

Abstract
Plasma membrane proteins that are exposed on the cell surface have important biological functions, such as signaling into and out of the cells, ion transport, and cell-cell and cell-matrix interactions. The expression level of many of the plasma membrane proteins involved in these key functions is altered on cancer cells, and these proteins may also be subject to post-translational modification, such as altered phosphorylation and glycosylation. Additional protein alterations on cancer cells confer metastatic capacities, and some of these cell surface proteins have already been successfully targeted by protein drugs, such as human antibodies, that have enhanced survival of several groups of cancer patients. The combination of novel analytical approaches and subcellular fractionation procedures has made it possible to study the plasma membrane proteome in more detail, which will elucidate cancer biology, particularly metastasis, and guide future development of novel drug targets. The technical advances in plasma membrane proteomics and the consequent biological revelations will be discussed herein. Many of the advances have been made using cancer cell lines, but because the main goal of this research is to improve individualized treatment and increase cancer patient survival, further development is crucial to direct analysis of clinically relevant patient samples. These efforts include optimized specimen handling and preparation as well as improved proteomics platforms. Identification of potentially useful proteomics-based biomarkers must be validated in larger, well defined retrospective and prospective clinical studies, and these combined efforts should result in identification of biomarkers that will greatly improve early detection, prognosis, and prediction of treatment response.
During the past two decades, there has been a growing interest in systems biology approaches toward discovering new biomarkers that may allow early diagnosis, prognosis, classification of disease subtypes, prediction of treatment response, and identification of potential targets for drug therapy. For most of these applications, a single marker is likely insufficient for stratification, and a panel of markers, the so-called molecular profiles or biosignatures, can be more informative. Such biomarker profiles can be identified at different molecular levels, such as DNA, RNA, microRNA, and protein, and include a plethora of different modifications from DNA deletions/amplifications and gene and microRNA expression alterations to post-translational modifications (PTMs)1 of proteins. A significant portion of the biomarker discovery efforts using -omics approaches has been in the area of cancer, and several markers are already in routine clinical practice, such as K-Ras mutation and HER-2 amplification. In particular, global gene expression analysis has been extensively utilized, and the cancer management results are currently being translated into clinical tests, such as MammaPrint and Oncotype DX. Proteome analysis of cancer tissue samples as well as identification of potential protein biomarkers within serum or plasma is also a growing field. The direct analysis of protein, the functional unit of the cell, using proteomics analysis has several advantages over indirect measures, such as transcriptomics analysis, despite requiring more tissue and being more time-consuming. Proteomics technologies for analysis of clinical specimens are rapidly being refined. A particular area of interest is the cell surface proteome because it is easily accessible and may serve as an ideal target for novel protein drugs. We focus herein on the most recent proteomics developments in the cancer biomarker discovery area with a particular focus on plasma membrane (PM) proteins.
PLASMA MEMBRANE PROTEINS
Membranes play a critical role in cell structure by providing a physical barrier between the cell, the environment, and the various subcellular compartments. The cell surface membrane, or PM, encloses the cell and maintains the essential boundaries between the cytoplasm and the extracellular environment. PM proteins constitute ∼50% of the mass of the PM and exhibit essential specific or multidisciplinary functions (1–4). Membrane-associated proteins include proteins physically embedded in the lipid bilayers, such as ion channels and transmembrane proteins, and proteins anchored to the membrane that sense external signals, transport specific molecules, and connect the membrane to the cytoskeleton, the extracellular matrix (ECM), and adjacent cells (5). Thus, a number of PM proteins are permanently bound to the lipid bilayer as integral or peripheral proteins, whereas others associate to the membrane only briefly under specific conditions (6).
PM receptors are transmembrane proteins that bind hydrophilic signaling molecules that cannot cross the lipid bilayer and ultimately act as signal transducers to regulate cell processes. They can be subdivided into four main classes: G-protein-linked, enzyme-linked, ligand-gated ion channel-linked, and receptors of cellular adhesion. Thus, receptors are key molecules for cell survival and consequently primary therapeutic targets. Many receptors are found tightly associated with other proteins, and information gained from proteomics of receptor complexes in PMs can elucidate the mechanisms of drug action and possibly the mechanisms of diseases (7). Receptor-protein interactions are independent of protein synthesis, making proteomics the principal approach, as opposed to genomics and transcriptomics, for analyzing signaling and regulation of receptors (8, 9).
A current estimate indicates that 30% of all the predicted open reading frames in a typical genome encode membrane proteins and PM proteins in numbers that represent a significant portion of the human proteome. However, because PM proteins are low abundant compared with many soluble proteins, the overall fraction of PM proteins in a cell/tissue lysate is very low, making them difficult to study even with the recent advances in proteomics technologies (10–12).
PLASMA MEMBRANE PROTEIN ALTERATIONS ASSOCIATED WITH CANCER
The phenotypic changes associated with malignant transformation, including cell proliferation, adhesion, and migration, are often mediated or initiated by proteins associated with the PM, making these central in the biological process and potentially effective drug targets (13–16). The difference between the cancer cells and the normal cells from which the cancer cells mutated are often alterations in the expression level of particular PM proteins. This can be either higher expression of a certain receptor, such as HER-2, that can contribute to tumor cell growth when activated by circulating or locally produced ligands or down-regulation of certain adhesion molecules that allow the cells to detach from the primary tumor and spread (17). In many comparative proteomics MS studies of cancer cell lines, such as cancer versus normal cells or metastatic versus non-metastatic cancer cells, a cutoff of 2–3-fold difference is set to determine whether proteins, including PM proteins, are differently expressed in the two groups, whereas in other studies as little as 1.2–1.5-fold differences are considered a significant difference (18–20). Whether these alterations have biological meaning is difficult to evaluate, but smaller expression alterations in several interacting proteins may have additive effects. Confirmatory studies using different biochemical assays may elucidate this. In addition, the expression of the differently expressed PM proteins in biologically relevant clinical tissues is often not evaluated, although it is hypothesized that these proteins may be potential therapeutic targets or biomarkers. In some studies, however, the identified proteins were examined in detail, including evaluation of their expression in clinically relevant tissue samples (21–23).
More rarely, specific PM proteins that are expressed in normal cells are not expressed in the corresponding malignant cells. An example of this is down-regulation or lack of HLA class I antigen expression in some tumors. In other rare cases, proteins not normally expressed by most normal cells are expressed in the cancer cells, possibly representing oncofetal antigens, such as survivin (24), and cancer/testis antigens (25). These proteins are important in specific stages of fetal development but are not, with the exception of germ cells, expressed in normal adult tissues. Most of these cancer-specific antigens are intracellular proteins and not PM proteins (25).
Altered Post-translational Modification of Cancer Cells
Although the amino acid sequence of the PM proteins predominantly determines their three-dimensional structure, the PTMs, such as glycosylation and phosphorylation, of the proteins modulate their physical and chemical properties and thus their stability and molecular function. The diversity of PTMs expands the functional properties of key proteins in the cell and plays an important role in the processes of cell division, differentiation, growth, cell death, and interaction with stromal tissue for both normal and cancer cells (26). Several proteomics methods (described later in this review) for studying the various PTMs of proteins in complex biological systems have been developed, but the pattern of PTMs in normal and cancer cells at a given time and under given conditions remains to be systematically investigated (27).
Glycosylation of Cancer Cells
Among the various PTMs, glycosylation is the most common, being present in ∼50% of the total number of proteins (28). Cancer cells frequently display glycoproteins with increased branching of the glycan structures and/or altered expression levels compared with normal cells, and these alterations most often involve Asn (N)- and Thr/Ser (O)-linked glycans and proteoglycans (29). An increase in the branching creates additional sites for terminal sialic acid residues, negatively charged acidic sugars that can be recognized by lectins (30). The alterations can affect interactions between cancer cell-associated PM glycoproteins and endogenous lectins that subsequently may influence the metastatic potential of the cancer cell (31, 32). Enhanced expression of terminal α2–6-linked sialic acid on N-linked glycans and of sialyl-Lewis X on O-linked glycans (typically found on mucins) often correlates with poor prognosis (33–35). Although the glycan structure or expression level of many PM glycoproteins may be altered, alterations may also occur on secreted glycoproteins and serve as biomarkers for early detection of cancers (29, 36–38).
Glycan analysis can be challenging for several reasons. Unlike proteins, glycan biosynthesis is governed by a series of enzymes present in the cell and does not rely on an underlying template. As a result, the cell can reproduce a single amino acid sequence each time it synthesizes a specified protein, whereas the glycosylation pattern in cancer can be highly aberrant due to changes in the expression or activity of glycosyltransferases and glycosidases (e.g. sialyltransferases and fucosyltransferases) (39–41). Proteomics approaches to elucidate glycoprotein patterns have mainly focused on cell lines or serum/plasma from which the glycoproteins have been enriched followed by identification of glycan structures or deglycosylated peptide sequencing through MS analysis (42–45). Profiling glycans on invasive versus non-invasive breast cancer cell lines identified statistically significant differences in certain neutral, sialylated, and fucosylated structures, suggesting that these profiles may contain distinct glycan biomarkers that may correspond to glycan signatures of cancer (46). Well characterized N-glycosylation alterations in sera of breast cancer patients compared with disease-free controls include increased sialylation and fucosylation of glycan structures (47, 48). Profiling glycoproteins, without specific identification of the glycans, of premalignant versus malignant/metastatic cell lines identified a range of unique glycoproteins associated with breast cancer metastasis (49).
A complete characterization of altered glycoproteins between cancer versus normal cells requires identification of the protein, the glycan composition, and the glycosylation site. Characterization of the peptide or the glycan structure alone will not determine whether the alteration is due to a more abundant glycoprotein, more frequent occupation of a specific glycosylation site, or an alteration in the glycan structure alone where altered expression of glycosyltransferases is likely involved. Performing such a complete analysis is complicated by the complexity of biological samples, low levels and heterogenous structures of glycans, and lack of a single technique that can provide a full qualitative or quantitative structural analysis of glycoproteins or glycopeptides (50).
Mucins are a family of high molecular weight, heavily glycosylated proteins that are overexpressed in most carcinomas (51). Some mucins are PM-bound due to the presence of a hydrophobic membrane-spanning domain that favors retention in the PM, whereas most mucins are secreted and can serve as ligands for adhesion receptors, such as selectins, that promote the ability of tumor cells to interact with host platelets, leukocytes, and endothelial cells (29). MUC1, which is one of the PM mucins, is anchored to the apical surface of many epithelial cells. In breast, colon, and a number of other epithelial tumors, MUC1 is overexpressed and exhibits truncated O-glycan structures (52). MUC1 is the main component of Stimuvax®, a vaccine that stimulates the immune response of the patient against MUC1-expressing cancer cells, that is currently being evaluated in two phase III trials in non-small cell lung cancer and estrogen receptor (ER)-positive breast cancer (53).
