

Abstract
Human DNMT3A is responsible for de novo DNA cytosine methylation patterning during development. Here we show that DNMT3A methylates 5–8 CpG sites on human promoters before 50% of the initially bound enzyme dissociates from the DNA. Processive methylation is enhanced 3-fold in the presence of DNMT3L, an inactive homolog of DNMT3A, therefore providing a mechanism for the previously described DNMT3L activation of DNMT3A. DNMT3A processivity on human promoters is also regulated by DNA topology, where a 2-fold decrease in processivity was observed on supercoiled DNA in comparison with linear DNA. These results are the first observation that DNMT3A utilizes this mechanism of increasing catalytic efficiency. Processive de novo DNA methylation provides a mechanism that ensures that multiple CpG sites undergo methylation for transcriptional regulation and silencing of newly integrated viral DNA.
Keywords: DNA Methylation, DNA Methyltransferase, DNA Topology, Epigenetics, Gene Regulation, Protein-Protein Interactions, DNMT3A, DNMT3L, Processivity
Introduction
Methylation of DNA is an epigenetic modification involved in silencing of transposable elements, X-chromosome inactivation, genomic imprinting, and cellular differentiation (1, 2). In conjunction with other epigenetic processes such as histone modifications and interfering RNA, DNA methylation represents an essential component of the transcriptional regulation machinery (3). The deregulation of DNA methylation leads to diverse diseases including cancer and neurological disorders (4). Mammalian DNA methyltransferases (DNMTs)2 catalyze the transfer of a methyl group from S-adenosyl methionine (AdoMet) onto the 5′ position of cytosine at CpG sites, although the methylation of CpA dinucleotides occurs during the early stages of embryogenesis (5, 6). DNMT3A and the closely related DNMT3B carry out de novo methylation, laying down new DNA methylation patterns throughout the genome (7–9). Methylation patterns are then stably maintained over cell divisions by the maintenance methyltransferase DNMT1 (10, 11). Although 70–80% of CpG sites are methylated in the genome, some regions (CpG islands) are largely resistant to methylation, whereas others (retrotransposons and highly repetitive sequences) undergo extensive methylation (8). Determining how DNMT3A methylates an entire region in the genome is necessary to understand the temporal and spatial regulation of de novo methylation.
Many mechanisms that control de novo methyltransferase specificity have been proposed, including cis-acting signals in the DNA sequence (12), DNA topology (13), histone modifications (14), interactions between DNMTs (15), auxiliary proteins (16), or RNA molecules (17). However, the underlying mechanisms leading to the acquisition of new methylation patterns remain poorly understood. The methylation of multiple, proximal CpG sites during a single binding event (processive catalysis) by DNMT3A or DNMT3B may contribute to the localized acquisition of methylation marks.
The large family of DNA methyltransferases including both bacterial and mammalian enzymes displays highly variable degrees of processivity. The mammalian DNMT1 (18, 19) and bacterial methyltransferase SssI (20) are extremely processive, whereas other bacterial enzymes, including methyltransferase EcoRI (21), are entirely lacking in processivity. In this study, we investigated the enzymatic properties of the human de novo methyltransferase DNMT3A and determined its processivity using pulse-chase assays and kinetic modeling and determining kcat values for substrates with a varying number of CpG sites. We found that DNMT3A is processive, spreading methylation to distal CpG sites. DNMT3A processivity is modulated by DNA topology and protein-protein interactions. DNMT3L, a known activator of DNMT3A catalytic activity, is shown to be a processivity factor of DNMT3A. The data presented support a model of DNA methylation spreading through the action of processive catalysis.
EXPERIMENTAL PROCEDURES
Materials
The DNA used as substrates, poly(dI-dC) (∼1000 bp) (Sigma-Aldrich), poly(dA-dT) (∼1000 bp) (Sigma-Aldrich), GCbox2 (5′-GGGAATTCAAGGGGCGGGGCAATGTTAGGG-3′) duplex, and Gusch (5′-TCAAAAAGGATCTTCACCTAG-3′) duplex (non-CpG containing sequence), were purchased from Midland Certified Reagent Co. (Midland, TX). The recognition site for DNMT3A is underlined. Duplexes were prepared by annealing the complementary strands in a buffer containing 10 mm Tris-Cl, pH 8.0, 1 mm EDTA, and 50 mm NaCl, heating the DNA to 85 °C for 5 min, and slow-cooling the DNA. Annealing was confirmed by 12% PAGE analysis.
Bacterial plasmid pBR322 (New England Biolabs) was produced by transforming the plasmid in XL blue cells (New England Biolabs) (Escherichia coli, with no topoisomerase mutations), which were then grown in LB media overnight at 37 °C. The plasmid was extracted by a HiSpeed plasmid midi kit (Qiagen). The human promoters used as substrates were amplified from genomic DNA extracted from HeLa cells. PCR conditions include Taq DNA polymerase (10 units/100-μl reaction, New England Biolabs), dNTPs (0.2 mm), primers (0.5 μm), thermo buffer II (1×), glycerol (1.3%), and genomic DNA (200 ng/100 μl). Reactions were run for 5 min at 95 °C, and then Taq DNA polymerase was added followed by 30 cycles at 30 s at 94 °C, 30 s at 62 °C (EFS1), 53 °C (p21), 55 °C (p15), 63 °C (ODC), 55 °C (β-actin), 55 °C (Smarca), 55 °C (CDC), 55 °C (Survivin), 55 °C (H-cad), 53 °C (Xist), 1 min at 72 °C, then 10 min at 72 °C. The primers for the promoters are listed in supplemental Table 1. The PCR products were then used as the substrate for further amplification of each promoter. The sequences of the PCR products were confirmed by sequencing.
Plasmid pCpGL was kindly provided by Michael Rehli (22). The p21-pCpGL substrate was created by amplification of the p21 promoter, with PCR conditions including Phusion high fidelity DNA polymerase(2 unit/100 μl reaction, New England Biolabs), dNTPs (0.2 mm), primers (0.5 μm), HF buffer (1×), and genomic DNA (200 ng/100 μl). Reactions were run for 4 min at 98 °C followed by 35 cycles for 10 s at 98 °C, 25 s at 68 °C, 1 min at 72 °C, and then 10 min at 72 °C. Promoters were digested with HindIII and BamHI and then ligated into digested and gel-purified pCpGL. The inserted sequence was confirmed by sequencing. The pCpGL and p21-pCpGL substrates were produced by transforming into GT115-competent cells (InvivoGen) and then grown overnight in LB media at 37 °C. The plasmid was extracted using a HiSpeed plasmid midi kit (Qiagen). The linear p21-pCpGL was digested with BamHI endonuclease overnight at 37 °C and purified using a Qiagen PCR cleanup kit. The form of the substrates was confirmed by analyzes on 0.8% agarose gel stained with ethidium bromide. Bacterial plasmids prepared in this way have an average superhelical density of −0.06 (23).
