

Abstract
Co-amplification and co-overexpression of ErbB2 and Grb7 are frequently found in various cancers, including breast cancer. Biochemical and functional correlations of the two molecules have identified Grb7 to be a pivotal mediator downstream of ErbB2-mediated oncogenesis. However, it remains largely unknown how Grb7 is involve in the ErbB2-mediated tumorigenesis. In this study, we show that Grb7-mediated cell proliferation and growth are essential for the tumorigenesis that occurs in ErbB2-Grb7-overexpressing breast cancer cells. Intrinsically, EGF-induced de novo Grb7 tyrosine phosphorylation/activation recruits and activates Ras-GTPases and subsequently promotes the phosphorylation of ERK1/2, thereby stimulating tumor growth. Furthermore, we also found the anti-tumor effect could be synergized by co-treatment with Herceptin plus Grb7 knockdown in Sk-Br3 breast cancer cells. Our findings illustrate an underlying mechanism by which Grb7 promotes tumorigenesis through the formation of a novel EGFR-Grb7-Ras signaling complex, thereby highlighting the potential strategy of targeting Grb7 as an anti-breast cancer therapy.
Keywords: Adaptor Proteins, Breast Cancer, ERK, Growth Factors, Protein Phosphorylation, Ras, Receptor Tyrosine Kinase, ErbB2, Grb7, Tumorigenesis
Introduction
Growth factor receptor-bound protein-7 (Grb7)2 is the prototype of an emerging adaptor protein family that includes Grb10 and Grb14 and contains an N-terminal proline-rich region followed by a putative RA (Ras-associating) domain, a central PH (pleckstrin homology) domain, a BPS (Between the PH and SH2 domains), motif and a C-terminal SH2 domain (1, 2). Most of these domains are known to interact with a variegated spectrum of receptors and/or regulators, through which Grb7 is capable of activating versatile signal transduction pathways to modulate various cellular functions (1, 2). For instance, the SH2 domain of Grb7 is capable of binding to the phospho-tyrosine sites of receptor tyrosine kinases such as ErbB family receptors, PDGF receptor, Tek/Tie2, c-Kit, and EphB1, as well as cytoplasmic proteins such as SHPTP2, Shc, and FAK (focal adhesion kinase) (2). The PH domain is the calmodulin- or phosphoinositide-binding domain that is responsible for plasma membrane localization and is also accessible for phosphorylation by FAK (3, 4). Structural prediction and biochemical validation both support the direct interaction of the RA domain with Ras-GTPases, similar to the canonical ubiquitin-like folded Ras-binding domain of c-Raf and RalGDS (5). Using yeast two-hybrid screening, Tankyrase 2, an ankyrin repeat-containing poly(ADP-ribose) polymerase, was identified to interact with the N-terminal proline-rich region of Grb14 (6). However, the downstream signaling targets of the identified interactions and their corresponding functional consequences remain largely unknown.
Originally identified as an activated EGF receptor-binding partner, Grb7 has often been found to be co-amplified with ErbB2 in several cancers because both genes are located in the chromosome 17q12 (7). In fact, Grb7 is concurrently overexpressed with ErbB2 in about 30% of human breast cancers, and in these cases, the patients present a poor recurrence survival rate. Therefore, Grb7 is warranted to be used as an adverse prognostic marker for breast cancer (7). In accordance with this finding, Grb7 has been shown to promote tumor cell proliferation, survival, motility, and even angiogenesis, and the role of Grb7 in cell migration has been well documented (1, 2). For instance, the overexpression of Grb7 results in invasive and/or metastatic consequences in various cancers and tumor cells (7–9). The interaction of Grb7 with autophosphorylated FAK at Tyr-397 could also promote integrin-mediated cell migration in NIH 3T3 and CHO cells, whereas the overexpression of its SH2 domain, a dominant negative mutation of Grb7, has been shown to inhibit cell migration (10, 11). The recruitment and phosphorylation of Grb7 by EphB1 receptors were shown to enhance cell migration in an ephrin-dependent manner (12). In contrast, Grb7 knockdown by RNAi resulted in the inhibition of breast cancer motility (13). G7–18NATE, a selective Grb7-SH2 domain affinity cyclic peptide, was demonstrated to efficiently block the cell migration of tumor cells (14, 15). Nevertheless, it remains to be elucidated how the genetic and/or epigenetic variations in Grb7 link to various aspects of pathophysiological relevance, such as tumorigenesis, through the Grb7-mediated cellular functions.
In this study, we aimed to elucidate the role of Grb7 in the ErbB2 oncogenic tumorigenesis of breast cancer. Additionally, to understand the underlying mechanism(s) by which Grb7 promotes tumorigenesis of breast cancer, we evaluated the tumorigenic capability of Ras-ERK signaling in response to EGF-induced Grb7 phosphorylation/activation in Sk-Br3 cells. Meanwhile, the effect of targeting Grb7-mediated EGF-dependent tumorigenesis was examined.