Tumor markers in current clinical use, such as human chorionic gonadotropin-β, α-fetoprotein, carcinoembryonic antigen, prostate-specific antigen, HER-2, and mucins (e.g. CA 19.9, CA 125, and CA 15.3), are all glycoproteins that are either membrane-associated or secreted to the serum (37, 54, 55). Typically, the disease marker is the protein part and not the glycan moiety of the glycoprotein or mucin.
Phosphorylation of Cancer Cells
Protein phosphorylation of tyrosine, serine, and threonine amino acid residues is a reversible PTM and a key mechanism in the regulation of cellular signaling pathways that control biological processes. Protein phosphorylation is strictly controlled by the interplay between protein kinases and protein phosphatases (56, 57). Phosphoproteins are present at very low levels, and the phosphorylation process is a transient modification, enabling the cell to respond to cellular or environmental changes by phosphorylation/dephosphorylation (58). Abnormal protein phosphorylation is known to cause, or be a consequence of, many diseases, including cancer (59, 60).
Tyrosine kinases are among the most important oncogenes yet known. Receptor tyrosine kinases are found on the PM and bind to peptides, such as epidermal growth factor, platelet-derived growth factor, fibroblast growth factor, and insulin and insulin-like growth factor. Following ligand binding, the receptor tyrosine kinases undergo dimerization and autophosphorylation, resulting in phosphorylation of tyrosine sites in other proteins (61). Phosphorylation often occurs at multiple residues within a protein and, in most cases, by different protein kinases activated by diverse mechanisms, making proteomics analysis of these changes very challenging. Most phosphoproteomics studies involve the enrichment of phosphorylated proteins from tissue or cell lines prior to MS identification and quantification of protein phosphorylation sites (62, 63). Various new cancer drugs and drug candidates are aimed at protein kinase targets, such as epidermal growth factor receptor (EGFR)/HER-1 and HER-2 (64–66).
Plasma Membrane Proteins and Cancer Metastasis
Metastases are established through a complex set of events that is yet not fully elucidated but that requires detachment of single cells from the primary tumor, penetration of the tissue matrix, and migration to distant locations where they induce angiogenesis and undergo expansive growth (67). The metastatic process is highly dependent on interactions between tumor cell PM proteins and the microenvironment, and metastatic cells develop PM protein alterations that enable them to bind more strongly to corresponding ECM proteins, including laminins, collagens, and proteoglycans (67, 68). The interaction between PM proteins and the ECM activates signaling cascades that regulate gene expression, cytoskeletal organization, cell adhesion, and cell survival mechanisms, resulting in degradation of ECM by specific enzymes, such as the matrix metalloproteinases and the urokinase-type plasminogen activator. As a result, cancer cells become more invasive, migratory, and able to survive in different microenvironments (69).
Integrins, a diverse family of glycoproteins that form heterodimeric receptors for ECM molecules, comprise one of the primary links between tumor cells and the ECM and play essential roles in several steps of the metastatic process (70). The heterodimeric receptors are composed of an α and a β subunit; there are 18 α-subunits and eight β-subunits, giving rise to at least 24 different integrin heterodimers. In a recent quantitative proteomics study of PM protein alterations associated with the ability of disseminated breast tumor cells to establish lung metastasis, we demonstrated that integrin β1, αv, and α6 were more highly expressed on cells capable of generating metastasis. Immunohistochemical analysis of clinical breast cancer biopsies confirmed a significant correlation between high integrin β1 expression and poor outcome, measured as tumor spread or distant recurrence within a 10-year follow-up (22, 71). Chen et al. (72) also identified integrin β1 as being up-regulated in cancer when they performed quantitative proteomics profiling to compare cancerous and normal pancreatic tissues. The elevated level of integrin β1 in pancreatic cancers was confirmed by Western blots and immunohistochemistry. Using cell surface biotinylation, Kischel et al. (73) compared the profiles of PM proteins in the human breast cancer cell line MDA-MB-231 and a bone metastatic subclone. This strategy allowed the identification of several proteins, including αvβ3 integrins, that were up-regulated in the osteotropic cell line.
PROTEOMICS METHODS FOR STUDY OF PLASMA MEMBRANE PROTEINS
The complexity and concentration of individual proteins in the sample are crucial when performing proteomics analyses because abundant proteins may hinder the detection of more infrequent proteins, e.g. PM proteins. Separation of crude extracts according to the different biochemical features of the subcellular compartments and organelles increases the likelihood of detecting the more infrequent proteins. Proteins expressed at low levels may be enriched from larger volumes by selective fractionation, immunoprecipitation, chromatographic, or electrophoretic methods (74–77).
Proteomics Analysis of Cell Lines
Most strategies used for enrichment of PM proteins from cultured cells use either homogenization followed by membrane density separation or whole cell protein tagging followed by affinity purification. The former is usually initiated by a step in which the cells are incubated in a hypotonic buffer followed by mechanical homogenization and removal of nuclei and cell debris by centrifugation at low speeds (8, 23, 71, 78–80). The membranes, including the associated membrane proteins that are maintained in the supernatant, can be separated based on the different lipid-to-protein ratios in the different cellular membranes either in a discontinuous sucrose gradient or on a 35% sucrose cushion (8, 79). The latter approach was used to identify differentially expressed, functionally related proteins in three breast cancer cell lines versus a human mammary epithelial cell line (8). Alternatively, a crude membrane fraction containing all membrane types can be obtained by sedimenting the membranes by ultracentrifugation and separation by SDS-PAGE (23, 78). Membrane purification may also be achieved by combining different methods, such as sedimentation and a discontinuous sucrose gradient (80) or Percoll/sucrose density separation (71, 78); both strategies separate the membranes of different organelles according to their varying densities (81). We have obtained good results with PM protein separation using a Percoll/sucrose density gradient to compare a metastatic versus a non-metastatic cell line and identified 526 membrane proteins of which 16 exhibited altered expression between the two cell lines. Two of the proteins were also expressed at significantly higher levels in primary tumors that had spread within a 10-year follow-up period compared with tumors that had not (22, 71).
Enrichment of PM proteins using whole cell protein tagging is often based on a membrane-impermeable biotin labeling reagent followed by cell lysis and affinity purification using streptavidin-coated beads (18, 19, 21, 73). Other systems, including coating with cationic silica beads and purification through a density gradient, have also been used (82). Conn et al. (21) used biotinylation and streptavidin precipitation to isolate PM proteins from a fibrosarcoma cell line pair that differed 50–100-fold in their ability to intravasate and disseminate. They found that NCAM, JAM-C, and tissue factor were expressed at higher and TIMP-2 was expressed at lower levels in the highly invasive versus less invasive cell line. Other alternative purification approaches include lysing cells directly in the cell culture flask and sequentially recovering the basolateral cell membranes (83) or purifying subpopulations of PM proteins, such as glycoproteins, through lectin affinity purification (49). Wang et al. (49) identified a number of proteins that exhibited altered expression in a precancerous versus a malignant metastatic breast cancer cell line by extracting glycoproteins using lectin affinity columns.
Proteomics Analysis of Patient Material
Cancer cell lines are easy to handle and comprise a homogeneous and almost inexhaustible source of biological material, including proteins (84, 85). However, each cell line represents only one tumor unaffected by signals from the microenvironment, and the cells may have been subjected to clonal drift and in vitro selection, which may render them less representative of the tumor from which they originated (84–86). It is more clinically relevant, but less straightforward, to perform a proteomics analysis directly on patient tumor tissue from which proteins may be extracted immediately upon removal from the patient (87) or later from frozen or formalin-fixed, paraffin-embedded (FFPE) tissue (88–91).
Protein purification from tissue specimens can be achieved similarly to that of cell line material, although there are some important differences. Tissue, especially from solid tumors, is harder/more compact and requires harsher mechanical homogenization methods, e.g. various types of bead mill, rotor-stator, blade homogenizers, grinders, or ultrasonic disintegrators (92). The preparation protocols for the tissue samples should ensure reproducibility and minimize loss of polypeptides and bias of the composition while removing interfering compounds, such as aggregates, lipids, and carbohydrates, especially if the aim is to compare different samples. As described for cultured cells, enrichment strategies need to be used for PM proteins to identify and compare their levels in different tissue samples. Nielsen et al. (93) purified PM proteins from murine brain tissue using centrifugation and density separation, procedures similar to those described for cell line material, and by adding a reverse phase chromatography separation step, up to 60% of membrane proteins were identified. Ex vivo biotinylation offers another PM protein purification strategy. By injecting biotinylation solution into the blood vessels of surgically excised colon specimens from colon cancer patients, Conrotto et al. (94) were able to affinity purify cell surface proteins from the tumors and identified proteins primarily expressed in colon cancer and not in healthy tissues.
The experimental parameters must be carefully controlled to distinguish between true clinical differences and variations in sample collection and experimental setting or normal biological variability (95). Stringent sample collection, storage, preparation, and analysis are required, and several standardized protocols have been established (96). To avoid protein degradation, biological samples should be aliquoted and stored at −80 °C or lower, and protease inhibitors should be added as soon as possible (97, 98).
In addition to technical considerations, when performing proteomics analysis of clinical tumor samples, issues, such as tumor heterogeneity, percentage of necrotic tumor tissue, and percentage of tumor versus surrounding cells (connective tissue, blood vessels, and infiltrating immune cells), must be considered (99). When using tissue samples from patients with well defined medical histories and long term clinical follow-up, the conditions under which these historical samples have been stored must also be evaluated. Moreover, it is crucial to analyze an adequate number of tissues to ensure statistical validity; proteomics are time-consuming, and many studies use too few samples. Finally, the study design is crucial; an initial discovery phase to identify candidate proteomic markers associated with a particular disease condition should be followed by a validation study using an independent sample set. For this purpose, collections of FFPE or frozen tissues stored in biobanks around the world are especially useful because they permit studies with long term end points, such as recurrence of breast cancer that can take 10 or more years to manifest. The use of such archival tissue for proteomics analysis requires introduction of additional purification steps, e.g. to remove paraffin, which renders the proteins very hydrophobic and thus difficult to solubilize. Furthermore, both paraffin and formalin may give rise to additional peaks in the mass spectra obtained from such tissue. A few groups have successfully obtained useful proteomics data from analysis of FFPE tissue (88, 91, 100). Hood et al. (91) compared protein expression in prostate cancer and benign prostate hyperplasia tissue from the same patient using cells from different areas (i.e. cancer and hyperplasia) of a 10-μm-thick section of FFPE tissue block. The proteins were isolated using laser microdissection, digested directly in the deparaffinized tissue, and analyzed by linear ion trap MS. For quantification, the peptides were labeled with 18O and 16O, and 68 proteins were found to exhibit altered expression (91). Assessment of the cellular localization of the identified proteins revealed another important issue regarding MS analysis of archival tissue. The purification procedures often only contain few steps to compensate for the low amounts of protein available or protein degradation. Consequently, there is no PM protein enrichment step, and the percentages of identified membrane and PM proteins are often lower in studies using tissue than in studies using cell lines. Hood and Cheresh (71) identified ∼25% of membrane proteins from FFPE tissue compared with 66% in our recent cell line study.
