

Abstract
Disulfide formation in newly synthesized proteins entering the mammalian endoplasmic reticulum is catalyzed by protein disulfide isomerase (PDI), which is itself thought to be directly oxidized by Ero1α. The activity of Ero1α is tightly regulated by the formation of noncatalytic disulfides, which need to be broken to activate the enzyme. Here, we have developed a novel PDI oxidation assay, which is able to simultaneously determine the redox status of the individual active sites of PDI. We have used this assay to confirm that when PDI is incubated with Ero1α, only one of the active sites of PDI becomes directly oxidized with a slow turnover rate. In contrast, a deregulated mutant of Ero1α was able to oxidize both PDI active sites at an equivalent rate to the wild type enzyme. When the active sites of PDI were mutated to decrease their reduction potential, both were now oxidized by wild type Ero1α with a 12-fold increase in activity. These results demonstrate that the specificity of Ero1α toward the active sites of PDI requires the presence of the regulatory disulfides. In addition, the rate of PDI oxidation is limited by the reduction potential of the PDI active site disulfide. The inability of Ero1α to oxidize PDI efficiently likely reflects the requirement for PDI to act as both an oxidase and an isomerase during the formation of native disulfides in proteins entering the secretory pathway.
Keywords: Disulfide, Endoplasmic Reticulum (ER), Enzymes, Protein Folding, Protein Isomerase, Ero1 (ER Oxidase 1), Protein Disulfide Isomerase
Introduction
The process of disulfide formation occurring within secretory proteins entering the endoplasmic reticulum requires the combined activities of a protein disulfide isomerase (PDI)3 and Ero1 (1). In mammalian cells, PDI is one of a large family of enzymes that can exchange disulfides with substrate proteins (2). During this exchange, the active site of PDI, which contains a disulfide, becomes reduced and must then be reoxidized. This reoxidation is thought to be catalyzed by Ero1, which, following an internal disulfide exchange, can pass electrons onto molecular oxygen via enzyme-bound FAD (3). Hence, a relay system exists to allow the passage of electrons from the substrate protein to molecular oxygen via PDI and Ero1, resulting in the net formation of a disulfide in the substrate protein and the reduction of oxygen to hydrogen peroxide (3).
In addition to catalyzing the formation of disulfides in proteins, PDI also is involved in isomerization of non-native disulfides. These disulfides can be formed in substrate proteins during their normal folding pathway but must be resolved to allow folding to the native state (4). For this reaction to occur, the PDI active site must be in a reduced state. Although no enzymatic pathway has been found to reduce PDI, it is clear that the glutathione redox buffer in the endoplasmic reticulum can maintain PDI in a partially reduced state (5–7). By adjusting the redox buffer, it is possible for PDI to catalyze both oxidation and reduction reactions in vitro with model substrates (8). If similar reactions are catalyzed by PDI in the endoplasmic reticulum, the reduction potential of the active sites must be poised delicately both to allow the donation (to Ero1) and acceptance (from substrates) of electrons. If the reduction potential of the active site is too low, the disulfide will be quite stable, and PDI would be a poor oxidant; if it is too high, however, PDI would not be able to act as a reductant.
Classical PDIs contain four thioredoxin-homologous domains, two containing active sites, called the a and the a′ domains and two noncatalytic domains called the b and b′ domains (9) (see Fig. 1A). The b domain is thought to have primarily a structural role, whereas the b′ domain is thought to be involved in substrate binding (10). The active site domains are characterized by the presence of a CGHC motif, which shuttles from a dithiol to a disulfide form, allowing the exchange of a disulfide bond with its substrate. It has been demonstrated previously that Ero1α can preferentially oxidize the a′ domain of human PDI (11, 12) whereas Ero1p preferentially oxidizes the a domain of yeast PDI (13), leading to the question of how this specificity is achieved. Most recently, it has been shown that the minimal requirement for PDI to be oxidized by Ero1α is the presence of an active site domain C-terminal to the b′ domain, suggesting that the specificity of oxidation of PDI by Ero1α lies with the positioning of the active site domains (12). The implication is that there is a necessity for PDI to bind to Ero1α via its b′ domain, thereby aligning the active site motif next to the Ero1α active site disulfide to allow exchange to occur.
FIGURE 1.

