

Abstract
Kupffer cells (KCs) are thought to mediate hepatocyte injury via their production of proinflammatory cytokines and reactive oxygen species in response to stress. In this study, we depleted KCs from the liver to examine their role in total warm hepatic ischemia/reperfusion (I/R) injury with bowel congestion. We injected 8-wk-old C57BL/10J mice with liposome-encapsulated clodronate 48 h before 35 min of hepatic ischemia with bowel congestion, followed by 6 or 24 h of reperfusion. KC-depleted animals had a higher mortality rate than diluent-treated animals and a 10-fold elevation in transaminase levels that correlated with increases in centrilobular necrosis. There was extensive LPS binding to the endothelial cells, which correlated with an upregulation of endothelial adhesion molecules in the KC-depleted animals versus diluent-treated animals. There was an increase in the levels of proinflammatory cytokines in KC-depleted animals, and a concomitant decrease in IL-10 levels. When KC-depleted mice were treated with recombinant IL-10, their liver damage profile in response to I/R was similar to diluent-treated animals, and endothelial cell adhesion molecules and proinflammatory cytokine levels decreased. KCs are protective in the liver subjected to total I/R with associated bowel congestion and are not deleterious as previously thought. This protection appears to be due to KC secretion of the potent anti-inflammatory cytokine IL-10.
Kupffer cells (KCs) have long been implicated in the pathogenesis of hepatic ischemia/reperfusion (I/R) injury. They have been classified as the key cell type in this process via production of proinflammatory cytokines, activation of complement, and production of reactive oxygen species (1). More importantly, blockade of their activation through pharmacologic mechanisms has led to improvements in liver outcomes subsequent to I/R. Specifically, blockade of KC function with either GdCl3 or glycine decreased serum transaminase levels and ameliorated I/R injury (2). However, additional evidence has indicated that GdCl3 only impairs phagocytic activity of KC and abolishes expression of certain KC-specific markers, such as the KC receptor F4/80, ED1, and ED2, thereby allowing other KC functions to occur (3). Although overall hepatic damage is reduced, there is overexpression of TNF in GdCl3-treated livers because of increased stability of the mRNA transcript (4). Therefore, GdCl3 might merely cause a switch in KC phenotype.
The response of KCs to stress is thought to be biphasic, initiated by the secretion of proinflammatory factors such as TNF, IFN-γ, IL-6, and IL-1, and followed by a secondary secretion of anti-inflammatory mediators, such as IL-10 (5). In the timeline of I/R injury, KC activation has been thought to occur first, giving rise to endothelial activation and dysfunction. Next, endothelial cells, thought to be the least tolerant of the nonparenchymal cells to I/R, are activated, leading to the secretion of microcirculatory mediators and proinflammatory cytokines and the upregulation of adhesion molecules (5), ultimately resulting in hepatic damage. However, the precise mechanisms of the interaction between endothelial cells and KCs remain unclear.
It is clear that endothelial cells play a key role in I/R injury. It has been shown that inactivation of endothelin or blockade of its receptor lessens hepatic damage after I/R injury (6, 7). LPS increases levels of CD54/ intracellular adhesion molecule (ICAM)-1 on liver sinusoidal endothelial cells (LSECs), greatly influencing neutrophil adhesion, ultimately leading to increased hepatic damage following LPS challenge (8). In addition, the IL-10 secreted by KCs control the proinflammatory mediator release from LSECs in response to LPS challenge (9). To this end, IL-10 has been shown to reduce the incidence of hepatic injury after various harmful insults (10).
Clinically, levels of translocated endotoxin subsequent to bowel congestion have been shown to correlate with poor liver graft outcomes after transplantation or resection (11). In our total hepatic I/R model of transplantation that simulates this bowel congestion, we have seen that removal of translocated endotoxin with anti-LPS Abs improved outcomes following I/R (12). KCs are predominantly thought to deal with hepatic LPSt. By using a method to deplete KCs from the hepatic microenvironment using liposome encapsulated dichloromethylene biphosphonate [liposomal clodronate (LC)], we sought to examine the role of KCs in total hepatic warm I/R injury, with bowel congestion as a model of clinical liver transplantation.
Materials and Methods
Animals
Male, 8-wk-old C57BL/10J (The Jackson Laboratory, Bar Harbor, ME) mice were used in all experiments. Mice were housed 3–4 per cage in a temperature-controlled room (22–25°C) with a 12-h light-dark cycle. Water and food were available ad libitum. All experiments were performed under aseptic conditions in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Liposomal clodronate administration
Forty-eight hours prior to ischemia, animals were injected with 200 μl of the indicated doses of liposomal clodronate (dichloromethylene bisphosphonate, LC) i.p. LC was produced as previously described (13).
Total hepatic warm I/R
I/R was performed as previously described (14) with some modification. Mice were laparotomized and a sterile pediatric vessel loop was placed around the portal triad for 35 min to induce total hepatic ischemia and mesenteric congestion. After the loop was removed, the livers were reperfused for 6 or 24 h. At sacrifice, serum was collected by sterile cardiac puncture, and aliquoted into pyrogen-free, glass vials. Portions of the liver were placed in 10% neutral buffered formalin, and the remaining liver was snap-frozen in liquid nitrogen and stored at −80°C.
Serum alanine aminotransferase
Serum alanine aminotransferase (ALT) concentrations were measured with a Synchron LX20 system (Beckman Coulter, Fullerton, CA) and expressed as international units per liter (Clinical Laboratory Services, Medical University of South Carolina, Charleston, SC).
Centrilobular necrotic index grading
Formalin-fixed, paraffin-embedded samples were stained with H&E. Necrosis grading was quantified from H&E-stained slides as previously described with reference to the central vein (15). A grade of 0 indicated absent necrosis, 1 indicated individual hepatocyte dropout, 2 indicated small foci of missing hepatocytes up to 2–3 cells across, and 3 indicated confluent foci of hepatocyte dropout >3 cells across. Ten high-powered fields per section were analyzed in a blinded fashion.
Immunohistochemistry
Formalin-fixed, paraffin-embedded sections were incubated with primary Ab (ICAM-1, BD Biosciences, San Jose, CA, #550287; F4/80, Abcam #ab6640, Cambridge, MA; Escherichia coli LPS, Abcam #ab35654; SE-1, Novus Biologicals #NB110-68095, Littleton, CO) for 1 h after Ag retrieval using a heat induced (LPS, ICAM) or enzymatic (F4/80, SE-1) epitope retrieval method. Samples were incubated with biotinylated secondary Ab (Vector Laboratories, Burlingame, CA), followed by Vectastain ABC kit (Vector Laboratories) for an additional hour. Immunoperoxidase staining was performed with the diaminobenzidine substrate kit (Vector Laboratories). The specificity of immunostaining was demonstrated by omission of primary Ab. Sections were counter-stained with Harris hematoxylin and examined by light microscopy in a blinded fashion. When appropriate, positive cells were counted as a ratio of total cells in 10 high-powered fields per section.