The most prevalent method for analysis of frozen and fresh tumor tissue is two-dimensional gel electrophoresis combined with MALDI-TOF MS, but PM proteins are rarely identified using this technique (90, 101). Celis et al. (87) used this strategy to examine fresh mammary adipose tissue and corresponding fat interstitial fluid from 21 high risk breast cancer patients. A total of 359 unique proteins, including numerous signaling molecules, hormones, cytokines, and growth factors involved in a variety of biological processes, were identified. Sprung et al. (102) compared shotgun proteomics analyses of frozen versus FFPE specimens prepared from the same colon adenoma tissues. Following deparaffinization, rehydration, and tryptic digestion under mild conditions, analysis of the combined frozen and FFPE data showed a 92% overlap in the protein groups identified, suggesting that proteomics analysis could be performed on retrospective FFPE tissue. Furthermore, equally low (∼10%) amounts of PM proteins were identified.
Identification of Post-translational Modifications
Elucidating a full range of PTMs, such as phosphorylations and glycosylations, from highly complex biological systems is complicated by their low abundance and heterogeneity in the starting material. Most phospho- and glycoproteomics studies use MS as the most sensitive method for detection typically combined with an enrichment step or derivatization/tagging as phosphorylated and glycosylated peptides tend to display a decreased signal intensity in MS in the presence of non-modified peptides (41, 103, 104).
Glycosylation is the most complex, but also one of the most common, types of modification. Glycoproteomics usually includes enzymatic digestion of the glycoprotein-containing samples to generate peptides and glycopeptides. The glycopeptides are then enriched using selective chromatographic methods, typically using immobilized lectins, hydrophilic interaction LC, titanium dioxide (TiO2), or graphite (41, 49, 50, 105–108). Lectins, e.g. concanavalin A, differ in their specificity and selectivity toward glycan compositions, and serial lectin affinity chromatography with immobilized lectins will recover various subsets of glycopeptides from complex biological samples (104, 109, 110). Hydrophilic interaction chromatography makes use of the polar interactions between the hydroxy groups of glycans and the stationary phase, and in this method, the retention mechanism is governed by the size of the glycan, resulting in separating glycoforms with the same peptide moiety (111). Titanium dioxide is a very selective method for quantitative and qualitative assessment of sialic acid-containing peptides from complex peptide mixtures (45). Porous graphitized carbon is well suited for enriching glycopeptides with smaller peptide portions, but sufficient selectivity is not achieved with larger tryptic glycopeptides (112). The glycopeptides recovered by one or a combination of enrichment methods are then analyzed using MS (MALDI or ESI), which can be used to obtain spectra of intact glycoproteins, glycopeptides, or released glycans (26). However, individual glycoforms can only be resolved from peptides containing a single glycosylation site. The challenge with quantitative and qualitative analysis of glycopeptides is that it is not always possible to obtain glycopeptides with just one glycosylation site, and not all glycosylation sites are necessarily occupied by glycan moieties, whereas others may be partially occupied. Details of the glycan structure can be obtained by exoglycosidase digestion or mass spectrometric fragmentation (113, 114). A database of human N-linked glycosylation sites (UniPep) can be used for targeted biomarker identification, but O-linked protein glycosylations, which are often related to cancer, are not covered by the database (115).
The analysis of phosphorylation related to membrane proteins on cancer cells has been very challenging because of frequent alterations in phosphorylation patterns and inconsistent reproducibility. The methods of phosphoproteomics have recently been reviewed (56, 62, 116), but only a few studies have described the methodology on biologically relevant samples using a combination of isotopic or chemical labeling, enrichment of phosphopeptides by immobilized metal affinity chromatography, and MS to compare and sequence phosphopeptides presented by multiple cancer cell lines (62, 117–119).
Phosphoproteomics methods use MS analysis of fractions enriched in phosphopeptides by different ion exchange chromatographic techniques, including strong cation exchange, IMAC, and TiO2 (103, 120). Phosphospecific antibodies may also be used if tyrosine, serine, and threonine phosphorylations are specifically examined. A comprehensive study of protein phosphorylation should include the identification of phosphoproteins and sites of phosphorylation, identification of the kinases and phosphatases involved in the phosphorylation process, and a description of the biological events following the phosphorylation (103). Current methods for phosphopeptide enrichment, such as IMAC and TiO2 chromatography, provide varying degrees of selectivity and specificity (58). One of the major drawbacks of those strategies is the nonspecific binding of peptides containing acidic amino acids and the strong binding of multiphosphorylated peptides with IMAC and monophosphorylated peptides with TiO2. Multiphosphorylated peptides are generally suppressed in the ionization process to a higher extent than monophosphorylated peptides, and thus fractionation of mono- and multiphosphorylated peptides through sequential elution from immobilized metal affinity chromatography overcomes this obstacle (121). Thingholm et al. (122) described an efficient method that combines PM protein fractionation with TiO2-based phosphopeptide enrichment in a cell model of human mesenchymal stem cells; they assigned 703 unique phosphorylation sites in 376 phosphoproteins.
Protein Quantification Methods
Because most disease-associated markers are not exclusively expressed in either the disease or the “healthy” state, quantification of protein expression differences must be included in marker identification strategies. Such quantitative proteomics approaches include comparison of proteins expressed in specific subcellular components, such as the PM as well as their PTMs (123). Several quantitative profiling studies of PM proteins on different cell types and at specific differentiation or disease stages have identified novel molecular markers that may be recognized by monoclonal antibodies and other protein molecules (124–126). The optimal quantification strategy for proteins and PTMs is independent of subcellular origin; however, some strategies may be advantageously used when examining tissue specimen, whereas others may be more effective when examining cell lines.
The quantification strategies used in combination with MS-based proteomics are often based on the introduction of stable isotopes into the samples, which can be done either by metabolic, chemical, or proteolytic labeling. The most widely used metabolic labeling strategy is stable isotope labeling by amino acids in cell culture (SILAC). SILAC is simple and powerful because the label is introduced prior to protein purification but can mainly be applied to cells in culture. Several of the previously cited studies have used SILAC to identify PM proteins differentially expressed between two cell lines (18, 19, 22, 23, 71), and the method was carefully reviewed by Mann (127).
Quantitative proteomics using chemical and proteolytic labels is, in contrast to SILAC, sensitive to variations in protein purifications between the compared samples because the labels are introduced after protein purification. On the other hand, labeling following protein purification enables labeling of material without metabolic activity, such as PM proteins obtained from surgically excised frozen tissue specimens. Examples of chemical and proteolytic labels include ICAT (cleavable isotope-coded affinity tags), iTRAQ (stable isotope-tagged amine-reactive reagents), and 18O (128–132). ICAT consists of a reactive group, which reacts with cysteine residues; a linker containing the stable isotopes; and a biotin tag for purification of labeled peptides. The specificity for cysteines and the subsequent affinity purification reduce the complexity of the peptide mixtures but may also eliminate peptides of special interest (129, 133). Ramus et al. (134) developed a strategy using ICAT in the presence of high concentrations of SDS and urea followed by electrophoresis and enzymatic digestion to obtain quantitative data from the highly hydrophobic membrane proteins of murine embryonic stem cells. ICAT was also used by Pawlik et al. (135) to compare nipple aspirate fluid from patients with early breast cancer versus healthy controls. They found a higher content of vitamin D-binding protein in nipple aspirate fluid in breast cancer versus healthy controls, but the menopausal status of the two groups differed significantly, which may have influenced the results.
In the iTRAQ system, the tags react with the N termini of the peptide and lysine residues, thus tagging all peptides. The iTRAQ tags are designed to make the derivatized peptides isobaric and chromatographically indistinguishable and yield a signature of reporter ions in MS/MS mode (131). Han et al. (136) reported a strategy for PM proteins that combines gel-assisted digestion, iTRAQ labeling, and LC-MS/MS for characterization of differentially expressed proteins of kidney cell plasma membranes from wild-type versus PKD1 knock-out mice. More than 100 proteins showed at least 2-fold up- or down-regulation (136). Rajcevic et al. (137) focused on membrane-associated proteins in glioma xenografts transplanted in rats and profiled protein expression during the progression from an invasive to an angiogenic phenotype by iTRAQ combined with two-dimensional LC and MALDI-TOF/TOF MS. They extracted 1460 quantifiable proteins (137). Ho et al. (138) used the iTRAQ labeling system to identify seven novel breast cancer metastasis-associated proteins in four cell lines with different metastatic potential and verified the differential expression of four of the proteins on 50 matched invasive and metastatic breast carcinomas.
Heavy oxygen (18O) can be introduced into peptides through proteolytic labeling by digesting the proteins in the presence of H218O using trypsin, Lys-C, or Glu-C, which introduces one or two 18O molecules into the peptides (132). This technique is simple and works well with small amounts of sample, but the major drawback is that the labeling is not homogeneous, i.e. sometimes one 18O is incorporated, whereas at other times, two 18O molecules are incorporated (139). Kristiansen et al. (140) used 18O labeling to compare proteins exhibiting altered expression between frozen xenografts derived from two human cholangiocarcinomas and frozen normal human biliary tract tissue. Chi et al. (141) used 18O labeling in combination with laser capture microdissection to identify proteins expressed more highly in oral cancer versus adjacent non-tumor tissue. Stockwin et al. (142) performed a quantitative proteomics analysis of PM from hypoxia-adapted murine B16F10 melanoma using differential 18O stable isotopic labeling and multidimensional liquid chromatography-tandem MS. The analysis resulted in quantitative information for 2433 proteins (142).
An alternative to the stable isotope-based strategies is the label-free approach, which is based on ion intensity quantification either by counting the fragment spectra of the peptides identifying the different proteins or by measuring the chromatographic peaks of the peptides. This technique is cheaper to perform but requires the experiment to be repeated more times and required more robust computing power and algorithms. Röwer et al. (143) applied label-free protein quantification on tissue samples from healthy breast and tumor to generate an invasive ductal breast carcinoma proteome signature consisting of 60 protein entries. Proteins from tumor tissue are most often quantified by either a label-free technique (144) or a postharvest labeling technique, e.g. chemical or enzymatic (135, 140). However, metabolic labeling strategies can also be adapted for quantification of proteins in tissue samples (145, 146). For example, Sihlbom et al. (146) used proteins derived from SILAC-labeled cells as internal standards to study the regeneration process in murine brain tissue. This method could potentially also be combined with PM protein purification to quantify and compare PM proteins within tissue specimens as depicted in Fig. 1.