DNA concentrations are given in base pairs unless noted otherwise. Concentrations were determined by the absorbance at 260 nm, using the following molar absorptive coefficients: poly(dI-dC), 6.9 mm−1 cm−1; poly(dA-dT), 6.6 mm−1 cm−1; and for promoters and plasmids, 6.8 mm−1 cm−1 for the number of base pairs and Gusch 0.4123 m−1 cm−1 and GCbox2 0.3038 m−1 cm−1 for the number of molecules. Unlabeled AdoMet was purchased from Sigma-Aldrich; S-[methyl-3H]adenosyl-l-methionine was purchased from GE Healthcare.
Expression Constructs
The plasmids used for E. coli recombinant protein expression include pET28a-hDNMT3ACopt and pET28a-hDNMT3A_catalytic_domain (Δ1–611) as described in Purdy et al. (24). pTYB1-3L used to express hDNMT3L was kindly provided by Frederic Chedin (University of California Davis).
Protein Expression
Full-length DNMT3A and DNMT3L were expressed in E. coli strain Rosetta2 (DE3) pLysS (from Novagen), and the DNMT3A catalytic domain was expressed in E. coli strain ER2566 (New England Biolabs) that had previously been transformed with the pLysS plasmid (Novagen). Cell cultures were grown in 2× YT media at 37 °C to an A600 nm of 0.9 (DNMT3A), 0.4 (DNMT3L), and 0.7 (DNMT3A catalytic domain). The temperature was lowered for full-length DNMT3A and DNMT3A catalytic domain to 28 °C, and expression was induced by the addition of 1 mm isopropyl-β-d-thiogalactopyranoside (Fisher Scientific). Induction was 2 h for DNMT3A and DNMT3L and 4 h for the DNMT3A catalytic domain. Cells were harvested by centrifugation and frozen at −80 °C.
Full-length DNMT3A Purification
DNMT3A was purified from cells lysed in 50 mm Tris-HCl, 500 mm NaCl, pH 7.5, with 1 mm EDTA, 0.5 mm DTT, 0.2% (v/v) Triton X-100, 0.4 mm PMSF, and 1% (v/v) protease inhibitor mixture (Sigma-Aldrich) with a French press in the presence of 25 μg/ml DNase I and then clarified by centrifugation. The supernatant was diluted to 300 mm NaCl and loaded onto a Biorex-70 column (Bio-Rad). The column was washed with 50 mm Tris-Cl, 300 mm NaCl, pH 7.0, with 1 mm EDTA, 0.5 mm DTT, 0.1% Triton X-100, 0.2 mm PMSF, and 1 μm pepstatin A (BioRex wash buffer) followed by the same buffer but with 400 mm NaCl. The protein was eluted in BioRex wash buffer containing 800 mm NaCl (and no EDTA). The eluant was brought to 40 mm imidazole (nickel wash buffer) and loaded on a 2-ml His·Bind resin (Novagen), which was washed with nickel wash buffer followed by a short wash of the same buffer with no Triton X-100. Protein was eluted with nickel wash buffer (400 mm NaCl, 400 mm imidazole, and no Triton X-100). The protein was then dialyzed into storage buffer (50 mm Tris-Cl, 200 mm NaCl, 1 mm EDTA, 20% (v/v) glycerol, pH 7.2, with 0.5 mm DTT) and stored at −80 °C.
Purification of DNMT3A Catalytic Domain
The catalytic domain (Δ1–611) was purified as described in Purdy et al.(24). Briefly the catalytic domain was purified by a DEAE column and a nickel-Sepharose column followed by a BioRex-70 column and dialyzed into storage buffer.
Purification of DNMT3L
DNMT3L was purified from cells lysed by French press in nickel buffer (20 mm Tris-HCl, pH 7.5, 500 mm EDTA, 40 mm imidazole, with 0.2% Triton X-100, 10% glycerol) with 25 μg/ml DNase I and 1 mm PMSF and then clarified by centrifugation. Supernatant was loaded onto a nickel-nitrilotriacetic acid agarose (Qiagen). The column was washed with nickel buffer and eluted with nickel buffer with 500 mm imidazole. The eluted protein was dialyzed against chitin binding buffer (20 mm Tris-HCL, pH 8.5, 200 mm NaCl, 0.2% Triton X-100, and 10% glycerol) and bound to the chitin resin. DNMT3L was cleaved from the bound chitin-binding protein using chitin cleavage buffer (20 mm Tris-HCl, pH 8.5, 500 mm NaCl, 100 mm dithiothreitol, 0.2% Triton X-100, and 10% glycerol). The eluted protein was dialyzed into storage buffer and frozen at −80 °C (25).
Protein purities of 85, 90, and 95% for full-length DNMT3A, DNMT3A catalytic domain, and DNMT3L, respectively, were estimated by densitometry of Coomassie Brilliant Blue-stained SDS-PAGE gels. Protein concentrations were determined by using calculated (26) 280 nm extinction coefficients, which were within 20% of Bradford assay results using bovine serum albumin as a standard. The total concentration was calculated by multiplying the total protein concentration by the fraction of purity.
Methylation Assays
Reactions were carried out at 37 °C in a buffer of 50 mm KH2PO4/K2HPO4, 1 mm EDTA, 1 mm DTT, 0.2 mg/ml BSA, 20 mm NaCl, 2 μm AdoMet (a 6 Ci/mmol mix of unlabeled and [3H]methyl-labeled) at pH 7.8. Aliquots of 20 μl were removed from a larger reaction. Reactions were quenched by the addition of 500 μm unlabeled AdoMet and digestion with 50 μg/ml proteinase K at 37 °C for 5 min (27). Samples were spotted onto Whatman DE81 filters and then washed, dried, and counted as described previously (28).
kcat Values
kcat values were determined as described previously except with DNMT3A at 150 nm and 50 mm Tris-HCl, pH 7.8, in place of phosphate. For each substrate, the kcat value was obtained by varying the substrate concentration (10 concentrations per substrate between 0 and 50 μm bp). Each reaction was initiated with DNA, run for 1 h at 37 °C, and quenched as stated above. The data were fit to Michaelis-Menten or substrate inhibition equations using Prism Version 4.0 (GraphPad Software). kcat values were calculated by dividing the Vmax values by the amount of active enzyme in the reaction. The kcat values for each substrate were compared with the number of CpG sites and the size of the promoters, and data were fit to a linear regression using the Prism Version 4.0 program.
Processivity Assays
Processivity assays were carried out with DNMT3A or the DNMT3A-catalytic domain at 50 nm. The reaction was initiated following a 3-min preincubation at 37 °C by the addition of substrate DNA: poly(dI-dC) (5 μm), pBR322 (10 μm), ESF1 (10 μm), or p21-pCpGL (10 μm). The addition of 200 μm plasmid pCpGL (lacks CpG sites (22) after 20 min (unless otherwise noted)) forms the “chase” condition. The addition of this same chase DNA at the start of the reaction concurrent with DNA substrate addition provides a control.