Confocal microscopy
Paraffin-embedded sections were incubated as above. In the case of double labeling, incubation was done first with anti-LPS overnight, followed by labeling with Alexa-Fluor 488 Avidin (Invitrogen, Carlsbad, CA). Next, the second primary labeling was done with either SE-1 or F4/80, followed by labeling with Alexa-Fluor 555 Avidin (Invitrogen). Nuclear staining was obtained by incubation with TO-PRO-III Iodide (Invitrogen). Samples were visualized on a Leica TCS SP2 AOBS (Leica Microsystems, Deer-field, IL) confocal microscope and analyzed with Leica confocal software, version 2.61.
Quantitative real time RT-PCR
Total RNA was extracted from liver samples by RNA-Bee reagent (Tel-Test, Friendswood, TX). The mRNA coding for TNF, IL-1β, IL-6, IL-10, e-selectin, p-selectin, ICAM-1, VCAM-1, and GAPDH were quantified by SYBR Green two-step reverse-transcription (RT) PCR. Briefly, 1 mg of total RNA from each sample was used for RT with the Transcriptor first strand cDNA synthesis kit (Roche, Indianapolis, IN). PCR reaction mixture was then prepared with the use of SYBR Green PCR Master Mix (FastStart SYBR Green Master Mix; Roche, Indianapolis, IN). Table I lists the sequences of the primers. Thermal cycling conditions were 10 min at 95°C followed by 50 cycles of 95°C for 15 s and 60°C for 30 s on a Roche LightCycler 480. Each gene expression was normalized to GAPDH mRNA and calculated relative to baseline control using the comparative Ct method.
Table I.
Sequences of the primers for SYBR Green real-time RT-PCR
| Target | Forward Primers (5′–3′) | Reverse Primers(5′–3′) |
|---|---|---|
| E-selectin | AGCTACCCATGGAACACGAC | CGCAAGTTCTCCAGCTGTT |
| P-selectin | ATGCCTGGCTACTGGACACT | CTTCATCGCACATGAACTGG |
| ICAM-1 | GGCTGGCATTGTTCTCTAA | TTCAGAGGCAGGAAACAGG |
| VCAM-1 | CCCAAACAGAGGCAGAGTGT | CAGGATTTTGGGAGCTGGTA |
| IL-1β | CAACCAACAAGTGATATTCTCC | GATCCACACTCTCCAGCTGCA |
| IL-6 | ACAACCACGGCCTTCCCTACTT | CACGATTTCCCAGAGAACATGTG |
| IL-10 | ACAGGAGAAGGGACGCCAT | GAAGCCCTACAGACGAGCTCA |
| TNF | CATCTTCTCAAAATTCGAGTGACAA | TGGGAGTAGACAAGGTACAACCC |
| GAPDH | TTCACCACCATGGAGAAGGC | GGCATGGACTGTGGTCATGA |
IL-10 treatment
When applicable, ~30 min prior to I/R, animals were injected i.v. via the dorsal tail vein with 1 μg of IL-10 (BD Biosciences #550070), diluted in injectable saline.
IL-10 ELISA
Total protein was extracted from 50 mg of hepatic tissue using CelLytic MT Cell Lysis Reagent (Sigma-Aldrich, St. Louis, MO) according to the manufacturer's instructions. Total protein levels were normalized using a bicinchoninic acid protein assay, and these concentrations were used for normalization of IL-10 content. IL-10 protein levels were determined using the mouse IL-10 Quantikine ELISA Kit (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions. Results are presented as fold change over baseline at each time point.
Statistical analysis
All values are expressed as mean±SEM. An α value of 0.05 was established as the limit for statistical significance. For a single pairwise comparison, a two-tailed t test was used when samples were normally distributed. For multiple comparisons of normally distributed data, a one-way ANOVA with Tukey-Kramer posthoc analysis was used. For histologic analysis and samples that were not normally distributed, a Mann-Whitney U test was used. For multiple independent groups, a Kruskal Wallis nonparametric comparison was used, with a Tamhane's test for posthoc analysis using SPSS (Chicago, IL) statistical software.
Results
LC dose response study in the depletion of KCs
Animals were treated with increasing doses of LC for 48 h and liver KC levels were determined. LC decreased KC levels ~35% with a dose of 0.035 mg, and escalating doses further decreased KC levels, as assessed by F4/80 immunohistochemistry. Doses >0.35 mg achieved >98% depletion (Fig. 1). These depletion levels were maintained throughout the reperfusion period, as assessed by immunohistochemistry.
FIGURE 1.
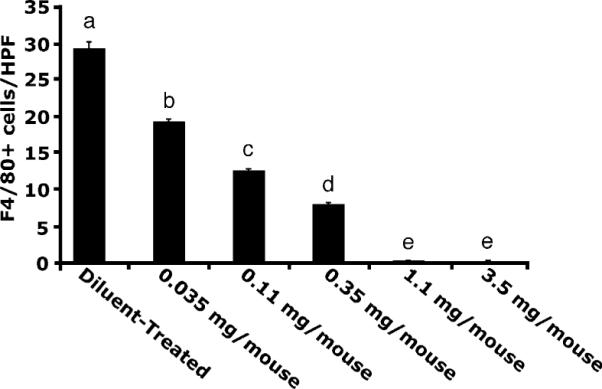
Dose response for KC depletion via LC. Various doses of LC were administered along a logarithmic curve; 0.035 mg caused ~35% reduction, and escalating doses further decreased KC levels. Doses >1.1 mg caused >98% depletion of KCs. Means with different lettered labels (a–e) within each group are significantly different from each other (p < 0.05). Data are expressed as mean ± SEM; n = 5–7, as measured by F4/80 immunohistochemistry.
KC depletion decreases survival and increases liver damage and cell death after I/R
Pretreatment with LC decreased animal survival following 35 min of total hepatic ischemia and 24 h of reperfusion (~55% versus ~95% in diluent treated controls; Fig. 2). Whereas an LC dose of 0.35 mg tended to decrease animal survival, only doses of 1.1 mg or higher, which deplete >98% of KCs, decreased the survival rate. In our model, animals that survive the initial 24 h period typically do not die of I/R.
FIGURE 2.