Fig. 1.

Proteins derived from SILAC-labeled cell line can be used to provide internal standard for quantification of plasma membrane proteins obtained from tissue specimens.
Other Methods for Identification and Quantification of Membrane Proteins
Methods based on principles other than MS are also used for biomarker discovery and include antibody arrays and subtractive immunization. Subtractive immunization allows scanning of the entire cell surface proteome and identification of molecules that exhibit altered expression between two cell types (147). Concurrently, this method gives rise to monoclonal antibodies for further characterization of the identified proteins. The strategy is based on subtractive immunization of mice. Following immunization with one cell line, the mice are treated with a cytotoxic agent, which eliminates the stimulated B cells. Subsequently, the mice are immunized with the second cell line, giving rise to an immune response to those proteins that exhibit altered expression between the two cell lines (Fig. 2).
Fig. 2.
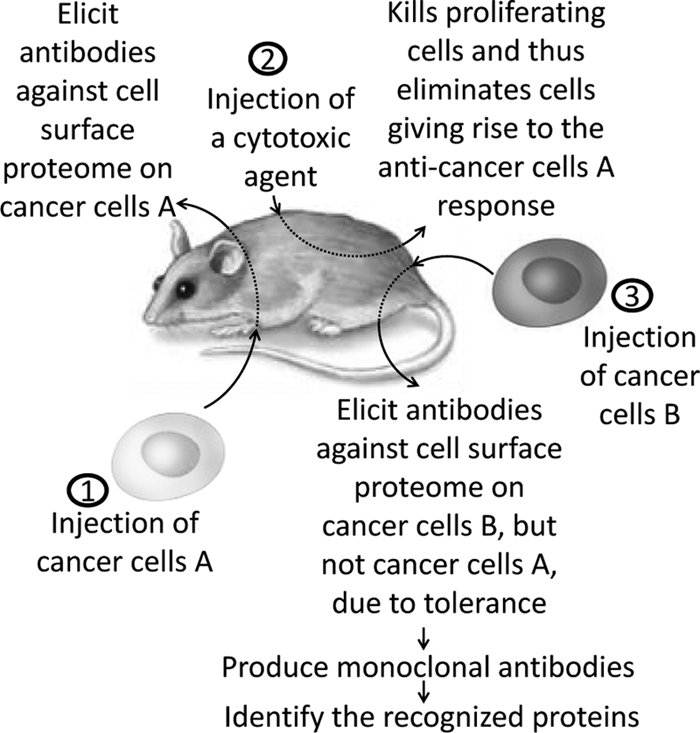
Proteomic scanning of PM using subtractive immunization strategy in mice. An antibody response toward PM proteins predominantly expressed on cell line B compared with cell line A is elicited and can subsequently be cloned, allowing identification of PM proteins associated with particular phenotypes, e.g. PM proteins exhibiting altered expression in a metastatic cancer cell line compared with the corresponding isogenic non-metastatic cancer cell line.
APPROCHES FOR VALIDATION OF CLINICAL PROTEOMIC BIOMARKERS
High throughput proteomics, such as MS, is an excellent tool for biomarker discovery, but for further validation of a limited number of the identified proteins, including PM proteins, other high throughput methods, such as tissue arrays, antibody arrays, or ELISAs, may be advantageous. An exception is the use of MS to monitor selected peptides, e.g. using QconCAT, for identification and quantification of a set of predefined representative peptides.
Tissue Microarray
Tissue microarrays (TMAs), which allow simultaneous immunohistochemical analysis of tens to hundreds of samples on a single glass slide, have become an attractive validation strategy and also are sometimes described as a proteomics technique (148, 149). This type of validation of potential novel biomarkers, including PM proteins, relies on access to large numbers of biological samples, e.g. biopsies of primary tumors and metastases collected and stored for research at hospitals (150). Different cylindrical core sizes of tissue can be used ranging from 0.6 to 2 mm. More reliable assessment of the expression of a given protein includes two cores from a given tumor in the same block, either randomly selected or from predefined areas, such as the invasive front and a central area of the tumor. The readout of these TMAs, which should be based on predefined criteria, depends on the protein in question. In some instances, the readout is a simple positive/negative result, whereas for some other markers, particular features are evaluated, such as the staining intensity, percentage of positive tumor cells, and subcellular localization. The classical breast cancer markers ER, progesterone receptor, and HER-2/neu are typical examples of how different criteria define whether a clinical sample is positive or negative (151). The readout of these assays often requires a highly skilled pathologist, and it may be beneficial to have two independent pathologists score the arrays to limit subjectivity. To make the readout more objective, different instruments have been developed for automated quantitative analysis of TMA (152–154).
Antibody Arrays
Antibody arrays are used to detect the presence of a specific set of proteins in a given sample (serum, tissue homogenates, etc.) and are generated by spotting antibodies on a solid surface. They are the protein analog of cDNA arrays but are technically more difficult to make because proteins, including antibodies, are more complex in their composition, protein folding, denaturation, aggregation, and multimerization (155, 156). The availability of specific antibodies in sufficient amounts is also a challenge. A particular problem related to PM proteins is that they may have altered conformation when they are no longer in the context of the PM, which might render them unrecognizable by the antibodies. Antibody arrays are useful for high throughput analysis of candidate biomarkers in patient samples (155, 157). However, the method has a limited role in discovery-based identification of novel biomarkers where other platforms, such as MS, are better suited (155, 156).
QconCAT
The QconCAT strategy enables absolute quantification of 10–25 preselected peptides in a complex protein sample by using stable isotope-labeled peptides as reference. The strategy relies on the absolute quantification method AQUA in which a stable isotope-labeled synthetic internal standard peptide, which mimics a peptide produced during proteolysis of the target protein, is introduced at a known concentration to the sample prior to digestion. AQUA has been shown to be useful for quantification of both proteins and post-translational modifications (158). The QconCAT peptides are designed as a concatemer, i.e. a chain of DNA sequences. The concatemer is expressed in e.g. Escherichia coli, and the resulting polypeptide, consisting of concatenated peptides, is metabolically labeled with stable isotopes similarly to SILAC. By spiking a protein sample with a known amount of the polypeptide prior to digestion, the absolute content of the peptides of interest can be determined by MS (159, 160). The drawbacks of this method include a number of “trial and error” steps prior to selecting the correct peptides for each protein followed by purification and determination of the protein concentration.
CURRENT STATUS OF CLINICAL BIOMARKER DISCOVERY
Early Detection of Cancer
Clearly, the earlier a primary tumor is detected, the better the prognosis and response to treatment, including the possibility of a complete cure. However, current methods for early detection are insufficient, and most tumors are first detected at a late stage in which the tumor size is extensive and has invaded other tissues or spread to other organs. Identification of new methods for early detection is therefore of the utmost importance, and proteomics analysis of body fluids, such as serum, spinal fluid, saliva, or urine, is a promising area. In addition to the detection of early stage primary tumors, such tests may also be useful to follow cancer patients that are deemed clinically cured but for whom recurrence may occur. Some cancer-associated PM proteins or fragments thereof are released into the plasma/serum in increased amounts in patients with cancers. This can occur by exterior protein cleavage, membrane sloughing, or cell lysis, and these proteins may represent unique markers that might be useful for e.g. predicting disease progression (161–165).
The extracellular domains of the two cancer-associated PM receptors HER-2/neu and EGFR are proteolytically released upon receptor activation and can be detected in serum. The value of serum HER-2/neu and EGFR levels has been evaluated in different cancers with somewhat conflicting results. Importantly, soluble EGFR is also present in healthy individuals, and one study found serum levels to be significantly higher in normal individuals than in patients with primary breast cancer (166). In another study, a small subgroup of patients with decreased serum EGFR was identified and shown to exhibit significantly reduced survival compared with patients with normal serum EGFR levels. However, no significant differences in objective response rate, time to progression, or time to treatment failure were observed (167). Another breast cancer study found that the serum EGFR and serum HER-2/neu levels at the onset of metastatic disease were not associated with overall survival, and no correlation was observed between expression of EGFR in primary tumors and EGFR serum levels (168). In contrast, HER-2/neu serum levels at the time of metastatic disease correlated with HER-2/neu expression in the primary tumors as determined by immunohistochemistry (168). Serum HER-2/neu therefore seems somewhat more promising and has been shown to be a useful marker for early prediction of probability of response, progression-free survival, and overall survival in patients with advanced breast cancer treated with metronomic chemotherapy (167). An elevated serum HER-2/neu level as measured after 2 months of chemotherapy treatment was significantly associated with reduced long term clinical benefit. Serum HER-2/neu was also found to predict response to trastuzumab-based therapies as individuals who did not achieve a significant decline in serum HER-2/neu levels had decreased benefit from trastuzumab-based therapy (169). Many of these findings need confirmation in other independent cohorts; however, overall, they seem promising.
Other fragments of serum PM proteins often detected in sera of cancer patients include soluble forms of PM glycoprotein CD44 that functions as a receptor for hyaluronan and is involved in cell-cell and cell-matrix adhesion, cell migration, and signaling. CD44 is found in several isoforms, including a standard CD44 (CD44s) and different variable forms (CD44v). CD44 may be shed or released into circulation by proteolytic enzymatic mechanisms. Alternative splicing of CD44 and aberrant levels of soluble CD44v in the serum of cancer patients have been correlated to tumor progression and metastasis in different tumors, including breast, colon, and head, and neck cancer (170). Soluble CD44v can be modified by different glycotopes, some of which have been found to be highly expressed in patients with cancers, whereas other glycotopes are frequently found in patients with benign disorders (171).
Prognostic Markers
Different sources of patient tissue, such as the tumor itself, serum, or other bodily fluids, may be used for prediction of prognosis, although currently most studies have focused on serum. In a recent proteomics study aimed at identifying prognostic serum biomarkers of breast cancer, preoperative serum samples obtained from 48 breast cancer patients and 28 controls were used to generate MALDI-TOF MS protein profiles (172). Among 533 common peaks, the investigators identified 72 peaks exhibiting statistically significant intensity differences between cases and controls. They subsequently constructed a diagnostic rule based on these 72 mass values with cross-validated sensitivity and specificity of ∼85%. Although the study yielded relatively high sensitivity and specificity, which are in the same range as those of a few other protein profiling studies in breast cancer where sensitivities and specificities were reported (173, 174), these values are still too low for direct clinical implementation of these methods. In addition, cross-center validation to verify the clinical significance of the results is needed. The identities and general applicability of these markers were not defined in this study. Although some of these profiles can distinguish between cancer and healthy controls, the discriminating peaks/proteins may not be cancer-related but rather a sign of inflammation, which may be seen in multiple disease conditions. This limits their utility as a stand-alone tumor marker, but they may still be useful in a multimarker panel for early detection of cancer.
