistical significance was set at p < 0.05.
RESULTS
IFN-γ-induced Autophagy and Autophagy-regulated Cellular Inflammatory Responses
In IFN-γ-treated Atg5−/− MEFs, but not in WT MEFs, there was no induction of autophagy, which is characterized by microtubule-associated protein light chain 3 (LC3; Atg8) conversion (Fig. 1A), an autophagic characteristic of LC3 lipidation (13). EGFP-LC3 transfection showed that in Atg5−/− MEFs there was no IFN-γ-induced autophagosome formation, which is characterized by EGFP-LC3 aggregation in the cytoplasm (Fig. 1B). We found, using time-lapse fluorescence microscopy, that IFN-γ induced transient autophagy flux in MEFs without inducing cell death (data not shown).
FIGURE 1.

IFN-γ-induced autophagy was critical for cellular inflammation. A, Western blotting was used to determine LC3 conversion in IFN-γ (10 ng/ml)-treated WT and Atg5−/− MEFs. β-Actin was the internal control. Data are representative of three individual experiments. B, confocal fluorescence microscopic observation of EGFP-LC3 punctate formation 6 h after IFN-γ (10 ng/ml) treatment in WT and Atg5−/− MEFs is shown. Scale bars, 20 μm. C, luciferase reporter assay of IRF1 promoter transactivation 1 h after IFN-γ (10 ng/ml) treatment in WT and Atg5−/− MEFs is shown. D, Griess reagent was used to detect the generation of nitrite 48 h after IFN-γ (10 ng/ml) treatment. E and F, ELISA was used to measure RANTES and IP-10 production 24 h after IFN-γ (10 ng/ml) treatment. Data, obtained from triplicate cultures, are means ± S.D. (error bars). *, p < 0.05.
We next investigated whether autophagy is required for IFN-γ-elicited cellular responses, including the transactivation of IRF1, a directly responsive transcription factor for IFN-γ signaling, for the expression of proinflammatory mediators, and for antiproliferation and antiviral replication (9). A luciferase reporter assay showed that IFN-γ, autophagy-dependently, induced IRF1 promoter transactivation (p = 0.012) (Fig. 1C). Atg5 deficiency extensively inhibited IFN-γ-induced proinflammatory responses, namely, inducible nitric-oxide synthase expression (data not shown), nitrite generation (p = 0.038) (Fig. 1D), RANTES (p = 0.015) (Fig. 1E), and IP-10 production (p = 0.0005) (Fig. 1F). Furthermore, Atg5 deficiency significantly suppressed the IFN-γ-induced inhibition of cell proliferation in MEFs (p = 0.036) (supplemental Fig. S1A). However, we found that IFN-γ-treated MEFs were not cytotoxic (data not shown). During an HSV-1 infection, autophagy is required to increase viral recognition, the subsequent induction of viral degradation, and the presentation of endogenous viral antigens on major histocompatibility complex class I molecules (5). In Atg5−/− MEFs, HSV-1 replication was significantly higher (p = 0.0037) (supplemental Fig. S1B). IFN-γ pretreatment totally blocked HSV-1 replication. Notably, Atg5 deficiency suppressed IFN-γ-inhibited viral replication.
Treating MEFs (Fig. 2A) or mouse macrophage RAW264.7 cells (Fig. 2B) with a noncytotoxic dose of 3-MA, a known inhibitor of class III phosphatidylinositol 3-kinase and autophagy, significantly reduced IFN-γ-induced nitrite, RANTES, and TNF-α expression (p < 0.001). These results demonstrated that autophagy is crucial for IFN-γ-induced inflammatory responses.
FIGURE 2.