LC-treated animals have increased mortality. Increasing doses of LC cause increasing mortality after I/R. However, only doses >1.1 mg cause significant decreases in survival (*p < 0.05). Data are expressed as mean ± SEM; n = 12–15.
Hepatocyte injury was assessed via serum ALT levels. Increasing doses of LC had no effect on ALT levels in the absence of I/R (Fig. 3A). After 24 h of reperfusion, ALT levels increased 3-fold in animals pretreated with LC (1.1 mg) and subjected to I/R, compared with animals subjected to I/R alone. To further explore the time course of ALT release, ALT levels were measured at 6 and 24 h after I/R. At 6 h, ALT levels increased 10-fold in LC-treated mice subjected to I/R compared with diluent-treated mice subjected to I/R (Fig. 3B). Similar to Fig. 3A, ALT levels at 24 h in the LC-treated animals had decreased, but still remained elevated at 24 h versus diluent-treated animals.
FIGURE 3.
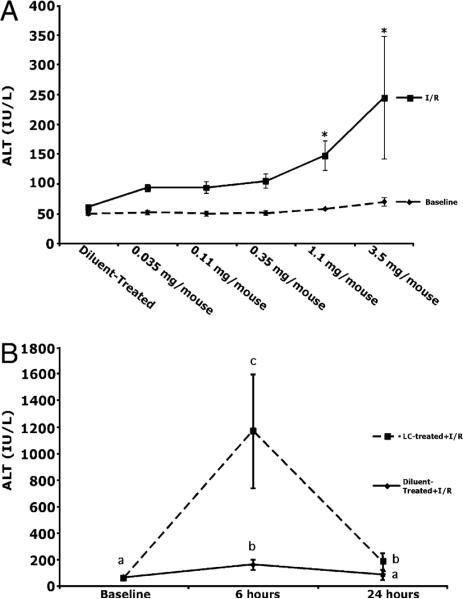
LC-treatment prior to I/R increases hepatocellular damage. A, LC treatment in the absence of I/R caused no increase in hepatocellular damage. Animals with >98% KC depletion had significantly increased ALT values at 24 h. B, To further evaluate ALT levels, animals were sacrificed at 6 and 24 h. ALT levels were elevated 10-fold in LC-treated animals over diluent-treated animals and remained significantly elevated compared with diluent-treated animals at 24 h. Means with different lettered labels (a–c) within each group are significantly different from each other (p < 0.05). Data are expressed as mean ± SEM; n = 12–5. Each time point represents a separate group of animals.
To gain insight into the type and scope of hepatocyte injury, histologic sections were stained with H&E, and injury was graded on a scale of 0–3, as described previously (15). Increasing doses of LC alone had no effect on hepatocyte injury in the absence of I/R (Fig. 4A). Large, confluent zones of necrosis were noted around zone 3 (pericentral areas) in LC-treated (1.1 mg) animals subjected to I/R (Fig. 4B). This result has not been seen previously in our model or in the literature, because the diluent-treated animals had little to no significant necrosis present in this area or elsewhere after I/R (Fig. 4B). Scoring of the hepatocyte injury revealed a 2-fold increase in the necrotic index in diluent-treated animals subjected to I/R at 6 h, and this increase was maintained at 24 h (Fig. 4C). In LC-treated animals subjected to I/R, the necrotic index increased 8-fold over baseline (2.5-fold over diluent treatment) at 6 h and was maintained at 24 h. These results reveal that LC-treated animals exhibit more extensive hepatocyte injury as measured by ALT release and necrotic cell death at 6 and 24 h of reperfusion. Partially depleting doses of LC showed no significant increases in necrosis after I/R (Fig. 4A). These results provide evidence that a reserve of KCs exist in the liver and that KC depletion >70% results decreased animal survival and liver injury following I/R injury.
FIGURE 4.

Necrosis is increased in LC-treated animals. A, Necrosis significantly increased at LC doses >1.1 mg. B, Large, confluent areas of necrosis were present around central veins (zone 3, arrows) at 6 and 24 h of reperfusion in LC-treated animals, as demonstrated by H&E staining (original magnification ×40). Normal hepatic lobular architecture was present in diluent-treated animals. C, Grading of this necrosis revealed 8-fold elevations in necrosis at 6 h in KC-depleted animals versus 2-fold elevations in diluent-treated animals. This increase persisted at 24 h. Means with different lettered labels (a–c) within each group are significantly different from each other (p < 0.05). Data are expressed as mean ± SEM; n = 12–15.
LPS binds extensively to LSECs
To more fully investigate the damaged phenotype displayed in LC-treated animals subjected to I/R, we used confocal microscopy to examine liver binding of the gut-derived LPS after I/R. An LC dose of 1.1 mg was used for the remainder of the experiments. Prior to examining animals subjected to I/R, animals were injected with 2.5 mg/kg LPS to validate our technique. There was binding of LPS to the sinusoidal membranes, as demonstrated by colocalization of LPS with SE-1, a marker of LSECs (Supplemental Fig. 1). At baseline, there was no visible binding of LPS in either LC- or diluent-treated animals (Fig. 5A). After I/R, LPS bound to cells in a sinusoidal pattern at 6 and 24 h with no difference between LC- and diluent-treated mice. To confirm the binding to LSECs, double staining was performed using an Ab against LSECs (SE-1). The majority of the translocated LPS bound to LSECs, as visualized by the colocalized signal of SE-1 and LPS, in both the diluent- and LC-treated animals subjected to I/R (Fig. 5A). Using F4/80 staining, a marker of KC, we observed some colocalization of LPS with KCs in animals subjected to I/R (Fig. 5B). However, the preponderance of the LPS staining did not colocalize with the KC immunostain. These results reveal that LPS binds to KC after I/R, but the bulk of the translocated endotoxin binds to endothelial cells, an observation that is unchanged in the absence of KCs.
FIGURE 5.

Extensive colocalization of LPS with endothelial cells. A, LPS (green, Alexa Fluor 488) is extensively present in an endothelial pattern. SE-1 (red, Alexa Fluor 555) colocalizes extensively with LPS (yellow). No LPS translocation is present in baseline samples. B, There is colocalization of LPS with F4/80 (red, Alexa Fluor 555), but not as extensively as with the SE-1 signal. There is no colocalization of LPS and F4/80 in LC-treated animals. Representative of 3–5 animals per group. Original magnification ×63.