DNMT3L Assays
DNMT3L was tested for its activation of DNMT3A by varying DNMT3L from 12.5 to 200 nm with 50 nm DNMT3A and poly(dI-dC) at 25 μm. DNMT3A and DNMT3L were preincubated in reaction buffer with AdoMet for 1 h at 37 °C before starting the reaction with DNA. Reactions were run for 1 h and stopped as stated above, and methylation was counted. A 1:1 ratio (50 nm) with the 1-h preincubation (with AdoMet) gave the maximum amount of activity, which was used for all DNMT3L assays. The -fold stimulation was calculated by product formed by DNMT3A with DNMT3L divided by product formed by DNMT3A without DNMT3L. Activation by DNMT3L on DNMT3A on poly(dI-dC) and GCbox2 was carried out with DNA at 20 μm, and the reaction was run for 1 h. Four replicates were used in each reaction to obtain the error bars shown in Figs. 1, 3, 4, and 5 (mean S.D.). Processivity assays, which include DNMT3L, had the same conditions (buffer, enzyme, and DNA concentrations) as the DNMT3A processivity assays, which included the preincubation of DNMT3L with DNMT3A as stated above.
FIGURE 1.

DNMT3A is processive on a variety of substrates, as shown using a pulse-chase assay. A, poly(dI-dC) (5 μm bp). B, EFS1 human promoter (10 μm bp). C, bacterial supercoiled plasmid pBR322 (10 μm bp). D and E, the p21 human promoter in the pCpGL plasmid, linearized (20 μm bp) (D) and supercoiled (20 μm bp) (E). Substrate was added at time 0 to start the reaction. Full-length DNMT3A is at 50 nm, and AdoMet is at 2 μm. ■ = only substrate; ● = substrate and then 200 μm bp pCpGL at 20 min; ▴ = substrate and 200 μm bp pCpGL at the start of the reaction.
FIGURE 3.

Commitment for processivity. A, processivity assays were performed by adding chaser DNA (pCpGL) at different times into the reaction. Reactions were run with full-length DNMT3A at 50 nm, AdoMet at 2 μm, substrate DNA (poly(dI-dC)) at 5 μm, and chaser DNA (pCpGL) at 200 μm. The timing of adding chaser DNA varied in each reaction, as indicated by arrows. Enzyme was preincubated with AdoMet for 3 min before DNA addition (similar results were seen by preincubating with DNA, data not shown). ■ = poly(dI-dC) at 5 μm; ● = poly(dI-dC) at 5 μm with 200 μm pCpGL at 20 min; ♦ = poly(dI-dC) at 5 μm and 200 μm pCpGL at 15 min, □ = poly(dI-dC) at 5 μm and 200 μm pCpGL at 10 min; ◇ = poly(dI-dC) at 5 μm and 200 μm pCpGL at 5 min, ▴ = poly(dI-dC) at 5 μm and 200 μm pCpGL at start of reaction. B, schematic of the actions of an enzyme when chaser DNA is added at different times.
FIGURE 4.

The N terminus of DNMT3A is not needed for processive catalysis. A, the pulse-chase processivity assay was preformed on the catalytic domain of DNMT3A. Chaser DNA (pCpGL) was added 20 min into the reaction. Enzyme was at 50 nm, AdoMet was at 2 μm, substrate DNA (poly(dI-dC)) was at 5 μm, and chaser DNA was at 200 μm. ■ = poly(dI-dC) at 5 μm, ● = poly(dI-dC) at 5 μm and 200 μm pCpGL at 25 min, ▴ = poly(dI-dC) at 5 μm and 200 μm pCpGL at the start of reaction. B, schematic of the domains of DNMT3A, DNMT3A catalytic domain, and DNMT3L.
FIGURE 5.

DNMT3L is a processivity factor of DNMT3A. A, DNMT3L activates DNMT3A, where DNMT3L is varied from 12.4 to 200 nm, and DNMT3A is at 50 nm. B, comparing DNMT3L activation on DNMT3A on a multiple recognition substrate poly(dI-dC) (25 μm) and a single site substrate GCbox2 (25 μm) in saturating conditions. C and D, processive assay same as in Fig. 1 but including DNMT3L. DNMT3A and DNMT3L (50 nm each) were preincubated for 1 h in reaction buffer with AdoMet at 2 μm followed by DNA addition at time 0. C, substrate, poly(dI-dC) (5 μm). D, the p21-pCpGL plasmid, supercoiled, was used as the substrate (20 μm). ■ = only substrates; ● = substrate and then 200 μm pCpGL added at 20 min; ▴ = substrate and 200 μm pCpGL at start of reaction. E and F, average number of catalytic events on a substrate before dissociation (n½ value), with and without DNMT3L; error bars are S.D. between three independent reactions.
Processivity Data Analyses
The data were fit to a non-linear regression (one phase decay). Data fit separately before the experimental reaction had chaser DNA added (time 0–20 min) and after chaser DNA was added (20–260 min), using Prism Version 4.0 (GraphPad Software). Error bars in Figs. 1, 3, 4, and 5 represent the mean S.D. of at least three replicates. The number of turnovers is calculated by product formed (nm) divided by catalytic active sites (nm) (total protein concentration times the percentage of active sites (18%)). The percentage of active sites (18%) was determined as described previously in Purdy et al. (24). Inactive DNMT3A that can bind DNA should not affect the assays or the measurement of processivity because the substrate DNA is in great excess to the total amount of enzyme (active and non-active enzyme).
The change in product formation between the non-chase and chase experiment is related as shown in Equation 1.
where P′chase is the product formed after chaser DNA is added and P′non-chase is the product formed without chaser for the same give time. r is a ratio of the product formed in the chase experiment when compared with non-chase experiment, providing a measure of the enzyme dissociation from the DNA.
Modeling of Processivity Data
The processivity profiles were analyzed using the non-linear regression package in Mathematica (Wolfram Inc.). The results were reported as the best fit values ± S.E. The processivity can be schematically described as
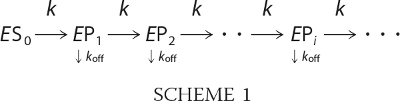 |
Every k is a turnover rate constant for methylation, and every koff is the dissociation rate constant for the enzyme-DNA complex. EPi represents the enzyme that has processed i steps, and ES0 is the initial enzyme-DNA complex. ES0 is equal to the total active enzyme concentration in the assay because the enzyme and the DNA concentration are well above the dissociation constant for the enzyme-DNA complex. ES0 is calculated by the amount of total enzyme multiplied by the percentage of active enzyme.
The following equations, modified from those described in Svedruzić and Reich (19), were used.
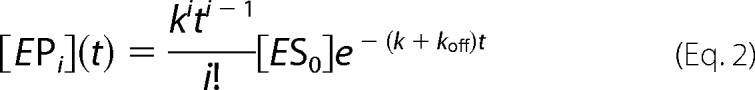 |
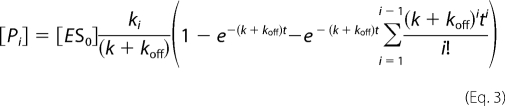 |
The equation for product formed in a processive reaction is a sum of all processive steps (i = 1 to n) and is equal to
 |
k and koff were modeled using Equation 3. The processivity probability (p) was determined using
 |
The probability for the processive step n is equal to
where Pn of 0.5 is equal to n½, which is the average number of catalytic turnovers before half of the enzymes are dissociated from the original substrate.