MATERIALS AND METHODS
Reagents
Protein A-Sepharose 4B, fibronectin, 5′-bromo-2-deoxyuridine (BrdU), and monoclonal α-BrdU antibody were purchased from Sigma-Aldrich. Herceptin (Trastuzumab) was obtained from Roche Applied Science (Genentech Inc.). Lipofectamine 2000TM and Dulbecco's modified Eagle's medium (DMEM) were obtained from Invitrogen. The α-phosphotyrosine monoclonal antibody PY99, α-HA, the polyclonal α-Grb7, α-HA, α-GFP, α-Ras, α-EGFR, α-ErbB2, and α-actin antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The polyclonal α-ERK1/2, α-pERK (pT202/pY204), α-p38 MAPK, α-phospho-p38 MAPK (Thr-180/Tyr-182), α-SAPK/JNK, α-phospho-SAPK/JNK (Thr-183/Tyr-185), α-AKT, α-pS473-AKT, α-STAT3, and α-pY705-STAT3 antibodies were purchased from Cell Signaling (Danvers, MA). The PD98059 was from Calbiochem (Darmstadt, Germany), and the EGF was from Millipore (Billerica, MA).
Cell Culture
Sk-Br3 human breast cancer cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS). NIH 3T3 cells were cultured in DMEM containing 10% calf serum. Cell transfection was carried out by Lipofectamine 2000TM according to the manufacturer's instructions. Grb7 knockdown cells were generated by lentiviral infection of shGrb7-12, shGrb7-13, or their combination as described previously (16). Cells infected with shLuc (lentiviruses expressing a small hairpin fragment of luciferase gene) was used as a control for all of the gene knockdown experiments in this study.
BrdU Incorporation Assay
After serum starvation for 24 h in DMEM with 0.5% FBS, cells were incubated for 24 h in DMEM containing 10% FBS and 100 μm BrdU. To monitor the BrdU incorporation rate, cells were subjected to immunofluorescent staining as described previously (17). Cells were then counted in multiple fields and scored for BrdU-positive staining in each independent experiment.
Tumor Growth in SCID Mice
For the in vivo tumorigenesis assay, a suspension of 5 × 106 Sk-Br3 cells with or without Grb7 knockdown, through infection with lentiviruses encoding either Grb7 shRNA (shGrb7-12 and shGrb7-13) or luciferase shRNA (shLuc), was injected subcutaneously into the right flanks of 8-week-old NOD.CB17-PrkdcscidSCID mice. Tumor volumes and numbers were measured on the 21st day after injection; they were excised, photographed, and weighed.
Soft Agar Assay
Anchorage-independent growth was examined using soft agar assays as described previously (16). We supplemented the cells twice a week with DMEM containing 10 ng/ml EGF, and for some experiments, PD98059 (20 μm) or lentivirus harboring shLuc, shGrb7 (shGrb7-12 and shGrb7-13), or RasG12V was added 2 days before the assays. After the 2nd week of treatment, colony numbers were scored using a phase-contrast microscope at 20× magnification. Images were captured using Image-Pro Plus software under a light microscope (model IX71, Olympus, Japan) equipped with a charge-coupled device camera (DP70, Olympus, Japan).
GST-(Raf)-RBD Pulldown Assay
The Ras-binding domain of Raf- or Grb7-tagged glutathione S-transferase (GST) fusion protein (GST-Raf-RBD or GST-RBD; GST-RA-WT/GST-RA-R134L, respectively) was precoupled with glutathione-Sepharose 4B according to the manufacturer's instructions and then incubated with Sk-Br3 cell lysates for 90 min at 4 °C to detect GTP-bound Ras-GTPases as described previously (18). The amount of bound, activated Ras-GTP in the pulldowns and total Ras in lysates was visualized by immunoblotting with Ras antibodies.
Co-immunoprecipitation and Western Blot Analysis
Proteins were extracted as described and subjected to co-immunoprecipitation and/or Western blotting (4), and proteins were detected using chemiluminescence.
Anti-tumor Assays by Herceptin
Stable pool Sk-Br3 cells infected by shLuc or shGrb7 (combination of shGrb7-12 and shGrb7-13) (105 cells/well) were seeded into 24-multiwell dishes (Corning) and treated with 2.5 μg/ml Herceptin (Trastuzumab, Genentech) every 24 h; untreated cells served as controls. At 1, 2, and 3 days after Herceptin treatment, the live cells were measured by hemocytometer. The representative cell images of each treatment were photographed by a light microscope (Model IX71, Olympus, Japan) equipped with a charge-coupled device camera (DP71, Olympus, Japan).
RESULTS
Grb7 Is Involved in the Tumorigenesis of Breast Cancer
Concurrent with the high level of ErbB2, Grb7 expression in the Sk-Br3 breast cancer cells was also elevated, which is consistent with the commonly found co-amplification of both genes in a variety of cancers, including breast cancer (19). To assess how Grb7, in the context of ErbB2 overexpression, participates in the tumorigenesis of breast cancer, we employed the Sk-Br3 breast cancer cell line for further investigations. A lentivirus-based Grb7 shRNA approach for Grb7 gene knockdown, which we had previously established (16), also resulted in an effective knockdown efficacy for Grb7 protein but did not affect the amounts of EGFR and ErbB2 in Sk-Br3 cells (Fig. 1A). It is noteworthy that Grb7 tyrosine phosphorylation was obviously occurring in Sk-Br3 cells, implying that ErbB-mediated oncogenesis through Grb7 may occur in the cells.
