

Abstract
Oxidative stress can induce cytotoxicity in neurons, which plays an important role in the etiology of neuronal damage and degeneration. The present study seeks to determine the cellular and biochemical mechanisms underlying resveratrol’s protective effect against oxidative neuronal death. The cultured HT22 cells, an immortalized mouse hippocampal neuronal cell line, were used as an in vitro model, and the oxidative stress and neurotoxicity in these neuronal cells were induced by exposure to high concentrations of glutamate. Resveratrol strongly protected HT22 cells from glutamate-induced oxidative cell death. Resveratrol’s neuroprotective effect was independent of its direct radical-scavenging property, but instead was dependent on its ability to selectively induce the expression of mitochondrial superoxide dismutase (SOD2), and subsequently, reduce mitochondrial oxidative stress and damage. The induction of the mitochondrial SOD2 by resveratrol was mediated through the activation of the PI3K/Akt and GSK-3β/β-catenin signaling pathways. Taken together, the results of this study show that up-regulation of the mitochondrial SOD2 by resveratrol represents an important mechanism for its protection of neuronal cells against oxidative cytotoxicity resulting form mitochondrial oxidative stress.
Keywords: Resveratrol, Superoxide dismutase, Oxidative stress, HT22, PI3K/Akt signaling pathway
INTRODUCTION
Glutamate is an endogenous excitatory neurotransmitter, and it is estimated that this neurotransmitter is used by as many as one-third of the synapses in the central nervous system [1]. At high concentrations, glutamate is neurotoxic, and glutamate-induced neuronal death contributes to the development of neurodegenerative diseases [1,2]. Two common pathways of glutamate neurotoxicity have been described. One is the excitotoxic pathway that is mediated by ionotropic glutamate receptors [3,4]. The mechanism of excitotoxicity has been extensively characterized, and it is believed that transient Ca2+ fluxes lead to alterations in calcium homeostasis, increases in the levels of reactive oxygen species (ROS), and ultimately cell death. The second distinct pathway in glutamate toxicity does not involve glutamate receptors, but rather involves a glutamate/cystine antiporter, which is required for the delivery of cystine into neuronal cells [5]. Inhibition of cystine uptake by high concentrations of extracellular glutamate leads to imbalance in cellular cysteine homeostasis, reduction in cellular glutathione levels, and accumulation of ROS. Glutamate-induced oxidative toxicity has been described in neuronal cell lines [5-9], primary neuronal cultures [10], and oligodendrocytes [11]. This oxidative neuronal death pathway is thought to contribute importantly to neuronal injury and degeneration in many brain disorders [1].
HT22 cells, an immortalized mouse hippocampal cell line, have been widely used as an in vitro model for elucidating the mechanism of oxidative stress-induced neurotoxicity [5,12,13]. HT22 cells lack functional ionotropic glutamate receptor [8], thus excluding excitotoxicity as a cause for glutamate-triggered cell death. HT22 cells are similar to undifferentiated neuronal stem cells, and express neuron-specific enolase and neurofilament proteins [14]. Because these cells divide rapidly in culture and lack ionotropic glutamate receptors, they do not exhibit the morphology of neurons. A number of studies have shown that glutamate at high concentrations could induce oxidative stress and subsequently cell death in cultured HT22 cells by inhibiting cystine uptake, which results in decreased intracellular glutathione levels and ultimately oxidative stress and cell death [5,12,13]. Our recent study showed that the oxidative stress elicited by glutamate treatment could induce, in a time-dependent manner, both necrosis and apoptosis in cultured HT22 cells [15].
In recent years, several dietary phenolic compounds, such as resveratrol, caffeic acid and vitamin E, were found to have a protective effect in cultured neuronal cells against the oxidative cytotoxicity of glutamate [16-18] and hydrogen peroxide [19,20]. This neuroprotective effect is generally thought to be due to the direct antioxidant and free radical-scavenging properties of these dietary compounds. Using resveratrol (trans-3,4′,5-trihydroxystilbene) as an example, which is a well-known phenolic compound abundant in our food sources [21-23], we further investigated in this study the mechanism of its neuroprotective effect in cultured HT22 mouse hippocampal cells. Our results show that the neuroprotective effect of resveratrol is largely independent of its direct antioxidant activity, rather it protects HT22 neuronal cells from glutamate-induced oxidative cytotoxicity by inducing the expression of the mitochondrial superoxide dismutase 2 (SOD2) via activation of the PI3K/Akt GSK-3β/β-catenin signaling pathways.
MATERIALS AND METHODS
Materials
Resveratrol, Dullbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), and trypsin-EDTA solution (containing 0.5 g/L trypsin and 0.2 g/L EDTA) were purchased from Sigma Chemical Co. (St. Louis). Resveratrol was dissolved in 200-proof ethanol and stored at −20°C. The antibiotics solution (containing 10,000 U/mL penicillin and 10 mg/mL streptomycin) was obtained from Gibco (Invitrogen, Grand Island, NY). The inhibitors of mitogen-activated protein kinases (MAPKs) and PI3K used in this study were obtained from Calbiochem (La Jolla, CA), and they were dissolved in dimethyl sulfoxide (DMSO) at a stock concentration of 10 mM.
Cell culture
Glutamate-sensitive HT22 murine hippocampal neuronal cells (a gift from Dr. David Schubert, Salk Institute, La Jolla, CA) were maintained in DMEM supplemented with 10% (v/v) FBS and antibiotics (penicillin-streptomycin) and incubated at 37°C under 5% CO2. Cells were sub-cultured once every 2 days. To study the protective effect of resveratrol on glutamate-induced neuronal death, cells were seeded in 96-well plates at a density of 5000 cells per well, and 5 wells were used for each treatment group. The stock solutions of glutamate (1 M in DMEM without serum) and resveratrol (100 mM in 200-proof ethanol) were diluted in the culture medium immediately before addition to each well at the desired final concentrations, and the treatment usually lasted for 24 hours. To test the effect of the inhibitors of MAPKs and PI3K, cells were pre-treated with each of the inhibitors (at 1.25-5 μM) for 2 hours before the addition of glutamate and/or resveratrol. Cells in the control group were treated with vehicle only.
MTT assay
For determining cell viability, the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used. MTT (10 μL, at 5 mg/mL) was added to each well at a final concentration of 500 μg/mL, and the mixture was further incubated for 1 hour at 37°C, and the liquid in the wells was removed thereafter. DMSO (100 μL) was then added to each well, and the absorbance was read with a UV max microplate reader (Molecular Device, Palo Alto) at 560 nm. The relative cell density was expressed as percentage of the control that was not treated with glutamate or reveratrol.
Flow cytometric analysis
After treatment with glutamate and/or resveratrol, cells were harvested by trypsinization and washed once with phosphate-buffered saline (PBS, pH 7.4). After centrifugation, cells were stained with annexin-V and propidiun iodide (PI) using the annexin V-FITC apoptosis detection kit (BD Biosciences, San Jose, CA) for analysis of the translocation of phosphatidylserine (PS) from inner to outer leaflets of the plasma membrane. For annexin-V and PI double staining, the procedure was performed according to the instructions of the manufacturers. For analysis of mitochondrial membrane potential (MMP), cell pellets were resuspended in 1 mL cell culture medium containing 25 nM DiOC6(3) (Molecular Probes, Eugene, OR) and incubated at 37°C for 15 minutes. After centrifugation, cells were resuspended in PBS. For analysis of relative mitochondria density, cell pellets were resuspended in 1 mL culture medium containing 500 nM MitoTracker Red FM (Molecular Probes) and incubated at 37°C for 15 minutes. After centrifugation, cells were resuspended in PBS. All flow cytometric analyses were performed using a flow cytometer (model BD LSR II, BD Bioscience).
Western blotting
Cells were washed with PBS, and then suspended in 100 μL of lysis buffer (20 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, protease inhibitor cocktail, 2 mM Na3VO4, and 10 mM NaF, pH 7.5). The protein concentration was determined using the Bio-Rad protein assay (Bio-Rad, Hercules, CA). An equal amount of proteins was loaded in each lane. Proteins were separated using 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and electrically transferred to a polyvinylidene difluoride membrane (Bio-Rad). After blocking the membrane with 5% skim milk, target proteins were immunodetected using specific antibodies. All primary antibodies were obtained from Cell Signaling Technology (Beverly, MA) except the anti-phospho JNK1/2 (Thr183/Tyr185) specific antibody (from Biosource, Camarillo, CA) and the anti-SOD1 and anti-SOD2 specific antibodies (from Santa Cruz Biotechnology, Santa Cruz, CA). Specific antibody for the phospho-p44/42 MAP kinase (Thr202/Tyr204) was used as the anti-phospho-ERK1/2 antibody and the antibody for phospho-p38 MAP kinase (Thr180/Tyr182) was used as the anti-phospho-p38 antibody. The horseradish peroxidase (HRP)-conjugated anti-rabbit IgG was applied as the secondary antibody, and bands were detected using Amersham ECL Plus Western blotting detection reagents (GE Healthcare, Piscataway, NJ).