Prediction of Benefit to Given Treatment
Very few studies have used discovery-based proteomic approaches, such as MS, to develop predictive marker profiles. In breast cancer, the expression of ER, as measured by immunohistochemistry, is predictive of response to adjuvant endocrine treatments with tamoxifen and aromatase inhibitors (175, 176). Although tamoxifen is of great benefit for many patients, recurrence occurs in ∼30% of patients within 15 years despite adequate treatment (177). Thus, there is a need to identify a marker that indicates whether a given patient would benefit from endocrine treatment or whether chemotherapy should be added. Umar et al. (178) performed proteomics analysis of clinical primary ER-positive breast cancer samples from patients that had either objective response or disease progression following tamoxifen treatment of metastatic disease. For the marker discovery phase, laser-captured microdissected cancer cells from each of the two groups of patients were pooled and compared using nano-LC-FTICR-MS. Sample pooling limits the number of samples that need to be analyzed but also makes it impossible to determine whether the increase of a given protein is intense in just one sample or a more moderate increase in all the samples. The verification phase of such studies is thus very important. Here, the authors used whole tissue sections from the patients who were already included in the discovery phase and processed them individually using targeted MS/MS (178). The authors verified 47 of the initial 100 proteins that exhibited altered expression between the two groups. Only a few of these were membrane proteins. The expression of the top candidate, extracellular matrix metalloproteinase inducer, was validated in an independent patient cohort using TMAs and shown to be expressed more highly in therapy-resistant versus -sensitive tumors.
CONCLUSION
PM proteins have attracted significant attention as biomarkers for disease diagnosis, prognosis, monitoring, and treatment benefit prediction. In addition, they may serve as potential targets for drug therapy, a fact already proven as two-thirds of all drugs targeting proteins are directed against PM proteins. However, PM proteins are hydrophobic and possess a variety of PTMs, which require more sophisticated proteomics methods for their separation and identification. Although most proteomics studies of PM proteins are currently using cell lines, newer proteomics strategies for PM biomarker identification in clinical tissue samples, such as tumors, are being developed. Future effort should focus on optimizing these strategies and performing MS-based proteomics on clinically well characterized tissue samples. Study design is essential to effectively address the clinical problem in question, including selection of proper patient materials and sufficient numbers of samples stored under optimal conditions. These efforts require close collaborations between translational researchers, biostatisticians, pathologists, and clinicians. Moving markers from the discovery phase to daily clinical practice is also a crucial area. It is a cumbersome process that requires marker verification, validation and assay development, and testing of large numbers of clinical samples, but these efforts should determine the clinical utility of proteomics in cancer patient care.
Acknowledgments
We thank M. K. Occhipinti-Bender for editorial assistance.
* This work was supported by the Danish Cancer Society, the Danish Research Council, The Danish Strategic Research Council, A Race against Breast Cancer, the Danish Centre for Translational Breast Cancer Research, and a Centre of Excellence, the Sino-Danish Breast Cancer Research Centre.
1 The abbreviations used are:
- PTM
- post-translational modification
- PM
- plasma membrane
- ECM
- extracellular matrix
- FFPE
- formalin-fixed, paraffin-embedded
- ER
- estrogen receptor
- EGFR
- epidermal growth factor receptor
- SILAC
- stable isotope labeling by amino acids in cell culture
- iTRAQ
- isobaric tag for relative and absolute quantitation
- TMA
- tissue microarray
- AQUA
- absolute quantification.
REFERENCES
- 1.Mouritsen O. G., Bloom M. (1993) Models of lipid-protein interactions in membranes. Annu. Rev. Biophys. Biomol. Struct. 22, 145–171 [DOI] [PubMed] [Google Scholar]
- 2.Mukherjee S., Maxfield F. R. (2004) Membrane domains. Annu. Rev. Cell Dev. Biol. 20, 839–866 [DOI] [PubMed] [Google Scholar]
- 3.Guidotti G. (1972) Membrane proteins. Annu. Rev. Biochem. 41, 731–752 [DOI] [PubMed] [Google Scholar]
- 4.Bretscher M. S., Raff M. C. (1975) Mammalian plasma membranes. Nature 258, 43–49 [DOI] [PubMed] [Google Scholar]
- 5.Zheng Y. Z., Foster L. J. (2009) Biochemical and proteomic approaches for the study of membrane microdomains. J. Proteomics 72, 12–22 [DOI] [PubMed] [Google Scholar]
- 6.Escribá P. V., González-Ros J. M., Goñi F. M., Kinnunen P. K., Vigh L., Sánchez-Magraner L., Fernández A. M., Busquets X., Horváth I., Barceló-Coblijn G. (2008) Membranes: a meeting point for lipids, proteins and therapies. J. Cell. Mol. Med. 12, 829–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kabbani N. (2008) Proteomics of membrane receptors and signaling. Proteomics 8, 4146–4155 [DOI] [PubMed] [Google Scholar]
- 8.Patwardhan A. J., Strittmatter E. F., Camp D. G., 2nd, Smith R. D., Pallavicini M. G. (2005) Comparison of normal and breast cancer cell lines using proteome, genome, and interactome data. J. Proteome Res. 4, 1952–1960 [DOI] [PubMed] [Google Scholar]
- 9.Cox J., Mann M. (2007) Is proteomics the new genomics? Cell 130, 395–398 [DOI] [PubMed] [Google Scholar]
- 10.Krogh A., Larsson B., von Heijne G., Sonnhammer E. L. L. (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 [DOI] [PubMed] [Google Scholar]
- 11.Wallin E., von Heijne G. (1998) Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 7, 1029–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pike L. J. (2009) The challenge of lipid rafts. J. Lipid Res. 50, S323–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harvey S., Zhang Y., Landry F., Miller C., Smith J. W. (2001) Insights into a plasma membrane signature. Physiol. Genomics 5, 129–136 [DOI] [PubMed] [Google Scholar]
- 14.Landry Y., Gies J. P. (2008) Drugs and their molecular targets: an updated overview. Fundam. Clin. Pharmacol. 22, 1–18 [DOI] [PubMed] [Google Scholar]
- 15.Dorsam R. T., Gutkind J. S. (2007) G-protein-coupled receptors and cancer. Nat. Rev. Cancer 7, 79–94 [DOI] [PubMed] [Google Scholar]
- 16.Gschwind A., Fischer O. M., Ullrich A. (2004) The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat. Rev. Cancer 4, 361–370 [DOI] [PubMed] [Google Scholar]
- 17.Swanton C., Futreal A., Eisen T. (2006) Her2-targeted therapies in non-small cell lung cancer. Clin. Cancer Res. 12, 4377s–4383s [DOI] [PubMed] [Google Scholar]
- 18.Qiu H., Wang Y. (2008) Quantitative analysis of surface plasma membrane proteins of primary and metastatic melanoma cells. J. proteome Res. 7, 1904–1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aggelis V., Craven R. A., Peng J., Harnden P., Cairns D. A., Maher E. R., Tonge R., Selby P. J., Banks R. E. (2009) Proteomic identification of differentially expressed plasma membrane proteins in renal cell carcinoma by stable isotope labelling of a von Hippel-Lindau transfectant cell line model. Proteomics 9, 2118–2130 [DOI] [PubMed] [Google Scholar]
- 20.Gou L. T., Tong A. P., Chen L. J., Tang M. H., Chen B., Liang S. F., Huang C., Wei Y. Q. (2008) Comparative plasma membrane-associated proteomics of immortalized human hepatocytes. Biochemistry 73, 1200–1206 [DOI] [PubMed] [Google Scholar]
- 21.Conn E. M., Madsen M. A., Cravatt B. F., Ruf W., Deryugina E. I., Quigley J. P. (2008) Cell surface proteomics identifies molecules functionally linked to tumor cell intravasation. J. Biol. Chem. 283, 26518–26527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leth-Larsen R., Lund R., Hansen H. V., Laenkholm A. V., Tarin D., Jensen O. N., Ditzel H. J. (2009) Metastasis-related plasma membrane proteins of human breast cancer cells identified by comparative quantitative mass spectrometry. Mol. Cell. Proteomics 8, 1436–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang X., Zhao J., Hajivandi M., Wu R., Tao J., Amshey J. W., Pope R. M. (2006) Quantification of membrane and membrane-bound proteins in normal and malignant breast cancer cells isolated from the same patient with primary breast carcinoma. J. proteome Res. 5, 2632–2641 [DOI] [PubMed] [Google Scholar]
- 24.Falleni M., Pellegrini C., Marchetti A., Oprandi B., Buttitta F., Barassi F., Santambrogio L., Coggi G., Bosari S. (2003) Survivin gene expression in early-stage non-small cell lung cancer. J. Pathol. 200, 620–626 [DOI] [PubMed] [Google Scholar]
- 25.Simpson A. J., Caballero O. L., Jungbluth A., Chen Y. T., Old L. J. (2005) Cancer/testis antigens, gametogenesis and cancer. Nat. Rev. Cancer 5, 615–625 [DOI] [PubMed] [Google Scholar]
- 26.Larsen M. R., Trelle M. B., Thingholm T. E., Jensen O. N. (2006) Analysis of posttranslational modifications of proteins by tandem mass spectrometry. BioTechniques 40, 790–798 [DOI] [PubMed] [Google Scholar]
- 27.Jensen O. N. (2006) Interpreting the protein language using proteomics. Nat. Rev. Mol. Cell Biol. 7, 391–403 [DOI] [PubMed] [Google Scholar]
- 28.Apweiler R., Hermjakob H., Sharon N. (1999) On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta 1473, 4–8 [DOI] [PubMed] [Google Scholar]
