

Abstract
Atherosclerosis is promoted by a combination of hypercholesterolemia and vascular inflammation. The function of Angiopoietin-2 (Ang-2), a key regulator of angiogenesis, in the maintenance of large vessels is unknown. A single systemic administration of adenoviral Ang-2 (AdAng-2) to apoE-/- mice fed a Western diet significantly reduced atherosclerotic lesion size (~40%) and oxidized LDL and macrophage content of the plaques. Nitric oxide (NO) synthase (NOS) inhibitor abolished these beneficial effects of Ang-2. In endothelial cells, eNOS activation per se inhibited LDL oxidation and Ang-2 stimulated NO release in a Tie2-dependent manner to decrease LDL oxidation. These findings demonstrate a novel atheroprotective role for Ang-2 where endothelial cell function is compromised and suggest that growth factors, which stimulate NO release without inducing inflammation, could offer atheroprotection.
Keywords: Angiopoietin-2, Atherosclerosis, Endothelial cells, LDL cholesterol, Nitric oxide, Nitric oxide synthases
Introduction
The endothelium plays a vital role in the prevention of atherosclerosis, yet the molecular mechanisms that confer atheroprotection are poorly understood.1 Angiopoietin-2 (Ang-2), a context-dependent agonist/antagonist for the Tie2 receptor, promotes vascular regression or facilitates vascular endothelial growth factor (VEGF)-driven angiogenesis.2 Despite the extensive studies of Ang-2 in the microvasculature, its role in atherosclerosis is unknown. Ang-2 in high concentrations induces robust phosphorylation of Tie23 and prevents endothelial cell apoptosis4 through activation of the pro-survival Akt pathway, which can phosphorylate eNOS.4, 5 Stressed endothelial cells express high levels of Ang-2, which acts as an autocrine Tie2 agonist to bolster Akt activity.6 As loss of eNOS activity is an established contributor to endothelial dysfunction7 and endothelium-derived NO plays a vital role in the prevention of atherosclerosis,8 we explored whether Ang-2 could suppress atherosclerosis by stimulating NO release in apoE-/- mice. In this report we show that overexpression of Ang-2 reduces lesion size, macrophage accumulation and oxidized LDL content of plaques in atherosclerosis-prone apoE-/- mice. The protective effect of Ang-2 was abolished by NOS inhibition. Our investigation demonstrates a novel function for Ang-2 as an atheroprotective mediator, which inhibits atherosclerosis through the activation of eNOS to reduce LDL oxidation.
Material and methods
The expanded Materials and Methods section in the online supplement, available at http//:circres.ahajournals.org provides a description of the apoE-/- mouse model of atherosclerosis, adenoviruses, immunohistochemical, NO and LDL oxidation methods.
Results and Discussion
Ang-2 suppresses atherosclerosis in apoE null mice
After three weeks on a Western diet, eight-week old apoE-/- mice received a single systemic administration of Ang-2 adenovirus (AdAng-2; 1.0 × 1010 pfu) or control adenovirus (AdEV) and sacrificed four weeks later. All mice developed atherosclerotic lesions along the proximal aortic wall and at the valve cusps. Circulating levels of Ang-2 were detected only in the AdAng-2-treated group (mean ± SD 12.8 ± 5.6 μg/ml; Online Figure I), and there were no significant differences in the body weight or plasma lipid levels between the two groups (Online Table I). These findings suggest that the beneficial effect of Ang-2 was not due to suppression of appetite. AdAng-2-treated mice showed a highly significant reduction in atherosclerotic plaque lesion size compared with control mice receiving AdEV (0.269 ± 0.04 mm2 vs. 0.437±0.05 mm2; n=10/group; P< 0.01; Figure 1A). Lesion size in a separate group of uninfected animals (0.547±0.05 mm2; n=3) was similar to AdEV-treated mice indicating that adenoviral infection per se did not increase atheroclerosis. Histological analysis revealed a decrease in CD11b-positive macrophages/monocytes (Mø) in the plaque (middle panel, Figure 1B). Immunohistochemical analysis of malondialdehyde–lysine (MDA2), a marker of oxidized LDL,9 revealed reduced oxidized LDL staining in aortic sections of AdAng-2-treated animals (bottom panel, ox-LDL, Figure 1B). Staining for CD31 showed that the aortic endothelium remains intact in the AdAng-2 treated mice. This is in marked contrast to the effect of Ang-2 promoting endothelial cell detachment reported in a three-dimensional culture model.10 It is possible that acute effects of Ang-2 may be deleterious, but long-term treatment may be protective. Indeed, prolonged exposure of endothelial cells to Ang-2 induces a robust phosphorylation of Tie2,3 a key pro-survival signal.6
Figure 1. Ang-2 reduces atherosclerotic plaque formation, LDL oxidation and macrophage accumulation in apoE-/- mice.