FIGURE 1.

Grb7 is essential for Sk-Br3 breast cancer cell growth in vitro and in vivo. A, Grb7 expression and phosphorylation were markedly decreased by infection with two different lentivirus-bearing shGrb7 knockdowns, shGrb7-12 and shGrb7-13. WCL, whole cell lysates. B, Sk-Br3 cells with (shGrb7-12 or shGrb7-13) or without (shLuc or shGrb7+Grb7) knockdown of Grb7 were subjected to a measurement of cell proliferation by BrdU incorporation in the presence of 10 ng/ml EGF after serum starvation for 24 h in DMEM with 0.5% FBS. C, approximately 5 × 104 Sk-Br3 cells with or without Grb7 knockdown (shGrb7-12 or shGrb7-13 or combination) or re-expressing wild type Grb7 in Grb7 knockdown cells (combination of shGrb7-12 and shGrb7-13) were subjected to a soft agar assay in the presence of EGF (10 ng/ml) to examine the capability for anchorage-independent growth of these tumor cells. D, Sk-Br3 cells with (combination of shGrb7-12 and shGrb7-13) or without (shLuc) Grb7 knockdown were implanted into 8-week-old SCID mice. Tumor volumes and numbers were measured at the 21st day after injection. Data are represented as the mean ± S.E. of at least three independent experiments, or nine mice for each. N.S., nonsignificance.
BrdU incorporation assays were conducted to determine whether Grb7 is able to control cell proliferation in Sk-Br3 cells. When compared with cells infected by control viruses, Sk-Br3 cells with Grb7 knockdown by shGrb7 lentiviral infection exhibited a significant decrease in BrdU incorporation (Fig. 1B). A reversal in the reduction of cell proliferation resulting from the Grb7 knockdown was observed after ectopic reintroduction of wild type Grb7 (Fig. 1B). These results suggest the role of Grb7 in the regulation of tumor growth.
To directly investigate the impact of Grb7 on the tumorigenicity of breast cancer, Sk-Br3 cells with and without Grb7 overexpression were subjected to soft agar assays. By measuring the number of tumor foci under each condition, we observed a marked decrease in the number of foci in shGrb7-infected cells when compared with that of control shLuc-infected cells (Fig. 1C). Indeed, re-expression of wild type Grb7 in the knockdown cells reversed the attenuation, highlighting the involvement of Grb7 in the tumorigenesis of breast cancer. In accordance with the in vitro result, Grb7 knockdown resulted in a significant decrease in the tumor growth of SCID mice when compared with that of the shLuc-infected Sk-Br3 cells in xenografts (Fig. 1D). Taken together, our data provide comprehensive evidence for the participation of Grb7 in breast cancer tumorigenesis.
Downstream Targets of EGF-induced Grb7 Signaling
To uncover the mechanisms by which Grb7 participates in the tumorigenesis of breast cancer, we examined several downstream signaling targets that are crucial for tumorigenesis, including STAT3, AKT, and MAPKs (i.e. ERK1/2, JNK, and p38). In comparison with the changes in the activity of these signal proteins between cells with and without Grb7 knockdown, we found that phospho-STAT3, phospho-AKT, and phospho-ERK1/2, but not phospho-JNK or phospho-p38, were related to the presence of Grb7 upon EGF stimulation (Fig. 2, A and B). These data are consistent with the putative regulatory roles of these signal mediators in tumor invasion, survival, and proliferation, all of which are important for tumorigenesis and progression (20). Hence, these data also indicate that Grb7 is a pivotal signal mediator, controlling a variety of signal transduction pathways in the regulation of tumor development and/or progression. In light of the Ras activation of MAPKs, including ERK1/2, being essential for tumorigenesis, we promptly tested the regulatory role of Grb7 in the Ras-ERK signaling cascade. Our results displayed a concomitant decrease of active Ras and phospho-ERK1/2 in the cells without Grb7 overexpression when compared with cells retaining Grb7 overexpression (Fig. 3A). In contrast, in a Grb7 reconstitution experiment in NIH3T3 cells (which have no endogenous Grb7 expression), we found a positive relationship between Grb7 tyrosine phosphorylation and the Ras-ERK signaling cascade in response to EGF stimulation (Fig. 3B). These data indicate an intimate signaling link for breast cancer development via an EGF-Grb7-Ras-ERK axis.
FIGURE 2.

EGF-induced Grb7-mediated downstream signaling. A and B, cell lysates from the shLuc- or shGrb7-infected (shGrb7-12 or shGrb7-13) Sk-Br3 cells were subjected to Western blotting with various antibodies, as indicated, to examine the potential downstream signaling targets of EGF-induced Grb7 activation. WCL, whole cell lysates; p indicates phosphorylation.
FIGURE 3.