Schematic depicting the assay of the redox status of protein thiols using unlabeled and isotopically labeled alkylating agents. The domain structure depicting the various thioredoxin-like domains is outlined in A. B, the protein redox status is first frozen by acid precipitation. To determine whether cysteines are in a free thiol or disulfide form, the samples are treated with iodoacetate under denaturing conditions. Proteins are then separated by SDS-PAGE, the protein band is excised, and an in-gel reduction followed by alkylation with an isotopically labeled bromoacetate carried out. An in-gel digest followed by MALDI-TOF is performed to determine the mass of the individual peptides. Peptides that contain cysteines that were in the dithiol form will be increased in mass by 2 Da per cysteine.
The activity of Ero1 toward its substrates is complicated by the additional requirement to initially reduce regulatory disulfides within Ero1 itself. Activation is required because both yeast Ero1 (Ero1p) and mammalian Ero1 (Ero1α) are inactivated by the formation of noncatalytic disulfides (11, 14, 15). The initial reduction and thereby activation of the enzyme may be brought about by the substrate itself. However, the Ero1α noncatalytic disulfides have a reduction potential of −280 mV, which is significantly below the previously determined reduction potential of the PDI active site disulfides (∼−180 mV) (16). In addition, the activity of Ero1p and Ero1α toward PDI is low when measured in vitro, compared with a non-natural substrate such as thioredoxin (11, 15). These results could reflect either the inability of the PDI active sites to reduce the regulatory disulfides within Ero1 or the possibility that Ero1 can only slowly oxidize PDI.
To study the ability of Ero1 to oxidize PDI in more detail, we have developed a technique to simultaneously measure the redox state of both active sites of PDI. This technique is based on the differential alkylation of the active site cysteines present as either a dithiol or disulfide (see Fig. 1B). We have used this approach to determine the equilibrium constant for the individual active sites using a glutathione redox buffer and shown that their reduction potentials are quite similar. In addition, we have assayed the direct oxidation of the individual active sites of PDI by Ero1α. The results show that the intrinsic activity of Ero1α toward PDI in vitro is limited by the reduction potential of the PDI active sites.
EXPERIMENTAL PROCEDURES
Plasmid Construction and Mutagenesis
Constructs expressing His-tagged PDI and ΔS1 and ΔS2 mutants were obtained from Lloyd Ruddock (Oulu, Finland). Histidine to proline mutations within the remaining catalytic CGHC domains of ΔS1 and ΔS2 mutants of PDI were created using QuikChange site-directed mutagenesis with Pfu DNA polymerase (Stratagene, Santa Clara, CA).
Protein Purification
Ero1α, PDI WT, and PDI mutant proteins were expressed and purified as described previously (11).
Reduction Potential Determination of PDI
Recombinant PDI (1 μm) was incubated for 16 h in 50 mm Tris-HCl, pH 7.5, 1 mm EDTA (thoroughly purged with nitrogen), supplemented with varying concentrations of GSSG (0.2–1 mm) and GSH (0.1–0.3 mm). PDI was precipitated from the reaction by addition of TCA to 10% (v/v), and the protein pellet was washed in ice-cold acetone before resuspension in 200 mm Tris-HCl buffer, pH 8.0, containing 6 m urea, 0.5% (w/v) SDS, and 50 mm iodoacetic acid (Sigma). Samples were incubated at 37 °C for 2 h in the dark. Samples were separated by SDS-PAGE, and gels were stained with Coomassie Blue. The PDI band was excised from the gel, washed twice with 50% (v/v) acetonitrile in 25 mm ammonium bicarbonate, dehydrated by incubation with acetonitrile, and dried by vacuum centrifugation. Gel slices were rehydrated in 10 mm DTT, 25 mm ammonium bicarbonate and incubated at 56 °C for 1 h. Gel slices were incubated with 50 mm 1,2-13C2-bromoacetic acid (Cambridge Isotopes), in 100 mm ammonium bicarbonate at 37 °C for 2 h in the dark. Gel slices were washed and then incubated alternately with ammonium bicarbonate and acetonitrile twice each and again dried by vacuum centrifugation. Wild type PDI gel slices were rehydrated with 5 ng/μl trypsin (Promega) in 25 mm ammonium bicarbonate and incubated overnight at 37 °C. Mutant PDI gel slices were rehydrated with 5 ng/μl trypsin and 10 ng/μl Glu-C (Sigma) in 100 mm Tris-HCl buffer, pH 8.5, and incubated overnight at 37 °C.