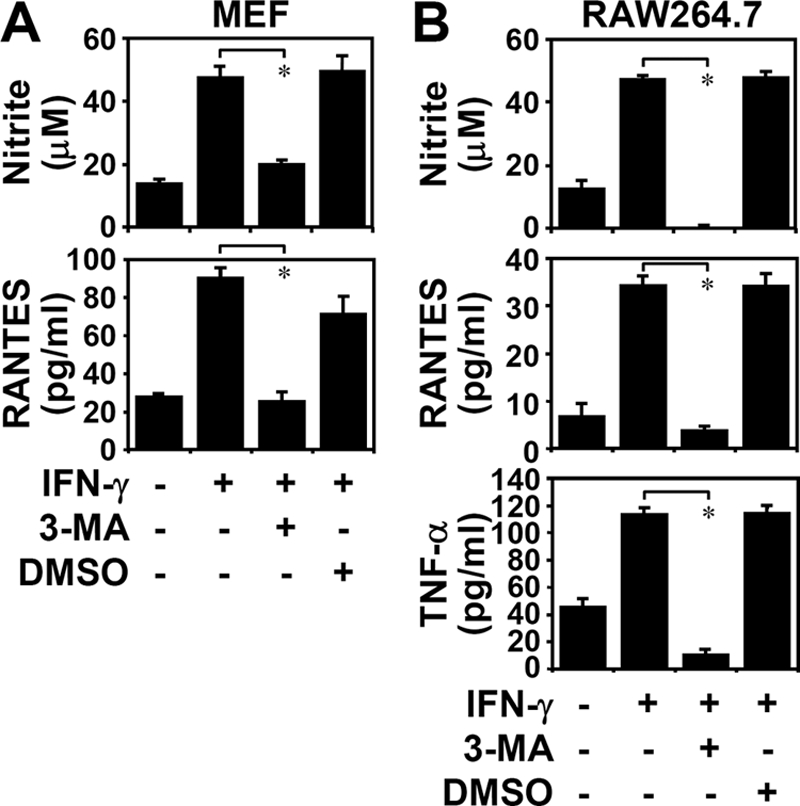
Inhibiting autophagy reduced IFN-γ-induced inflammation. Griess reagent was used to detect nitrite generation 48 h after IFN-γ (10 ng/ml) treatment in WT MEFs (A) and RAW264.7 cells (B) pretreated for 0.5 h with (+) and without (−) 3-MA (1 mm). ELISA was used to measure RANTES and TNF-α 24 h after treatment. Dimethyl sulfoxide (DMSO) was the control. Data, obtained from triplicate cultures, are means ± S.D. (error bars). *, p < 0.05.
IFN-γ Autophagy-dependently Activated Jak2
To investigate why autophagy is required for IFN-γ bioactivities, we examined how it regulates IFN-γ signaling. Immunostaining and then flow cytometry showed that the IFN-γ receptors IFNGR1 and IFNGR2 (Fig. 3A) were expressed normally in WT and Atg5−/− MEFs. Additionally, IFN-γ treatment did not change IFNGR expression (data not shown). Because Jak2 is activated upon ligand binding and is then followed by the formation of the IFNGR2·Jak2 complex (9, 14, 15), we examined, by inducing the formation of the IFNGR2·Jak2 complex, whether autophagy is required for Jak2 activation. A co-immunoprecipitation analysis showed that the Atg5 deficiency did not cause a defective IFNGR2·Jak2 complex to form (Fig. 3B). These results indicated an independent role for autophagy in promoting the complex formation of IFNGR2·Jak2. We then checked the activation of Jak2-STAT1, the major intracellular signaling pathway for IFN-γ (9). The Atg5 deficiency suppressed IFN-γ-induced Jak2 phosphorylation (Tyr1007/Tyr1008) (Fig. 3C). We next examined the activation of the tyrosine kinases Jak1 and Pyk2, two downstream molecules of Jak2 (9, 14), to confirm this. Atg5 deficiency reduced Jak1 phosphorylation (Tyr1022/Tyr1023) (Fig. 3C) and Pyk2 phosphorylation (Tyr402) (data not shown). Although we showed that autophagy is necessary for Jak2 activation, the mechanism is still unclear. The expression of Jak2 slightly decreased in Atg5-deficient MEFs; however, the mechanisms are also still unknown.
FIGURE 3.

Effects of autophagy on the expression of IFN-γ receptors and the activation of Jak2 signaling. A, flow cytometry was used to detect the expression of IFNGR1 and IFNGR2 using specific antibodies in WT and Atg5−/− MEFs. The percentages of positive cells are shown. B, after 0.25 h of IFN-γ (10 ng/ml) treatment, co-immunoprecipitation (IP) was used to detect Jak2 interaction with IFNGR2. Western blotting (IB) was used to determine the expression of Jak2 and IFNGR2. H, heavy chain; L, light chain; NB, nonspecific binding. C, after IFN-γ (10 ng/ml) treatment, Western blotting was used to determine the time kinetic phosphorylation of Jak2 (Tyr1007/Tyr1008) and Jak1 (Tyr1022/Tyr1023). β-Actin was the internal control. Data are representative of three individual experiments.