Measurement of ROS and mitochondrial superoxide formation
ROS were detected using 2′,7′-dichlorofluorescin diacetate (H2-DCF-DA; Sigma), and the mitochondrial superoxide generation was determined using the MitoSOX Red (Molecular Probes), a mitochondrial superoxide-specific indicator, according to the manufacturer’s protocols. HT22 cells were seeded at 2 × 105 cells/well in 6-well plates. Twenty-four hours later, cells were treated with 4 mM glutamate and 10 μM resveratrol for 8 hours, and then 10 μM H2-DCF-DA or 5 μM MitoSOX Red was added to each well. After incubation for 20 minutes at 37°C, the liquid was removed and then PBS was added. Multiple viewing fields were photographed under a fluorescence microscope (AXIO, Carl Zeiss Corporation, Germany). The experiment was repeated three times, and similar observations were made. One representative figure for each treatment group that best represents the average changes is shown.
Analysis of cells by transmission electron microscopy
Cells were harvested using Trypsin-EDTA and fixed in 2% glutaraldehyde for 4 hours, and centrifuged to form pellets. Sample preparation was carried out according to the previous method [24]. Briefly, the pellets were rinsed in 0.1 M cacodylate buffer (purchased from Electron Microscopy Sciences [EMS], Hatfield, PA) and post-fixed in 1% osmium tetroxide (EMS). Cell pellets were dehydrated through a graded series of ethanol and then passed through a propylene oxide twice and lastly placed in propylene oxide/Embed 812 resin (EMS) overnight for infiltration, and then polymerized in a 60°C oven overnight. Then sections were cut on a Leica UCT ultra microtome at 80 nm using a Diatome diamond knife. Sections were contrasted with uranyl acetate and Sato’s lead citrate (EMS), and viewed and photographed on a JEOL 100CXII TEM at 60 KV (J.E.O.L. Ltd., Tokyo, Japan).
siRNA experiments
siRNA duplexes targeting the mouse PI3K p110α (Cat #: sc-39128), β-catenin(sc-29210), and scrambled non-targeting siRNA(sc-37007) were purchased from Santa Cruz Biotechnology. The cells were seeded 12 hours before transfection and reached a density of 30% confluence at the time of transfection. Then, 40 nM of the siRNA duplex was transfected using Lipofectamine 2000 (Invitrogen) according to the protocols of the manufacturers. Forty-eight hours after transfection, cells were processed for Western blotting of protein levels and MTT assay of cell viability.
Construction of SOD1 or SOD2 stable knock-down in HT22 cells
The control shRNA plasmid (sc-108060), the SOD1 shRNA plasmid (sc-36522-SH), and the SOD2 shRNA plasmid (sc-41656-SH) were purchased from Santa Cruz Biotechnology. The control shRNA plasmid encoded a scrambled shRNA sequence that would not lead to specific degradation of any known cellular mRNA. The SOD1 shRNA plasmid and the SOD2 shRNA plasmid carried the puromycin resistance gene for the purpose of isolating shRNA plasmid DNA-transfected cells. Transfection of shRNA plasmid was carried out using Lipofectamine 2000 (Invitrogen) according to manufacturers’ protocols.
Construction of the SOD2 stable overexpression in HT22 cells
The pEGFP-N1/SOD2 plasmid was a generous gift provided by Dr. Sonia Flores, at the Division of Pulmonary Sciences and Critical Care Medicine, University of Colorado, Denver, CO, USA. HT22 cells were transfected with the pEGFP-N1/SOD2 plasmid as described by Connor et al. [25] using Lipofectamine 2000. Although the pEGFP-N1/SOD2 plasmid had a neomycin-resistant gene, HT22 cells were also strongly resistant to neomycin. Therefore, we could not use neomycin for selection of transfected cells. To establish the stably-transfected cells, we collected the GFP-positive cells using FACS Aria II (BD Bioscience). After three times of cell sorting, the population of GFP-positive cells was increased to approximately 77%. Then, the transfected cells were seeded in 96-well culture plate at 1 cell per well. After 2-week culture, single colony was harvested for determination of the SOD2GFP fusion protein level by Western blotting.
Analysis of SOD activity
Mitochondria and cytosol were fractionated using the Mitochondria/Cytosol Fractionation Kit (BioVision). The amount of proteins was determined using the Bio-Rad protein assay (Bio-Rad). The SOD activity was determined using the Superoxide Dismutase Activity Assay Kit (Bio Vision). Relative SOD activity was normalized according to the protein content and shown as percentage of the SOD activity present in control cells.
Reproducibility of experiments and statistical analysis
All quantitative data and experiments described in this study were repeated at least three times. Most of the data were presented as mean ± S.D. of multiple independent experiments. Statistics were analyzed with one-way ANOVA followed by multiple comparisons with Dunnett’s test (SPSS software). P < 0.05 or P < 0.01 was used to denote as statistically significant or statistically very significantly, respectively.
RESULTS
Resveratrol protects HT22 cells against glutamate-induced oxidative cytotoxicity
When HT22 cells were cultured in the presence of increasing concentrations of glutamate (2, 4, 6, 8 and 10 mM) for 24 hours, it decreased cell viability in a concentration-dependent manner (Fig. 1A). The presence of 4 mM glutamate reduced cell viability over 80%. The presence of resveratrol alone (at 1, 5, 10 and 20 μM) caused a weak but concentration-dependent decrease in cell viability (MTT assay), which was due to a transient, non-cytotoxic S-phase delay induced by resveratrol [26]. Co-treatment of HT22 cells with glutamate and resveratrol reduced glutamate-induced cell death in a concentration-dependent manner (Fig. 1A). While resveratrol at 20 μM prevented the death of HT22 cells induced by varying concentrations (2–10 mM) of glutamate, at 10 μM it effectively prevented cell death induced by ≤4 mM glutamate. We, therefore, selected 4 mM glutamate and 10 μM resveratrol to further investigate the mechanism underlying resveratrol’s neuroprotective effect.
FIGURE 1. Resveratrol (RES) prevents HT22 cells from undergoing glutamate-induced apoptosis.
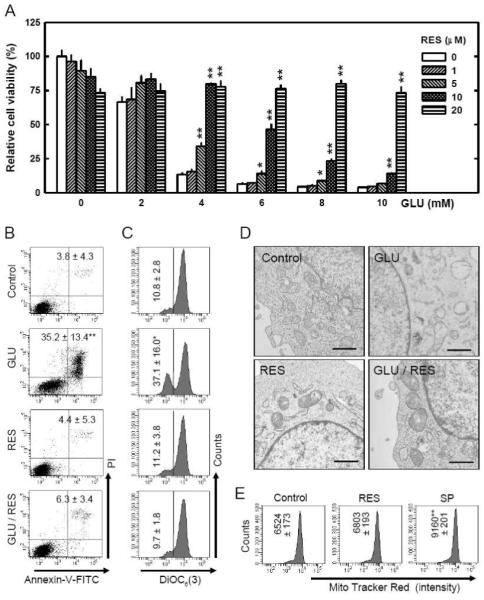
A. HT22 cells were treated with resveratrol and/or glutamate at indicated concentrations for 24 hours. The MTT assay was used to determine cell viability. B and C. HT22 cells were treated with resveratrol (10 μM) and/or glutamate (4 mM) for 24 hours, and then stained with annexin-V and PI (B) or DiOC6(3) (C) as described in the Materials and Methods section, and then analyzed using a flow cytometer. The values as listed in the panels are the mean ± SD for the annexin-V/PI double positive cell population obtained from three separate experiments (B), or the mean ± SD for the DiOC6(3)-negative cell population, also from three separate experiments (C). D. HT22 cells were treated with resveratrol (10 μM) and/or glutamate (4 mM) for 12 hours, and then mitochondrial morphological changes were analyzed using the transmission electron microscopy as described in the Materials and Methods section. Scale bar = 1 μm. E. HT22 cells were treated with resveratrol (10 μM) or 50 mM sodium pyruvate (SP) for 24 hours and then stained with the MitoTracker Red FM as described in the Materials and Methods section, and then analyzed using a flow cytometer. Each value is the mean ± SD of three separate measurements. *P < 0.05, **P < 0.01 versus respective controls.
First, we investigated changes in plasma membrane asymmetry (using the annexin-V and PI double staining method) to quantify the population of dead cells. With this method, cells stained single positive for PI are considered mostly necrotic cells, and cells stained single-positive for annexin-V are considered mostly early apoptotic cells, but cells that are stained double-positive are either necrotic or apoptotic [27-29]. We found that treatment of HT22 cells with 4 mM glutamate alone for 24 hours increased the number of annexin-V and PI double-stained cells (which are dead cells), but co-treatment of these cells with glutamate plus resveratrol markedly reduced this population of cells (Fig. 1B). Notably, treatment with resveratrol alone did not have an appreciable effect on plasma membrane asymmetry; this observation was consistent with the cell viability data as shown in Fig. 1A, thus confirming that resveratrol itself was not cytotoxic.