Peptides were extracted from gel slices by incubation with 50% (v/v) acetonitrile and 0.2% (v/v) formic acid for 30 min at room temperature. Samples were analyzed by MALDI-TOF mass spectrometry.
Kinetic Analysis of PDI Oxidation by Ero1
Reduced PDI was generated by incubation of the protein with 50 mm DTT at room temperature for 1 h. DTT was removed by buffer exchange through a PD10 column (GE Healthcare) into 50 mm Tris-HCl buffer, pH 7.5, containing 1 mm EDTA. Reduced PDI (50 μm) and Ero1 (2 μm) were mixed and incubated in 50 mm Tris-HCl buffer, pH 7.5, containing 1 mm EDTA at room temperature. At various time points, aliquots were removed, and TCA was added to a final concentration of 10% (v/v). TCA-precipitated proteins were washed twice with ice-cold acetone and resuspended in 200 mm Tris-HCl buffer, pH 8 containing 6 m urea, 0.5% (w/v) SDS, and 50 mm iodoacetic acid and incubated at 37 °C for 2 h in the dark. Proteins were processed to assess redox status as described for reduction potential determination of PDI.
Ratiometric Measurement of Reduced/Oxidized PDI by MALDI-TOF Mass Spectrometry
Mass spectra were acquired on an Ultraflex II time-of-flight mass spectrometer (Bruker Daltonics, Bremen, Germany) equipped with LIFT capability. All acquisitions were performed in positive ion reflectron mode, and the 355 nm wavelength smartbeam laser operated at 100 Hz. Mass spectra were acquired from 2000 laser shots with ions detected over an m/z range of 800–4000. Matrix ions were deflected up to m/z 650. For MS/MS spectra, ions were detected over an m/z range of 40–3620.
To calculate the fraction of reduced active site, we needed to take into consideration the contributions of naturally occurring isotopes. The active site peptides derived from oxidized protein will have a mass that is 4 Da greater than peptide derived from the reduced peptide as the 1,2-13C2-bromoacetic acid increases the mass by 2 Da, and there are two cysteines per PDI active site peptide. The ratio of the area under the fifth and the first isotope peak for the fully reduced peptide was determined by examination of multiple spectra. This ratio was used to calculate the contribution made by the reduced peptide to the signal from the oxidized peptide. This contribution was then subtracted from the signal for the oxidized active site peptide to provide a normalized value. If the area under the first isotopic peak = r and the normalized area under the oxidized active site peptide peak = ox, then the fraction of reduced PDI peptide in each sample was defined as r/(r + ox). The equilibrium constant Keq for the redox reaction was calculated according to Equation 1 assuming two molecules of glutathione are required to reduce the two cysteines in the PDI active site.
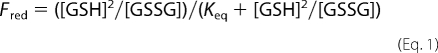 |
where Fred is the fraction of reduced PDI active site. The standard reduction potential for the active site disulfide could then be derived from the Nernst equation (E′0(PDI) = E′0(GSH) − (RT/nF) ln(Keq)) using the calculated Keq from Equation 1, T = 25 °C, n = 2, and the standard reduction potential of GSH = −240 mV.
RESULTS
Determining the Redox Status of PDI Active Sites
To determine the fraction of PDI active sites in either the dithiol or disulfide form, we needed to rapidly freeze the redox status. Previous work measuring the redox status of PDI has demonstrated that acid quenching followed by alkylation of free thiol under denaturing conditions is necessary to prevent changes to the redox status during sample preparation (17). The redox status of the active sites can then be measured by separation and quantification of the reduced and oxidized form by radiolabeling (16) or by HPLC (17). The major drawback to this approach is that the redox status of proteins with more than one active site disulfide cannot be assayed due to a lack of separation of the various forms by HPLC. This has meant that the redox status of the individual active sites of PDI have been only measured using individual a or a′ domains or using mutant PDI where either the a or a′ domains have been mutated.