IFN-γ Autophagy-dependently Induced STAT1 Activation
Western blotting showed that, in addition to regulating Jak2, the Atg5 deficiency inhibited IFN-γ-induced STAT1 phosphorylation (Tyr701) as well as the expression of IFN-inducible proteins, such as IRF1 and SOCS1 (Fig. 4A), both of which are up-regulated via a STAT1-regulated pathway (9). Western blotting (Fig. 4A) and flow cytometry showed that endogenous STAT1 expression was lower in Atg5−/− MEFs (supplemental Fig. S2A). We also examined the effect of proteasome, which regulates STAT1 stabilization (16). Treatment with MG132, a typical proteasome inhibitor, did not reverse STAT1 down-regulation (supplemental Fig. S2B). Others (9, 17) have speculated that STAT1 expression is regulated by a mechanism involving Jak2-STAT1-IRF1. Silencing Atg5 in HMEC-1 cells using lentivirus-based shRNA reconfirmed the role of Atg5 by showing that a lack of Atg5 caused defects in IFN-γ-induced STAT1 phosphorylation (Tyr701) but not protein expression (Fig. 4B). Our results indicated that the Atg5 deficiency inhibits IFN-γ bioactivities primarily by interfering with Jak2-STAT1 activation.
FIGURE 4.
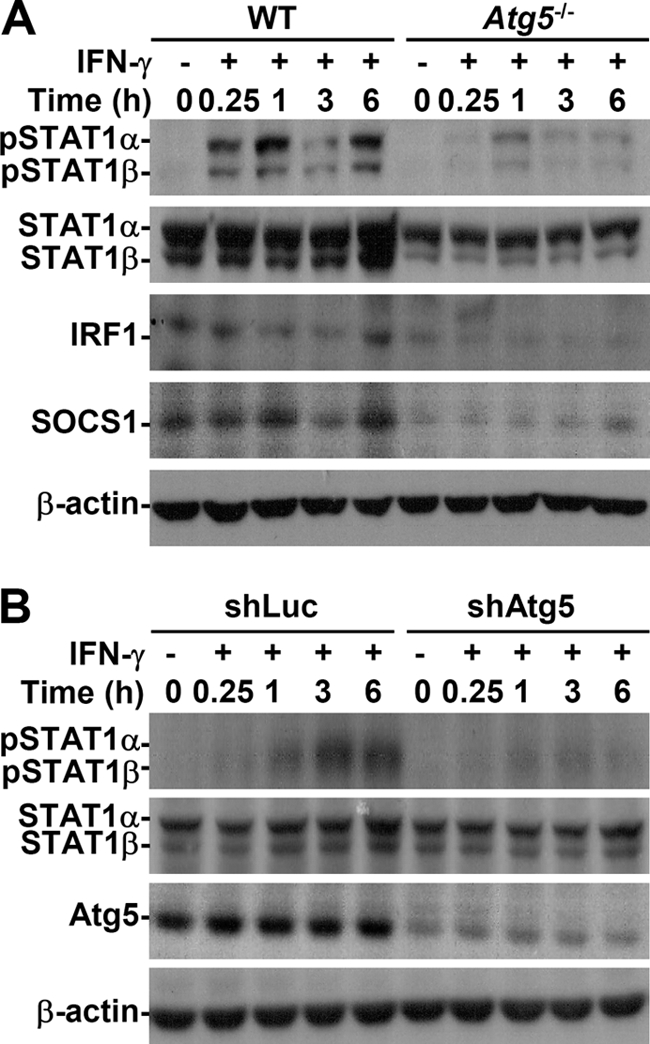
Autophagy was important for IFN-γ-activated STAT1. A, after IFN-γ (10 ng/ml) treatment, Western blotting was used to determine the time kinetic phosphorylation of STAT1α/β (Tyr701) as well as IRF1 and SOCS1 expression in WT and Atg5−/− MEFs. B, HMEC-1 cells were pretreated with or without lentivirus-based short hairpin Atg5 RNA (shAtg5) or control luciferase-shRNA (shLuc) transfection and then treated with IFN-γ (10 ng/ml) for the indicated times. Western blotting was used to determine the expression of phosphorylation of STAT1α/β (Tyr701) and Atg5. β-Actin was the internal control. Data are representative of three individual experiments.
Atg5 has a novel E3 ligase-like activity for protein lipidation after being in a complex with Atg12, which is activated by Atg7 (18); therefore, we confirmed the effects of autophagy on IFN-γ signaling. In Atg7−/− MEFs, LC3 conversion (Fig. 5A), IFN-γ-induced phosphorylation of Jak2 (Tyr1007/Tyr1008) and STAT1 (Tyr701) (Fig. 5B), and the generation of nitrite (p = 0.025) (Fig. 5C), IP-10 (p = 0.015) (Fig. 5D), and RANTES (p < 0.05) (data not shown) were inhibited. Endogenous STAT1 expression was low in Atg7−/− MEFs, as it was in Atg5−/− MEFs. In addition, 3-MA treatment also decreased IFN-γ-activated STAT1 in Atg7−/− MEFs (data not shown). These results confirmed that autophagy is important in IFN-γ-activated Jak2-STAT1 and cellular inflammatory responses.