Our recent study showed that mitochondrial dysfunction is an essential earlier event in glutamate-induced oxidative toxicity [15]. Hence, we further investigated the effect of resveratrol on glutamate-induced changes in mitochondrial function and morphology. First, we determined the changes of mitochondrial membrane potential (MMP) by using DiOC6(3), a cell-permeable, green-fluorescent dye that is selective for the mitochondria of live cells [30]. Treatment with 4 mM glutamate alone reduced the MMP. While resveratrol alone did not have an appreciable effect on MMP, co-treatment with resveratrol almost completely prevented glutamate-induced MMP reduction (Fig. 1C). To examine the changes of mitochondrial morphology, we applied the transmission electron microscopy analysis. After treatment of HT22 cells with 4 mM glutamate, most mitochondria were found to be swollen, and some of them lost cristae. However, co-presence of resveratrol with glutamate mostly abrogated these mitochondrial morphological changes (representative images are shown in Fig. 1D). Lastly, to determine whether resveratrol also alters mitochondria density in HT22 cells, the MitoTracker Red FM staining method was used. As shown Fig. 1E, treatment of HT22 cells with 10 μM resveratrol for 24 hours did not significantly alter the mitochondria density in these cells. In comparison, treatment of these cells with 50 mM sodium pyruvate, an agent known to increase mitochondria biogenesis and density in some cells [31], increased mitochondria density by 40.4% (P < 0.05).
Resveratrol inhibits ROS accumulation
Many earlier studies have shown that resveratrol possesses direct radical-scavenging activity [32]. Because ROS accumulation is a hallmark of glutamate-induced oxidative cell death in HT22 cells [17], next we determined whether resveratrol could affect glutamate-induced intracellular ROS accumulation. When H2-DCF-DA was used as a ROS-sensitive fluorescence indicator, the accumulation of the ROS in HT22 cells was markedly elevated by glutamate treatment, reaching peak levels at approximately 6 to 8 hours after treatment, but co-presence of resveratrol with glutamate abrogated ROS accumulation (Fig. 2A, DCFDA). Furthermore, we have also examined the mitochondrial superoxide generation following glutamate treatment. Similar to the accumulation of intracellular ROS, mitochondrial superoxide generation was elevated following glutamate treatment, and the co-presence of resveratrol with glutamate markedly reduced the mitochondrial superoxide generation (Fig. 2A, MitoSOX Red).
FIGURE 2. Resveratrol (RES) inhibits ROS formation.

A. HT22 cells were treated with glutamate (4 mM) and resveratrol (10 μM) for 8 hours followed by incubation for 20 minutes with a fluorescent dye 2′,7′-dichlorodihydrofluorescin diacetate (H2-DCF-DA) at 10 μM (for detection of total cellular ROS) or MitoSOX Red at 5 μM (for detection mitochondrial ROS). Accumulation of cellular and mitochondrial ROS was observed and photographed using a fluorescence microscope (×200). B. HT22 cells were treated with resveratrol and/or hydrogen peroxide at indicated concentrations for 24 hours. The MTT assay was used to determine cell viability. C. HT22 cells were treated with α-tocopherol and/or hydrogen peroxide at indicated concentrations for 24 hours. The MTT assay was used to determine cell viability. D. HT22 cells were pre-treated with resveratrol (10 μM) for 6 or 8 hours and then resveratrol was removed immediately before addition of glutamate. Cells were incubated for 24 hours with glutamate in the absence of resveratrol. Cell viability was determined by using the MTT assay. Each value is mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 versus respective controls.
Unexpectedly, resveratrol could not protect HT22 cells against hydrogen peroxide-induced cytotoxicity (Fig. 2B), whereas α-tocopherol, a well-known antioxidant, effectively protected these cells from hydrogen peroxide-induced cell death (Fig. 2C). This observation prompted us to consider the possibility that the protective effect of resveratrol against glutamate-induced oxidative toxicity may not be attributable to its direct antioxidant property. To test of this hypothesis, we pretreated HT22 cells with resveratrol for 6 or 8 hours, and resveratrol was then removed prior to addition of glutamate. We found that the 6- or 8-hour pre-treatment with resveratrol effectively protected these cells from glutamate cytotoxicity (i.e., subsequent glutamate exposure for 24 hours) (Fig. 2D). This observation supports our hypothesis.
Role of MAPKs and PI3K/Akt signaling pathways in mediating resveratrol’s protection against glutamate cytotoxicity
Earlier studies showed that resveratrol could alter the activity of MAPKs and PI3K/Akt signaling molecules [33-41], which are regulated by phosphorylation. In the present study, we examined the roles of these signaling molecules in mediating glutamate-induced neuronal death and resveratrol’s protective effect in HT22 cells by investigating their activation (phosphorylation). As shown in Fig. 3, after 12-hour treatment with glutamate alone, c-jun N-terminal kinase 1/2 (JNK), extracellular signaling-regulated protein kinase (ERK), and p38 were significantly activated (phosphorylated). However, when the cells were co-treated with resveratrol, these changes were abrogated. Next, we used selective inhibitors of these signaling proteins to further probe their functional roles. HT22 cells were first pretreated with each of the inhibitors and then cultured in the presence of glutamate with or without resveratrol. As shown in Fig. 4A (MTT assay) and Fig. 4B (annexin-V and PI double staining), SP600125 (an inhibitor of JNK-1, -2 and -3), SB202190 (an inhibitor of p38 MAPK-β), and U0126 (an inhibitor of MEK1/2) each significantly reduced glutamate-induced cell death in a concentration-dependent manner. By contrast, resveratrol’s protective effect was strongly diminished in a concentration-dependent manner by LY294002 (a PI3K inhibitor). In addition, we found that wortmannin (another PI3K inhibitor) also inhibited resveratrol’s neuroprotective effect, but LY303511 (a structural analog of LY294002 but with no PI3K-inhibiting activity) exerted no inhibition against resveratrol’s neuroprotective effect (Fig. 4A). Taken together, these results suggest that while the MAPK signaling pathways (JNK, p38 and ERK) are involved in glutamate-induced oxidative cell death, activation of the PI3K/Akt signaling pathway is specifically associated with resveratrol’s neuroprotective effect.
FIGURE 3. Activation of JNK, p38 and ERK signaling pathways after glutamate and/or resveratrol treatment.

HT22 cells were treated with glutamate (4 mM) and/or resveratrol (10 μM). After incubation for the indicated length of time, cell extracts were subjected to SDS-PAGE and immunoblotting with antibodies specific for phospho-JNK (Thr183/Tyr185), phospho-p38 (Thr180/Tyr182), and phospho-ERK (Thr202/Tyr204). Membranes were stripped and re-probed for total-JNK, total-p38, or total-ERK. The ratio of phosphorylated protein was calculated from three independent experiments (shown under each immunoblot of the phosphorylated protein). The relative protein level for the phosphorylated protein was calculated according to the densitometry reading of each band, which was then normalized according to the densitometry reading for the corresponding total protein level. The control group was arbitrarily set at 1.o for ease of comparison.
FIGURE 4. Role of JNK, p38, ERK and PI3K signaling pathways in mediating glutamate-induced cell death and the protective effect of resveratrol (RES).

A. HT22 cells were pre-treated with each of the inhibitors at 0-5 μM concentrations (SP600125 as a JNK inhibitor; SB202190 as a p38 inhibitor; U0126 as an ERK inhibitor; LY294002 and wortmannin as PI3K inhibitors) for 2 hours and then incubated with glutamate (4 mM) and/or resveratrol (10 μM) for additional 24 hours. In this experiment, LY303511, a structural analog of LY294002 but with no PI3K-inhibiting activity, was used as a negative control (0-10 μM) for LY294002. Cell viability was analyzed using the MTT assay. Each value is mean ± SD of three separate experiments. *P < 0.05, **P < 0.01 versus respective controls. B. HT22 cells were pre-treated with inhibitors (5 μM) for 2 hours and then incubated with glutamate (4 mM) and resveratrol (10 μM) for additional 24 hours. Cells were stained with annexin-V-FITC and PI as described in the Materials and Methods section, and then analyzed using a flow cytometer. These analyses were repeated multiple times, and similar observations were made. A representative data set was shown.