KC control activation of LSEC
Because of the widespread binding of LPS to the LSECs, we investigated endothelial cell activation through observation of endothelial cell adhesion molecule expression. ICAM-1 is an endothelial cell marker that is upregulated in times of stress, such as I/R, and under conditions of LPS administration (16). ICAM-1 levels were slightly elevated in diluent-treated animals receiving I/R at both 6 and 24 h after reperfusion, as measured by immunohistochemical analysis (Fig. 6A). Levels of ICAM-1 were similarly upregulated at 6 h in LC-treated animals and markedly upregulated at 24 h compared with diluent-treated animals. To further investigate endothelial activation, we performed real-time RT-PCR to examine levels of multiple endothelial activation markers and cell adhesion molecules (for primer sequences, see Table I). mRNA levels of P-selectin increased 6-fold at 6 h in both diluent- and LC-treated animals subjected to I/R (Fig. 6B). These levels decreased to basal levels at 24 h and were similar in both groups. Levels of E-selectin were not elevated in either group at 6 h after I/R, but were upregulated in LC-treated animals at 24 h, whereas levels in diluent-treated animals declined. Levels of ICAM-1 mRNA were upregulated in LC-treated animals at both time points after I/R as compared with diluent-treated animals that increased 2-fold. Levels of VCAM-1 were upregulated at 6 h in LC-treated animals and decreased to basal levels at 24 h after I/R. VCAM-1 levels in diluent-treated animals did not change at 6 h, but decreased at 24 h below basal levels. There was no significant upregulation of any of the markers at baseline, indicating that LC alone did not stimulate activation marker expression. These results reveal that in the absence of KC, there is an upregulation of endothelial cell adhesion molecules at both 6 and 24 h following I/R. This upregulation is greater than that seen when KCs are intact.
FIGURE 6.

Upregulation of endothelial cell adhesion molecules. A, Levels of ICAM-1 (as assessed by ICAM immunohistochemistry) are slightly upregulated at 6 h of reperfusion in LC-treated animals and diluent-treated controls (original magnification ×40). There is a significant upregulation in LC-treated animals at 24 h, whereas diluent-treated animals maintain a level of expression similar to 6 h animals. B, At 6 h, there is no increase in E-selectin in LC-treated and diluent-treated animals, whereas there is a significant increase in E-selectin levels in LC-treated animals at 24 h, and diluent-treated animals have decreased levels. Levels of P-selectin, are upregulated in both groups at 6 h and decline significantly at 24 h in both groups. Levels of endothelial cell markers, ICAM-1 and VCAM-1, are significantly upregulated at 6 h after I/R versus diluent-treated controls. This upregulation is maintained at 24 h. Means with different lettered labels (a–c) within each group are significantly different from each other (p < 0.05) and are shown as fold change after normalization to baseline. Data are expressed as mean ± SEM; n = 12–15.
KCs prevent cytokine dysregulation
One of the most potent anti-inflammatory molecules produced in the liver is IL-10, and KCs are thought to be the predominant hepatic producer of this cytokine (9). Endothelial cells can produce a variety of proinflammatory mediators, but produce little IL-10. To further investigate the inflammatory milieu in the liver following I/R in the presence and absence of KCs, we used real-time RT-PCR to examine transcription levels of important hepatic proinflammatory cytokines. There was a marked increased in IL-10 mRNA levels in diluent-treated animals at 6 and 24 h after I/R (Fig. 7). In contrast, IL-10 mRNA levels in LC-treated animals at 6 and 24 h did not increase following I/R. In diluent-treated animals, there was no change in TNF mRNA levels at 6 or 24 h after I/R. In contrast, LC-treated animals TNF mRNA levels increased 5-fold at 6 h and returned to baseline levels at 24 h after I/R. IL-6 mRNA levels did not change at 6 and 24 h after I/R. However, in LC-treated animals, IL-6 mRNA levels increased 4-fold at 6 h and remained elevated at 24 h after I/R. Although levels of IL-1β mRNA changed numerically, none of the groups were significantly elevated over the others. There was no change in any of the cytokines at baseline just prior to I/R, indicating that LC alone did not stimulate cytokine expression. These results reveal that the deletion of KCs can cause an upregulation in key proinflammatory cytokines that exceeds cytokine production seen in diluent-treated animals. There is no production of IL-10 in LC-treated animals, whereas diluent-treated animals show marked IL-10 production.
FIGURE 7.
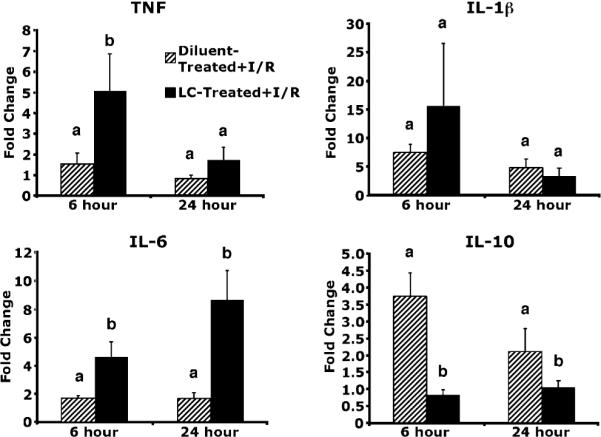
Cytokine upregulation in LC-treated animals. Levels of TNF and IL-6 were upregulated at 6 h of reperfusion in LC-treated animals versus diluent-treated controls. Levels of IL-6 remained upregulated at 24 h, whereas levels of TNF had returned to the level of the diluent-treated controls. Levels of IL-1b were not significantly upregulated in any of the groups. IL-10 levels were significantly downregulated in LC-treated animals at both 6 and 24 h of reperfusion. Means with different lettered labels (a, b) within each group are significantly different from each other (p < 0.05) and are presented as fold change after normalization to baseline. Data are expressed as mean ± SEM; n = 8–10.
IL-10 protein levels are decreased in LC-treated animals
To verify that the mRNA levels of IL-10 correlated with the protein production, an IL-10 ELISAwas performed on total hepatic protein. There was an increase in IL-10 levels at 6 h post reperfusion in diluent-treated animals, whereas LC-treated animals showed no such increase at 6 h (Fig. 8). These levels of IL-10 in diluent-treated animals continued to increase at 24 h, whereas the levels of IL-10 in LC-treated animals continued to remain at or below baseline levels. These results reveal that IL-10 protein levels correlate with mRNA levels, which both show that there is an increase in IL-10 levels after reperfusion in diluent-treated animals, whereas there is no production of IL-10 in LC-treated animals.
FIGURE 8.