Analysis of the Ability of Different DNA to Function as a Suitable Chase
The substrate was poly(dI-dC), and the assays were performed as described for the processivity assay, using the following as chaser DNA: poly(dA-dT) (200 μm bp) and Gusch (short 30-mer with no CpG sites (50 μm molecules)).
M.SssI Processivity Assay
M.SssI (0.157 units, New England Biolabs) processivity assay was carried out as for DNMT3A other then different conditions due to the faster turnover by M.SssI. Conditions were M.SssI (0.157 units, New England Biolabs, 1.4 nm), 5 μm DNA (poly(dI-dC)), and AdoMet at 8 μm in 1× reaction buffer (New England Biolabs buffer 2). Chaser DNA was pCpGL at 200 μm added at 6 min.
RESULTS
Full-length DNMT3A, DNMT3A catalytic domain, and DNMT3L were purified to 85, 90, and 95% purity, respectively (supplemental Fig. 1). The enzyme shows maximum activity on poly(dI-dC) DNA. With this substrate, DNMT3A has a turnover rate constant of 1.2 ± 0.3 h−1, where the turnover rate constant is defined as the maximal turnover rate divided by the active protein concentration. This value falls into the range reported previously for recombinantly expressed DNMT3A of 1.8–1.07 h−1 (5, 24, 25, 27, 29). We recently showed that DNMT3A preparations are only partially active (18%) (24). Taking this into consideration, the turnover rate constant with poly(dI-dC) becomes 6.7 h−1 (Table 1). During steady-state turnover, the enzyme retains greater than 75% activity over the 4-h methylation reaction (supplemental Fig. 2).
TABLE 1.
kcat values for DNMT3A with different substrates
DNMT3A at 50 nm for poly(dI-dC), pBR322 and EFS1 and 150 nm for all other substrates. AdoMet at 2 μm, substrate concentration were varied and product formation measured after one hour. The data for each substrate was fit to Michaelis-Menten or substrate inhibition equations, then kcat values were obtained by the Vmax values divided by the amount of active enzyme.
| Substrate | Form | No. of CpG | Size | kcata |
|---|---|---|---|---|
| bp | h−1 | |||
| poly(dI-dC) | Linear | ∼800 | ∼1900 | 5.9 ± 0.3 |
| pBR322 | Supercoiled | 329 | 4359 | 1.5 ± 0.2 |
| EFS1 | Linear | 85 | 936 | 1.2 ± 0.3 |
| p21-pCpGL | Linear | 37 | 4275 | 0.74 ± 0.16 |
| p21-pCpGL | Supercoiled | 37 | 4275 | 0.42 ± 0.06 |
| ß-Actin | Linear | 3 | 550 | 0.38 ± 0.08 |
| CDC25 | Linear | 3 | 278 | 0.40 ± 0.06 |
| ODC | Linear | 14 | 1268 | 0.43 ± 0.07 |
| p15 | Linear | 29 | 865 | 0.83 ± 0.04 |
| p21 | Linear | 39 | 808 | 0.92 ± 0.06 |
| Smarca | Linear | 21 | 516 | 0.59 ± 0.07 |
| Survivin | Linear | 26 | 918 | 0.52 ± 0.06 |
| Xist | Linear | 6 | 117 | 0.32 ± 0.05 |
| Gcbox2 | Linear | 1 | 21 | 0.21 ± 0.03 |
a kcat values were obtained by dividing Vmax values by the amount of active enzyme for each substrate.
Design of the Processivity Assay
The processivity assay determines the length of time the enzyme stays associated with its DNA substrate and the number of turnovers using a pulse-chase experiment. Product formation is measured by the amount of tritiated methyl groups transferred by the enzyme to the DNA in a given time. Our conditions involve a 100–200-fold excess of DNA over enzyme, unless otherwise noted. Thus, each DNA molecule will be occupied by a single DNMT3A enzyme and the enzyme is allowed to carry out catalysis on multiple sites along a single piece of DNA. After the given time periods, the reaction is challenged with a 40-fold excess of chaser DNA (a plasmid, pCpGL, lacking the recognition site for DNMT3A) when compared with substrate DNA. A processive enzyme will continue to methylate the original substrate, and the chaser DNA will have no, or a delayed impact, on DNA methylation. A non-processive enzyme will dissociate after one methyl transfer; thus, after the addition of chaser DNA, the enzyme will bind the excess chaser DNA, leading to an immediate decrease in methylation activity.
The effects on turnover with the addition of chaser DNA are compared with the activity of the enzyme without chaser. An additional control has both the substrate and the chaser DNA added to the enzyme at the start of the reaction to confirm that the chase DNA suppresses product formation. In the experimental reaction, chaser DNA is added 20 min into the methylation assay. The change in product formation between the non-chase and chase experiment is given as r (Equation 1) for an indication of how fast the enzyme disassociates from its substrate. An r of 1 indicates that the chase and non-chase have the same amount of product formed, showing no dissociation. An r value of 0 shows that no product is formed after chaser DNA is added, an indication of no processive catalysis. Processivity was also calculated for DNMT3A using kinetic modeling originally developed for helicase processivity studies (30, 31) and previously used for DNMT1 (19). The modeling is based on the shape of the curve for the non-chase experiment, which determines the methylation rate (k) and dissociation rate (koff) (Equation 4). A processivity value (p, Equation 5) was determined from the k and koff rates. The processivity values were used to calculate n½, which indicates the average numbers of catalytic turnovers before half of the enzymes are dissociated from the original substrate.
We tested three different chaser DNA sequences, pCpGL, poly(dA-dT), and Gusch (a small 21-nucleotide oligonucleotide with no CpG sites). pCpGL is a 3872-bp plasmid lacking the recognition site for DNMT3A (CG). We found that poly(dA-dT) at 40-fold excess of the substrate DNA (poly(dI-dC)) did not decrease product formation when added at the start of the reaction (supplemental Fig. 4); Gusch also had little effect on product formation, with less than 10% decrease in product formation. DNMT3A had limited methylation on pCpGL (kcat 0.11 ± 0.03 h−1), and when mixed at 40-fold excess poly(dI-dC), it eliminated >90% of product formation. Similar results were previously seen with the highly processive M.SssI (20). The significant decrease in DNMT3A activity in the presence of CpG-lacking DNA suggests that DNMT3A can interact with DNA devoid of CpG residues but may not interact with substrates lacking cytosines (poly(dA-dT) or Gusch).
M.SssI Processivity
The processivity assay was first tested on a known processive DNA methyltransferase M.SssI (20) with the same recognition sequence as DNMT3A. The pulse-chase assay showed that adding in excess chaser DNA did not decrease product formation, and modeling showed an n½ of 346, demonstrating as expected that M.SssI is highly processive (supplemental Fig. 3).