Resveratrol increases SOD2 protein levels through PI3K/Akt and GSK-3β/β-catenin signaling pathways
To determine how the activation of PI3K/Akt by resveratrol resulted in increased antioxidant capacity in HT22 cells, we sought to determine the involvement of the PI3K/Akt and GSK-3β/β-catenin signaling pathways. β-Catenin is a component in the Wnt signaling pathway, and in the inactive state, GSK-3β constitutively phosphorylates β-catenin which is then ubiquitinated and degradated by proteasomes. However, in the active state, GSK-3β is inactivated (by phosphorylation) which permits β-catenin to go into the cell nucleus which will interact with transcription factors and regulate gene expressions (reviewed by Liu et al. [42]). As shown in Fig. 4A, Akt was phosphorylated (activated) within the first 15 minutes following resveratrol treatment, and returned to the basal level after about 4 hours. GSK-3β was also phosphorylated (inactivated) in a time-dependent manner following resveratrol treatment, and obviously, there was a short delay in GSK-3β phosphorylation when compared to Akt phosphorylation, which was consistent with the factor that GSK-3β phosphorylation is downstream of the Akt phosphorylation. Resveratrol treatment increased β-catenin levels in a time-dependent manner. SOD2 expression was also increased in a time-dependent manner following resveratrol treatment, but SOD1 expression level was not significantly altered.
To verify that these changes were the result of changes in the upstream PI3K/Akt activity, a parallel experiment using the PI3K inhibitor LY294002 was performed. Pre-treatment of cells with LY294002 (5 μM) suppressed these changes (phosphorylation of Akt and GSK-3β) elicited by resveratrol (Fig. 5A). Furthermore, LY294002 also reduced SOD2 induction by resveratrol (Fig. 5A). In separate experiments, we also compared the changes of GSK-3β, β-catenin, and SOD2 in HT22 cells treated with glutamate alone, resveratrol alone, or glutamate plus resveratrol. While treatment with glutamate alone did not appreciably affect GSK-3β inactivation (phosphorylation), β-catenin stabilization, and SOD2 induction, treatment with resveratrol alone or co-treatment with resveratrol plus glutamate each increased GSK-3β inactivation, β-catenin stabilization, and SOD2 induction (Fig. 5B). To further confirm the induction of SOD2 by resveratrol, we also determined the SOD enzyme activity in cytosolic and mitochondrial fractions, respectively. As shown in Fig 5C, an increase in the SOD enzyme activity was selectively seen in the mitochondrial fraction. These data also indicate that resveratrol selectively induces SOD2 expression.
FIGURE 5. Induction of SOD by resveratrol (RES) via PI3K/Akt and GSK-3β/β-catenin signaling pathways.
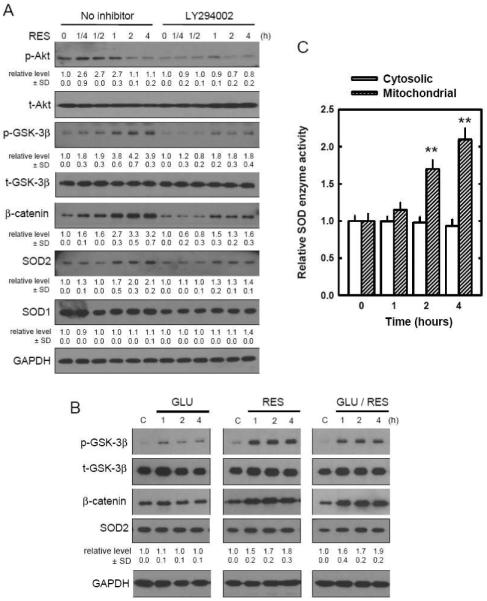
A. HT22 cells were pre-treated with 5 μM of LY294002 for 2 hours and then incubated with 10 μM of resveratrol for the indicated time. Cell extracts were subjected to SDS-PAGE and immunoblotting with antibodies specific for phospho-Akt(Ser 473), phospho-GSK-3β(Ser 9), β-catenin, SOD1, and SOD2. Membranes were stripped and re-probed for total-Akt, total-GSK-3β, or GAPDH (control). The relative protein levels for the phosphorylated Akt and GSK-3β were calculated according to their densitometry readings, which were then normalized according to the densitometry readings for the corresponding total protein levels. Similarly, the relative protein levels for β-catenin, SOD1 and SOD2 were normalized according to the GAPDH protein levels. The control group was arbitrarily set at 1.0 for ease of comparison. B. HT22 cells were treated with glutamate (4 mM) and/or resveratrol (10 μM) for indicated length of time. Cell extracts were subjected to SDS-PAGE and immunoblotting with antibodies specific for phospho-GSK-3β (Ser 9), β-catenin, and SOD2. Membranes were stripped and re-probed for total-GSK-3β and GAPDH (control). These analyses were repeated multiples, and similar observations were made. A representative data was shown. For the expression levels of SODs, the mean densitometry value ± SD was calculated from three separate experiments, with the mean of the control group arbitrarily set at 1.0. C. HT22 cells were treated with 10 μM resveratrol for the indicated hours. Mitochondrial and cytosolic fractions were isolated as described in Materials and Methods. The SOD enzymatic activity in each fraction was measured and normalized as a ratio relative to the control activity. Each value is mean ± SD of three independent experiments. **P < 0.01 versus respective controls.
It is known that β-catenin released from GSK-3β is stabilized and translocated into the nucleus to activate transcription factors involved in a number of genes, including SOD2 expression [43]. To provide evidence that PI3K/Akt and β-catenin were involved in resveratrol’s neuroprotective actions through the induction of SOD2, we conducted a series of experiments. First, we selectively suppressed the expression of PI3K p110α subunit using the siRNA approach. As shown in Fig. 6A, specific siRNA for PI3K p110α significantly reduced PI3K p110α protein levels, whereas the control siRNA (scrambled sequence) did not have this effect. Second, we compared the viability of HT22 cells with or without PI3K p110α knockdown. Forty-eight hours after transfection, HT22 cells were incubated with glutamate with or without resveratrol for 24 hours. Knockdown of PI3K p110α did not appreciably affect glutamate-induced neuronal death, but it significantly reduced resveratrol’s protection against glutamate-induced oxidative cytotoxicity (Fig. 6B). Third, we electively suppressed β-catenin expression using the siRNAs. As shown in Fig. 6C, transfection of siRNA targeting β-catenin suppressed β-catenin expression in HT22 cells. Resveratrol increased β-catenin and SOD2 expression in cells transfected with control siRNA, but not in cells transfected with β-catenin siRNA. Transfection with β-catenin siRNA also significantly suppressed resveratrol’s protection against glutamate-induced cell death (Fig. 6D). Lastly, to provide more definitive evidence for the essential role of SOD2 induction as a key mechanistic component for the protective actions of resveratrol against glutamate cytotoxicity, we constructed stable SOD1 and SOD2 knockdown cells (stably transfected with the SOD1 or SOD2 shRNA plasmid). SOD1 and SOD2 protein levels were partially reduced in cells transfected with the SOD1 and SOD2 shRNA plasmid, respectively (Fig. 7A). Transfection of SOD2 shRNA into HT22 cells abrogated resveratrol’s protective effect against glutamate-induced oxidative cell death, but this was not the case when the cells were transfected with SOD1 shRNA (Fig. 7B).
FIGURE 6. Suppression of protective effect of resveratrol (RES) in HT22 cells by transfection with siRNA selectively targeting PI3K p110α or β-catenin.
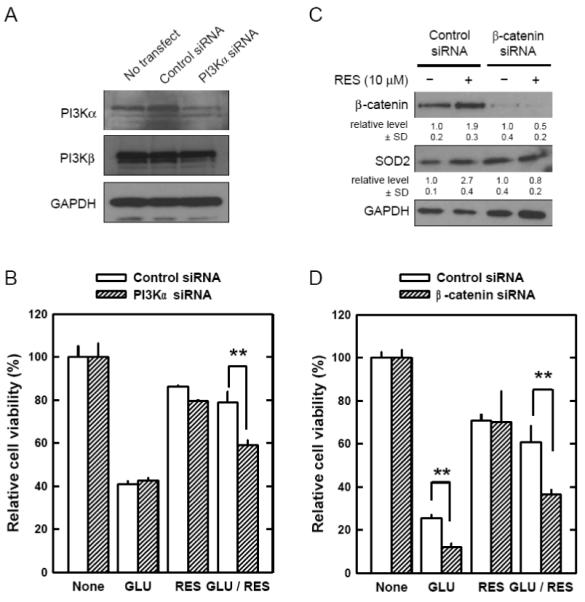
A. HT22 cells were transfected with control siRNA or PI3Kα siRNA, and 72 hours later cell extracts were prepared and subjected to SDS-PAGE and immunoblot analysis using antibodies specific for PI3K p110α, PI3K p110β, or GAPDH. B. HT22 cells were transfected with control siRNA or PI3Kα siRNA. After 48-hour incubation, cells were further incubated with glutamate (4 mM) and/or 10 μM resveratrol for additional 24 hours. Cell viability was determined using the MTT assay. C. HT22 cells were transfected with control siRNA or β-catenin siRNA, and 48 hours later cells were incubated with or without 10 μM resveratrol. After 24-hour incubation, cell extracts were prepared and subjected to SDS-PAGE and immunoblot analysis using antibodies specific for β-catenin, SOD2, or GAPDH. The relative protein levels for β-catenin and SOD2 were calculated according to their densitometry reading, which was normalized according to the corresponding reading for the GAPDH protein band. The corresponding groups without resveratrol treatment were arbitrarily set at 1.0. D. HT22 cells were transfected with control siRNA or β-catenin siRNA. After 48-hour incubation, cells were further incubated with glutamate (4 mM) and/or 10 μM resveratrol for additional 24 hours. Cell viability was determined using the MTT assay. Each value is mean ± SD from three independent experiments. ** P < 0.01 versus respective controls.