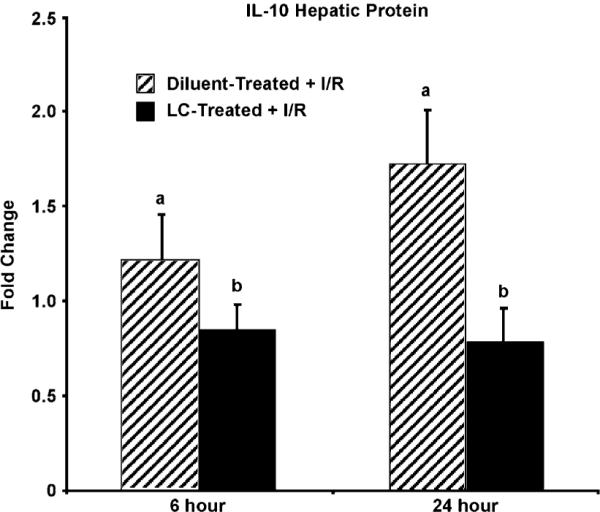
IL-10 protein levels are decreased in the livers of LC-treated animals. ELISA analysis shows that diluent-treated animals have increased production of IL-10 protein at 6 h, continuing to 24 h after reperfusion. In contrast, LC-treated animals failed to show an increase in IL-10 protein levels after reperfusion. Means with different lettered labels (a, b) within each group are significantly different from each other (p < 0.05) and are presented as fold change after normalization to baseline. Data are expressed as mean ± SEM; n = 8–10.
IL-10 treatment prevents I/R damage in LC-treated animals
To determine the importance of IL-10 in KC protective mechanisms and I/R-induced hepatic injury, we pretreated (30 min) animals with murine recombinant IL-10 (1 μg) or diluent and subjected the animals to I/R injury. As discussed above, LC-treated animals had a survival rate of ~50–55%, and diluent treated animals had a survival rate of 100% following I/R (Fig. 8). LC-treated animals pretreated with IL-10 had an improved survival rate of 88%, which was not different from diluent-treated animals. In addition, LC-treated animals pretreated with IL-10 prevented ALT release at 6 h and decreased centrilobular necrosis following I/R compared with animals treated with LC alone.
IL-10 treatment prevents endothelial activation and blunts cytokine release
To show that IL-10 can inhibit the activation of LSEC after I/R in KC-depleted animals, we measured real-time PCR levels of endothelial markers and cytokine levels at 6 h after I/R. Animals treated with diluent plus IL-10 exhibited a decrease in ICAM-1 levels, but VCAM-1, TNF, and IL-6 levels did not change in either diluent-treated group (Fig. 9). The 5- and 3-fold increases in ICAM-1 and VCAM-1 levels, respectively, were decreased to diluent-treated levels in animals treated with LC plus IL-10. Likewise, the 5- and 4-fold increases in TNF and IL-6 levels, respectively, were decreased to diluent-treated levels in animals treated with LC plus IL-10 (Fig. 10). Thus, treatment with IL-10 in KC-depleted animals can return cytokine and adhesion molecule production to diluent-treated levels.
FIGURE 9.

IL-10 treatment improves survival and decreases hepatic damage. A, Animals treated with recombinant IL-10 and LC had a significantly improved mortality rate versus animals treated with LC alone. B, Similarly IL-10/LC-treated animals had a decreased release of ALT versus LC-treated animals at 6 h of reperfusion. Animals treated with either LC or LC plus IL-10 had elevated levels of ALT as compared with diluent-treated animals at 24 h. C, Animals treated with diluent or LC plus IL-10 had a 2-fold increase in necrosis at 6 h that persisted to 24 h. Animals treated with LC alone had a 6-fold increase in necrosis at 6 h that persisted to 24 h. Means with different lettered labels (a–c) within each group are significantly different from each other (p < 0.05). Data are expressed as mean ± SEM; n = 8–10.
FIGURE 10.

IL-10 treatment decreases endothelial activation and cytokine production at 6 h after I/R. A, ICAM-1 levels were decreased in animals treated with diluent plus IL-10 versus those treated with diluent alone at 6 h. Animals treated with LC showed a 5-fold increase that was reduced to diluent-treated levels in animals treated with LC plus IL-10. VCAM-1 levels were not upregulated in either diluent-treated group, but were 3-fold elevated in LC-treated animals. Animals treated with LC plus IL-10 showed reduced production that was still elevated compared with animals treated with diluent plus IL-10. B, Levels of TNF and IL-6 were not elevated in the diluent-treated group or in animals treated with LC plus IL-10. Levels of TNF and IL-6 were upregulated 5- and 4.5-fold in LC-treated animals, respectively. Means with different lettered labels (a–c) within each group are significantly different from each other (p < 0.05) and are presented as fold change as normalized to baseline. Data are expressed as mean ± SEM; n = 8–10.
Discussion
The series of experiments presented in this study addressed the role of KCs in the pathogenesis of total warm hepatic I/R with bowel congestion. It was previously thought that KCs played a central role in the detrimental effects of reperfusion, because they release numerous toxic mediators (1) and inhibition of their function using GdCl3 or glycine improved outcomes after I/R (17). We have used a different pharmacologic approach that results in KC depletion, and we show that extensive KC elimination led to a decrease in animal survival and an increase in liver injury, as measured by ALT and indices of necrosis. In addition, there was marked binding of LPS to LSECs that led to their increased activation in the absence of KCs, as shown by an increase in endothelial adhesion molecules. With this, we saw an increase in hepatic proinflammatory cytokines owing to the decrease in the hepatic levels of IL-10. This damage, LSEC activation and inflammatory state were reduced to diluent-treated levels by the administration of exogenous IL-10.
An important note here is that our model of total hepatic I/R with bowel congestion differs from other models involving isolated lobar hepatic ischemia. To more closely mimic the setting of liver transplantation, where portal outflow obstruction and bowel congestion are unavoidable, we incorporated this variable into our model. In models of 70% partial ischemia, portal outflow is maintained. Although this model elegantly allows the study of hepatic I/R alone, we find it to be less relevant to the clinical scenario of hepatic transplantation. Previously, we have seen negligible death and liver damage in lean animals in our I/R model (14, 18). However, after the depletion of KCs, we observed a nearly 50% decrease in the survival of lean animals after I/R. This finding would suggest that KCs play a role in promoting animal survival after hepatic I/R.
Transaminase release further suggests that hepatic damage contributed to overall mortality, because transaminase levels in the absence of KCs were nearly 10-fold greater than in diluent-treated animals at 6 h of reperfusion. This damage occurs early (6 h) and is transient because ALT levels at 24 h returned closer to baseline. The degree of hepatocellular necrosis in LC-treated animals mirrored the transaminase increase. At both 6 and 24 h of reperfusion, there were large areas of zone 3 necrosis and bridging necrosis that were not present in KC-competent animals. These data strengthen the hypothesis that KCs protect the liver from damage after I/R, promoting overall animal survival, and demonstrate that KCs act within the first 6 h of reperfusion to protect the hepatocytes from plasma membrane injury and necrosis.