To overcome this problem, we developed a differential alkylation approach (Fig. 1B). Here, the initial redox state of the active site is first trapped by acid quenching, and then any free thiols present are modified by carboxymethylation with iodoacetate under denaturing conditions. The resulting preparation is then subjected to SDS-PAGE, and the protein band is excised. In-gel reduction followed by carboxymethylation with 13C2-labeled bromoacetate is then carried out to isotopically label cysteine residues that were present as disulfides in the original sample. For each cysteine that originally formed a disulfide, there will be an increased peptide mass of 2 Da. The gel slices are then subjected to proteolysis, and the masses of the resulting peptides are identified by MALDI-TOF. Each PDI active site peptide contains two cysteine residues. The ratio of unlabeled (mass = n) to isotopically labeled (mass = n + 4) carboxymethylated peptides can be calculated to quantify the fraction of the peptide that was originally in the dithiol or disulfide form. As the PDI active site peptides have different masses, this approach allows us to simultaneously measure the redox status of each active site in the intact PDI molecule. The identity of each of the active site peptides was verified by MS/MS MALDI-TOF using the LIFT facility to provide a fragmentation pattern for the individual peptides.
We applied the differential alkylation approach to measure the reduction potential of the individual PDI active sites. PDI was equilibrated in GSH/GSSG buffer with varying reduction potentials, and the proteins were processed as described above. The nonisotopically labeled active site peptides have a mass of 1915 and 1944 Da for the a′ and a domain, respectively. In a redox buffer that is oxidizing (−92 mV), both the PDI active sites are in a disulfide form as evidenced by their monoisotopic peak at 1919 and 1948 (Fig. 2). The PDI active sites are progressively more reduced as the redox buffer becomes more reducing as evidenced by the appearance of a monoisotopic peak at 1915 and 1944. The redox status of the noncatalytic cysteines present in the b′ domain of PDI remained in a free thiol status following equilibration in all GSH/GSSG buffers as judged by their peptide masses of 861 and 963.
FIGURE 2.

Redox status of wild type PDI after equilibration in various GSH/GSSG buffers. Purified reduced PDI was incubated with a variety of GSH/GSSG buffers with the indicated reduction potentials for 16 h. The mass of the two individual active site peptides was then determined by MALDI-TOF following the protocol outlined in the legend to Fig. 1.
When the fraction of reduced PDI active site is plotted against the ratio [GSH]2:[GSSG] (Fig. 3B), the Keq can be calculated (Table 1). It is clear that the a and a′ domain active sites have very similar Keq values, which equate to reduction potentials of −162.7 ± 2.9 and −169.4 ± 2.3 mV, respectively. The fact that these values are quite similar would suggest that any difference in reactivity of the active site disulfides during disulfide exchange reactions is unlikely to be due to differences in reduction potential.
FIGURE 3.

Equilibrium constant measurements for wild type PDI active sites and CGHC to CGPC active site mutants. A, the domain structure of PDI indicating the active site mutants used in this study. B, equilibrium constant measurements for wild type PDI a and a′ active sites and the a HP and a′ HP mutants. The fraction of PDI active site reduced following incubation with each GSH/GSSG buffer was determined as in Fig. 2 and plotted versus [GSH]2/[GSSG].
TABLE 1.
Equilibrium constant and reduction potential of PDI active site disulfides
| Peptide |
||||
|---|---|---|---|---|
| WT PDI a domain | WT PDI a′ domain | PDI ΔS2 a HP | PDI ΔS1 a′ HP | |
| Peptide (m/z) | 1944 | 1915 | 1287 | 1327 |
| Keq (mm) | 2.64 ± 0.05 | 4.35 ± 0.07 | 413.3 ± 19.8 | 348.9 ± 69.8 |
| Reduction potential E′0 | −162.7 ± 2.9 | −169.4 ± 2.3 | −228.9 ± 0.6 | −225.9 ± 2.6 |
Can Ero1α Directly Oxidize PDI in the Absence of Added GSH?
The ability of either Ero1p or Ero1α to oxidize PDI has been demonstrated previously using an oxygen consumption assay (11, 15). However, for this assay, to provide a measurable change in the amount of oxygen consumed, it was necessary to include GSH in the reaction presumably to continuously reduce any oxidized PDI. The inclusion of GSH in the assay prevents any evaluation of the ability of Ero1α to directly oxidize PDI. To determine whether Ero1α can directly oxidize PDI in the absence of GSH, we assayed the redox status of both active sites of wild type PDI following incubation with Ero1α (Fig. 4B). In the absence of GSH the a′ and the a domain were oxidized, albeit with a slow turnover rate (∼ 0.14 min−1). In the presence of GSH, there was little oxidation of either active site of PDI (Fig. 4C) suggesting that the activity seen during the oxygen consumption assay is maintained by the rapid reduction of PDI by GSH. The alternative explanation that GSH is oxidized directly by Ero1α is unlikely, as previous work has shown that GSH was only consumed in the presence of Ero1p when PDI and reduced ribonuclease also are present (18). These results demonstrate that when GSH is not present, PDI is a substrate for Ero1α at least in vitro.