FIGURE 7. Role of mitochondrial SOD2 levels in modulating the protective effect of resveratrol (RES) in HT22 cells.
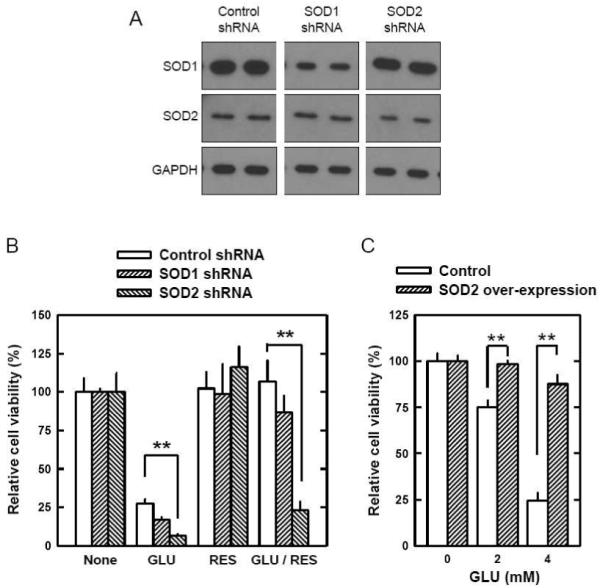
A. HT22 cells were stably transfected with the shRNA plasmid for SOD1, SOD2, or the control plasmid as described in the Materials and Methods section. Cell extracts from SOD1 shRNA, SOD2 shRNA and control cells were subjected to SDS-PAGE and immunoblotting with antibodies specific for SOD1, SOD2 and GAPDH as a control. B. Control, SOD1 and SOD2 shRNA cells were treated with glutamate (4 mM) and/or resveratrol (10 μM) for 24 hours. Cell viability was determined by using the MTT assay. C. HT22 cells that were stably transfected with a SOD2 expression plasmid or a mock plasmid (described in Methods) were treated with glutamate at indicated concentrations for 24 h. Cell viability was determined using the MTT assay. Each value is mean ± SD from three independent experiments. **P < 0.01 versus respective controls.
Furthermore, we also showed that stable over-expression of the mitochondrial SOD2 alone (in the absence of resveratrol) would provide a similar protection against glutamate-induced oxidative neuronal death in cultured HT22 cells. As shown in Fig. 7C, the glutamate-induced oxidative cell death was significantly suppressed in cultured HT22 cells that stably over-expressed the mitochondrial SOD2. In the SOD2-over-expressing cells, the cytosolic and mitochondrial SOD activities are 116.8 ± 5.5% and 201.4 ± 12.2%, respectively, compared to the SOD activity in the corresponding cellular fractions from mock plasmid-transfected cells (described in a previous study [44]).
DISCUSSION
The results of our present study showed that resveratrol could strongly protect HT22 cells from glutamate-induced oxidative cytotoxicity by removing intracellular ROS. To determine whether resveratrol removed ROS directly (i.e., through its direct antioxidant activity) or indirectly (i.e., through the induction of cellular antioxidant capacity), we conducted two parallel experiments: One sought to determine whether co-presence of resveratrol with hydrogen peroxide could protect against hydrogen peroxide-induced cell death, and the other sought to determine whether prior treatment of cells with resveratrol could prevent subsequent cell death induced by glutamate (in the absence of resveratrol). We found that when a relatively high concentration (20 μM) of resveratrol was present, it failed to protect HT22 cells against the cytotoxicity induced by 500 or 1000 μM hydrogen peroxide (Fig. 2B). Notably, an earlier study reported that resveratrol exerted a significant protective effect in cerebral cortical astrocytes against the cytotoxicity induced by a lower concentration (100 μM) of hydrogen peroxide [20]. These data suggest that the direct antioxidant activity of resveratrol is not strong enough for the protection against the cytotoxicity induced by high concentrations of hydrogen peroxide. In comparison, under another experimental condition where HT22 cells were only pretreated with resveratrol for 6-8 hours and afterwards they were exposed to glutamate (in the absence of resveratrol) for an additional 24 hours, these cells were still effectively protected from glutamate-induced oxidative cell death (Fig. 2C). Taken together, these results suggest that resveratrol’s direct radical-scavenging activity is not a major contributing factor in its protection against glutamate-induced neurotoxicity in cultured HT22 cells as observed in this study. Therefore, we hypothesized that resveratrol may stimulate relevant signaling pathway(s) that subsequently increases the antioxidant capacity in HT22 cells and reduce glutamate-induced oxidative cytotoxicity.
To probe the signaling pathway(s) that are targeted by resveratrol, we focused on testing a mechanistic hypothesis that activation of the PI3K/Akt signaling pathway by resveratrol may lead to inactivation of glycogen synthase kinase 3β (GSK-3β), stabilization and nuclear translocation of β-catenin, and subsequently, increased expression of the antioxidant enzymes (such as SODs). This hypothesis was formulated on the basis of some recent studies suggesting that resveratrol augmented cellular antioxidant defense capacity through intracellular signaling pathways including the Akt signaling pathway [19], and that long-term exposure of human cells to resveratrol resulted in up-regulation of SOD [45]. We found that treatment of HT22 cells with resveratrol alone or resveratrol plus glutamate resulted in Akt activation, GSK-3β inactivation, and β-catenin stabilization in a time-dependent manner, and subsequently, increased expression of SOD2 protein, whereas treatment with glutamate alone did not markedly affect these molecules (Fig. 5). The effects of resveratrol on these signaling molecules were suppressed by treatment with a PI3K inhibitor (LY294002), and this suppressive effect paralleled the alteration of the protective effect of resveratrol on cell viability (Fig. 4). To provide further evidence, we demonstrated that selective knockdown of PI3K (the p110α subunit) or β-catenin each attenuated the protective effect of resveratrol (Fig. 6). Here it is of note that the inhibition efficiency of PI3K-p110α siRNA appeared to be relatively lower than that of the PI3K inhibitor LY294002. This was likely due to the fact that the siRNA targeting the PI3K-p110α catalytic subunit would not affect other PI3K subunits, such as the p110β subunit (Fig. 6A), which might also partially contribute to the PI3K-mediated signaling activity. Whereas in the case of LY294002, it could non-discriminatively inhibit the activity of different PI3K isoforms by binding to the ATP-binding site [46,47], and thus may abrogate the neuroprotective effect of resveratrol more effectively. Consistent with our observation of a neuroprotective role of the PI3K/Akt–GSK-3β/β-catenin signaling pathway, Alvarez and coworkers also reported earlier [48] that the Wnt-3a ligand, which activates the GSK-3β/β-catenin [49-51], protected hippocampal neurons from amyloid-β-peptide-induced apoptosis. Similarly, it was recently suggested that the neuroprotective effect of fibroblast growth factor-1 against glutamate-induced cell death may also involve the inactivation of GSK-3β via activation of the PI3K/Akt [52].
The SOD family is presently known to include the cytosolic SOD1, mitochondrial SOD2, and extracellular SOD3 [53,54]. While the SOD1 and SOD2 jointly contribute to reducing the burden of intracellular ROS, recent studies have shown that SOD2 plays a more important role in neuronal cells against oxidant-induced mitochondrial oxidative stress and cytotoxicity [55-57]. The results of our present study showed that resveratrol exerts a strong protection against glutamate-induced mitochondrial oxidative stress and alterations of mitochondrial morphology and functions. To provide definitive evidence concerning the role of the mitochondrial SOD2 in resveratrol’s protection against glutamate-induced oxidative neurotoxicity, we constructed SOD1 and SOD2 knockdown HT22 cells by stably transfecting these cells with specific shRNA plasmids targeting SOD1 and SOD2, respectively. As we expected, cells with a stable knockdown of SOD2, but not SOD1, became selectively unresponsive to resveratrol’s protective effect (Fig. 7B). Although the total induction of SOD2 by resveratrol was relatively modest (~2-fold) (Fig. 5), its effect on neuroprotection was very strong. Additional experiments using the over-expression approach (Fig. 7C) confirmed that a ~2-fold increase in SOD2 expression could effectively prevent HT22 cells from glutamate-induced oxidative cytotoxicity. Here it is of interest to note that the induction of SOD2 levels occurs very rapidly, within 1 h following resveratrol treatment (Fig. 5). This is not surprising since earlier studies have shown that the transcriptional activation and protein synthesis of some genes can take place in 15 min [58, 59]. It is possible that the HT22 cells may have a built-in mechanism for the rapid induction of SOD2 because it is such a vitally important enzyme for cell survival following mitochondrial oxidative insults.