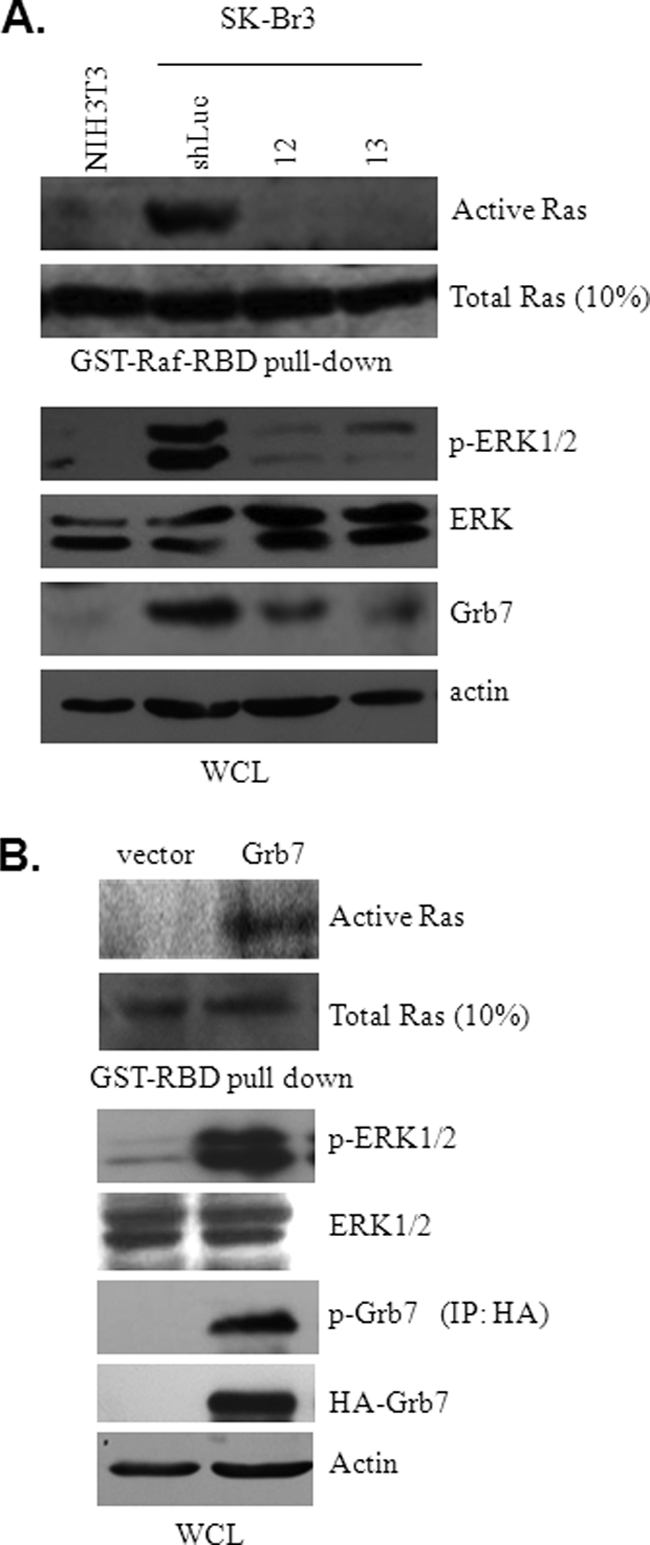
Grb7-mediated Ras activation is essential for ERK phosphorylation. A, cell lysates from the shLuc- or shGrb7-infected (shGrb7-12 or shGrb7-13) Sk-Br3 cells were subjected to GST-Raf-RBD pulldown assays and/or Western blotting with various antibodies, as indicated. There is undetectable endogenous Grb7 in NIH3T3 cells that was used as a control. p indicates phosphorylation. B, cell lysates from NIH3T3 cells transfected with either pKH3-Grb7 or vector alone and then subjected to a GST-Raf-RBD pulldown followed by Western analysis. For all experiments, cells were serum-starved for 24 h and stimulated by 10 ng/ml EGF for 15 min prior to collection of the cell lysates. IP, immunoprecipitation; WCL, whole cell lysate. p indicates phosphorylation.