Previously, we demonstrated that the LPS bolus that occurs subsequent to the anhepatic phase of transplant as a result of bowel congestion is integral in the damaging effects of I/R (12). Thus, we examined LPS binding in the liver via immunohistochemistry and confocal microscopy following I/R. We observed no binding of LPS to KCs or endothelial cells under basal conditions. However, following I/R, LPS binding occurred in endothelial cells and to a lesser extent in KCs. TLR4 is extensively present on KCs, and these cells respond exquisitely to LPS stimulation via production of proinflammatory cytokines (19) and IL-10 (9). However, the hepatic response to LPS in the absence of KCs is unknown. We know that TLR4 is present on endothelial cells (20, 21) and is the main receptor for LPS. Therefore, we would expect that endothelial response would play a role in the pathology of I/R. In addition, endothelial cells play a role in the hepatic tolerance to LPS, because they can modulate TLR4 activation without altering receptor expression (8). In addition, scavenger receptor A has been shown to be important in LPS removal from the circulation, thus avoiding TLR4 stimulation and upregulation (8). Thus, we have shown that LPS associates strongly with endothelial cells after I/R. Although this association occurs similarly in KC-depleted and competent animals, in the absence of KC the endothelial cells appear to play a larger role than was previously acknowledged.
LPS has been shown to directly upregulate levels of P-selectin and ICAM-1 (22), and the levels of ICAM-1 and VCAM-1 have been shown to correlate with the levels of tissue injury in other organs independent of cell migration (23). In addition, it has been shown that TNF can upregulate surface expression of ICAM-1 (16), and that ICAM-1 knockout mice are protected from endotoxin-mediated septic shock (24). We observed that P-selectin and ICAM-1 mRNA levels increase following 6 h of I/R and that ICAM-1 is elevated at 24 h. LC treatment plus I/R further increased ICAM-1 levels and increased VCAM-1 levels 6 h after I/R. At 24 h, only E-selectin and ICAM-1 levels remained elevated in animals treated with LC plus I/R. Thus, there is early and sustained endothelial cell adhesion molecule production. These activated endothelial cells appear to be capable of exerting a negative role after I/R in the absence of KC monitoring. In summary, KCs exert their hepatic protective effect by preventing the early increases in ICAM-1 and VCAM-1 and endothelial cell activation, potentially leading to inflammatory cell infiltration.
Endothelial cells have recently been implicated as important producers of cytokines, both in a proinflammatory setting and a regenerative one (25). Our data indicate that in the absence of KC, there is a significant imbalance in the production of cytokines. Specifically, we saw overproduction of TNF and IL-6 at 6 h of reperfusion. This was accompanied by a concomitant decreased production of IL-10. KCs are well known to be the predominant hepatic producers of IL-10, a potent anti-inflammatory cytokine, and their absence led to a dramatic drop in hepatic levels of this molecule. We suggest for the first time that the absence of KCs, and their production of IL-10, led to an unchecked proinflammatory response in our model of warm hepatic I/R, potentially by endothelial cells.
Pretreatment of LC-treated animals with IL-10 restored survival and decreased hepatic damage to minimal, wild-type levels. Previous data have shown that decreased endogenous IL-10 can cause increased inflammatory disorders, such as Crohn's disease, or autoimmunity, whereas increased endogenous levels can cause susceptibility to infection (26). In addition, IL-10 pretreatment decreased ICAM-1, VCAM-1, TNF, and IL-6 in mice depleted of KCs and subjected to I/R. IL-10 has been shown to destabilize TNF mRNA, thus promoting downregulation of the proinflammatory response (27). In addition to preventing proinflammatory cytokine persistence, IL-10 has also been shown to have direct effects on the inhibition of the production of adhesion molecules, specifically ICAM-1 (28). When administered in vitro to endothelial cells, IL-10 has been shown to downregulate ICAM-1 and VCAM-1 (29). Finally, IL-10−/− mice have been shown to have increased colonic levels of ICAM-1 and VCAM-1 (30), and the IL-10 genotype is predictive of the levels of adhesion molecules in patients (31). These results provide strong evidence that IL-10 production by KCs following I/R injury blocks hepatocyte necrosis, endothelial cell activation, and the proinflammatory state.
KC-derived IL-10 has been shown to be critical in other models of liver disease as well. Specifically, LC-depleted animals were shown to have decreased regenerative capacity after partial hepatectomy because of an unbalanced cytokine milieu (4). In contrast to our results, however, models of partial hepatectomy show decreased inflammatory cytokine production, whereas we show increases. This finding is likely due to the predominant inflammatory component in our model, compared to the LPS-free regenerative stimulus in a partial hepatectomy model. IL-10−/− mice have been shown to have greatly increased susceptibility to liver injury in multiple models, including acetaminophen hepatotoxicity (32), small-for-size liver grafts (33), and I/R injury (34). This is in line with our results demonstrating that animals treated with LC plus IL-10 had reduced levels of cytokine secretion versus animals treated with LC alone.
Finally, studies have shown that pretreatment of animals with IL-10 neutralizing Abs increases hepatic injury sustained after I/R. As early as 6 h after reperfusion, there is increased production of IL-1b, increased hepatic necrosis, and increased release of malonyldialdehyde and ALT in animals pretreated with anti–IL-10 Abs. In addition, the removal of IL-10 results in a blunting of the protective effects of ischemic preconditioning, as shown by increases in parameters of liver injury as compared with animals not pretreated with neutralizing Abs (35). Additional recent studies have shown that adenoviral transfection of donor animals with human IL-10 can significantly prevent necrotic and apoptotic I/R injury as compared with animals transfected with lacZ (36).
We have shown that, contrary to previous reports, KCs appear to play a protective role in the liver subjected to total hepatic I/R with bowel congestion. Their removal dramatically increases hepatocellular damage and decreases animal survival. In addition, removal of KCs causes overactivation of endothelial cells and an increase in proinflammatory cytokines. There is also a concomitant decrease in IL-10 levels in LC-treated mice. When animals are treated with IL-10, hepatic damage is dramatically decreased, as is endothelial cell activation, indicating that KC-derived IL-10 plays an essential role in mediating damage after hepatic I/R and keeping the inflammatory milieu in balance.
Acknowledgments
We thank Kathy Haines for support in running our laboratory, Margaret Kelly for technical assistance, the Medical University of South Carolina Hollings Cancer Center Cell, and Molecular Imaging Shared Resource.