It is expected that the induction of SOD2 would affect the cellular production of hydrogen peroxide [60]. Since cells with a higher level of SOD2 are better protected against mitochondrial superoxide-mediated oxidative cytotoxicity, it is suggested that the accumulation of mitochondrial superoxide likely is more cytotoxic than the accumulation of hydrogen peroxide when they are present at equimolar concentrations. The formed hydrogen peroxide is then further concerted to water. Here it is of note that since the total intracellular GSH level is markedly reduced due to inhibition of cystine uptake by glutamate, the glutathione peroxidase (GPx) pathway may play a less important role in the conversion of hydrogen peroxide to water [44]. In comparison, heme oxygenase 1 (HO-1) may play a more important role in the detoxification. This suggestion is supported by a recent study showing that HO-1 is induced in HT22 cells following exposure to hydrogen peroxide [61]. Biochemically, the neuroprotective actions of HO-1 are attributable to the formation of biliverdin and bilirubin during heme degradation, both of which can serve as ROS scavengers [62,63].
Consistent with the crucial role of intracellular ROS in glutamate-induced oxidative neurotoxicity, earlier studies have shown that the MAPK pathways are activated following glutamate-induced oxidative stress and cell death in cultured HT22 cells [64]. This observation was confirmed in the present study. We found that JNK, p38, and ERK were activated (phosphorylated) at 12 hours after glutamate treatment (Fig. 3). This delay in the activation of MAPKs agreed with the time-dependent accumulation of intracellular ROS as reported in earlier studies showing that marked ROS accumulation occurred at 6 to 8 hours after glutamate treatment [13,15]. In addition, we found that treatment with resveratrol alone or in combination with glutamate suppressed the activation (phosphorylation) of JNK, p38, and ERK. The suppression of their activation is an important element of the neuroprotective actions of resveratrol in HT22 cells because pharmacological inhibition of JNK, p38 and ERK could confer the same degrees of protection in these cells. It should also be noted that although MAPK inhibitors and resveratrol both exert comparable neuroprotective efficacy in cultured HT22 cells, the mechanisms of their actions are different. While the MAPK inhibitors exert their neuroprotection by directly inhibiting these kinases, resveratrol exerts its protection by removing the intracellular ROS, and subsequently, abrogating ROS-mediated activation of MAPKs.
Kainic acid (KA), an agonist for a subtype of ionotropic glutamate receptor, has been commonly used to induce excitotoxicity [65,66]. An earlier study showed that KA can cause a decrease in glutathione levels in the hippocampus, which subsequently increases mitochondrial superoxide production, oxidative DNA damage, and apoptosis in the hippocampal CA1 and CA3 neurons [67]. These oxidative neuronal changes were partially protected by resveratrol administration [68]. Notably, an earlier study reported that dietary resveratrol intake elicited a similar degree of up-regulation of SOD2 in the mouse brain as we have seen in cultured HT22 cells [69]. Therefore, the findings of our present study provide a mechanistic explanation for the neuroprotective effect of resveratrol against both excitotoxicity and oxidative cytotoxicities.
The overall conclusion of our present study is depicted in Fig. 8. Briefly, when present at high concentrations, extracellular glutamate can induce oxidative stress in HT22 cells through inhibition of cystine uptake and subsequent depletion of the intracellular glutathione levels (discussed in ref. [5]). Resveratrol can activate the PI3K/Akt signaling pathway, and the activated Akt then inactivates GSK-3β and stabilizes β-catenin. The stabilized β-catenin will translocate into the nuclei and bind to TCF family transcription factors or FOXO to activate the expression of target genes, including SOD2. The induced SOD2 catalyzes the conversion of the mitochondrial superoxide to hydrogen peroxide, which is then further detoxified to water by the HO-1/biliverdin and bilirubin systems [61]. The observations made in the present study suggest that dietary or pharmacologic up-regulation of the mitochondrial antioxidant SOD2 via activation of the PI3K/Akt and GSK-3β/β-catenin pathways may represent a viable clinical strategy for the prevention of oxidative neuronal damage and degeneration.
FIGURE 8. Schematic illustration of the mechanism underlying the protective effect of resveratrol (RES) against glutamate-induced cell death in HT22 cells.
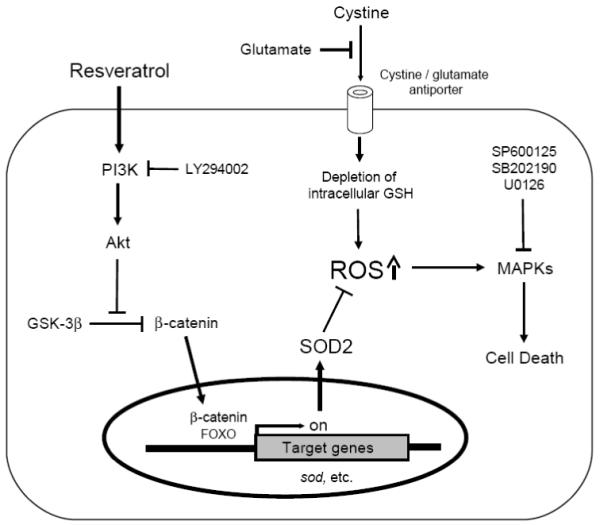
High concentrations of extracellular glutamate induce oxidative stress as a result of inhibition of cystine uptake and subsequently depletion of intracellular glutathione [5]. Elevated levels of intracellular ROS activate MAPK signaling pathways and subsequently induce cell death. Resveratrol (RES) can activate the PI3K/Akt signaling pathways, and the activated Akt subsequently inactivates GSK-3β and stabilizes β-catenin. The stabilized β-catenin translocates into the nuclei, binds to TCF family transcription factors or FOXO, and induces the expression of target genes, including SOD2. SOD2 proteins quench ROS that are accumulated in the mitochondria as a result of glutathione depletion. Collectively, resveratrol confers strong protection against glutamate-induced oxidative stress and neuronal cell death in HT22 cells.
ACKNOWLEDGMENTS
We would like to thank Dr. Joyce Slusser for technical assistance with the flow cytometric analysis and Stephanie C. Bishop for proof-reading of the manuscript. This work was supported, in part, by an NIH grant (ES015242). Some of the analytical and imaging instruments employed in this study are part of the COBRE core facility that is supported by an NIH Grant P20RR021940 from the National Center for Research Resources.