FIGURE 4.

Direct oxidation of PDI active sites by Ero1α. The PDI domain structure of the ΔS1 and ΔS2 construct is shown (A). Direct oxidation of the a and a′ active site of PDI (50 μm) by Ero1α (2 μm) in the absence (B) and presence (C) of added GSH. Direct oxidation of ΔS1 (D) and ΔS2 PDI (E; 50 μm) by Ero1α (2 μm) in the presence and absence of GSH (10 mm).
Previously, Ero1α was shown to be unable to oxidize the a domain in the absence of the a′ domain using an oxygen electrode assay (11). To determine whether the specificity toward the two active sites could be reproduced in our differential alkylation assay, we analyzed the activity of Ero1α toward PDI mutants where either active site CGHC motif was mutated to AGHA (Fig. 4A). The ΔS1 protein only has an active a′ domain, and the ΔS2 protein only has an active a domain. The results clearly show that only the ΔS1 protein, i.e. the a′ domain, was oxidized in the absence of GSH (Fig. 4, D and E). These results suggest that the a domain is not directly oxidized by Ero1α but can become oxidized by an internal disulfide exchange within PDI.
We then tested the idea that there is a requirement to reduce the regulatory disulfides in Ero1α to ensure specific oxidation of the a′ domain. Within Ero1α, at least two regulatory disulfides are formed between the first active site cysteines (Cys94 and Cys99) and noncatalytic cysteines (Cys104 and Cys131) to form both a Cys94–131 and a Cys99–104 linkage (Fig. 5A). To test whether preventing these regulatory disulfides from forming might change Ero1α specificity, we created an Ero1α protein with cysteine 104 and 131 mutated to alanine. It should be noted that there is an additional potential regulatory disulfide that forms in Ero1α between cysteine 85 and 391. We were unable to prepare a stable C85A/C391A mutant. We assayed the ability of the C104A/C131A mutant to oxidize the active sites in wild type PDI (Fig. 5B). Both active sites were oxidized by the C104A/C131A mutant giving rise to approximately twice the level of oxidized PDI than with the wild type Ero1α. To determine whether in this experiment oxidation of the a domain was due to an internal disulfide exchange between the a′ and a domain, we also assayed the ability of the C104A/C131A mutant to oxidize the ΔS2 protein (Fig. 5C). In contrast to the situation with the wild type Ero1α, the a domain was now oxidized with a similar rate to the a′ domain. These results demonstrate that the specificity of Ero1α toward the two active sites of PDI is in part dependent upon the presence of the regulatory disulfides. The fact that the a domain of PDI can be oxidized even when it is positioned N-terminal to the b′ domain also indicates that it is not simply the positioning of the PDI active site domains, which accounts for their differential oxidation (12).
FIGURE 5.

Direct oxidation of PDI active sites by deregulated Ero1α. A, schematic depicting WT and deregulated (C104A/C131A) Ero1α. Numbers indicate cysteine residues, and lines indicate predicted disulfide linkages in the two proteins. B, and C, direct oxidation of the a or a′ active site of WT PDI (50 μm) by deregulated Ero1α (2 μm). D, direct oxidation of ΔS1 PDI (50 μm) by deregulated Ero1α (2 μm).
What Is the Influence of the PDI Active Site Reduction Potential on Its Ability to Act as a Substrate for Ero1α?
The low turnover rate of PDI during its oxidation by Ero1α contrasts with a much faster rate with the nonphysiological substrate thioredoxin (∼40–60 min−1) (11, 12). The difference in rate could reflect the much lower reduction potential of the thioredoxin active site (−270 mV) (19). Such a low reduction potential would make thioredoxin more able to reduce the regulatory disulfides in Ero1α and more able to donate electrons to the active site of Ero1α. To test whether the reduction potential of the PDI active site limits its oxidation by Ero1α, we mutated the individual active sites from CGHC to CGPC. The mutated active site is now identical to the thioredoxin active site, which should lower the reduction potential of the active site disulfide. We created the mutations in either the ΔS1 or ΔS2 PDI constructs to ensure that we only assessed the ability of Ero1α to oxidize the individual active sites and to prevent any possible intramolecular disulfide exchange or competition between the active sites (Fig. 3A). The proteins were designated ΔS1 a′ HP or ΔS2 a HP to indicate the change in the a′ or a domain, respectively.