DNMT3A Is Processive on a Variety of Substrates and Is Inhibited by Supercoiling
Using poly(dI-dC), DNMT3A carries out multiple rounds of catalysis before dissociating. 78% of the product formed in the absence of any chase DNA is formed with the chase reaction at 90 min (Fig. 1A) (r = 0.78 ± 0.01) (Table 2). Modeling the non-chase data revealed that DNMT3A on poly(dI-dC) has a processivity value (p) of 0.98 and an n½ of 27, indicating that 27 turnovers occur before 50% of the enzyme is dissociated from the poly(dI-dC) substrate (Table 3).
TABLE 2.
Chase Reaction Results
The difference in product formation between the non-chase and chase experiment is compared using Eq. 1 (r). r is the difference in product formation between the chase and non-chase experiment as an indication on how fast the enzyme is dissociating from its substrate.
| Proteins | Substrate | r at 90 mina | r at 240 minb |
|---|---|---|---|
| DNMT3A | Supercoiled p21 | 0.53 | 0.56 |
| DNMT3A | Linear p21 | 0.89 | 0.67 |
| DNMT3A/DNMT3L | Supercoiled p21 | 0.93 | 0.89 |
| DNMT3A | EFS1 | 0.86 | 0.77 |
| DNMT3A | pBR322 | 0.62 | 0.56 |
| DNMT3A | Poly(dI-dC) | 0.78 ± 0.04 | 0.63 ± 0.03 |
| DNMT3A/DNMT3L | Poly(dI-dC) | 0.84 ± 0.01 | 0.84 ± 0.06 |
a r = difference in product formation at 90 min.
b r = difference in product formation at 240 min.
TABLE 3.
Processivity values for DNMT3A from kinetic modeling
Best fit values for methylation rates (k), substrate dissociation rates (koff), (Eq. 4). All rates are given as the best fit value ± asymptotic standard error. The processivity probability p was calculated according to Equation 5. The n½ values (Eq. 6) represent a number of processive steps for the given reaction before 50% of the enzyme is dissociated of the substrate DNA.
| Proteins | Substrate | ka | koffb | pc | n½d |
|---|---|---|---|---|---|
| h−1 | h−1 | ||||
| DNMT3A | pBR322-supercoiled | 7.4 ± 0.40 | 1.0 ± 0.11 | 0.88 | 5.3 |
| DNMT3A | EFS1-linear | 4.8 ± 0.18 | 0.52 ± 0.05 | 0.90 | 6.6 |
| DNMT3A | p21-pCpGL-supercoiled | 1.2 ± 0.03 | 0.22 ± 0.02 | 0.85 | 4.2 |
| DNMT3A | p21-pCpGL-linear | 1.6 ± 0.08 | 0.14 ± 0.03 | 0.92 | 8.1 |
| DNMT3A/DNMT3Le | p21-pCpGL-supercoiled | 9.0 ± 0.59 | 0.48 ± 0.06 | 0.95 | 13 |
| DNMT3A | Poly(dI-dC)-linear | 11 ± 0.58 | 0.29 ± 0.04 | 0.98 | 27 |
| DNMT3A/DNMT3L | Poly(dI-dC)-linear | 18 ± 0.66 | 0.14 ± 0.02 | 0.99 | 91 |
| DNMT3A catalytic domain | Poly(dI-dC)-linear | 11 ± 0.80 | 0.25 ± 0.06 | 0.98 | 30 |
a Best fit values for methylation rate (k) ± standard error calculated according to Eq. 4.
b Best fit values for substrate dissociation rate (koff) ± standard error calculated according to Eq. 4.
c Processivity probability (p) was calculated according to Eq. 5.
d Represents the number of processive steps for the given reaction before 50% of the enzyme is dissociated for the substrate DNA (n½), calculated according to Eq. 6.
e DNMT3L preincubated with DNMT3A at a 1:1 ratio.
We then used biologically relevant DNA substrates for additional tests of DNMT3A processivity. These include the EFS1 human promoter, amplified from HeLa genomic DNA, which contains a CpG island with 85 CpG sites in a 936-bp linear fragment (Fig. 1B), and supercoiled pBR322 (Fig. 1C), a bacterial plasmid that has 329 CpG sites in the 4359-bp plasmid. DNMT3A shows processive catalysis on both substrates with an r value of 0.62 for pBR322 and 0.86 for EFS1 at 90 min. Modeling the non-chase data showed the same trend as the chase data. EFS1 has a processive value (p) of 0.90 and an n½ of 6.6, and pBR322 has a p value of 0.88 and an n½ of 5.3.
The impact of supercoiling was also examined with the human p21 promoter (CpG island with 39 CpG sites in a 530-bp promotor) amplified from HeLa genomic DNA and then inserted into the pCpGL (p21-CpGL) plasmid in the linear and supercoiled form. As shown in Fig. 1E, the supercoiled plasmid shows a decrease in activity shortly after the addition of the chaser DNA (r = 0.53) when compared with the linear plasmid (r = 0.89) (Fig. 1D). Modeling showed that the enzyme is twice as processive on linear DNA (n½ of 8.1 when compared with an n½ of 4.2) than the supercoiled form. This is caused by the enzyme dissociating from the substrate twice as quickly on the supercoiled DNA, a koff of 0.22 ± 0.02 h−1 when compared with a koff of 0.14 ± 0.03 h−1 for the linear DNA. The rate of methylation (k) remains largely unaltered, 1.2 ± 0.03 h−1 for supercoiled when compared 1.6 ± 0.08 h−1 for linear DNA (Table 3).
The Increase in kcat with the Number of CpG Sites Is Consistent with Processive Catalysis
A processive enzyme does not dissociate from its product between catalytic turnovers, which results in rate enhancement when the reaction rate is limited by product dissociation steps. Therefore, substrates with larger numbers of recognition sites have higher kcat values for a processive enzyme. For example, DNMT3A is most active with poly(dI-dC) (multiple site substrate) and has significantly lower kcat values on a single site substrate (Table 1). To systematically examine the relationship between kcat and the number of CpG sites, we investigated 10 human promoters (Table 1). These promoters contain CpG islands with varying densities of CpG sites. Fig. 2, C, D, and E, show representative Michaelis-Menten graphs. A positive linear correlation was found between kcat values and the number of CpG sites in the promoters with an R2 value of 0.85 (Fig. 2A). This correlation is not seen when comparing the length of DNA to kcat values, where R2 is equal to 0.45 (Fig. 2B). This is further evidence that DNMT3A is a processive enzyme, where the rate of product formation increases with the number of CpG sites in the DNA substrate.
FIGURE 2.