This work was supported by National Institutes of Health Grant 1R01DK069369.
Abbreviations used in this paper
- ALT
alanine aminotransferase
- ICAM
intracellular adhesion molecule
- I/R
ischemia/reperfusion
- KC
Kupffer cell
- LC
liposomal clodronate
- LSEC
liver sinusoidal endothelial cell
Footnotes
Disclosures The authors have no financial conflicts of interest.
References
- 1.Montalvo-Jave EE, Escalante-Tattersfield T, Ortega-Salgado JA, Piña E, Geller DA. Factors in the pathophysiology of the liver ischemia-reperfusion injury. J. Surg. Res. 2008;147:153–159. doi: 10.1016/j.jss.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giakoustidis DE, Iliadis S, Tsantilas D, Papageorgiou G, Kontos N, Kostopoulou E, Botsoglou NA, Gerasimidis T, Dimitriadou A. Blockade of Kupffer cells by gadolinium chloride reduces lipid peroxidation and protects liver from ischemia/reperfusion injury. Hepatogastroenterology. 2003;50:1587–1592. [PubMed] [Google Scholar]
- 3.Hardonk MJ, Dijkhuis FW, Hulstaert CE, Koudstaal J. Heterogeneity of rat liver and spleen macrophages in gadolinium chloride-induced elimination and repopulation. J. Leukoc. Biol. 1992;52:296–302. doi: 10.1002/jlb.52.3.296. [DOI] [PubMed] [Google Scholar]
- 4.Meijer C, Wiezer MJ, Diehl AM, Schouten HJ, Schouten HJ, Meijer S, van Rooijen N, van Lambalgen AA, Dijkstra CD, van Leeuwen PA. Kupffer cell depletion by CI2MDP-liposomes alters hepatic cytokine expression and delays liver regeneration after partial hepatectomy. Liver. 2000;20:66–77. doi: 10.1034/j.1600-0676.2000.020001066.x. [DOI] [PubMed] [Google Scholar]
- 5.Farmer D, Amersi F, Kupiec-Weglinski JW, Busuttil RW. Current Status of Ischemia and Reperfusion Injury in the Liver. Transplant. Rev. 2000;14:106–126. [Google Scholar]
- 6.Urakami A, Todo S, Zhu Y, Zhang S, Jin MB, Ishizaki N, Shimamura T, Totsuka E, Subbotin V, Lee R, Starzl TE. Attenuation of ischemic liver injury by monoclonal anti-endothelin antibody, AwETN40. J. Am. Coll. Surg. 1997;185:358–364. [PMC free article] [PubMed] [Google Scholar]
- 7.Farmer DG, Kaldas F, Anselmo D, Katori M, Shen XD, Lassman C, Kaldas M, Clozel M, Busuttil RW, Kupiec-Weglinski J. Tezosentan, a novel endothelin receptor antagonist, markedly reduces rat hepatic ischemia and reperfusion injury in three different models. Liver Transpl. 2008;14:1737–1744. doi: 10.1002/lt.21621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uhrig A, Banafsche R, Kremer M, Hegenbarth S, Hamann A, Neurath M, Gerken G, Limmer A, Knolle PA. Development and functional consequences of LPS tolerance in sinusoidal endothelial cells of the liver. J. Leukoc. Biol. 2005;77:626–633. doi: 10.1189/jlb.0604332. [DOI] [PubMed] [Google Scholar]
- 9.Knolle P, Schlaak J, Uhrig A, Kempf P, Meyer zum Büschenfelde KH, Gerken G. Human Kupffer cells secrete IL-10 in response to lipopolysaccharide (LPS) challenge. J. Hepatol. 1995;22:226–229. doi: 10.1016/0168-8278(95)80433-1. [DOI] [PubMed] [Google Scholar]
- 10.Santucci L, Fiorucci S, Chiorean M, Brunori PM, Di Matteo FM, Sidoni A, Migliorati G, Morelli A. Interleukin 10 reduces lethality and hepatic injury induced by lipopolysaccharide in galactosamine-sensitized mice. Gastroenterology. 1996;111:736–744. doi: 10.1053/gast.1996.v111.pm8780580. [DOI] [PubMed] [Google Scholar]
- 11.Filos KS, Kirkilesis I, Spiliopoulou I, Scopa CD, Nikolopoulou V, Kouraklis G, Vagianos CE. Bacterial translocation, endotoxaemia and apoptosis following Pringle manoeuvre in rats. Injury. 2004;35:35–43. doi: 10.1016/s0020-1383(03)00288-2. [DOI] [PubMed] [Google Scholar]
- 12.Fiorini RN, Shafizadeh SF, Polito C, Rodwell DW, Cheng G, Evans Z, Wan C, Belden S, Haines JK, Birsner J, et al. Anti-endotoxin monoclonal antibodies are protective against hepatic ischemia/reperfusion injury in steatotic mice. Am. J. Transplant. 2004;4:1567–1573. doi: 10.1111/j.1600-6143.2004.00549.x. [DOI] [PubMed] [Google Scholar]
- 13.Van Rooijen N, Sanders A. Kupffer cell depletion by liposome-delivered drugs: comparative activity of intracellular clodronate, propamidine, and ethylenediaminetetraacetic acid. Hepatology. 1996;23:1239–1243. doi: 10.1053/jhep.1996.v23.pm0008621159. [DOI] [PubMed] [Google Scholar]
- 14.Evans ZP, Ellett JD, Schmidt MG, Schnellmann RG, Chavin KD. Mitochondrial uncoupling protein-2 mediates steatotic liver injury following ischemia/reperfusion. J Biol Chem. 2007;283:8573–8579. doi: 10.1074/jbc.M706784200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neil DA, Hubscher SG. Are parenchymal changes in early post-transplant biopsies related to preservation-reperfusion injury or rejection? Transplantation. 2001;71:1566–1572. doi: 10.1097/00007890-200106150-00014. [DOI] [PubMed] [Google Scholar]
- 16.Ohira H, Ueno T, Torimura T, Tanikawa K, Kasukawa R. Leukocyte adhesion molecules in the liver and plasma cytokine levels in endotoxin-induced rat liver injury. Scand. J. Gastroenterol. 1995;30:1027–1035. doi: 10.3109/00365529509096349. [DOI] [PubMed] [Google Scholar]
- 17.Giakoustidis D, Papageorgiou G, Iliadis S, Giakoustidis A, Kostopoulou E, Kontos N, Botsoglou E, Tsantilas D, Papanikolaou V, Takoudas D. The protective effect of alpha-tocopherol and GdCl3 against hepatic ischemia/reperfusion injury. Surg. Today. 2006;36:450–456. doi: 10.1007/s00595-005-3162-9. [DOI] [PubMed] [Google Scholar]