We first determined the consequence of the CGPC mutation on the reduction potential of the individual PDI active sites. As expected, there was a significant increase in the Keq for both active sites (Fig. 3B and Table 1) equating to a decrease in the reduction potential to −228.9 ± 0.6 mV and −225.9 ± 2.6 mV for the a and a′ domain, respectively. Although these values are still not as low as thioredoxin, the effect will be to enhance the ability of the active site to act as a reductase. Changing the a domain to CGPC has been shown previously to reduce the activity of PDI, but it is still much more active than thioredoxin as an oxidase (20). Hence, although the change in active site residue creates the same consensus sequence as thioredoxin, there are still significant differences between these two proteins in terms of reduction potential and ability to act as oxidoreductases.
We next determined the ability of these active site mutants to be oxidized by Ero1α. Following incubation with wild type Ero1α, there was a dramatic increase in the oxidation of both the CGPC a and a′ mutants (Fig. 6, A and B). The a domain was now oxidized by wild type Ero1α and the increase in turnover rate for the a′ domain was ∼12 fold (1.65 min−1). Despite the clear increase in rate the activity of Ero1α toward its physiological substrate in the absence of GSH is still considerably less than the activity toward thioredoxin. The results presented here confirm that Ero1α is able to access the a domain of PDI to catalyze oxidation and that the oxidation of PDI is limited due to the relatively high reduction potential of its active site disulfides.
FIGURE 6.

The oxidation of PDI by Ero1α is increased when the reduction potential of the active site disulfide is reduced. All assays were performed with 50 μm PDI (various mutants as depicted) and 2 μm wild type Ero1α. Samples were processed for mass spectrometry as described in the legend to Fig. 1.
DISCUSSION
We have described here an approach to assay the redox status of proteins based on differential alkylation of thiols with unlabeled and isotopically labeled cysteine-specific alkylating agents. The ability to assay simultaneously the redox status of different disulfides in the same protein enabled us to determine both the reduction potential of the PDI active sites and the ability of Ero1α to oxidize the individual active sites. Our measurements for the Keq of PDI a and a′ domains (2.6 and 4.4 mm) are similar to the value obtained previously (3 mm) using an approach that probably measures a composite value for both active sites (16). In addition, the values determined for the isolated a and a′ domains (0.7 and 1.9 mm) (17) also are quite similar to our results, which show that there are only minor changes to the redox properties of the active site domains when in the intact protein and when in isolation. Such similarity of reduction potential between the active sites suggests that differences in the stability of the disulfide are an unlikely explanation for their differential oxidation by Ero1α.
We have shown here that the specificity of Ero1α toward the two active sites of PDI is determined in part by the presence of regulatory disulfides in Ero1α. Such a result could be explained if the a′ domain is required to initially reduce the regulatory disulfide in Ero1α prior to oxidation of further PDI molecules by the Ero1α active site. For each regulatory disulfide reduced, one molecule of PDI will be oxidized, so this reaction would give rise to a low level of oxidized PDI. A single turnover of Ero1α (2 μm) could not explain the amount of PDI that does become oxidized (∼40 μm); however, the reduced regulatory disulfide in Ero1α could potentially become reoxidized by an internal disulfide exchange, allowing further PDI molecules to be oxidized. Arguing against the idea that PDI simply is being oxidized during the reduction of Ero1α regulatory disulfides is the fact that both PDI active sites are more efficiently oxidized by the C104A/C131A mutant of Ero1α. In addition, PDI only reduces the regulatory disulfides in a fraction of the total Ero1 when incubated in the absence of GSH (11).
It has been suggested previously that the a′ domain mainly acts as an oxidase, whereas the function of the a domain is as an isomerase (21). The suggestion was based on the preferential oxidation of the a′ domain of yeast PDI by Ero1p and the inhibition of oxidation of the a domain in the presence of the substrate protein ribonuclease. More recent data suggest that Ero1p actually preferentially oxidizes the a domain of yeast PDI (13). The difference in the reports was suggested to be due to a difference in the method of preparation of reduced PDI for the assays (13). The presence of a noncatalytic disulfide in the a domain of yeast PDI may have been reduced in the study by Kulp et al. (21), which remains oxidized in the report by Vitu et al. (13). Such a complication is not relevant to this study, however, as there are only two noncatalytic cysteines in human PDI, both in the b′ domain, and both of which are present as free thiols. Measurement of the reduction potentials of the individual active sites of yeast PDI indicate that, in contrast to human PDI, they differ significantly with the a domain having the lower reduction potential (13). Hence, with yeast PDI, Ero1p preferentially oxidizes the active site with the lower reduction potential.