The number of CpGs in a promoter increases the rate of product formation. The turnover rate was calculated for DNMT3A on substrates with a varying CpG sites. A, kcat values were compared with the number of CpG sites in the DNA substrate. Each point is the kcat value for the human promoters and synthesized substrate with one CpG site. The inset includes poly(dI-dC). The regression line shows R2 = 0.85, with R2 = 0.98 for the inset. B, comparing the length of DNA with kcat values shows little correlation (R2 = 0.45). C–E, substrate inhibition plots for the PCR-amplified human promoters, which are representative plots for obtaining Vmax data. Vmax values were divided by the amount of active enzyme in the reaction to obtain kcat values.
Commitment to Processive Catalysis
We sought to determine whether DNMT3A processivity requires an initial period to assemble an enzyme-DNA complex that displays processive catalysis (commitment). The addition of chaser DNA after only 5 and 10 min of DNMT3A catalysis results in very little processive catalysis. In contrast, when chaser DNA was added at 15 min, there was an increase in processive catalysis but still less than when chaser DNA was added at least 20 min into the reaction. These experiments involved the preincubation of enzyme with AdoMet, but similar results were obtained by preincubating with DNA followed by AdoMet addition (data not shown). DNMT3A requires an initial period with AdoMet and DNA to assemble a processive complex, as diagrammed in Fig. 3B. The data show that the enzyme does not show the same processive behavior if challenged before one turnover occurs. DNMT3A requires an initial period to assemble a processive complex.
The DNMT3A Catalytic Domain Is Processive
The N-terminal domain of DNMT3A is not required for enzyme activity (32). We recently showed that the N-terminal PWWP domain in DNMT3A binds DNA, which may play a role in the processive mechanism of the enzyme (33). The DNMTs are diagrammed in Fig. 4B. The truncated enzyme lacking 525 residues of the N terminus shows processive catalysis (Fig. 4A). The processivity of the catalytic domain compares with the full-length protein, with an n½ of 30 turnovers when compared with 27 for the full-length protein. Previous studies found DNMT1 and DNMT3B were also processive without the full N-terminal domain (18, 32).
DNMT3L Increases DNMT3A Processivity
DNMT3L, a catalytically inactive homolog of DNMT3A, is necessary for methylation of imprinted genes (34). This is thought to involve the recruitment and activation of DNMT3A. DNMT3L activates DNMT3A in vitro 4-fold (16, 25) (Fig. 5A). How DNMT3L activates DNMT3A is not understood despite high resolution structures of the intact DNMT3L and a complex of the catalytic domains of DNMT3L and DNMT3A (15). One possible mechanism for DNMT3L activation is through increasing DNMT3A processivity, which would increase the efficiency of multiple methylations.
The mechanism of DNMT3L activation of DNMT3A was tested by comparing the activation on a single site substrate (GCbox2) with multiple site substrate (poly(dI-dC)). As shown in Fig. 5B, DNMT3L activates DNMT3A 1.5–2-fold when on a single site substrate and 4–5-fold on the multiple site substrate. The effect of DNMT3L on DNMT3A processivity was further tested by using the pulse-chase processivity assays (Fig. 5, C and D). The majority of DNMT3A remains bound to the initial substrate, staying associated through the 240 min of the reaction, over 40 turnovers. DNMT3A with DNMT3L on poly(dI-dC) has an r of 0.84 ± 0.06 h−1 when compared with an r of 0.63 ± 0.03 h−1 for DNMT3A by itself. Modeling the non-chase reaction of DNMT3L with DNMT3A on poly(dI-dC) shows that 91 turnovers would occur before 50% of the enzyme is dissociated from the substrate, when compared with 27 for the DNMT3A reaction in the absence of DNMT3L. The effect of DNMT3L on DNMT3A processivity was further shown using p21-pCpGL as the substrate, which showed an n½ of 13 when DNMT3L is present when compared with an n½ of 4.2 with only DNMT3A (Fig. 5D). Thus, DNMT3L increases DNMT3A processivity by 3-fold on both substrates tested. DNMT3L in addition activates 2–3-fold more on a multiple site substrate when compared with a single site substrate. DNMT3L increases kcat for DNMT3A on both substrates and decreases koff selectively with poly(dI-dC) (Table 3).
DISCUSSION
How new epigenetic marks such as DNA methylation and histone modifications are acquired remains a fundamental unanswered question. A functional understanding of the key enzymes known to carry out these processes is needed to fully appreciate how de novo changes occur. The mechanism investigated here is whether DNMT3A has the ability to methylate multiple CpG sites upon binding to DNA (diagrammed in Fig. 6). Processive methylation could explain how methylation spreading occurs on newly integrated viral DNA (35). Similarly, the recruitment of DNMT3A to a particular promoter followed by processive catalysis ensures that multiple CpG sites undergo methylation to induce transcriptional silencing (36). The regulation of processive methylation by interacting proteins or changing the form of the DNA provides another mechanism to control de novo methylation.
FIGURE 6.

Model of de novo methylation. A depiction of DNMT3A with DNMT3L carrying out either processive or non-processive catalysis is shown.
We provide three independent pieces of evidence that the full-length DNMT3A is processive. First, the excellent correlation between the number of CpG sites and kcat (Fig. 2) is expected for a processive enzyme where product release is rate-limiting; in contrast, a distributive enzyme will not show this behavior. Second, processivity experiments without any chase DNA (Fig. 1, A–E) show the anticipated curvature in product formation resulting from the partitioning of enzyme following each cycle of methylation. Kinetic modeling of these data was used to determine the relative contributions toward processivity deriving from the dissociation rate versus catalytic events (Table 3). This analytical approach, originally developed for helicase processivity studies (30, 31), was used to show that the maintenance DNMT1 is highly processive (19). The full-length DNMT3A is shown to be processive on diverse substrates using this assay (Fig. 1, A–E). On human promoters, between 5 and 8 methylation events are observed per binding event. The DNMT3A catalytic domain shows similar levels of processivity as the full-length protein, suggesting that the PWWP domain and other parts of the N-terminal domain are not essential for processivity (Fig. 4).
The chase assays provide further evidence for the inherent processivity of the full-length and catalytic domain of DNMT3A (Figs. 1, 3, and 4). The addition of chaser DNA after one turnover results in less than 25% decrease in product formation, whereas the addition of the same chaser at the start of the reaction causes more than 90% of the activity to be lost. These data indicate that the majority of enzyme is not dissociating from the substrate and that it carries out multiple rounds of catalysis on the same piece of DNA. However, no processivity was observed when DNMT3A was challenged by chaser DNA before one turnover (Fig. 3), showing that DNMT3A requires an initial period to assemble a processive complex.
Our demonstration that DNMT3A is processive with human promoters is consistent with several whole genome methylation analyses (37–39), which showed that human promoters have bimodal distributions of methylated and unmethylated sequences for total and individual subclones. The majority of promoters were either less than 10% methylated or greater than 80% methylated (37). Processive methylation by DNMT3A could certainly contribute to this bimodal distribution.
Our findings show that DNMT3A processivity can be modulated by DNA topology (Fig. 1, B–E) and protein-protein interactions (Fig. 5), which provide new avenues whereby de novo methylation may be regulated in the cell. The compaction of DNA has been proposed to alter DNMT function, and nucleosomes, a topologically strained form of DNA, are poor DNMT substrates (13, 40–42). We suggest that alterations to DNA topology regulate DNMT3A processivity, which in turn controls which CpG sites are methylated. This model is supported by chromatin remodelers being essential for DNA methylation in many model organisms (14, 43).