EGF-induced Grb7 Phosphorylation Enables Recruiting and Activating Ras-ERK Signaling
To reveal mechanistic insights for Grb7 underlying the activation of Ras during the tumorigenesis of breast cancer, various Grb7 mutants were subjected to an examination of their ability to trigger Ras activity. These mutants include two tyrosine phosphorylation-deficient mutants, Y188F and Y338F; R458L, with a defect in the phospho-tyrosine binding capacity of its SH2 domain; F511R, with a dimerization defect; and Y188E, a phospho-tyrosine mimic. As shown in Fig. 4A, to promote Ras activity and subsequently ERK1/2 phosphorylation, the tyrosine phosphorylation, at least on Tyr-188 and/or Tyr-338, of Grb7 is necessary. Consistent with the hypothesis that the important role for Grb7 activation/phosphorylation is mediated by receptor phospho-tyrosine motif recruitment and/or dimerization through its SH2 domain (21), our data show a failure to promote Ras activity by R458L and F511R mutants of Grb7. Additionally, the activation of Ras by Grb7 was demonstrated to be EGF-dependent (Fig. 4B) but FAK-independent (Fig. 4C) in Sk-Br3 cells. Furthermore, in line with the direct interactions between Grb7 and activated Ras-GTPases that have been described previously (22), a point mutation at Arg-134 to Lys (i.e. R134L) within the RA domain of Grb7 was incapable of promoting Ras activity (Fig. 5A), implicating an important role for the RA domain of Grb7 participating in the Ras activation. Noticeably, this R134L mutant remains tyrosine phosphorylation in response to EGF stimulation, indicating an additional requirement for Grb7 promoting Ras activation (Fig. 5A, bottom panels). Similar to Grb10 and Grb14 (21), we found that the RA domain of Grb7 enables Grb7 directly interacting with Ras by the in vitro GST pulldown assay but that the R134L mutation of RA domain lost the capability to bind with Ras (Fig. 5B). Furthermore, the activity of Ras and subsequent ERK1/2 activation were diminished in association with the overexpression of the RA domain of Grb7 in a dose-dependent manner (Fig. 5C), further supporting the Ras-associating role of Grb7 through this featured domain. These results provide a physical and signaling interaction between Grb7 and Ras. Based on the above results, a novel complex comprised of EGFR, pY338 Grb7, and active Ras was observed in response to EGF stimulation in Sk-Br3 cells (Fig. 5D), which also requires tyrosine phosphorylation and dimerization of Grb7. Taken together, our results highlight a profound role in mediating EGF-dependent signaling in Sk-Br3 breast cancer cells.
FIGURE 4.

EGF-induced Grb7 dimerization and phosphorylation are required for Ras-ERK activation. A, Sk-Br3 cells were transfected with either wild type Grb7 or mutants, as indicated, to determine the effect on Ras activation using a GST-Raf-RBD pulldown assay and the phosphorylation of ERK1/2 in the presence of 10 ng/ml EGF. WCL, whole cell lysates; p indicates phosphorylation. B, NIH3T3 cells were transfected with wild type Grb7, Y338F, or vector alone in the presence or absence of EGF stimulation and were then subjected to a GST-Raf-RBD pulldown assay. C, NIH3T3 cells were transfected with wild type Grb7, the F511R mutant, or vector alone or in combination with FAK or without FAK and were then subjected to a GST-Raf-RBD pulldown assay. WCL, whole cell lysate.
FIGURE 5.

Ras activation through direct interaction with the RA domain of Grb7 is crucial for EGF-induced ERK phosphorylation in Sk-Br3 breast cancer cells. A, Sk-Br3 cells were transfected with either wild type Grb7 or mutants, as indicated, to determine the effect on Ras activation using a GST-Raf-RBD pulldown assay and the phosphorylation of Grb7 and its mutants in the presence of 10 ng/ml EGF. WCL, whole cell lysates; p indicates phosphorylation. B, cell lysates from 293 cells with or without overexpressing the constitutively active form RasV12G were incubated with recombinant GST fusion protein containing the wild type RA domain of Grb7 or harboring an R134L point mutation. The GST pulldown assay was performed to determine the binding capability for the Grb7 RA domain and its R134L mutant with the constitutively active form RasV12G. C, Sk-Br3 cells overexpressed different amounts of the RA domain of Grb7, and the effects on Ras activity and ERK phosphorylation were determined by a GST-RBD pulldown assay and Western blot analyses, respectively. D, EGF-stimulated cell lysates from Sk-Br3 cells transfected with either wild type Grb7 or F511R were subjected to a GST pulldown followed by Western blot analyses to determine the association among EGFR, Grb7, and activated Ras.
EGFR-Grb7-Ras-ERK Signaling Is Essential for the Tumorigenesis of Sk-Br3 Breast Cancer Cells
Next, we sought the involvement of the EGFR-Grb7-Ras-ERK signaling axis in the tumorigenesis of breast cancer. Sk-Br3 cells with or without Grb7 expression were subjected to soft agar assays stimulated by EGF (Fig. 6). In agreement with this signaling scenario, we found that foci formation of the Sk-Br3 cells was impaired upon Grb7 knockdown regardless of the presence or absence of PD98059, an ERK1/2 signaling inhibitor. In contrast, overexpression of a constitutively active Ras (RasG12V) resulted in an increase of tumor foci formation; however, it did not do so if PD98059 was present. These data indicate that Grb7 plays an important role in the regulation of tumorigenesis for breast cancer through the formation of novel EGFR-Grb7-Ras complexes and the activation of the Ras-ERK signaling axis in response to the EGF-mediated tyrosine kinase-activated cascade.
FIGURE 6.