When the redox status of the individual active sites of mammalian PDI was measured in vivo, it was discovered that both active sites were oxidized equally (22). The oxidized a domain could be generated by either an internal exchange of disulfides between the a′ and a domains or as a product of the net reduction of substrate disulfides. We saw clear evidence of a disulfide exchange between the two active sites over the time course of our experiments, as the a domain became oxidized when the a′ domain was intact but did not become oxidized by Ero1α when the a′ domain was mutated.
Our results demonstrate that altering the reduction potential of the PDI active sites can determine the efficiency of their oxidation by Ero1α. It is highly likely that this increased turnover is a consequence of an increased ability to donate electrons to the Ero1α active site. Clearly, the activity of Ero1α toward PDI is limited, at least in our in vitro assay, by the relatively high reduction potential of the PDI active sites. Previously, we have shown that the regulatory disulfides in Ero1α collectively have a relatively low reduction potential, which would ensure that they are only poorly reduced by PDI (11). Such a disparity in reduction potentials argues for additional proteins being involved in the reduction and activation of Ero1α in vivo. Alternatively, the inefficient oxidation of PDI by Ero1α may be necessary to ensure that the intracellular pool of PDI is not overly oxidized, ensuring it can act as a reductase during the resolution of non-native disulfides. In addition, if Ero1 efficiently oxidized PDI in the presence of GSH, then a futile cycle of oxidation and reduction of PDI could be established, ultimately leading to the generation of hydrogen peroxide (3). The requirement for tight regulation of Ero1 to prevent excessive hydrogen peroxide production may have led to the relatively inefficient oxidation of PDI.
An alternative explanation for the lack of efficient direct oxidation of PDI by Ero1α could be that there are alternative substrates for this enzyme in vivo. It is clear that substrate-trapping mutants of several PDI family members can form mixed disulfides with Ero1α (23). Evidence for PDI being a substrate for Ero1α and its mammalian homologue Ero1β in vivo has come from studies on the overexpression of these proteins in mammalian cells (14). In both cases, overexpression led to an increased oxidation of PDI. The increased oxidation of PDI in these studies could be the result of a direct oxidation by Ero1 or by disulfide exchange with other PDI family members, which are themselves oxidized by Ero1α.
Recently, it has been shown that the mammalian Ero1s are nonessential (24). The pathway for disulfide formation in mice lacking both Ero1α and Ero1β was investigated, and remarkably, the mice were viable though compromised in their ability to secrete some disulfide-containing proteins. The viability of the Ero1-deficient mice is in stark contrast to the essential function of Ero1 in Saccharomyces cerevisiae (25, 26) but is consistent with the viability of Ero1-deficient Drosophila melanogaster (27). Hence, it would appear that the Ero1s are essential to ensure disulfide formation in lower eukaryotes but not in higher eukaryotes; although there is a requirement for the Ero1s to correct fold particular proteins, alternative pathways for disulfide formation may exist to compensate in their absence.
In summary, we have shown that the reduction potential of the PDI active sites is delicately poised to ensure oxidation by Ero1α ensuring that PDI remains only partially oxidized in vivo. The presence of primarily reduced PDI in the ER allows the enzyme to continue to act as a reductase or isomerase (6). The dependence on the reduction potential of the active site for activity demonstrates that, although the specificity of Ero1α is for disulfides within thioredoxin domains, the efficiency of oxidation will vary considerably within the various members of this protein family.
Acknowledgment
We greatly appreciate the gift of cDNA for PDI wild type and mutants from Lloyd Ruddock (University of Oulu, Finland).
This work was supported by Wellcome Trust Grants 074081 and 088053.
- PDI
- protein disulfide isomerase.