The number of proteins shown to interact with DNMT3A (and DNMT3B) continues to expand (44), although in only rare cases (16, 45, 46) have these interactions been shown to be direct. DNMT3A-interacting proteins include chromatin regulators (NCAPG, SMARCA, and ZNF2238) and ATP-dependent chromatin-remodeling enzyme (LSH and hSNF2H) (45, 46). LSH is proposed to facilitate DNMT processivity on nucleosomal DNA (46, 47).
The consequences on DNMT3A function resulting from protein-protein interactions remain poorly understood. Despite offering insight into the physical interaction between DNMT3L and DNMT3A, the co-crystal structure evokes no insight into how DNMT3L results in a 3–5-fold activation of DNMT3A activity (15). DNMT3L does not enhance the binding of DNMT3A to DNA (16) but causes a slight increase in AdoMet affinity (25). Recent work suggests that DNMT3L does not play a significant role in guiding DNMT3A to unmodified or modified nucleosomes (48). Knock-out of DNMT3L is lethal in mice when transmitted through the maternal germ line, and mutant males show reactivation of retrotransposons and extreme meiotic defects (49).
Our results (Fig. 6) provide evidence that DNMT3L is a processivity factor for DNMT3A. The underlying mechanism involves both an enhancement of catalysis and a stabilization of the DNMT3A-DNA complex. Our data support previous models that protein interactions increase the processivity of de novo methyltransferases (47). The activation by DNMT3L (Fig. 5A) is less pronounced on a single site substrate when compared with a multisite substrate because the former is incapable of revealing processive catalysis. The DNMT3A-DNMT3L interface is suggested to stabilize cofactor binding (16) as well as an active-site loop conformation (15). We suggest that the increase in stability of the DNMT3A-DNA complex in the presence of DNMT3L (Fig. 6, Table 3) may derive from alterations in the DNMT3A-DNA interface. It is intriguing to consider that the lethality of the DNMT3L knock-outs may in part derive from changes in DNMT3L modulation of DNMT3A processivity.
The modulation of processivity by interacting proteins occurs with many enzymes (50–53). Eukaryotic DNA polymerases become highly processive through their interactions with processivity factors, including clamp protein proliferating cell nuclear antigen, which encircles duplex DNA and interacts with the DNA polymerase (54). To our knowledge, all known processivity factors interact with the substrate of the enzyme (50–53). The mechanism of DNMT3L-mediated enhancement of DNMT3A processivity is not known but appears to be novel because DNMT3L is not thought to interact directly with DNA. The heterotetrameric co-crystal structure of the carboxyl-terminal domains of DNMT3A and DNMT3L lacks DNA; however, DNA from the M.HhaI-DNA co-crystal structure has been modeled into the DNMT3A structure and shows that neither of the DNMT3L subunits are likely to directly interact with DNA (15) (PDB: 2QRV, 3EEO).
Two prior reports suggested that the murine form of Dnmt3a does not act processively (5, 32). However, these studies were carried out in ways that preclude such interpretation. The initial report using full-length Dnmt3a reports a kcat of 0.05 h−1, yet the processivity studies were limited to time regimes under 90 min. Without the opportunity to carry out multiple rounds of catalysis, the processivity of an enzyme cannot be revealed. In subsequent work proffered as further evidence of a distributive mechanism using the catalytic domain of the murine Dnmt3a, the processivity assays used 50 nm DNA and 2 μm enzyme. Such single turnover conditions are incapable of revealing enzymatic processivity because multiple enzymes are binding to the same DNA molecules. The level of sequence relatedness of the murine and human enzymes (95% identity) suggests that the murine enzyme, like the human DNMT3A, is also processive.
Our finding that DNMT3A acts processively extends our understanding of this critical developmental regulator and biomedically relevant enzyme. Furthermore, the modulation of the processivity of DNMT3A by DNMT3L, DNA topology, or interactions with other cellular proteins or nucleic acids considerably expand the potential for regulation of the enzyme.
Supplementary Material
Acknowledgments
We are grateful to Zelijko Svedruzić for help in modeling the processivity data. We thank Michael Arensman for help in creating p21-pCpGL. We are also thankful to Douglas Matje for critical review of the manuscript. The pCpGL was kindly provided by Michael Rehli, and the pTYB1-3L used to express hDNMT3L was kindly provided by Frederic Chedin.
This work was supported by funds from the Cancer Research Coordinating Committee (University of California).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–4 and Table 1.
- DNMT
- DNA methyltransferase
- AdoMet
- S-adenosyl methionine
- h
- human
- CpG
- cytosine and guanine separated by a phosphate.
REFERENCES
- 1.Bird A. (2002) Genes Dev. 16, 6–21 [DOI] [PubMed] [Google Scholar]
- 2.Reik W., Dean W., Walter J. (2001) Science 293, 1089–1093 [DOI] [PubMed] [Google Scholar]
- 3.Li E. (2002) Nat. Rev. Genet. 3, 662–673 [DOI] [PubMed] [Google Scholar]
- 4.Robertson K. D. (2005) Nature Reviews Genetics 6, 597–610 [DOI] [PubMed] [Google Scholar]
- 5.Gowher H., Jeltsch A. (2001) J. Mol. Biol. 309, 1201–1208 [DOI] [PubMed] [Google Scholar]
- 6.Toth M., Müller U., Doerfler W. (1990) J. Mol. Biol. 214, 673–683 [DOI] [PubMed] [Google Scholar]
- 7.Bestor T. H. (2000) Hum. Mol. Genet. 9, 2395–2402 [DOI] [PubMed] [Google Scholar]
- 8.Chen Z. X., Riggs A. D. (2005) Biochem. Cell Biol. 83, 438–448 [DOI] [PubMed] [Google Scholar]
- 9.Hsieh C. L. (1999) Mol. Cell. Biol. 19, 8211–8218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fatemi M., Hermann A., Pradhan S., Jeltsch A. (2001) J. Mol. Biol. 309, 1189–1199 [DOI] [PubMed] [Google Scholar]