ABBREVIATIONS
- SOD
superoxide dismutase
- ROS
reactive oxygen species
- PI
propidium iodide
- MAPK
mitogen-activated protein kinase
- JNK
c-jun N-terminal kinase
- ERK
extracellular signaling-regulated protein kinase
- PI3K
phosphatidylinositol-3 kinase
- GSK-3β
glycogen synthase kinase 3β
- MMP
mitochondrial membrane potential
- GLU
glutamate
- siRNA
small interfering RNA
- shRNA
short-hairpin RNA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- [2].Blandini F, Porter RH, Greenamyre JT. Glutamate and Parkinson’s desease. Mol. Neurobiol. 1996;12:73–94. doi: 10.1007/BF02740748. [DOI] [PubMed] [Google Scholar]
- [3].Choi DW. Glutamate neurotoxicity and diseases of nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- [4].Choi DW. Exitotoxic cell death. J. Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- [5].Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547–1558. doi: 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]
- [6].Froissard P, Duval D. Cytotoxic effects of glutamic acid on PC12 cells. Neurochem. Int. 1994;24:485–493. doi: 10.1016/0197-0186(94)90096-5. [DOI] [PubMed] [Google Scholar]
- [7].Davis JB, Maher P. Protein kinase C activation inhibits glutamate-induced cytotoxicity in a neuronal cell line. Brain Res. 1994;652:169–173. doi: 10.1016/0006-8993(94)90334-4. [DOI] [PubMed] [Google Scholar]
- [8].Maher P, Davis JB. The role of monoamine metabolism in oxidative glutamate toxicity. J. Neurosci. 1996;16:6394–6401. doi: 10.1523/JNEUROSCI.16-20-06394.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Murphy TH, Schnaar RL, Coyle JT. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J. 1990;4:1624–1633. [PubMed] [Google Scholar]
- [10].Erdö SL, Michler A, Wolff JR, Tytko H. Lack of excitotoxic cell death in serum-free cultures of rat cerebral cortex. Brain Res. 1990;526:328–332. doi: 10.1016/0006-8993(90)91241-8. [DOI] [PubMed] [Google Scholar]
- [11].Oka A, Belliveau MJ, Rosenberg PA, Volpe JJ. Vulnerability of oligodendroglia to glutamate: pharmacology, mechanisms, and prevention. J. Neurosci. 1993;13:1441–1453. doi: 10.1523/JNEUROSCI.13-04-01441.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tan S, Wood M, Maher P. Oxidative stress induces a form of programmed cell death with characteristics of both apoptosis and necrosis in neuronal cells. J. Neurochem. 1998;71:95–105. doi: 10.1046/j.1471-4159.1998.71010095.x. [DOI] [PubMed] [Google Scholar]
- [13].Tan S, Schubert D, Maher P. Oxitosis: A novel form of programmed cell death. Cerr. Top. Med. Chem. 2001;1:497–506. doi: 10.2174/1568026013394741. [DOI] [PubMed] [Google Scholar]
- [14].Morimoto BH, Koshland DE., Jr. Induction and expression of long- and short-term neurosecretory potentiation in a neural cell line. Neuron. 1990;5:875–880. doi: 10.1016/0896-6273(90)90347-i. [DOI] [PubMed] [Google Scholar]
- [15].Fukui M, Song JH, Choi JY, Choi HJ, Zhu BT. Mechanism of glutamate-induced necrosis and apoptosis in cultured HT22 cells. Eur. J. Pharmacol. 2009;617:1–11. doi: 10.1016/j.ejphar.2009.06.059. [DOI] [PubMed] [Google Scholar]
- [16].Moosmann B, Behl C. The antioxidant neuroprotective effects of estrogens and phenolic compounds are independent from their estrogenic properties. Proc. Natl. Acad. Sci. USA. 1999;96:8867–8872. doi: 10.1073/pnas.96.16.8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ishige K, Schubert D, Sagara Y. Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free. Radic. Biol. Med. 2001;30:433–446. doi: 10.1016/s0891-5849(00)00498-6. [DOI] [PubMed] [Google Scholar]
- [18].Miyamoto M, Murphy TH, Schnaar RL, Coyle JT. Antioxidants protect glutamate-induced cytotoxicity in a neuronal cell line. J. Pharmacol. Exp. Ther. 1989;250:1132–1140. [PubMed] [Google Scholar]
- [19].Chen CY, Jang JH, Li MH, Surh YJ. Resveratrol upregulates heme oxygenase-1 expression via activation of NF-E2-related factor 2 in PC12 cells. Biochem. Bioph. Res. Co. 2005;331:993–1000. doi: 10.1016/j.bbrc.2005.03.237. [DOI] [PubMed] [Google Scholar]
- [20].Vieira de Almeida LM, Piñeiro CC, Leite MC, Brolese G, Leal RB, Gottfried C, Gonçalves CA. Protective effects of resveratrol on hydrogen peroxide induced toxicity in primary cortical astrocyte cultures. Neurochem. Res. 2008;33:8–15. doi: 10.1007/s11064-007-9399-5. [DOI] [PubMed] [Google Scholar]
- [21].Kopp P. Resveratrol, a phytoestrogen found in red wine. A possible explanation for the conundrum of the ‘French paradox’? Eur. J. Endocrinol. 1998;138:619–620. doi: 10.1530/eje.0.1380619. [DOI] [PubMed] [Google Scholar]
- [22].Sobolev VS, Cole RJ. Transe-resveratrol contain in commercial peanuts and peanut products. J. Agric. Food Chem. 1999;47:1435–1439. doi: 10.1021/jf9809885. [DOI] [PubMed] [Google Scholar]
- [23].Roldan A, Palacios V, Caro I, Perez L. Resveratrol content of Palomino fino grapes: influence of vintage and fungal infection. J. Agric. Food Chem. 2003;51:1464–1468. doi: 10.1021/jf020774u. [DOI] [PubMed] [Google Scholar]
- [24].Hanaichi T, Sato T, Iwamoto T, Malavasi-Yamashiro J, Hoshino M, Mizuno N. A stable lead by modification of Sato’s method. J. Electron Microsc. 1986;35:304–306. [PubMed] [Google Scholar]
- [25].Connor KM, Subbaram S, Regan KJ, Nelson KK, Mazurkiewicz JE, Bartholomew PJ, Aplin AE, Tai YT, Aguirre-Ghiso J, Flores SC, Melendez JA. Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J. Biol. Chem. 2005;280:16916–16924. doi: 10.1074/jbc.M410690200. [DOI] [PubMed] [Google Scholar]
- [26].Zhou R, Fukui M, Choi HJ, Zhu BT. Induction of a reversible, non-cytotoxic S-phase delay by resveratrol: implications for a mechanism of lifespan prolongation and cancer promotion. Br. J. Pharmacol. 2009;158:462–474. doi: 10.1111/j.1476-5381.2009.00268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Raynal P, Pollard HB. Annexins: The problem of assessing the biological role for a gene family of multifunctional calcium and phospholipids-binding proteins. Biochemica. Biophysica. Acta. 1994;1197:63–93. doi: 10.1016/0304-4157(94)90019-1. [DOI] [PubMed] [Google Scholar]
- [28].Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labeled Anexin V. J. Immunol. Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- [29].van Engeland M, Ramaekers FC, Schutte B, Reutelingsperger C. A novel assay to measure loss of plasma membrane asymmetry during apoptosis of adherant cells in culture. Cytometry. 1996;24:131–139. doi: 10.1002/(SICI)1097-0320(19960601)24:2<131::AID-CYTO5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- [30].Koning AJ, Lum PY, Williams JM, Wright R. DiOC6 staining reveals organelle structure and dynamics in living yeast cells. Cell Motil. Cytoskeleton. 1993;25:111–128. doi: 10.1002/cm.970250202. [DOI] [PubMed] [Google Scholar]
- [31].Wilson L, Yang Q, Szustakowski JD, Gullicksen PS, Halse R. Pyruvate induces mitochondrial biogenesis by a PGC-1α-independent mechanism. Am. J. Physiol.Cell Physiol. 2007;292:C1599–C1605. doi: 10.1152/ajpcell.00428.2006. [DOI] [PubMed] [Google Scholar]
- [32].Stojanovic S, Sprinz H, Brede O. Efficacy and mechanism of the antioxidant action of trans-resveratrol and its analogues in the radical liposome oxidation. Arch. Biochem. Biophys. 2001;391:79–89. doi: 10.1006/abbi.2001.2388. [DOI] [PubMed] [Google Scholar]
- [33].Clement MV, Hirpara JL, Chawdhury SH, Pervaiz S. Chemopreventive agent resveratrol, a natural product derived from grapes, triggers CD95 signaling-dependent apoptosis in human tumor cells. Blood. 2000;92:996–1002. [PubMed] [Google Scholar]
- [34].Gill C, Walsh SE, Morrissey C, Fitzpatrick JM, Watson RWG. Resveratrol sensitizes androgen independent prostate cancer cells to death-receptor mediated apoptosis through multiple mechanisms. Prostate. 2007;67:1641–1653. doi: 10.1002/pros.20653. [DOI] [PubMed] [Google Scholar]
- [35].Su JL, Lin MT, Hong CC, Chang CC, Shiah SG, Wu CW, Chen ST, Chau YP, Kuo ML. Resveratrol induces FasL-related apoptosis through Cdc42 activation of ASK1/JNK-dependent signaling pathway in human keukemia HL-60 cells. Carcinogenesis. 2005;26:1–10. doi: 10.1093/carcin/bgh220. [DOI] [PubMed] [Google Scholar]
- [36].Nakagawa H, Kiyozuka Y, Uemura Y, Senzaki H, Shikata N, Hioki K, Tsubura A. Resveratol inhibits human breast cancer cells growth and may mitigate the effect of linoleic acid, a potent breast cancer cell stimulator. J. Cancer Res. Clin. Oncol. 2001;127:258–264. doi: 10.1007/s004320000190. [DOI] [PubMed] [Google Scholar]