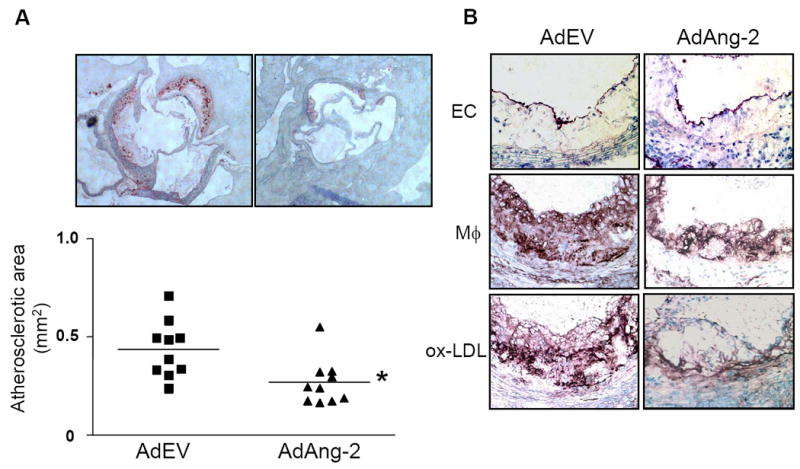
ApoE-/- mice maintained on a Western diet were administered AdAng-2 or control empty virus (AdEV). A, Atherosclerotic lesions in the aortic valves were stained with oil red O and the results expressed as the mean plaque area ± SEM. Ang-2 significantly reduced the mean plaque area (*P <0.01) compared with AdEV-treated apoE-/- mice. B, Immunohistochemical analysis showed that CD31-positive endothelium (EC) remained intact, and CD11b-positive macrophages (Mø) and malondialdehyde–lysine/MDA2 (ox-LDL) staining was reduced in Ang-2 treated mice.
Ang-2 induces NO release from endothelial cells
Stimulation of human umbilical vein endothelial cells (HUVEC) with Ang-2 resulted in a concentration-dependent release of NO (Figure 2A), which was inhibited by Tie2 neutralizing antibodies and a Tie2 blocking peptide (Figure 2B) demonstrating this effect is Tie2-dependent. Although VEGF and Ang1 can induce NO release,11 both can recruit inflammatory cells12, 13 In addition, VEGF increased plaque formation double deficient apoE/apoB100 mice,14 and Ang-1 failed to protect against the development of rat cardiac allograft arteriosclerosis.15 This paradox may be explained by the fact that unlike VEGF and Ang-1, Ang-2 has little effect on monocyte migration (Online Figure II). This ability of Ang-2 to stimulate NO release without promoting inflammatory cell recruitment gives it the characteristics of an atheroprotective factor.
Figure 2. Ang-2 suppresses LDL oxidation and stimulates NO release via Tie2 activation.