FIGURE 5.

Autophagy was required for IFN-γ-activated Jak2-STAT1 and inflammation. Western blotting was used to determine LC3 conversion (A) and the time kinetic phosphorylation of Jak2 (Tyr1007/Tyr1008) and STAT1α/β (Tyr701) (B) in IFN-γ (10 ng/ml)-treated WT and Atg7−/− MEFs. β-Actin was the internal control. Data are representative of three individual experiments. C, Griess reagent was used to detect nitrite generation 48 h after IFN-γ (10 ng/ml) treatment. D, ELISA was used to measure IP-10 production 24 h after IFN-γ (10 ng/ml) treatment. Data, obtained from triplicate cultures, are means ± S.D. (error bars). *, p < 0.05.
Autophagy Negatively Regulated ROS, Which Facilitated IFN-γ Signaling
Autophagy inhibition caused an abnormal accumulation of ROS (19, 20). Without autophagy, intracellular ROS-generating mitochondria accumulate because they are deregulated. In WT MEFs with IFN-γ transiently autophagy, imaging analysis showed that IFN-γ triggered autophagosome formation, which was co-localized with ROS-generating mitochondria (Fig. 6A). These results indicated that IFN-γ-induced autophagy may down-regulate the expression of damaged mitochondria, which generate ROS. Western blotting (Fig. 6B, top) and PCR (Fig. 6B, middle) of mitochondrial markers COX IV and COX I expression, respectively, and MitoTracker Green staining followed by flow cytometry (Fig. 6B, bottom) confirmed the higher expression of mitochondria in Atg5−/− MEFs. Confocal microscopic analysis showed that intracellular ROS had accumulated in mitochondria, particularly in Atg5−/− MEFs (data not shown). CM-H2DCFDA-stained Atg5−/− MEFs showed a high intracellular oxidative state (p = 0.031) (Fig. 6C). Inhibiting autophagy with 3-MA treatment also caused ROS generation (data not shown). These results indicated that autophagy negatively regulates the generation of ROS in mitochondria.
FIGURE 6.

ROS generation was deregulated in the absence of autophagy and negatively regulated IFN-γ-induced STAT1 activation. A, confocal fluorescence microscopic observation of the co-localization (Merge, yellow) of EGFP-LC3 punctate formation (green) and MitoSOX Red staining (red) in WT MEFs with or without IFN-γ (10 ng/ml) treatment for 3 h. 3D, three-dimensional. Scale bars, 20 μm. B, Western blotting, PCR, and flow cytometry, with COX IV antibodies, COX I primer, and MitoTracker Green staining, respectively, were used to detect mitochondria expression in WT and Atg5−/− MEFs. The ratios of mitochondrial DNA COX I to the total genomic DNA GAPDH were calculated based on the intensities of their PCR product in agarose gel. MFI, mean fluorescence intensity. C, CM-H2DCFDA staining and a microplate reader were used to measure ROS generation. Data, obtained from triplicate cultures, are means ± S.D. (error bars). *, p < 0.05. D, Western blotting was used to determine IFN-γ (10 ng/ml)-induced phosphorylation of STAT1α/β (Tyr701) in WT MEFs 0.25 h after they had been pretreated with (+) and without (−) caffeic acid phenethyl ester (CAPE, 25 μm) or H2O2 (10 mm) for 0.5 h. β-Actin was the internal control. Data are representative of three individual experiments. E, with (+) and without (−) H2O2 pretreatment for 0.5 h, generation of nitrite in IFN-γ (10 ng/ml)-treated WT MEFs was detected using Griess reagent at 48 h. Data, obtained from triplicate cultures, are means ± S.D. (error bars). *, p < 0.05.
We therefore investigated the role of ROS in IFN-γ-activated STAT1 and cellular inflammation. We first showed that the antioxidant caffeic acid phenethyl ester up-regulated IFN-γ-activated STAT1 (Fig. 6D, left) and that exogenous H2O2 totally inhibited IFN-γ-activated STAT1 (Fig. 6D, right). Furthermore, H2O2 also inhibited IFN-γ-induced nitrite generation (p < 0.001) (Fig. 6E). These results indicated that an autophagy deficiency deregulates ROS, thereby suppressing IFN-γ-activated STAT1 and cellular inflammation.