- [37].Aziz MH, Nihal M, Fu VX, Jarrard DF, Ahmad N. Resveratrol-caused apoptosis of human prostate carcinoma LNCaP cells in mediated via modulation of phosphatidilinositol 3′-kinase/Akt pathway and Bcl-2 family proteins. Mol. Cancer Ther. 2006;5:1335–1341. doi: 10.1158/1535-7163.MCT-05-0526. [DOI] [PubMed] [Google Scholar]
- [38].Pozo-Guisado E, Merino JM, Mulero-Navarro S, Lorenzo-Benayas MJ, Centeno F, Alvarez-Barrientos A, Fernandez-Salguero PM. Resveratrol-induced apoptosis in MCF-7 human breast cancer cells involves a caspase-independent mechanism with down-regulation of Bcl-2 and NF-κB. Int. J. Cancer. 2005;115:74–84. doi: 10.1002/ijc.20856. [DOI] [PubMed] [Google Scholar]
- [39].Alkhalaf M. Resveratrol-induced growth inhibition in MDA-MB-231 breast cancer cells is associated with mitogen-activated protein kinase signaling and protein translation. Eur. J. Cancer Pre. 2007;16:334–341. doi: 10.1097/01.cej.0000228413.06471.4c. [DOI] [PubMed] [Google Scholar]
- [40].She QB, Bode AM, Ma WY, Chen NY, Dong Z. Resveratrol-induced activation of p53 and apoptosis is mediated by extracellular-signal-regulated protein kinase and p38 kinase. Cancer Res. 2001;61:1604–1610. [PubMed] [Google Scholar]
- [41].She QB, Huang C, Zhang Y, Dong Z. Involvement of c-jun NH2-terminal kinases in Resveratrol-induced activation of p53 and apoptosis. Mol. Carcinogen. 2002;33:244–250. doi: 10.1002/mc.10041. [DOI] [PubMed] [Google Scholar]
- [42].Liu X, Rubin JS, Kimmel AR. Rapid, Wnt-induced changes in GSK3beta associations that regulate beta-catenin stabilization are mediated by Galpha proteins. Curr. Biol. 2005;15:1989–1997. doi: 10.1016/j.cub.2005.10.050. [DOI] [PubMed] [Google Scholar]
- [43].Essers MAG, de Vries-Smits LMM, Barker N, Polderman PE, Burgering BMT, Korswagen HC. Functional interaction between β-catenin and FOXO in oxidative stress signaling. Science. 2005;308:1181–1184. doi: 10.1126/science.1109083. [DOI] [PubMed] [Google Scholar]
- [44].Fukui M, Zhu BT. Mitochondrial superoxide dismutase SOD2, but not cytosolic SOD1, plays a critical role in protection against glutamate-induced oxidative stress and cell death in HT22 neuronal cells. Free Radic. Biol. Med. 2010;48:821–830. doi: 10.1016/j.freeradbiomed.2009.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Robb EL, Page MM, Wiens BE, Stuart JA. Molecular mechanisms of oxidative stress resistance induced by resveratrol: Specific and progressive induction of MnSOD. Biochem. Bioph. Res. Co. 2008;367:406–412. doi: 10.1016/j.bbrc.2007.12.138. [DOI] [PubMed] [Google Scholar]
- [46].Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J. Biol. Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- [47].Walker EH, Pacold ME, Perisic O, Stephens L, Hawkins PT, Wymann MP, Williams RL. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell. 2000;6:909–919. doi: 10.1016/s1097-2765(05)00089-4. [DOI] [PubMed] [Google Scholar]
- [48].Alvarez AR, Godoy JA, Mullendorff K, Olivares GH, Bronfman M, Inestrosa NC. Wnt-3α overcomes β-amyloid toxicity in rat hippocampal neurons. Exp. Cell Res. 2004;297:186–196. doi: 10.1016/j.yexcr.2004.02.028. [DOI] [PubMed] [Google Scholar]
- [49].Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Control of β-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- [50].Moon RT, Bowerman B, Boutros M, Perrimon N. The promise and Perils of Wnt signaling through β-catenin. Science. 2002;296:1644–1646. doi: 10.1126/science.1071549. [DOI] [PubMed] [Google Scholar]
- [51].Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates JR, 3rd., Nusse R. Wnt proteins are lipid-modified and can act as stem cell frowth factors. Nature. 2003;423:448–452. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- [52].Hashimoto M, Sagara Y, Langford D, Everall IP, Mallory M, Everson A, Digicaylioglu M, Masliah E. Fibroblast growth factor 1 regulates signaling via the glycogen synthase kinase-3β pathway. J. Biol. Chem. 2002;277:32985–32991. doi: 10.1074/jbc.M202803200. [DOI] [PubMed] [Google Scholar]
- [53].Halliwell B, Gutteridge JMC. In Free Radical in Biology and Medicine. Oxford, UK: 1995. Protection against oxidants in biological systems: the superoxide theory of oxygen toxicity; pp. 86–187. [Google Scholar]
- [54].Silva JP, Shabalina IG, Dufour E, Petrovic N, Backlund EC, Hultenby K, Wibom R, Nedergaard J, Cannon B, Larsson NG. SOD2 overexpression: Enhanced mitochondrial tolerance but absence of effect on UCP activity. EMBO J. 2005;24:4061–4070. doi: 10.1038/sj.emboj.7600866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ookawara T, Imazeki N, Matsubata O, Kizaki T, Oh-Ishi S, Nakao C, Sato Y, Ohno H. Tissue distribution of immunoreactive mouse extracellular superoxide dismutase. Am. J. Physiol. Cell Physiol. 1998;275:840–847. doi: 10.1152/ajpcell.1998.275.3.C840. [DOI] [PubMed] [Google Scholar]
- [56].Hinerfeld D, Traini MD, Weinberger RP, Cochran B, Doctrow SR, Harry J, Melov S. Endogenous mitochondrial oxidative stress: neurodegeneration, proteomic analysis, specific respiratory chain defects, and efficacious antioxidant therapy in superoxide dismutase 2 null mice. J. Neurochem. 2004;88:657–667. doi: 10.1046/j.1471-4159.2003.02195.x. [DOI] [PubMed] [Google Scholar]
- [57].Vincent AM, Russell JW, Sullivan KA, Backus C, Hayes JM, McLean LL, Feldman EL. SOD2 protects neurons from injury in cell culture and animal models of diabetic neuropathy. Exp. Neurol. 2007;208:216–227. doi: 10.1016/j.expneurol.2007.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Horovitz-Fried M, Cooper DR, Patel NA, Cipok M, Brand C, Bak A, Inbar A, Jacob AI, Sampson SR. Insulin rapidly upregulates protein kinase Cδ gene expression in skeletal muscle. Cell Signal. 2006;18:183–193. doi: 10.1016/j.cellsig.2005.04.004. [DOI] [PubMed] [Google Scholar]
- [59].Horovitz-Fried M, Brutman-Barazani T, Kesten D, Sampson SR. Insulin increases nuclear protein kinase Cδ in L6 skeletal muscle cells. Endocrinology. 2008;149:1718–1727. doi: 10.1210/en.2007-1572. [DOI] [PubMed] [Google Scholar]
- [60].Buettner GR, Ng CF, Wang M, Rodgers VG, Schafer FQ. A new paradigm: manganese superoxide dismutase influences the production of H2O2 in cells and thereby their biological state. Free Radic. Biol. Med. 2006;41:1338–1350. doi: 10.1016/j.freeradbiomed.2006.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kaizaki A, Tanaka S, Ishige K, Numazawa S, Yoshida T. The neuroprotective effect of heme oxygenase (HO) on oxidative stress in HO-1 siRNA-tansfected HT22 cells. Brain Res. 2006;1108:39–44. doi: 10.1016/j.brainres.2006.06.011. [DOI] [PubMed] [Google Scholar]
- [62].Otterbein LE, Choi AM. Heme oxygenase: colors of defense against cellular stress. Am. J. Physiol. Lung Cell Mol. Physiol. 2000;279:1029–1037. doi: 10.1152/ajplung.2000.279.6.L1029. [DOI] [PubMed] [Google Scholar]
- [63].Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 2000;235:1043–1045. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- [64].Stanciu M, Wang Y, Kentor R, Burke N, Watkins S, Kress G, Reynolds I, Klann E, Angiolieri MR, Johnson JW, DeFranco DB. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J. Biol. Chem. 2000;275:12200–12206. doi: 10.1074/jbc.275.16.12200. [DOI] [PubMed] [Google Scholar]
- [65].Sun AY, Chen YM. Oxidative stress and neurodegenerative disorders. J. Biomed. Sci. 1998;5:401–414. doi: 10.1007/BF02255928. [DOI] [PubMed] [Google Scholar]
- [66].Dawson R, Jr., Beal MF, Bondy SC, Di Monte DA, Isom GE. Excitotoxins, aging, and environmental neurotoxins: implications for understanding human neurodegenerative diseases. Toxicol. Appl. Pharmacol. 1995;134:1–17. doi: 10.1006/taap.1995.1163. [DOI] [PubMed] [Google Scholar]
- [67].Wang Q, Yu S, Simonyi A, Sun GY, Sun AY. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol. Neurobiol. 2005;31:3–16. doi: 10.1385/MN:31:1-3:003. [DOI] [PubMed] [Google Scholar]
- [68].Wang Q, Yu S, Simonyi A, Rottinghaus G, Sun GY, Sun AY. Resveratrol protects against neurotoxicity induced by kainic acid. Neurochem. Res. 2004;29:2105–2112. doi: 10.1007/s11064-004-6883-z. [DOI] [PubMed] [Google Scholar]
- [69].Robb EL, Winkelmolen L, Visanji N, Brotchie J, Stuart JA. Dietary resveratrol administration increases MnSOD expression and activity in mouse brain. Biochem Biophys Res Commun. 2008;372:254–259. doi: 10.1016/j.bbrc.2008.05.028. [DOI] [PubMed] [Google Scholar]