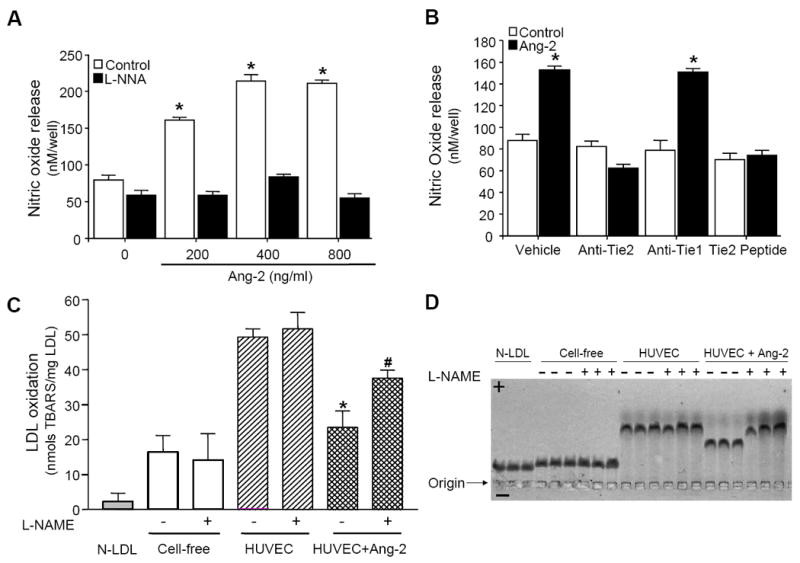
A, Ang-2-mediated NO release in HUVEC was inhibited by 0.5 mM NG-nitro-L-arginine (L-NNA). B, HUVEC were pretreated with either Tie2 (anti-Tie2; 5 μg/ml), or Tie1 (anti-Tie2; 5 μg/ml) neutralizing antibodies or Tie2 blocking peptide (Tie2 peptide; 0.5 mM) prior to incubation with Ang-2 (400 ng/ml) for 1 hour and NO release quantified. Results are the mean (±SEM) of three independent experiments (n = 9). C and D, HUVEC were incubated in serum-free medium containing 100 μg/ml LDL, 500 ng/ml of Ang-2 and/or 100 μM L-NAME for 16 hours. Oxidative modification of LDL was assessed using: C, TBARS assay (data represents the mean ± SEM; *P <0.01 vs. control, #P<0.05 vs. HUVEC+Ang-2 without L-NAME) and D, the relative electrophoretic mobility of LDL.
Ang-2 inhibits endothelial-mediated LDL oxidation
Oxidized LDL reduces endothelial function,16 however, it is unknown whether NO per se can inhibit LDL oxidation in a cellular context. Therefore, we assessed NO production and LDL oxidation in porcine aortic endothelial cells (PAEC) expressing constitutively active eNOSS1177D (PAEC/eNOSS1177D) or control cells (PAEC/pcDNA) using thiobarbituric acid reactive substances (TBARS) assay. PAEC/eNOSS1177D produced significantly more NO (Online Figure IIIA) and reduced LDL oxidation compared with control cells (Online Figure IIIB, P <0.01); an effect that was prevented by NOS inhibition indicating that NO inhibits cellular LDL oxidation.
The observed reduction in tissue LDL oxidation in Ang-2-treated animals prompted us to examine whether Ang-2 could suppress LDL oxidation by endothelial cells in vitro. In the absence of cells, native LDL oxidation was low (16.9 ± 4.8 nmol TBARS/mg) whereas in the presence of HUVEC, LDL oxidation increased significantly (49.5 ± 2.75 nmol TBARS/mg; Figure 2C). Ang-2 dramatically inhibited LDL oxidation in HUVEC as demonstrated by a 57% reduction in the formation of TBARS (23.5 ± 4.7 nmol TBARS/mg; p<0.01, n=4; Figure 2C), which was partially abolished by L-NAME. The relative electrophoretic mobility of modified LDL was used to confirm these findings (Figure 2D). In contrast to Ang-2, Ang-1 had no effect on LDL oxidation (Online Figure IV). These results demonstrate that the antiatherogenic property of Ang-2 may, in part, be due to its ability to inhibit LDL oxidation via a NO-dependent mechanism.
Ang-2-mediated atheroprotection is NO-dependent
To investigate if the protective effect of Ang-2 in apoE-/- mice requires NO, L-NAME was administered in the drinking water one day after adenoviral injection. In the absence of L-NAME, AdAng-2-treated mice demonstrated a significant reduction in lesion area compared with AdEV-treatment (0.141 ± 0.01 mm2 vs. 0.222 ± 0.03 mm2; n=8-10/group; P< 0.01; Figure 3A) as before (Figure 1). Inhibition of NOS had no significant effect on lesion size in AdEV-treated mice but abrogated the beneficial effects of Ang-2 (Figure 3B). Furthermore, AdAng-2 significantly decreased MOMA-2-positive macrophages in the lesions, an effect that was also inhibited by L-NAME (Figure 3A and C). Although lesion size was only quantified histologically without supporting en face staining, these results demonstrate that the effects of Ang-2 on lesion size were reproducible demonstrating the utility of this method for quantification of these early stage lesions.