- 29.Fuster M. M., Esko J. D. (2005) The sweet and sour of cancer: glycans as novel therapeutic targets. Nat. Rev. Cancer 5, 526–542 [DOI] [PubMed] [Google Scholar]
- 30.Lehmann F., Tiralongo E., Tiralongo J. (2006) Sialic acid-specific lectins: occurrence, specificity and function. Cell. Mol. Life Sci. 63, 1331–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blomme B., Van Steenkiste C., Callewaert N., Van Vlierberghe H. (2009) Alteration of protein glycosylation in liver diseases. J. Hepatol. 50, 592–603 [DOI] [PubMed] [Google Scholar]
- 32.Kim Y. J., Varki A. (1997) Perspectives on the significance of altered glycosylation of glycoproteins in cancer. Glycoconj. J. 14, 569–576 [DOI] [PubMed] [Google Scholar]
- 33.Seales E. C., Jurado G. A., Brunson B. A., Wakefield J. K., Frost A. R., Bellis S. L. (2005) Hypersialylation of beta1 integrins, observed in colon adenocarcinoma, may contribute to cancer progression by up-regulating cell motility. Cancer Res. 65, 4645–4652 [DOI] [PubMed] [Google Scholar]
- 34.Hedlund M., Ng E., Varki A., Varki N. M. (2008) alpha 2–6-Linked sialic acids on N-glycans modulate carcinoma differentiation in vivo. Cancer Res. 68, 388–394 [DOI] [PubMed] [Google Scholar]
- 35.Varki N. M., Varki A. (2007) Diversity in cell surface sialic acid presentations: implications for biology and disease. Lab. Invest. 87, 851–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dube D. H., Bertozzi C. R. (2005) Glycans in cancer and inflammation-potential for therapeutics and diagnostics. Nat. Rev. Drug Discov. 4, 477–488 [DOI] [PubMed] [Google Scholar]
- 37.Peracaula R., Barrabés S., Sarrats A., Rudd P. M., de Llorens R. (2008) Altered glycosylation in tumours focused to cancer diagnosis. Dis. Markers 25, 207–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Block T. M., Comunale M. A., Lowman M., Steel L. F., Romano P. R., Fimmel C., Tennant B. C., London W. T., Evans A. A., Blumberg B. S., Dwek R. A., Mattu T. S., Mehta A. S. (2005) Use of targeted glycoproteomics to identify serum glycoproteins that correlate with liver cancer in woodchucks and humans. Proc. Natl. Acad. Sci. U.S.A. 102, 779–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohyama C. (2008) Glycosylation in bladder cancer. Int. J. Clin. Oncol. 13, 308–313 [DOI] [PubMed] [Google Scholar]
- 40.Hägglund P., Bunkenborg J., Elortza F., Jensen O. N., Roepstorff P. (2004) A new strategy for identification of N-glycosylated proteins and unambiguous assignment of their glycosylation sites using HILIC enrichment and partial deglycosylation. J. Proteome Res. 3, 556–566 [DOI] [PubMed] [Google Scholar]
- 41.Taylor A. D., Hancock W. S., Hincapie M., Taniguchi N., Hanash S. M. (2009) Towards an integrated proteomic and glycomic approach to finding cancer biomarkers. Genome Med. 1, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mechref Y., Hussein A., Bekesova S., Pungpapong V., Zhang M., Dobrolecki L. E., Hickey R. J., Hammoud Z. T., Novotny M. V. (2009) Quantitative serum glycomics of esophageal adenocarcinoma and other esophageal disease. J. Proteome Res. 8, 2656–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ito H., Kuno A., Sawaki H., Sogabe M., Ozaki H., Tanaka Y., Mizokami M., Shoda J., Angata T., Sato T., Hirabayashi J., Ikehara Y., Narimatsu H. (2009) Strategy for glycoproteomics: identification of glyco-alteration using multiple glycan profiling tools. J. Proteome Res. 8, 1358–1367 [DOI] [PubMed] [Google Scholar]
- 44.Vercoutter-Edouart A. S., Slomianny M. C., Dekeyzer-Beseme O., Haeuw J. F., Michalski J. C. (2008) Glycoproteomics and glycomics investigation of membrane N-glycosylproteins from human colon carcinoma cells. Proteomics 8, 3236–3256 [DOI] [PubMed] [Google Scholar]
- 45.Larsen M. R., Jensen S. S., Jakobsen L. A., Heegaard N. H. (2007) Exploring the sialiome using titanium dioxide chromatography and mass spectrometry. Mol. Cell. Proteomics 6, 1778–1787 [DOI] [PubMed] [Google Scholar]
- 46.Goetz J. A., Mechref Y., Kang P., Jeng M. H., Novotny M. V. (2009) Glycomic profiling of invasive and non-invasive breast cancer cells. Glycoconj. J. 26, 117–131 [DOI] [PubMed] [Google Scholar]
- 47.Kyselova Z., Mechref Y., Kang P., Goetz J. A., Dobrolecki L. E., Sledge G. W., Schnaper L., Hickey R. J., Malkas L. H., Novotny M. V. (2008) Breast cancer diagnosis and prognosis through quantitative measurements of serum glycan profiles. Clin. Chem. 54, 1166–1175 [DOI] [PubMed] [Google Scholar]
- 48.Arnold J. N., Saldova R., Hamid U. M., Rudd P. M. (2008) Evaluation of the serum N-linked glycome for the diagnosis of cancer and chronic inflammation. Proteomics 8, 3284–3293 [DOI] [PubMed] [Google Scholar]
- 49.Wang Y., Ao X., Vuong H., Konanur M., Miller F. R., Goodison S., Lubman D. M. (2008) Membrane glycoproteins associated with breast tumor cell progression identified by a lectin affinity approach. J. Proteome Res. 7, 4313–4325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Selby D. S., Larsen M. R., Calvano C. D., Jensen O. N. (2008) Identification and characterization of N-glycosylated proteins using proteomics. Methods Mol. Biol. 484, 263–276 [DOI] [PubMed] [Google Scholar]
- 51.Hollingsworth M. A., Swanson B. J. (2004) Mucins in cancer: protection and control of the cell surface. Nat. Rev. Cancer 4, 45–60 [DOI] [PubMed] [Google Scholar]
- 52.Burchell J. M., Mungul A., Taylor-Papadimitriou J. (2001) O-Linked glycosylation in the mammary gland: changes that occur during malignancy. J. Mammary Gland Biol. Neoplasia 6, 355–364 [DOI] [PubMed] [Google Scholar]
- 53.Sangha R., Butts C. (2007) L-BLP25: a peptide vaccine strategy in non small cell lung cancer. Clin. Cancer Res. 13, s4652–4654 [DOI] [PubMed] [Google Scholar]
- 54.Perkins G. L., Slater E. D., Sanders G. K., Prichard J. G. (2003) Serum tumor markers. Am. Fam. Physician 68, 1075–1082 [PubMed] [Google Scholar]
- 55.Shariat S. F., Karam J. A., Margulis V., Karakiewicz P. I. (2008) New blood-based biomarkers for the diagnosis, staging and prognosis of prostate cancer. BJU Int. 101, 675–683 [DOI] [PubMed] [Google Scholar]
- 56.Chong P. K., Lee H., Kong J. W., Loh M. C., Wong C. H., Lim Y. P. (2008) Phosphoproteomics, oncogenic signaling and cancer research. Proteomics 8, 4370–4382 [DOI] [PubMed] [Google Scholar]
- 57.Sefton B. M., Shenolikar S. (2001) Overview of protein phosphorylation. Curr. Protoc. Protein Sci. Chapter 13, Unit 13.1 [DOI] [PubMed] [Google Scholar]
- 58.Thingholm T. E., Jensen O. N., Larsen M. R. (2009) Analytical strategies for phosphoproteomics. Proteomics 9, 1451–1468 [DOI] [PubMed] [Google Scholar]
- 59.Castellvi J., Garcia A., Ruiz-Marcellan C., Hernández-Losa J., Peg V., Salcedo M., Gil-Moreno A., Ramon y Cajal S. (2009) Cell signaling in endometrial carcinoma: phosphorylated 4E-binding protein-1 expression in endometrial cancer correlates with aggressive tumors and prognosis. Hum. Pathol. 40, 1418–1426 [DOI] [PubMed] [Google Scholar]
- 60.McArdle L., Rafferty M., Maelandsmo G. M., Bergin O., Farr C. J., Dervan P. A., O'Loughlin S., Herlyn M., Easty D. J. (2001) Protein tyrosine phosphatase genes downregulated in melanoma. J. Invest. Dermatol. 117, 1255–1260 [DOI] [PubMed] [Google Scholar]
- 61.Schlessinger J. (2000) Cell signaling by receptor tyrosine kinases. Cell 103, 211–225 [DOI] [PubMed] [Google Scholar]
- 62.Ashman K., Villar E. L. (2009) Phosphoproteomics and cancer research. Clin. Transl. Oncol. 11, 356–362 [DOI] [PubMed] [Google Scholar]
- 63.Larsen M. R., Thingholm T. E., Jensen O. N., Roepstorff P., Jørgensen T. J. (2005) Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol. Cell. Proteomics 4, 873–886 [DOI] [PubMed] [Google Scholar]
- 64.Scaltriti M., Verma C., Guzman M., Jimenez J., Parra J. L., Pedersen K., Smith D. J., Landolfi S., Ramon y Cajal S., Arribas J., Baselga J. (2009) Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene 28, 803–814 [DOI] [PubMed] [Google Scholar]
- 65.Capdevila J., Elez E., Macarulla T., Ramos F. J., Ruiz-Echarri M., Tabernero J. (2009) Anti-epidermal growth factor receptor monoclonal antibodies in cancer treatment. Cancer Treat. Rev. 35, 354–363 [DOI] [PubMed] [Google Scholar]
- 66.Moran M. F., Tong J., Taylor P., Ewing R. M. (2006) Emerging applications for phospho-proteomics in cancer molecular therapeutics. Biochim. Biophys. Acta 1766, 230–241 [DOI] [PubMed] [Google Scholar]
- 67.Chambers A. F., Groom A. C., MacDonald I. C. (2002) Metastasis: Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2, 563–572 [DOI] [PubMed] [Google Scholar]
- 68.Dowling P., Walsh N., Clynes M. (2008) Membrane and membrane-associated proteins involved in the aggressive phenotype displayed by highly invasive cancer cells. Proteomics 8, 4054–4065 [DOI] [PubMed] [Google Scholar]
- 69.Bidard F. C., Pierga J. Y., Vincent-Salomon A., Poupon M. F. (2008) A “class action” against the microenvironment: do cancer cells cooperate in metastasis? Cancer Metastasis Rev. 27, 5–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hood J. D., Cheresh D. A. (2002) Role of integrins in cell invasion and migration. Nat. Rev. Cancer 2, 91–100 [DOI] [PubMed] [Google Scholar]
- 71.Lund R., Leth-Larsen R., Jensen O. N., Ditzel H. J. (2009) Efficient isolation and quantitative proteomic analysis of cancer cell plasma membrane proteins for identification of metastasis-associated cell surface markers. J. Proteome Res. 8, 3078–3090 [DOI] [PubMed] [Google Scholar]