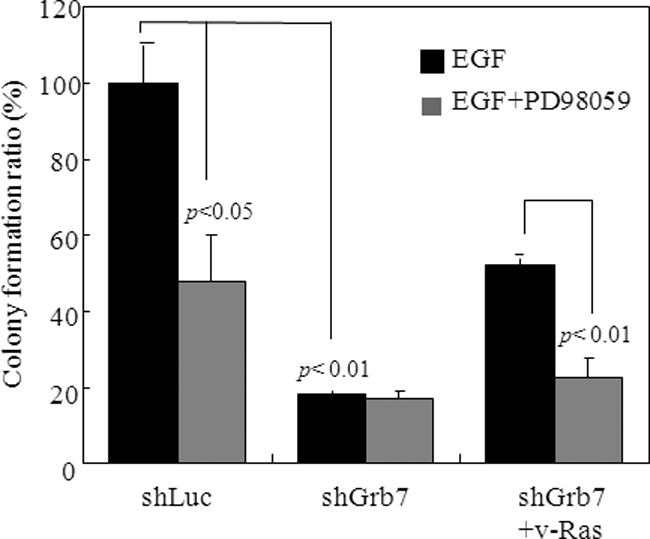
EGF-induced Grb7-mediated Ras-ERK activation is essential for tumorigenesis in breast cancer. Sk-Br3 cells with or without Grb7 knockdown (combination of shGrb7-12 and shGrb7-13) or expressing the constitutively active form RasV12G were subjected to soft agar assays in the presence of EGF (10 ng/ml) in combination with PD98059 or without PD98059 to examine the effect on the anchorage-independent growth of tumor cells. Data are represented as the mean ± S.E. of at least three independent experiments.
Lastly, we examined the anti-cancer effect by co-treatment with Grb7 knockdown and Herceptin, an inhibitory antibody targeting ErbB2, in Sk-Br3 breast cancer cells. The cell number of Sk-Br3 cells was very significantly diminished in combined treatment with Herceptin and Grb7 knockdown when compared with that of Grb7 knockdown alone or Herceptin treatment only (Fig. 7, A and B). In agreement with this, several signaling events crucial for tumorigenesis were suppressed by the combined treatment, including tyrosine phosphorylation of ErbB2, pY338 Grb7, and pY397FAK, Ras activation, and tyrosine phosphorylation of phospho-ERK1/2 and phospho-AKT (Fig. 7C). In fact, some of these examined signal molecules could be formed as a complex critical for the tumorigenesis of Sk-Br3 breast cancer cells as revealed above. Hence, these data shed new light for Grb7 as a therapeutic target in ErbB2-overexpressing breast cancer.
FIGURE 7.

Synergized anti-tumor effect by co-treatment with Herceptin and Grb7 knockdown. A, cell numbers of Sk-Br3 breast cancer cells treated with shLuc, Herceptin, shGrb7 (shGrb7-12 and shGrb7-13), and shGrb7 plus Herceptin were measured at the 1st, 2nd, and 3rd day after the treatments. B, representative images of Sk-Br3 cells treated with shLuc, Herceptin, shGrb7, and combination were photographed and shown. C, cell lysates from the various treatments, shLuc, Herceptin, and Herceptin plus shGrb7 in Sk-Br3 breast cancer cells were subjected to Western blotting with various antibodies, as indicated. WCL, whole cell lysates; p indicates phosphorylation.
DISCUSSION
A number of past observations have highlighted that Grb7, residing in the genomic neighborhood of ErbB2, is co-amplified and co-expressed with ErbB2 and that these expression patterns correlate with the malignant relevance of human cancers, including breast cancer (7, 19). An RNAi-based strategy for the functional dissection of DNA amplicons has provided evidence that co-amplified Grb7 can indeed contribute to tumor phenotypes and malignancy (13). Consistent with these findings, our results show further support for the participation of Grb7 in breast cancer development as seen by our observation that Grb7 knockdown impacted the in vitro and in vivo tumorigenicity of Sk-Br3 breast cancer cells. In support of its role in tumorigenesis, Grb7 knockdown resulted in the impairment of tumor cell proliferation and anchorage-independent growth, in an EGF-dependent manner, providing further underlying mechanisms for Grb7 in the ErbB2-positive breast cancers. Intriguingly, we noticed that de novo Grb7 dimerization, tyrosine phosphorylation, and direct Ras association through its RA domain are required for EGF-dependent Ras activation and subsequent ERK phosphorylation, indicating that the Grb7-mediated signaling, although downstream of EGF signaling, is more intimately relevant to breast tumor formation and/or development. In addition to being an adverse prognostic factor in many cancers (23), we and others have shown that Grb7 participates in cell motility through its association with FAK, D3-phosphoinositides, EphB1, and calmodulin as well as in angiogenesis, inferring a role for Grb7 in tumor progression as well (16). It would be interesting to further investigate whether Grb7 controls these cellular functions in various contexts of breast cancer.