ROS-activated SHP2 Reduced IFN-γ Signaling in Autophagy-deficient Cells
SOCS1 and SHP2 are important for the IFN-γ feedback that inactivates the Jak2-STAT1 signaling pathway (9, 10). In Atg5−/− MEFs, endogenous SOCS1 was low, which suggested that it independently and negatively regulated Jak2-STAT1 in autophagy-deficient MEFs (Fig. 4A). Excessive ROS inhibit IFN-γ-induced Jak2-STAT1 activation in neurons (21), and ROS positively activate SHP2 (22). We therefore hypothesized that ROS-mediated SHP2 activation is critical for inhibiting IFN-γ-activated STAT1 in autophagy-deficient cells. Western blotting, used to analyze the feedback regulatory role of SHP2, showed that shRNA inhibited SHP2 expression (Fig. 7). We showed that shSHP2 clone 2 (shSHP2–2) was the best clone for SHP2-knockdown expression. Exogenous H2O2 totally inhibited IFN-γ-activated STAT1 (Fig. 6D, right), which showed that inhibiting IFN-γ-activated STAT1 in autophagy-deficient cells is dependent upon ROS-mediated SHP2 activation (Fig. 7A). IFN-γ-treated Atg5−/− SHP2 knockdown MEFs showed that STAT1 was reactivated (Fig. 7B). Furthermore, the SHP2 inhibitor NSC-87877 reversed NO production in IFN-γ-stimulated Atg5−/− MEFs (p = 0.001) (supplemental Fig. S3). These findings indicated that autophagy negatively regulates ROS-activated SHP2, which, in turn, facilitates both IFN-γ-induced STAT1 activation and cellular inflammation.
FIGURE 7.

ROS-regulated SHP2 inhibited IFN-γ-activated STAT1 in the absence of autophagy. A, Western blotting was used to determine IFN-γ (10 ng/ml)-induced phosphorylation of STAT1α/β (Tyr701) in WT MEFs 0.25 h after they had been pretreated with (+) and without (−) SHP2-shRNA clone 2 (shSHP2–2) or control luciferase-shRNA (shLuc) transfection and then with H2O2 (10 mm) for 0.5 h. B, after IFN-γ (10 ng/ml) treatment with (+) and without (−) SHP2-shRNAs (shSHP2) or control (shLuc) transfection, Western blotting was used to determine the phosphorylation of STAT1α/β (Tyr701) in Atg5−/− MEFs. shSHP2-1, inefficiently silenced; shSHP2-2, efficiently silenced. β-Actin was the internal control. Data are representative of three individual experiments.
DISCUSSION
We found that IFN-γ induced transient autophagy flux without cytotoxicity, which is consistent with previous studies (1, 6, 11). We also provide evidence that autophagy significantly increases not only antimycobacteria (1, 2), but also IFN-γ-induced IRF1 expression, proinflammation, antiproliferation, and antiviral replication. We provide evidence that IFN-γ-induced autophagy inhibits ROS and SHP2, sustains Jak2-STAT1 activation, and facilitates IFN-γ-induced cellular inflammation, three functions not reported elsewhere in the literature. We hypothesize that transient autophagy regulates the homeostasis for IFN-γ signaling, especially on the feedback regulation of SHP2.
Excessive ROS inhibit IFN-γ-induced Jak2-STAT1 activation in neurons (21). In astrocytes and BCR/ABL-chronic myelogenous leukemia, ROS and ROS-regulated signaling positively regulate SHP2 (22). Current studies (19, 20) show that an Atg5 deficiency facilitates the accumulation of destructive mitochondria, after which ROS accumulate and up-regulate Toll-like receptor-mediated IL-1β. Consistent with these results, we confirmed that ROS-generating mitochondria accumulated in MEFs with an autophagy deficiency. Furthermore, confocal microscopic observation showed that autophagosomes and ROS-generating mitochondria had co-localized in IFN-γ-stimulated MEFs. We hypothesize that IFN-γ induces autophagy-mediated ROS down-regulation. Because we found that autophagy is essential for IFN-γ-induced Jak2-STAT1 activation in Atg5−/−, Atg7−/−, and shAtg5-treated HMEC-1, we conclude that ROS-regulated SHP2 inhibits Jak2-STAT1. Therefore, we hypothesize that autophagy negatively regulates the mechanisms that allow the regulation of ROS generation after the eradication of destructive mitochondria. However, the mechanisms through which ROS activates SHP2 are still unclear.