- 11.Pradhan S., Bacolla A., Wells R. D., Roberts R. J. (1999) J. Biol. Chem. 274, 33002–33010 [DOI] [PubMed] [Google Scholar]
- 12.Howell C. Y., Steptoe A. L., Miller M. W., Chaillet J. R. (1998) Mol. Cell. Biol. 18, 4149–4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takeshima H., Suetake I., Shimahara H., Ura K., Tate S., Tajima S. (2006) J Biochem. 139, 503–515 [DOI] [PubMed] [Google Scholar]
- 14.Cedar H., Bergman Y. (2009) Nat. Rev. Genet. 10, 295–304 [DOI] [PubMed] [Google Scholar]
- 15.Jia D., Jurkowska R. Z., Zhang X., Jeltsch A., Cheng X. (2007) Nature 449, 248–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suetake I., Shinozaki F., Miyagawa J., Takeshima H., Tajima S. (2004) J. Biol. Chem. 279, 27816–27823 [DOI] [PubMed] [Google Scholar]
- 17.Morris K. V. (2008) Curr. Top. Microbiol. Immunol. 320, 211–224 [DOI] [PubMed] [Google Scholar]
- 18.Vilkaitis G., Suetake I., Klimasauskas S., Tajima S. (2005) J. Biol. Chem. 280, 64–72 [DOI] [PubMed] [Google Scholar]
- 19.Svedruzić Z. M., Reich N. O. (2005) Biochemistry 44, 14977–14988 [DOI] [PubMed] [Google Scholar]
- 20.Renbaum P., Razin A. (1992) FEBS Lett. 313, 243–247 [DOI] [PubMed] [Google Scholar]
- 21.Surby M. A., Reich N. O. (1996) Biochemistry 35, 2201–2208 [DOI] [PubMed] [Google Scholar]
- 22.Klug M., Rehli M. (2006) Epigenetics 1, 127–130 [DOI] [PubMed] [Google Scholar]
- 23.Rickwood D. (1993) in DNA Topology, pp. 17–44, Oxford University Press, Oxford UK [Google Scholar]
- 24.Purdy M. M., Holz-Schietinger C., Reich N. O. (2010) Arch Biochem. Biophys. 498, 13–22 [DOI] [PubMed] [Google Scholar]
- 25.Kareta M. S., Botello Z. M., Ennis J. J., Chou C., Chédin F. (2006) J. Biol. Chem. 281, 25893–25902 [DOI] [PubMed] [Google Scholar]
- 26.Gill S. C., von Hippel P. H. (1989) Anal. Biochem. 182, 319–326 [DOI] [PubMed] [Google Scholar]
- 27.Suetake I., Miyazaki J., Murakami C., Takeshima H., Tajima S. (2003) J Biochem. 133, 737–744 [DOI] [PubMed] [Google Scholar]
- 28.Peterson S. N., Reich N. O. (2006) J. Mol. Biol. 355, 459–472 [DOI] [PubMed] [Google Scholar]
- 29.Aoki A., Suetake I., Miyagawa J., Fujio T., Chijiwa T., Sasaki H., Tajima S. (2001) Nucleic Acids Res. 29, 3506–3512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Porter D. J., Short S. A., Hanlon M. H., Preugschat F., Wilson J. E., Willard D. H., Jr., Consler T. G. (1998) J. Biol. Chem. 273, 18906–18914 [DOI] [PubMed] [Google Scholar]
- 31.Ali J. A., Lohman T. M. (1997) Science 275, 377–380 [DOI] [PubMed] [Google Scholar]
- 32.Gowher H., Jeltsch A. (2002) J. Biol. Chem. 277, 20409–20414 [DOI] [PubMed] [Google Scholar]
- 33.Qiu C., Sawada K., Zhang X., Cheng X. (2002) Nat. Struct. Biol. 9, 217–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bourc'his D., Xu G. L., Lin C. S., Bollman B., Bestor T. H. (2001) Science 294, 2536–2539 [DOI] [PubMed] [Google Scholar]
- 35.Orend G., Kuhlmann I., Doerfler W. (1991) J. Virol. 65, 4301–4308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turker M. S. (2002) Oncogene 21, 5388–5393 [DOI] [PubMed] [Google Scholar]
- 37.Eckhardt F., Lewin J., Cortese R., Rakyan V. K., Attwood J., Burger M., Burton J., Cox T. V., Davies R., Down T. A., Haefliger C., Horton R., Howe K., Jackson D. K., Kunde J., Koenig C., Liddle J., Niblett D., Otto T., Pettett R., Seemann S., Thompson C., West T., Rogers J., Olek A., Berlin K., Beck S. (2006) Nat. Genet. 38, 1378–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grunau C., Hindermann W., Rosenthal A. (2000) Hum. Mol. Genet. 9, 2651–2663 [DOI] [PubMed] [Google Scholar]
- 39.Strichman-Almashanu L. Z., Lee R. S., Onyango P. O., Perlman E., Flam F., Frieman M. B., Feinberg A. P. (2002) Genome Res. 12, 543–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gowher H., Stockdale C. J., Goyal R., Ferreira H., Owen-Hughes T., Jeltsch A. (2005) Biochemistry 44, 9899–9904 [DOI] [PubMed] [Google Scholar]
- 41.Okuwaki M., Verreault A. (2004) J. Biol. Chem. 279, 2904–2912 [DOI] [PubMed] [Google Scholar]
- 42.Robertson A. K., Geiman T. M., Sankpal U. T., Hager G. L., Robertson K. D. (2004) Biochem. Biophys. Res. Commun. 322, 110–118 [DOI] [PubMed] [Google Scholar]
- 43.Freitag M., Hickey P. C., Khlafallah T. K., Read N. D., Selker E. U. (2004) Mol. Cell 13, 427–434 [DOI] [PubMed] [Google Scholar]
- 44.Hervouet E., Vallette F. M., Cartron P. F. (2009) Epigenetics 4, 487–499 [DOI] [PubMed] [Google Scholar]
- 45.Fuks F., Hurd P. J., Deplus R., Kouzarides T. (2003) Nucleic Acids Res. 31, 2305–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Myant K., Stancheva I. (2008) Mol. Cell. Biol. 28, 215–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Athanasiadou R., de Sousa D., Myant K., Merusi C., Stancheva I., Bird A. (2010) PLoS ONE 5, e9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y., Jurkowska R., Soeroes S., Rajavelu A., Dhayalan A., Bock I., Rathert P., Brandt O., Reinhardt R., Fischle W., Jeltsch A. (2010) Nucleic Acids Res. 38, 4246–4253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Webster K. E., O'Bryan M. K., Fletcher S., Crewther P. E., Aapola U., Craig J., Harrison D. K., Aung H., Phutikanit N., Lyle R., Meachem S. J., Antonarakis S. E., de Kretser D. M., Hedger M. P., Peterson P., Carroll B. J., Scott H. S. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 4068–4073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berezuk M. A., Schroer T. A. (2007) Traffic 8, 124–129 [DOI] [PubMed] [Google Scholar]
- 51.Kato H., Sumimoto H., Pognonec P., Chen C. H., Rosen C. A., Roeder R. G. (1992) Genes Dev. 6, 655–666 [DOI] [PubMed] [Google Scholar]
- 52.Jeruzalmi D., O'Donnell M., Kuriyan J. (2002) Curr. Opin. Struct. Biol. 12, 217–224 [DOI] [PubMed] [Google Scholar]
- 53.Wang F., Podell E. R., Zaug A. J., Yang Y., Baciu P., Cech T. R., et al. (2007) Nature 445, 506–510 [DOI] [PubMed] [Google Scholar]
- 54.Bloom L. B. (2006) Crit. Rev. Biochem. Mol. Biol. 41, 179–208 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.