REFERENCES
- 1.Sevier C. S., Kaiser C. A. (2008) Biochim. Biophys. Acta. 1783, 549–556 [DOI] [PubMed] [Google Scholar]
- 2.Ellgaard L., Ruddock L. W. (2005) EMBO Rep. 6, 28–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gross E., Sevier C. S., Heldman N., Vitu E., Bentzur M., Kaiser C. A., Thorpe C., Fass D. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 299–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jansens A., van Duijn E., Braakman I. (2002) Science 298, 2401–2403 [DOI] [PubMed] [Google Scholar]
- 5.Chakravarthi S., Bulleid N. J. (2004) J. Biol. Chem. 279, 39872–39879 [DOI] [PubMed] [Google Scholar]
- 6.Jessop C. E., Bulleid N. J. (2004) J. Biol. Chem. 279, 55341–55347 [DOI] [PubMed] [Google Scholar]
- 7.Molteni S. N., Fassio A., Ciriolo M. R., Filomeni G., Pasqualetto E., Fagioli C., Sitia R. (2004) J. Biol. Chem. 279, 32667–32673 [DOI] [PubMed] [Google Scholar]
- 8.Freedman R. B. (1995) Curr. Opin. Struct. Biol. 5, 85–91 [DOI] [PubMed] [Google Scholar]
- 9.Freedman R. B., Hirst T. R., Tuite M. F. (1994) Trends Biochem. Sci. 19, 331–336 [DOI] [PubMed] [Google Scholar]
- 10.Nguyen V. D., Wallis K., Howard M. J., Haapalainen A. M., Salo K. E., Saaranen M. J., Sidhu A., Wierenga R. K., Freedman R. B., Ruddock L. W., Williamson R. A. (2008) J. Mol. Biol. 383, 1144–1155 [DOI] [PubMed] [Google Scholar]
- 11.Baker K. M., Chakravarthi S., Langton K. P., Sheppard A. M., Lu H., Bulleid N. J. (2008) EMBO J. 27, 2988–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L., Li S. J., Sidhu A., Zhu L., Liang Y., Freedman R. B., Wang C. C. (2009) J. Biol. Chem. 284, 199–206 [DOI] [PubMed] [Google Scholar]
- 13.Vitu E., Kim S., Sevier C. S., Lutzky O., Heldman N., Bentzur M., Unger T., Yona M., Kaiser C. A., Fass D. (2010) J. Biol. Chem. 285, 18155–18165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Appenzeller-Herzog C., Riemer J., Christensen B., Sørensen E. S., Ellgaard L. (2008) EMBO J. 27, 2977–2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sevier C. S., Qu H., Heldman N., Gross E., Fass D., Kaiser C. A. (2007) Cell 129, 333–344 [DOI] [PubMed] [Google Scholar]
- 16.Lundström J., Holmgren A. (1993) Biochemistry 32, 6649–6655 [DOI] [PubMed] [Google Scholar]
- 17.Darby N. J., Creighton T. E. (1995) Biochemistry 34, 16770–16780 [DOI] [PubMed] [Google Scholar]
- 18.Tu B. P., Ho-Schleyer S. C., Travers K. J., Weissman J. S. (2000) Science 290, 1571–1574 [DOI] [PubMed] [Google Scholar]
- 19.Krause G., Holmgren A. (1991) J. Biol. Chem. 266, 4056–4066 [PubMed] [Google Scholar]
- 20.Lu X., Gilbert H. F., Harper J. W. (1992) Biochemistry 31, 4205–4210 [DOI] [PubMed] [Google Scholar]
- 21.Kulp M. S., Frickel E. M., Ellgaard L., Weissman J. S. (2006) J. Biol. Chem. 281, 876–884 [DOI] [PubMed] [Google Scholar]
- 22.Appenzeller-Herzog C., Ellgaard L. (2008) Antioxid. Redox. Signal. 10, 55–64 [DOI] [PubMed] [Google Scholar]
- 23.Jessop C. E., Watkins R. H., Simmons J. J., Tasab M., Bulleid N. J. (2009) J. Cell Sci. 122, 4287–4295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zito E., Chin K. T., Blais J., Harding H. P., Ron D.J. Cell Biol. 188, 821–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pollard M. G., Travers K. J., Weissman J. S. (1998) Mol. Cell 1, 171–182 [DOI] [PubMed] [Google Scholar]
- 26.Frand A. R., Kaiser C. A. (1998) Mol. Cell 1, 161–170 [DOI] [PubMed] [Google Scholar]
- 27.Tien A. C., Rajan A., Schulze K. L., Ryoo H. D., Acar M., Steller H., Bellen H. J. (2008) J. Cell Biol. 182, 1113–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]