IFN-γ-elicited cellular responses such as antiproliferation are critical for tumor suppression through Jak2-STAT1-mediated induction of anticancer proteins (9). Actually, IFN-γ resistance is generally involved in viral escape and tumor escape (23, 24). BCR/ABL-chronic myelogenous leukemia hijacks ROS signaling, which interrupts the IFN-γ-mediated defense against the progression of cancer. We hypothesize that in IFN-γ-resistant leukemia, the absence of autophagy and the presence of higher levels of ROS and SHP2 generally permit tumors to elude the IFN-γ defense against cancer progression. Unlike U937 (acute myelogenous leukemia) cells, K562 (chronic myelogenous leukemia) cells were resistant to IFN-γ-induced STAT1 activation (supplemental Fig. S4A). Notably, IFN-γ treatment did not cause autophagy in K562 cells. Consistent with previous reports (25, 26), inactivating the mammalian target of rapamycin increased STAT1 activation. In addition, the combination of IFN-γ and CCI-779, a rapamycin analog that inhibits the mammalian target of rapamycin, increases anticancer activities through apoptosis (27). We also showed that rapamycin treatment increased STAT1 activation in IFN-γ-stimulated MEFs (supplemental Fig. S5) and K562 cells (supplemental Fig. S4B). Our findings imply that the effects of rapamycin, a typical inducer of autophagy, through autophagy-facilitated STAT1 are also involved. For IFN-γ resistance in tumorigenesis, preconditioning targeted cells for autophagy may provide strategies in IFN-γ-based antileukemia therapy. Furthermore, SHP2 is both a regulator of BCR/ABL-chronic myelogenous leukemia progression and a target for anticancer treatment (26). However, the post-translational regulation of SHP2 is still unclear until ROS are involved. Our findings indicate that the interference of activated SHP2 with STAT1 may be what allows K562 cells to escape from IFN-γ (supplemental Fig. S4C). IFN-γ induces transient autophagy. We hypothesize that the induction of IFN-γ resistance is caused by an autophagy deficiency. Ensuring that autophagy is induced or that SHP2 is inhibited may be a way to guarantee that IFN-γ protects cells against the progression of cancer.
Many current studies (4–6, 8, 9, 28) report that an autophagic mechanism is critical for innate and adaptive immune defense because it promotes the elimination of intracellular pathogens directly and indirectly through major histocompatibility complex molecules. They also report that IFN-γ synergistically facilitates autophagic antimicrobial control. We hypothesize that autophagy not only facilitates the recognition and elimination of intracellular pathogens but also the expression of antigens to these pathogens, which is supported by our findings in the present study, but that autophagy is also essential for IFN-γ-activated Jak2-STAT1 and bioactivities. This hypothesis and the molecular mechanisms that underlie it require additional investigation, as does confirmation of the probability that IFN-γ-induced autophagy is involved in the host defense against infection and disease in more than just the Jak-STAT signaling pathway (29).
Irgs and protein kinase R mediate IFN-γ-induced autophagy (6, 11, 12); however, the mechanisms are not currently known. We previously found (30) that glycogen synthase kinase-3β regulates Jak2-STAT1 by inactivating SHP2. Our preliminary data in the present study confirmed that IFN-γ induces glycogen synthase kinase-3β-dependent autophagy (data not shown). It is of great interest to investigate the associations among glycogen synthase kinase-3β, autophagy, ROS, and SHP2 in IFN-γ signaling and bioactivities. We hypothesize that, in addition to IFN-γ-induced autophagy, autophagy-regulated ROS and SHP2 regulate cellular functions in response to autophagic stimuli.
Supplementary Material
Acknowledgments
We thank Dr. T. Yoshimori for critical discussion on this study and S. L. Lu and H. F. Cheng for technical assistance on shRNA and PCR.
This work was supported by Grants NHRI-EX99-9917NC and NSC 96-2320-B-006-018-MY3 from the National Health Research Institutes and the National Science Council, respectively, Taiwan, and Landmark Project C020 of National Cheng Kung University, Taiwan.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5.
- IFNGR
- IFN-γ receptor
- Atg
- autophagy protein
- EGFP
- enhanced green fluorescent protein
- HMEC
- human microvascular endothelial cell
- IP
- IFN-inducible protein
- IRF
- IFN regulatory factor
- Irg
- IFN-inducible immunity-related GTPase
- LC
- light chain
- 3-MA
- 3-methyladenine
- MEF
- mouse embryonic fibroblast
- ROS
- reactive oxygen species
- SHP
- Src homology-2 domain-containing phosphatase
- SOCS
- suppressor of cytokine signaling.