- 72.Chen R., Yi E. C., Donohoe S., Pan S., Eng J., Cooke K., Crispin D. A., Lane Z., Goodlett D. R., Bronner M. P., Aebersold R., Brentnall T. A. (2005) Pancreatic cancer proteome: the proteins that underlie invasion, metastasis, and immunologic escape. Gastroenterology 129, 1187–1197 [DOI] [PubMed] [Google Scholar]
- 73.Kischel P., Guillonneau F., Dumont B., Bellahcène A., Stresing V., Clézardin P., De Pauw E. A., Castronovo V. (2008) Cell membrane proteomic analysis identifies proteins differentially expressed in osteotropic human breast cancer cells. Neoplasia 10, 1014–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garbis S., Lubec G., Fountoulakis M. (2005) Limitations of current proteomics technologies. J. Chromatogr. A 1077, 1–18 [DOI] [PubMed] [Google Scholar]
- 75.Zhao Y., Zhang W., Kho Y., Zhao Y. (2004) Proteomic analysis of integral plasma membrane proteins. Anal. Chem. 76, 1817–1823 [DOI] [PubMed] [Google Scholar]
- 76.Blonder J., Goshe M. B., Moore R. J., Pasa-Tolic L., Masselon C. D., Lipton M. S., Smith R. D. (2002) Enrichment of integral membrane proteins for proteomic analysis using liquid chromatography and tandem mass spectrometry. J. Proteome Res. 1, 351–360 [DOI] [PubMed] [Google Scholar]
- 77.Josic D., Clifton J. G. (2007) Mammalian plasma membrane proteomics. Proteomics 7, 3010–3029 [DOI] [PubMed] [Google Scholar]
- 78.Kristensen D. B., Brønd J. C., Nielsen P. A., Andersen J. R., Sørensen O. T., Jørgensen V., Budin K., Matthiesen J., Venø P., Jespersen H. M., Ahrens C. H., Schandorff S., Ruhoff P. T., Wisniewski J. R., Bennett K. L., Podtelejnikov A. V. (2004) Experimental Peptide Identification Repository (EPIR): an integrated peptide-centric platform for validation and mining of tandem mass spectrometry data. Mol. Cell. Proteomics 3, 1023–1038 [DOI] [PubMed] [Google Scholar]
- 79.Mannová P., Fang R., Wang H., Deng B., McIntosh M. W., Hanash S. M., Beretta L. (2006) Modification of host lipid raft proteome upon hepatitis C virus replication. Mol. Cell. Proteomics 5, 2319–2325 [DOI] [PubMed] [Google Scholar]
- 80.Bartee E., McCormack A., Früh K. (2006) Quantitative membrane proteomics reveals new cellular targets of viral immune modulators. PLoS Pathog. 2, e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Righetti P. G., Castagna A., Antonioli P., Boschetti E. (2005) Prefractionation techniques in proteome analysis: the mining tools of the third millennium. Electrophoresis 26, 297–319 [DOI] [PubMed] [Google Scholar]
- 82.Hör S., Ziv T., Admon A., Lehner P. J. (2009) Stable isotope labeling by amino acids in cell culture and differential plasma membrane proteome quantitation identify new substrates for the MARCH9 transmembrane E3 ligase. Mol. Cell. Proteomics 8, 1959–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rahbar A. M., Fenselau C. (2005) Unbiased examination of changes in plasma membrane proteins in drug resistant cancer cells. J. Proteome Res. 4, 2148–2153 [DOI] [PubMed] [Google Scholar]
- 84.Vargo-Gogola T., Rosen J. M. (2007) Modelling breast cancer: one size does not fit all. Nat. Rev. Cancer 7, 659–672 [DOI] [PubMed] [Google Scholar]
- 85.Chen E. I., Yates J. R., 3rd (2007) Cancer proteomics by quantitative shotgun proteomics. Mol. Oncol. 1, 144–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lacroix M., Leclercq G. (2004) Relevance of breast cancer cell lines as models for breast tumours: an update. Breast Cancer Res. Treat. 83, 249–289 [DOI] [PubMed] [Google Scholar]
- 87.Celis J. E., Moreira J. M., Cabezón T., Gromov P., Friis E., Rank F., Gromova I. (2005) Identification of extracellular and intracellular signaling components of the mammary adipose tissue and its interstitial fluid in high risk breast cancer patients: toward dissecting the molecular circuitry of epithelial-adipocyte stromal cell interactions. Mol. Cell. Proteomics 4, 492–522 [DOI] [PubMed] [Google Scholar]
- 88.Hwang S. I., Thumar J., Lundgren D. H., Rezaul K., Mayya V., Wu L., Eng J., Wright M. E., Han D. K. (2007) Direct cancer tissue proteomics: a method to identify candidate cancer biomarkers from formalin-fixed paraffin-embedded archival tissues. Oncogene 26, 65–76 [DOI] [PubMed] [Google Scholar]
- 89.Kim J., Kim S. H., Lee S. U., Ha G. H., Kang D. G., Ha N. Y., Ahn J. S., Cho H. Y., Kang S. J., Lee Y. J., Hong S. C., Ha W. S., Bae J. M., Lee C. W., Kim J. W. (2002) Proteome analysis of human liver tumor tissue by two-dimensional gel electrophoresis and matrix assisted laser desorption/ionization-mass spectrometry for identification of disease-related proteins. Electrophoresis 23, 4142–4156 [DOI] [PubMed] [Google Scholar]
- 90.Niméus E., Malmström J., Johnsson A., Marko-Varga G., Fernö M. (2007) Proteomic analysis identifies candidate proteins associated with distant recurrences in breast cancer after adjuvant chemotherapy. J. Pharm. Biomed. Anal. 43, 1086–1093 [DOI] [PubMed] [Google Scholar]
- 91.Hood B. L., Darfler M. M., Guiel T. G., Furusato B., Lucas D. A., Ringeisen B. R., Sesterhenn I. A., Conrads T. P., Veenstra T. D., Krizman D. B. (2005) Proteomic analysis of formalin-fixed prostate cancer tissue. Mol. Cell. Proteomics 4, 1741–1753 [DOI] [PubMed] [Google Scholar]
- 92.Schmitt M., Mengele K., Schueren E., Sweep F. C., Foekens J. A., Brünner N., Laabs J., Malik A., Harbeck N. (2007) European Organisation for Research and Treatment of Cancer (EORTC) Pathobiology Group standard operating procedure for the preparation of human tumour tissue extracts suited for the quantitative analysis of tissue-associated biomarkers. Eur. J. Cancer 43, 835–844 [DOI] [PubMed] [Google Scholar]
- 93.Nielsen P. A., Olsen J. V., Podtelejnikov A. V., Andersen J. R., Mann M., Wisniewski J. R. (2005) Proteomic mapping of brain plasma membrane proteins. Mol. Cell. Proteomics 4, 402–408 [DOI] [PubMed] [Google Scholar]
- 94.Conrotto P., Roesli C., Rybak J., Kischel P., Waltregny D., Neri D., Castronovo V. (2008) Identification of new accessible tumor antigens in human colon cancer by ex vivo protein biotinylation and comparative mass spectrometry analysis. Int. J. Cancer 123, 2856–2864 [DOI] [PubMed] [Google Scholar]
- 95.Clarke W., Zhang Z., Chan D. W. (2003) The application of clinical proteomics to cancer and other diseases. Clin. Chem. Lab. Med. 41, 1562–1570 [DOI] [PubMed] [Google Scholar]
- 96.Diaz J. I., Cazares L. H., Semmes O. J. (2008) Tissue sample collection for proteomics analysis. Methods Mol. Biol. 428, 43–53 [DOI] [PubMed] [Google Scholar]
- 97.Rai A. J., Gelfand C. A., Haywood B. C., Warunek D. J., Yi J., Schuchard M. D., Mehigh R. J., Cockrill S. L., Scott G. B., Tammen H., Schulz-Knappe P., Speicher D. W., Vitzthum F., Haab B. B., Siest G., Chan D. W. (2005) HUPO Plasma Proteome Project specimen collection and handling: towards the standardization of parameters for plasma proteome samples. Proteomics 5, 3262–3277 [DOI] [PubMed] [Google Scholar]
- 98.Pieragostino D., Petrucci F., Del Boccio P., Mantini D., Lugaresi A., Tiberio S., Onofrj M., Gambi D., Sacchetta P., Di Ilio C., Federici G., Urbani A. (2010) Pre-analytical factors in clinical proteomics investigations: impact of ex vivo protein modifications for multiple sclerosis biomarker discovery. J. Proteomics 73, 579–592 [DOI] [PubMed] [Google Scholar]
- 99.Hondermarck H., Tastet C., El Yazidi-Belkoura I., Toillon R. A., Le Bourhis X. (2008) Proteomics of breast cancer: the quest for markers and therapeutic targets. J. Proteome Res. 7, 1403–1411 [DOI] [PubMed] [Google Scholar]
- 100.Patel V., Hood B. L., Molinolo A. A., Lee N. H., Conrads T. P., Braisted J. C., Krizman D. B., Veenstra T. D., Gutkind J. S. (2008) Proteomic analysis of laser-captured paraffin-embedded tissues: a molecular portrait of head and neck cancer progression. Clin. Cancer Res. 14, 1002–1014 [DOI] [PubMed] [Google Scholar]
- 101.Neubauer H., Clare S. E., Wozny W., Schwall G. P., Poznanovic S., Stegmann W., Vogel U., Sotlar K., Wallwiener D., Kurek R., Fehm T., Cahill M. A. (2008) Breast cancer proteomics reveals correlation between estrogen receptor status and differential phosphorylation of PGRMC1. Breast Cancer Res. 10, R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sprung R. W., Jr., Brock J. W., Tanksley J. P., Li M., Washington M. K., Slebos R. J., Liebler D. C. (2009) Equivalence of protein inventories obtained from formalin-fixed paraffin-embedded and frozen tissue in multidimensional liquid chromatography-tandem mass spectrometry shotgun proteomic analysis. Mol. Cell. Proteomics 8, 1988–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Paradela A., Albar J. P. (2008) Advances in the analysis of protein phosphorylation. J. Proteome Res. 7, 1809–1818 [DOI] [PubMed] [Google Scholar]
- 104.Tissot B., North S. J., Ceroni A., Pang P. C., Panico M., Rosati F., Capone A., Haslam S. M., Dell A., Morris H. R. (2009) Glycoproteomics: past, present and future. FEBS Lett. 583, 1728–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dalpathado D. S., Desaire H. (2008) Glycopeptide analysis by mass spectrometry. Analyst 133, 731–738 [DOI] [PubMed] [Google Scholar]
- 106.Cho W., Jung K., Regnier F. E. (2008) Use of glycan targeting antibodies to identify cancer-associated glycoproteins in plasma of breast cancer patients. Anal. Chem. 80, 5286–5292 [DOI] [PubMed] [Google Scholar]
- 107.Jung K., Cho W., Regnier F. E. (2009) Glycoproteomics of plasma based on narrow selectivity lectin affinity chromatography. J. Proteome Res. 8, 643–650 [DOI] [PubMed] [Google Scholar]