The first insight indicating that Grb7 presents an RA domain came from sequence analysis (24), which promptly suggested the intriguing possibility of a direct interaction between Grb7 members and Ras-GTPases. Subsequent biochemical validations showed that the RA domain of Grb7 was capable of binding to activated Ras, including N-Ras, K-Ras, and H-Ras, but was weakly bound to Rap1 and Rap2 (22). Furthermore, a recent study has shown that the membrane localization of Grb14, through its PH domain-phosphoinositide interaction, as well as the stabilized interplay between its RA and PH domains, is required for its interaction with GTP-bound N-Ras and subsequently to modulate ERK phosphorylation in response to insulin stimulation (21). Irrespective of the opposite biological effects attributed to different family member proteins, Grb7 has been substantially linked to the promotion of ERK phosphorylation in several studies, including ours (16, 25). Conceivably, this link could result from the activation of Ras activity mediated by Grb7, but a direct demonstration of this phenomenon has not yet been performed. In an attempt to define the signaling and functional relationship between Grb7 and Ras, using an in vitro pulldown assay, we showed that the RA domain of Grb7 enables Grb7 physically interacting with Ras in breast cancer cells. A single amino acid mutation within the RA domain of Grb7, R134L, lost the capability to bind with Ras, which is consistent with a recent report showing that the other Grb7 member proteins, Grb10 and Grb14, possess the same feature in a structural basis study (21). Therefore, as expected, our data showed that the overexpression of the Grb7 harboring the R134L mutation and the Grb7 knockdown in Sk-Br3 cells led to a remarkable reduction of GTP-bound Ras, accompanied by the decreased phosphorylation of ERK1/2. However, in contrast to wild type Grb7 or phospho-tyrosine mimic mutant (Y188E), the overexpression of Grb7 mutants conferring deficiency in tyrosine phosphorylation (Y188F and Y338F), dimerization (F511R), or receptor binding (R458L) failed to lead to increases in Ras activity and ERK1/2 phosphorylation. Collectively, our data indicate a direct signaling event through the Grb7-Ras-ERK axis in the regulation of various cellular functions that are attributed to tumorigenesis and/or malignancy development. In agreement with the above, we also provide evidence for the existence of the EGFR-Grb7-Ras complex in facilitating the activation of MAPK signaling in the context of EGF tyrosine kinase-activated cascades.
Serving as an adaptor protein, Grb7 enables the transmission of upstream signals into a diverse array of downstream signal transduction pathways corresponding to the variegated spectrum of the receptors/regulators (1, 2, 26). In Fig. 2, we have demonstrated that the phosphorylations of STAT3, AKT, and ERK1/2, but not JNK or p38, are altered in the Grb7 knockdown breast cells when compared with those of control cells, suggesting that these signaling molecules could act as downstream signaling targets in an EGF-Grb7 signaling axis. Indeed, the functional relevant processes modulated by these signaling targets are in concert with Grb7-mediated cellular functions, such as cell proliferation and survival. This relevance has been consistently demonstrated in several reports. For example, in breast cancer cells overexpressing both ErbB2 and Grb7, it has been reported that Grb7 is capable of modulating AKT phosphorylation at both Thr-308 and Ser-473 to facilitate tumor growth (25), correlating with the well characterized cell survival role of AKT. As mentioned above, ERK1/2 activation is likely directed through the recruitment of activated Ras by the RA domain of Grb7 in an EGF-induced fashion. Recently, the role of STAT3 has been reported to direct breast cancer cells toward an unexpected function: tumor invasion (27). Altogether, our findings shed novel insights on the role of Grb7 in a diverse array of oncogenesis-related pathways, such as the regulation of tumor formation and further progression in SkBr-3 cells, thereby highlighting the potential strategy of targeting Grb7 as an anti-breast cancer therapy.
In conclusion, in this study, by employing a breast cancer cell line, Sk-Br3, which co-amplifies and overexpresses ErbB2 and Grb7, and an established lentivirus-based Grb7 knockdown, we were able to analyze the tumorigenicity in Sk-Br3 cells with or without Grb7 overexpression. The in vitro and in vivo data suggested that Grb7 is required for the tumorigenicity of Sk-Br3 cells. Additionally, we found that Ras activation was marked by its interaction with Grb7 through its RA domain in an EGF-induced tyrosine phosphorylation manner, which concurrently resulted in an increase in ERK1/2 phosphorylation and subsequently promoted tumorigenesis in vivo. A combined treatment with anti-ErbB2 monoclonal antibody and Grb7 knockdown exhibited a synergistic effect on inhibition of Sk-Br3 cell growth, which provides a promising therapeutic approach on ErbB2-overexpressing breast cancer. Our results delineate the novel EGFR-Grb7-Ras-ERK axis in the tumorigenesis of Sk-Br3 cells and therefore implicate a potential therapeutic strategy through the targeting of Grb7 in the Grb7-positive subgroup of breast cancers.
Acknowledgments
We thank Dr. Shan-Li Wang for critical reading of the manuscript and helpful comments. We are grateful for excellent technical assistance from Ya-Ni Tsai and Fei-Neng Wang.
This work was supported by National Science Council Grant NSC-96-2311-B-002-023-MY3 and Frontier Research Grant of National Taiwan University 98R0311 (to T.-L. S.).
- Grb7
- growth factor receptor bound protein-7
- ErbB2
- avian erythroblastosis oncogene B-2
- Ras
- rat sarcoma
- EphB1
- ephrin type-B receptor 1
- PH
- pleckstrin homology
- SH2
- Src homology 2
- FAK
- focal adhesion kinase
- RBD
- Ras-binding domain
- EGFR
- EGF receptor
- SCID
- severe combined immunodeficiency.