REFERENCES
- 1.Delgado M. A., Elmaoued R. A., Davis A. S., Kyei G., Deretic V. (2008) EMBO J. 27, 1110–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deretic V. (2009) Curr. Opin. Immunol. 21, 53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Codogno P., Meijer A. J. (2005) Cell Death Differ. 12, (Suppl. 2), 1509–1518 [DOI] [PubMed] [Google Scholar]
- 4.Lee H. K., Lund J. M., Ramanathan B., Mizushima N., Iwasaki A. (2007) Science 315, 1398–1401 [DOI] [PubMed] [Google Scholar]
- 5.English L., Chemali M., Duron J., Rondeau C., Laplante A., Gingras D., Alexander D., Leib D., Norbury C., Lippé R., Desjardins M. (2009) Nat. Immunol. 10, 480–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gutierrez M. G., Master S. S., Singh S. B., Taylor G. A., Colombo M. I., Deretic V. (2004) Cell 119, 753–766 [DOI] [PubMed] [Google Scholar]
- 7.Levine B., Deretic V. (2007) Nat. Rev. Immunol. 7, 767–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mizushima N., Levine B., Cuervo A. M., Klionsky D. J. (2008) Nature 451, 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schroder K., Hertzog P. J., Ravasi T., Hume D. A. (2004) J. Leukoc. Biol. 75, 163–189 [DOI] [PubMed] [Google Scholar]
- 10.You M., Yu D. H., Feng G. S. (1999) Mol. Cell. Biol. 19, 2416–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Al-Zeer M. A., Al-Younes H. M., Braun P. R., Zerrahn J., Meyer T. F. (2009) PLoS One 4, e4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orvedahl A., Levine B. (2008) Autophagy 4, 280–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klionsky D. J., Abeliovich H., Agostinis P., Agrawal D. K., Aliev G., Askew D. S., Baba M., Baehrecke E. H., Bahr B. A., Ballabio A., Bamber B. A., Bassham D. C., Bergamini E., Bi X., Biard-Piechaczyk M., Blum J. S., Bredesen D. E., Brodsky J. L., Brumell J. H., Brunk U. T., Bursch W., Camougrand N., Cebollero E., Cecconi F., Chen Y., Chin L. S., Choi A., Chu C. T., Chung J., Clarke P. G., Clark R. S., Clarke S. G., Clavé C., Cleveland J. L., Codogno P., Colombo M. I., Coto-Montes A., Cregg J. M., Cuervo A. M., Debnath J., Demarchi F., Dennis P. B., Dennis P. A., Deretic V., Devenish R. J., Di Sano F., Dice J. F., Difiglia M., Dinesh-Kumar S., Distelhorst C. W., Djavaheri-Mergny M., Dorsey F. C., Dröge W., Dron M., Dunn W. A., Jr., Duszenko M., Eissa N. T., Elazar Z., Esclatine A., Eskelinen E. L., Fésüs L., Finley K. D., Fuentes J. M., Fueyo J., Fujisaki K., Galliot B., Gao F. B., Gewirtz D. A., Gibson S. B., Gohla A., Goldberg A. L., Gonzalez R., González-Estévez C., Gorski S., Gottlieb R. A., Häussinger D., He Y. W., Heidenreich K., Hill J. A., Høyer-Hansen M., Hu X., Huang W. P., Iwasaki A., Jäättelä M., Jackson W. T., Jiang X., Jin S., Johansen T., Jung J. U., Kadowaki M., Kang C., Kelekar A., Kessel D. H., Kiel J. A., Kim H. P., Kimchi A., Kinsella T. J., Kiselyov K., Kitamoto K., Knecht E., Komatsu M., Kominami E., Kondo S., Kovács A. L., Kroemer G., Kuan C. Y., Kumar R., Kundu M., Landry J., Laporte M., Le W., Lei H. Y., Lenardo M. J., Levine B., Lieberman A., Lim K. L., Lin F. C., Liou W., Liu L. F., Lopez-Berestein G., López-Otín C., Lu B., Macleod K. F., Malorni W., Martinet W., Matsuoka K., Mautner J., Meijer A. J., Meléndez A., Michels P., Miotto G., Mistiaen W. P., Mizushima N., Mograbi B., Monastyrska I., Moore M. N., Moreira P. I., Moriyasu Y., Motyl T., Münz C., Murphy L. O., Naqvi N. I., Neufeld T. P., Nishino I., Nixon R. A., Noda T., Nürnberg B., Ogawa M., Oleinick N. L., Olsen L. J., Ozpolat B., Paglin S., Palmer G. E., Papassideri I., Parkes M., Perlmutter D. H., Perry G., Piacentini M., Pinkas-Kramarski R., Prescott