Figure 3. Ang-2-mediated reduction in atherosclerotic plaque formation requires NO.
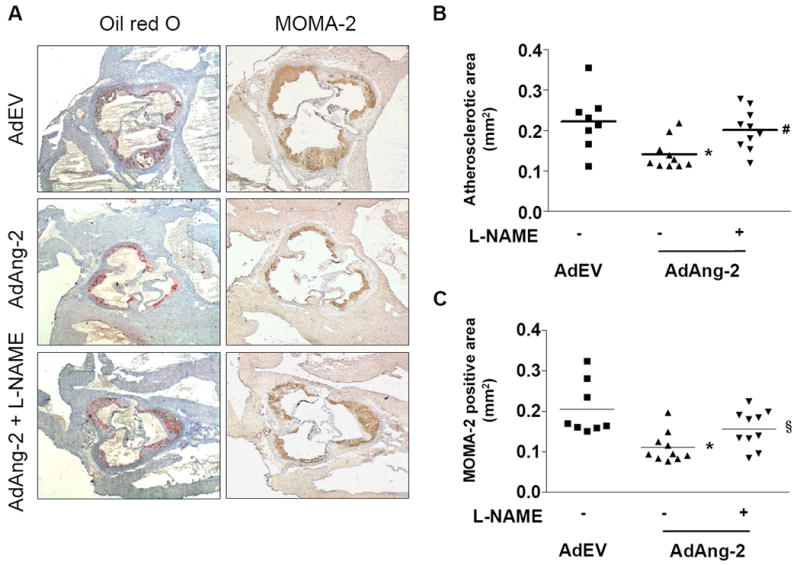
One day after administration of adenovirus, apoE-/- mice were treated with L-NAME. A, Representative images of plaques stained for neutral lipids (oil red O) and macrophage (MOMA-2) content. Quantification of B, mean plaque and C, MOMA-2 positive areas show that the atheroprotective effect of AdAng-2 is abolished following L-NAME treatment. Data are the mean area ± SEM; *P <0.01 vs. AdEV without L-NAME, §P<0.05 and #P<0.01 vs. AdAng-2 without L-NAME.
Taken together, this study demonstrates that NO per se suppresses LDL oxidation and Ang-2 inhibits atherosclerotic lesion development, in part, by reducing LDL oxidation and macrophage accumulation via endothelial NOS activation. These results are consistent with the contextual and concentration-dependent nature of Ang-23, 6 and indicate that Ang-2 may offer atheroprotection in a hypercholesterolemic environment when endothelial cell function is compromised. It will be important to establish whether the protective effects of the transient high circulating levels of Ang-2 observed in early atherosclerotic lesions persist and can be translated into long-term benefit.
Acknowledgments
We are grateful to Professor George Yancopoulos (Regeneron Pharmaceuticals, USA) for the angiopoietin reagents and Professor Stefanie Dimmeler (University of Frankfurt, Germany) for providing the various eNOS constructs. We thank Professor Joseph Witztum (University of California at San Diego, USA) for the MDA2 antibody.
Sources of funding This study was funded by the British Heart Foundation (PG/06/114) and Medical Research Council (G0601295 and G0700288) grants, the European Vascular Genomics Network supported by the European Community’s Sixth Framework Programme for Research Priority 1 “Life Sciences, Genomics and Biotechnology for Health” (contract LSHM-CT-2003-503254) and the NIH (HL70165) and Mid-Atlantic Affiliate of the American Heart Association (0355792U) grants.