REFERENCES
- 1.Han D. C., Shen T. L., Guan J. L. (2001) Oncogene 20, 6315–6321 [DOI] [PubMed] [Google Scholar]
- 2.Shen T. L., Guan J. L. (2004) Front. Biosci. 9, 192–200 [DOI] [PubMed] [Google Scholar]
- 3.Li H., Sánchez-Torres J., del Carpio A. F., Nogales-González A., Molina-Ortiz P., Moreno M. J., Török K., Villalobo A. (2005) Oncogene 24, 4206–4219 [DOI] [PubMed] [Google Scholar]
- 4.Shen T. L., Han D. C., Guan J. L. (2002) J. Biol. Chem. 277, 29069–29077 [DOI] [PubMed] [Google Scholar]
- 5.Depetris R. S., Hu J., Gimpelevich I., Holt L. J., Daly R. J., Hubbard S. R. (2005) Mol. Cell 20, 325–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lyons R. J., Deane R., Lynch D. K., Ye Z. S., Sanderson G. M., Eyre H. J., Sutherland G. R., Daly R. J. (2001) J. Biol. Chem. 276, 17172–17180 [DOI] [PubMed] [Google Scholar]
- 7.Lucas-Fernández E., García-Palmero I., Villalobo A. (2008) Curr. Genomics 9, 60–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Itoh S., Taketomi A., Tanaka S., Harimoto N., Yamashita Y., Aishima S., Maeda T., Shirabe K., Shimada M., Maehara Y. (2007) Mol. Cancer Res. 5, 667–673 [DOI] [PubMed] [Google Scholar]
- 9.Tanaka S., Sugimachi K., Kawaguchi H., Saeki H., Ohno S., Wands J. R. (2000) J. Cell. Physiol. 183, 411–415 [DOI] [PubMed] [Google Scholar]
- 10.Han D. C., Guan J. L. (1999) J. Biol. Chem. 274, 24425–24430 [DOI] [PubMed] [Google Scholar]
- 11.Han D. C., Shen T. L., Guan J. L. (2000) J. Biol. Chem. 275, 28911–28917 [DOI] [PubMed] [Google Scholar]
- 12.Han D. C., Shen T. L., Miao H., Wang B., Guan J. L. (2002) J. Biol. Chem. 277, 45655–45661 [DOI] [PubMed] [Google Scholar]
- 13.Kao J., Pollack J. R. (2006) Genes Chromosomes Cancer 45, 761–769 [DOI] [PubMed] [Google Scholar]
- 14.Porter C. J., Matthews J. M., Mackay J. P., Pursglove S. E., Schmidberger J. W., Leedman P. J., Pero S. C., Krag D. N., Wilce M. C., Wilce J. A. (2007) BMC Struct. Biol. 7, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanaka S., Pero S. C., Taguchi K., Shimada M., Mori M., Krag D. N., Arii S. (2006) J. Natl. Cancer Inst. 98, 491–498 [DOI] [PubMed] [Google Scholar]
- 16.Chu P. Y., Huang L. Y., Hsu C. H., Liang C. C., Guan J. L., Hung T. H., Shen T. L. (2009) J. Biol. Chem. 284, 20215–20226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen T. L., Guan J. L. (2001) FEBS Lett. 499, 176–181 [DOI] [PubMed] [Google Scholar]
- 18.Castro A. F., Rebhun J. F., Quilliam L. A. (2005) Methods 37, 190–196 [DOI] [PubMed] [Google Scholar]
- 19.Benusiglio P. R., Pharoah P. D., Smith P. L., Lesueur F., Conroy D., Luben R. N., Dew G., Jordan C., Dunning A., Easton D. F., Ponder B. A. (2006) Br. J. Cancer 95, 1689–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kreeger P. K., Lauffenburger D. A.Carcinogenesis (2010) 31, 2–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Depetris R. S., Wu J., Hubbard S. R. (2009) Nat. Struct. Mol. Biol. 16, 833–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodriguez-Viciana P., Sabatier C., McCormick F. (2004) Mol. Cell. Biol. 24, 4943–4954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nadler Y., González A. M., Camp R. L., Rimm D. L., Kluger H. M., Kluger Y. (2010) Ann. Oncol. 21, 466–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wojcik J., Girault J. A., Labesse G., Chomilier J., Mornon J. P., Callebaut I. (1999) Biochem. Biophys. Res. Commun. 259, 113–120 [DOI] [PubMed] [Google Scholar]
- 25.Bai T., Luoh S. W. (2008) Carcinogenesis 29, 473–479 [DOI] [PubMed] [Google Scholar]
- 26.Daly R. J. (1998) Cell Signal 10, 613–618 [DOI] [PubMed] [Google Scholar]
- 27.Guo W., Pylayeva Y., Pepe A., Yoshioka T., Muller W. J., Inghirami G., Giancotti F. G. (2006) Cell 126, 489–502 [DOI] [PubMed] [Google Scholar]