M., Proikas-Cezanne T., Raben N., Rami A., Reggiori F., Rohrer B., Rubinsztein D. C., Ryan K. M., Sadoshima J., Sakagami H., Sakai Y., Sandri M., Sasakawa C., Sass M., Schneider C., Seglen P. O., Seleverstov O., Settleman J., Shacka J. J., Shapiro I. M., Sibirny A., Silva-Zacarin E. C., Simon H. U., Simone C., Simonsen A., Smith M. A., Spanel-Borowski K., Srinivas V., Steeves M., Stenmark H., Stromhaug P. E., Subauste C. S., Sugimoto S., Sulzer D., Suzuki T., Swanson M. S., Tabas I., Takeshita F., Talbot N. J., Tallóczy Z., Tanaka K., Tanaka K., Tanida I., Taylor G. S., Taylor J. P., Terman A., Tettamanti G., Thompson C. B., Thumm M., Tolkovsky A. M., Tooze S. A., Truant R., Tumanovska L. V., Uchiyama Y., Ueno T., Uzcátegui N. L., van der Klei I., Vaquero E. C., Vellai T., Vogel M. W., Wang H. G., Webster P., Wiley J. W., Xi Z., Xiao G., Yahalom J., Yang J. M., Yap G., Yin X. M., Yoshimori T., Yu L., Yue Z., Yuzaki M., Zabirnyk O., Zheng X., Zhu X., Deter R. L. (2008) Autophagy 4, 151–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takaoka A., Tanaka N., Mitani Y., Miyazaki T., Fujii H., Sato M., Kovarik P., Decker T., Schlessinger J., Taniguchi T. (1999) EMBO J. 18, 2480–2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Igarashi K., Garotta G., Ozmen L., Ziemiecki A., Wilks A. F., Harpur A. G., Larner A. C., Finbloom D. S. (1994) J. Biol. Chem. 269, 14333–14336 [PubMed] [Google Scholar]
- 16.Tanaka T., Soriano M. A., Grusby M. J. (2005) Immunity 22, 729–736 [DOI] [PubMed] [Google Scholar]
- 17.Lehtonen A., Matikainen S., Julkunen I. (1997) J. Immunol. 159, 794–803 [PubMed] [Google Scholar]
- 18.Hanada T., Noda N. N., Satomi Y., Ichimura Y., Fujioka Y., Takao T., Inagaki F., Ohsumi Y. (2007) J. Biol. Chem. 282, 37298–37302 [DOI] [PubMed] [Google Scholar]
- 19.Saitoh T., Fujita N., Jang M. H., Uematsu S., Yang B. G., Satoh T., Omori H., Noda T., Yamamoto N., Komatsu M., Tanaka K., Kawai T., Tsujimura T., Takeuchi O., Yoshimori T., Akira S. (2008) Nature 456, 264–268 [DOI] [PubMed] [Google Scholar]
- 20.Tal M. C., Sasai M., Lee H. K., Yordy B., Shadel G. S., Iwasaki A. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 2770–2775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaur N., Lu B., Monroe R. K., Ward S. M., Halvorsen S. W. (2005) J. Neurochem. 92, 1521–1530 [DOI] [PubMed] [Google Scholar]
- 22.Park S. J., Kim H. Y., Kim H., Park S. M., Joe E. H., Jou I., Choi Y. H. (2009) Free Radic. Biol. Med. 46, 1694–1702 [DOI] [PubMed] [Google Scholar]
- 23.Randall R. E., Goodbourn S. (2008) J. Gen. Virol. 89, 1–47 [DOI] [PubMed] [Google Scholar]
- 24.Ikeda H., Old L. J., Schreiber R. D. (2002) Cytokine Growth Factor Rev. 13, 95–109 [DOI] [PubMed] [Google Scholar]
- 25.Fielhaber J. A., Han Y. S., Tan J., Xing S., Biggs C. M., Joung K. B., Kristof A. S. (2009) J. Biol. Chem. 284, 24341–24353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan G., Kalaitzidis D., Neel B. G. (2008) Cancer Metastasis Rev. 27, 179–192 [DOI] [PubMed] [Google Scholar]
- 27.Lee L., Sudentas P., Dabora S. L. (2006) Genes Chromosomes Cancer 45, 933–944 [DOI] [PubMed] [Google Scholar]
- 28.Jagannath C., Lindsey D. R., Dhandayuthapani S., Xu Y., Hunter R. L., Jr., Eissa N. T. (2009) Nat. Med. 15, 267–276 [DOI] [PubMed] [Google Scholar]
- 29.Gough D. J., Levy D. E., Johnstone R. W., Clarke C. J. (2008) Cytokine Growth Factor Rev. 19, 383–394 [DOI] [PubMed] [Google Scholar]
- 30.Tsai C. C., Kai J. I., Huang W. C., Wang C. Y., Wang Y., Chen C. L., Fang Y. T., Lin Y. S., Anderson R., Chen S. H., Tsao C. W., Lin C. F. (2009) J. Immunol. 183, 856–864 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.