Footnotes
Disclosures None
Subject Codes: [147] Growth factors/cytokines, [90] Lipid and lipoprotein metabolism, [95] Endothelium/vascular type/nitric oxide, [96] Mechanism of atherosclerosis/growth factors
References
- 1.Rakhit RD, Marber MS. Nitric oxide: an emerging role in cardioprotection? Heart. 2001;86:368–372. doi: 10.1136/heart.86.4.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, Yancopoulos GD, Wiegand SJ. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science. 1999;284:1994–1998. doi: 10.1126/science.284.5422.1994. [DOI] [PubMed] [Google Scholar]
- 3.Teichert-Kuliszewska K, Maisonpierre PC, Jones N, Campbell AI, Master Z, Bendeck MP, Alitalo K, Dumont DJ, Yancopoulos GD, Stewart DJ. Biological action of angiopoietin-2 in a fibrin matrix model of angiogenesis is associated with activation of Tie2. Cardiovasc Res. 2001;49:659–670. doi: 10.1016/s0008-6363(00)00231-5. [DOI] [PubMed] [Google Scholar]
- 4.Kim I, Kim JH, Moon SO, Kwak HJ, Kim NG, Koh GY. Angiopoietin-2 at high concentration can enhance endothelial cell survival through the phosphatidylinositol 3’-kinase/Akt signal transduction pathway. Oncogene. 2000;19:4549–4552. doi: 10.1038/sj.onc.1203800. [DOI] [PubMed] [Google Scholar]
- 5.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 6.Daly C, Pasnikowski E, Burova E, Wong V, Aldrich TH, Griffiths J, Ioffe E, Daly TJ, Fandl JP, Papadopoulos N, McDonald DM, Thurston G, Yancopoulos GD, Rudge JS. Angiopoietin-2 functions as an autocrine protective factor in stressed endothelial cells. Proc Natl Acad Sci U S A. 2006;103:15491–15496. doi: 10.1073/pnas.0607538103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heitzer T, Schlinzig T, Krohn K, Meinertz T, Munzel T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation. 2001;104:2673–2678. doi: 10.1161/hc4601.099485. [DOI] [PubMed] [Google Scholar]
- 8.Knowles JW, Reddick RL, Jennette JC, Shesely EG, Smithies O, Maeda N. Enhanced atherosclerosis and kidney dysfunction in eNOS(-/-)Apoe(-/-) mice are ameliorated by enalapril treatment. J Clin Invest. 2000;105:451–458. doi: 10.1172/JCI8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palinski W, Yla-Herttuala S, Rosenfeld ME, Butler SW, Socher SA, Parthasarathy S, Curtiss LK, Witztum JL. Antisera and monoclonal antibodies specific for epitopes generated during oxidative modification of low density lipoprotein. Arteriosclerosis. 1990;10:325–335. doi: 10.1161/01.atv.10.3.325. [DOI] [PubMed] [Google Scholar]
- 10.Scharpfenecker M, Fiedler U, Reiss Y, Augustin HG. The Tie-2 ligand angiopoietin-2 destabilizes quiescent endothelium through an internal autocrine loop mechanism. J Cell Sci. 2005;118:771–780. doi: 10.1242/jcs.01653. [DOI] [PubMed] [Google Scholar]
- 11.Babaei S, Teichert-Kuliszewska K, Zhang Q, Jones N, Dumont DJ, Stewart DJ. Angiogenic actions of angiopoietin-1 require endothelium-derived nitric oxide. Am J Pathol. 2003;162:1927–1936. doi: 10.1016/S0002-9440(10)64326-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marme D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood. 1996;87:3336–3343. [PubMed] [Google Scholar]
- 13.Lemieux C, Maliba R, Favier J, Theoret JF, Merhi Y, Sirois MG. Angiopoietins can directly activate endothelial cells and neutrophils to promote proinflammatory responses. Blood. 2005;105:1523–1530. doi: 10.1182/blood-2004-09-3531. [DOI] [PubMed] [Google Scholar]
- 14.Celletti FL, Waugh JM, Amabile PG, Brendolan A, Hilfiker PR, Dake MD. Vascular endothelial growth factor enhances atherosclerotic plaque progression. Nat Med. 2001;7:425–429. doi: 10.1038/86490. [DOI] [PubMed] [Google Scholar]
- 15.Nykanen AI, Pajusola K, Krebs R, Keranen MA, Raisky O, Koskinen PK, Alitalo K, Lemstrom KB. Common protective and diverse smooth muscle cell effects of AAV-mediated angiopoietin-1 and -2 expression in rat cardiac allograft vasculopathy. Circ Res. 2006;98:1373–1380. doi: 10.1161/01.RES.0000225987.52765.13. [DOI] [PubMed] [Google Scholar]
- 16.Steinberg D. Atherogenesis in perspective: Hypercholesterolemia and inflammation as partners in crime. Nat Med. 2002;8:1211–1217. doi: 10.1038/nm1102-1211. [DOI] [PubMed] [Google Scholar]