- 18.Ellett JD, Evans ZP, Fiorini JH, Fiorini RN, Haines JK, Schmidt MG, Chavin KD. The use of the Papworth cocktail is detrimental to steatotic livers after ischemia-reperfusion injury. Transplantation. 2008;86:286–292. doi: 10.1097/TP.0b013e31817b900f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su GL, Klein RD, Aminlari A, Zhang HY, Steinstraesser L, Alarcon WH, Remick DG, Wang SC. Kupffer cell activation by lipopolysaccharide in rats: role for lipopolysaccharide binding protein and toll-like receptor 4. Hepatology. 2000;31:932–936. doi: 10.1053/he.2000.5634. [DOI] [PubMed] [Google Scholar]
- 20.Chen LC, Gordon RE, Laskin JD, Laskin DL. Role of TLR-4 in liver macrophage and endothelial cell responsiveness during acute endotoxemia. Exp. Mol. Pathol. 2007;83:311–326. doi: 10.1016/j.yexmp.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu FL, You HB, Li XH, Chen XF, Liu ZJ, Gong JP. Glycine attenuates endotoxin-induced liver injury by downregulating TLR4 signaling in Kupffer cells. Am. J. Surg. 2008;196:139–148. doi: 10.1016/j.amjsurg.2007.09.045. [DOI] [PubMed] [Google Scholar]
- 22.Kamochi M, Kamochi F, Kim YB, Sawh S, Sanders JM, Sarembock I, Green S, Young JS, Ley K, Fu SM, Rose CE., Jr. P-selectin and ICAM-1 mediate endotoxin-induced neutrophil recruitment and injury to the lung and liver. Am. J. Physiol. 1999;277:L310–L319. doi: 10.1152/ajplung.1999.277.2.L310. [DOI] [PubMed] [Google Scholar]
- 23.Raeburn CD, Calkins CM, Zimmerman MA, Song Y, Ao L, Banerjee A, Harken AH, Meng X. ICAM-1 and VCAM-1 mediate endotoxemic myocardial dysfunction independent of neutrophil accumulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002;283:R477–R486. doi: 10.1152/ajpregu.00034.2002. [DOI] [PubMed] [Google Scholar]
- 24.Xu H, Gonzalo JA, St Pierre Y, Williams IR, Kupper TS, Cotran RS, Springer TA, Gutierrez-Ramos JC. Leukocytosis and resistance to septic shock in intercellular adhesion molecule 1-deficient mice. J. Exp. Med. 1994;180:95–109. doi: 10.1084/jem.180.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ping C, Xiaoling D, Jin Z, Jiahong D, Jiming D, Lin Z. Hepatic sinusoidal endothelial cells promote hepatocyte proliferation early after partial hepatectomy in rats. Arch. Med. Res. 2006;37:576–583. doi: 10.1016/j.arcmed.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 26.Mosser DM, Zhang X. Interleukin-10: new perspectives on an old cytokine. Immunol. Rev. 2008;226:205–218. doi: 10.1111/j.1600-065X.2008.00706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rai RM, Loffreda S, Karp CL, Yang SQ, Lin HZ, Diehl AM. Kupffer cell depletion abolishes induction of interleukin-10 and permits sustained overexpression of tumor necrosis factor alpha messenger RNA in the regenerating rat liver. Hepatology. 1997;25:889–895. doi: 10.1002/hep.510250417. [DOI] [PubMed] [Google Scholar]
- 28.Song S, Ling-Hu H, Roebuck KA, Rabbi MF, Donnelly RP, Finnegan A. Interleukin-10 inhibits interferon-gamma-induced intercellular adhesion molecule-1 gene transcription in human monocytes. Blood. 1997;89:4461–4469. [PubMed] [Google Scholar]
- 29.Krakauer T. IL-10 inhibits the adhesion of leukocytic cells to IL-1-activated human endothelial cells. Immunol. Lett. 1995;45:61–65. doi: 10.1016/0165-2478(94)00226-h. [DOI] [PubMed] [Google Scholar]
- 30.Kawachi S, Jennings S, Panes J, Cockrell A, Laroux FS, Gray L, Perry M, van der Heyde H, Balish E, Granger DN, et al. Cytokine and endothelial cell adhesion molecule expression in interleukin-10-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2000;278:G734–G743. doi: 10.1152/ajpgi.2000.278.5.G734. [DOI] [PubMed] [Google Scholar]
- 31.Kahraman S, Yilmaz R, Arici M, Altun B, Erdem Y, Yasavul U, Turgan C. IL-10 genotype predicts serum levels of adhesion molecules, inflammation and atherosclerosis in hemodialysis patients. J. Nephrol. 2006;19:50–56. [PubMed] [Google Scholar]
- 32.Bourdi M, Masubuchi Y, Reilly TP, Amouzadeh HR, Martin JL, George JW, Shah AG, Pohl LR. Protection against acetaminophen-induced liver injury and lethality by interleukin 10: role of inducible nitric oxide synthase. Hepatology. 2002;35:289–298. doi: 10.1053/jhep.2002.30956. [DOI] [PubMed] [Google Scholar]
- 33.Yang ZF, Ho DW, Ngai P, Lau CK, Zhao Y, Poon RT, Fan ST. Antiinflammatory properties of IL-10 rescue small-for-size liver grafts. Liver Transpl. 2007;13:558–565. doi: 10.1002/lt.21094. [DOI] [PubMed] [Google Scholar]
- 34.Dinant S, Veteläinen RL, Florquin S, van Vliet AK, van Gulik TM. IL-10 attenuates hepatic I/R injury and promotes hepatocyte proliferation. J. Surg. Res. 2007;141:176–182. doi: 10.1016/j.jss.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 35.Serafín A, Roselló-Catafau J, Prats N, Gelpí E, Rodés J, Peralta C. Ischemic preconditioning affects interleukin release in fatty livers of rats undergoing ischemia/reperfusion. Hepatology. 2004;39:688–698. doi: 10.1002/hep.20089. [DOI] [PubMed] [Google Scholar]
- 36.Li JQ, Qi HZ, He ZJ, Hu W, Si ZZ, Li YN, Li DB. Cytoprotective effects of human interleukin-10 gene transfer against necrosis and apoptosis induced by hepatic cold ischemia/reperfusion injury. J. Surg. Res. 2009;157:e71–e78. doi: 10.1016/j.jss.2009.03.004. [DOI] [PubMed] [Google Scholar]