- 108.Powlesland A. S., Hitchen P. G., Parry S., Graham S. A., Barrio M. M., Elola M. T., Mordoh J., Dell A., Drickamer K., Taylor M. E. (2009) Targeted glycoproteomic identification of cancer cell glycosylation. Glycobiology 19, 899–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yang Z., Hancock W. S. (2005) Monitoring glycosylation pattern changes of glycoproteins using multi-lectin affinity chromatography. J. Chromatogr. A 1070, 57–64 [DOI] [PubMed] [Google Scholar]
- 110.Drake R. R., Schwegler E. E., Malik G., Diaz J., Block T., Mehta A., Semmes O. J. (2006) Lectin capture strategies combined with mass spectrometry for the discovery of serum glycoprotein biomarkers. Mol. Cell. Proteomics 5, 1957–1967 [DOI] [PubMed] [Google Scholar]
- 111.Wuhrer M., Koeleman C. A., Hokke C. H., Deelder A. M. (2005) Protein glycosylation analyzed by normal-phase nano-liquid chromatography-mass spectrometry of glycopeptides. Anal. Chem. 77, 886–894 [DOI] [PubMed] [Google Scholar]
- 112.Koizumi K. (1996) High-performance liquid chromatographic separation of carbohydrates on graphitized carbon columns. J. Chromatogr. A 720, 119–126 [DOI] [PubMed] [Google Scholar]
- 113.Harvey D. J. (2005) Proteomic analysis of glycosylation: structural determination of N- and O-linked glycans by mass spectrometry. Expert Rev. Proteomics 2, 87–101 [DOI] [PubMed] [Google Scholar]
- 114.Kirmiz C., Li B., An H. J., Clowers B. H., Chew H. K., Lam K. S., Ferrige A., Alecio R., Borowsky A. D., Sulaimon S., Lebrilla C. B., Miyamoto S. (2007) A serum glycomics approach to breast cancer biomarkers. Mol. Cell. Proteomics 6, 43–55 [DOI] [PubMed] [Google Scholar]
- 115.Zhang H., Loriaux P., Eng J., Campbell D., Keller A., Moss P., Bonneau R., Zhang N., Zhou Y., Wollscheid B., Cooke K., Yi E. C., Lee H., Peskind E. R., Zhang J., Smith R. D., Aebersold R. (2006) UniPep—a database for human N-linked glycosites: a resource for biomarker discovery. Genome Biol. 7, R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cordwell S. J., Thingholm T. E. (2010) Technologies for plasma membrane proteomics. Proteomics 10, 611–627 [DOI] [PubMed] [Google Scholar]
- 117.Zarling A. L., Polefrone J. M., Evans A. M., Mikesh L. M., Shabanowitz J., Lewis S. T., Engelhard V. H., Hunt D. F. (2006) Identification of class I MHC-associated phosphopeptides as targets for cancer immunotherapy. Proc. Natl. Acad. Sci. U.S.A. 103, 14889–14894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim J. E., White F. M. (2006) Quantitative analysis of phosphotyrosine signaling networks triggered by CD3 and CD28 costimulation in Jurkat cells. J. Immunol. 176, 2833–2843 [DOI] [PubMed] [Google Scholar]
- 119.Wolf-Yadlin A., Hautaniemi S., Lauffenburger D. A., White F. M. (2007) Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc. Natl. Acad. Sci. U.S.A. 104, 5860–5865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Alcolea M. P., Kleiner O., Cutillas P. R. (2009) Increased confidence in large-scale phosphoproteomics data by complementary mass spectrometric techniques and matching of phosphopeptide data sets. J. Proteome Res. 8, 3808–3815 [DOI] [PubMed] [Google Scholar]
- 121.Thingholm T. E., Jensen O. N., Robinson P. J., Larsen M. R. (2008) SIMAC (sequential elution from IMAC), a phosphoproteomics strategy for the rapid separation of monophosphorylated from multiply phosphorylated peptides. Mol. Cell. Proteomics 7, 661–671 [DOI] [PubMed] [Google Scholar]
- 122.Thingholm T. E., Larsen M. R., Ingrell C. R., Kassem M., Jensen O. N. (2008) TiO(2)-based phosphoproteomic analysis of the plasma membrane and the effects of phosphatase inhibitor treatment. J. Proteome Res. 7, 3304–3313 [DOI] [PubMed] [Google Scholar]
- 123.Zhao Y., Lee W. N., Xiao G. G. (2009) Quantitative proteomics and biomarker discovery in human cancer. Expert Rev. Proteomics 6, 115–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hanash S. M., Pitteri S. J., Faca V. M. (2008) Mining the plasma proteome for cancer biomarkers. Nature 452, 571–579 [DOI] [PubMed] [Google Scholar]
- 125.Foster L. J., Zeemann P. A., Li C., Mann M., Jensen O. N., Kassem M. (2005) Differential expression profiling of membrane proteins by quantitative proteomics in a human mesenchymal stem cell line undergoing osteoblast differentiation. Stem Cells 23, 1367–1377 [DOI] [PubMed] [Google Scholar]
- 126.Van Hoof D., Dormeyer W., Braam S. R., Passier R., Monshouwer-Kloots J., Ward-van Oostwaard D., Heck A. J., Krijgsveld J., Mummery C. L. (2010) Identification of cell surface proteins for antibody-based selection of human embryonic stem cell-derived cardiomyocytes. J. Proteome Res. 9, 1610–1618 [DOI] [PubMed] [Google Scholar]
- 127.Mann M. (2006) Functional and quantitative proteomics using SILAC. Nat. Rev. Mol. Cell Biol. 7, 952–958 [DOI] [PubMed] [Google Scholar]
- 128.Gygi S. P., Rist B., Gerber S. A., Turecek F., Gelb M. H., Aebersold R. (1999) Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 17, 994–999 [DOI] [PubMed] [Google Scholar]
- 129.Li J., Steen H., Gygi S. P. (2003) Protein profiling with cleavable isotope-coded affinity tag (cICAT) reagents: the yeast salinity stress response. Mol. Cell. Proteomics 2, 1198–1204 [DOI] [PubMed] [Google Scholar]
- 130.Butler G. S., Dean R. A., Tam E. M., Overall C. M. (2008) Pharmacoproteomics of a metalloproteinase hydroxamate inhibitor in breast cancer cells: dynamics of membrane type 1 matrix metalloproteinase-mediated membrane protein shedding. Mol. Cell. Biol. 28, 4896–4914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ross P. L., Huang Y. N., Marchese J. N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S., Purkayastha S., Juhasz P., Martin S., Bartlet-Jones M., He F., Jacobson A., Pappin D. J. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 3, 1154–1169 [DOI] [PubMed] [Google Scholar]
- 132.Yao X., Freas A., Ramirez J., Demirev P. A., Fenselau C. (2001) Proteolytic 18O labeling for comparative proteomics: model studies with two serotypes of adenovirus. Anal. Chem. 73, 2836–2842 [DOI] [PubMed] [Google Scholar]
- 133.Zieske L. R. (2006) A perspective on the use of iTRAQ reagent technology for protein complex and profiling studies. J. Exp. Bot. 57, 1501–1508 [DOI] [PubMed] [Google Scholar]
- 134.Ramus C., Gonzalez de Peredo A., Dahout C., Gallagher M., Garin J. (2006) An optimized strategy for ICAT quantification of membrane proteins. Mol. Cell. Proteomics 5, 68–78 [DOI] [PubMed] [Google Scholar]
- 135.Pawlik T. M., Hawke D. H., Liu Y., Krishnamurthy S., Fritsche H., Hunt K. K., Kuerer H. M. (2006) Proteomic analysis of nipple aspirate fluid from women with early-stage breast cancer using isotope-coded affinity tags and tandem mass spectrometry reveals differential expression of vitamin D binding protein. BMC Cancer 6, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Han C. L., Chien C. W., Chen W. C., Chen Y. R., Wu C. P., Li H., Chen Y. J. (2008) A multiplexed quantitative strategy for membrane proteomics: opportunities for mining therapeutic targets for autosomal dominant polycystic kidney disease. Mol. Cell. Proteomics 7, 1983–1997 [DOI] [PubMed] [Google Scholar]
- 137.Rajcevic U., Petersen K., Knol J. C., Loos M., Bougnaud S., Klychnikov O., Li K. W., Pham T. V., Wang J., Miletic H., Peng Z., Bjerkvig R., Jimenez C. R., Niclou S. P. (2009) iTRAQ-based proteomics profiling reveals increased metabolic activity and cellular cross-talk in angiogenic compared with invasive glioblastoma phenotype. Mol. Cell. Proteomics 8, 2595–2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ho J., Kong J. W., Choong L. Y., Loh M. C., Toy W., Chong P. K., Wong C. H., Wong C. Y., Shah N., Lim Y. P. (2009) Novel breast cancer metastasis-associated proteins. J. Proteome Res. 8, 583–594 [DOI] [PubMed] [Google Scholar]
- 139.Ye X., Luke B., Andresson T., Blonder J. (2009) 18O stable isotope labeling in MS-based proteomics. Brief. Funct. Genomic. Proteomic. 8, 136–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kristiansen T. Z., Harsha H. C., Grønborg M., Maitra A., Pandey A. (2008) Differential membrane proteomics using 18O-labeling to identify biomarkers for cholangiocarcinoma. J. Proteome Res. 7, 4670–4677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chi L. M., Lee C. W., Chang K. P., Hao S. P., Lee H. M., Liang Y., Hsueh C., Yu C. J., Lee I. N., Chang Y. J., Lee S. Y., Yeh Y. M., Chang Y. S., Chien K. Y., Yu J. S. (2009) Enhanced interferon signaling pathway in oral cancer revealed by quantitative proteome analysis of microdissected specimens using 16O/18O labeling and integrated two-dimensional LC-ESI-MALDI tandem MS. Mol. Cell. Proteomics 8, 1453–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Stockwin L. H., Blonder J., Bumke M. A., Lucas D. A., Chan K. C., Conrads T. P., Issaq H. J., Veenstra T. D., Newton D. L., Rybak S. M. (2006) Proteomic analysis of plasma membrane from hypoxia-adapted malignant melanoma. J. Proteome Res. 5, 2996–3007 [DOI] [PubMed] [Google Scholar]
- 143.Röwer C., Vissers J. P., Koy C., Kipping M., Hecker M., Reimer T., Gerber B., Thiesen H. J., Glocker M. O. (2009) Towards a proteome signature for invasive ductal breast carcinoma derived from label-free nanoscale LC-MS protein expression profiling of tumorous and glandular tissue. Anal. Bioanal. Chem. 395, 2443–2456 [DOI] [PubMed] [Google Scholar]
- 144.Zhu Z., Boobis A. R., Edwards R. J. (2008) Identification of estrogen-responsive proteins in MCF-7 human breast cancer cells using label-free quantitative proteomics. Proteomics 8, 1987–2005 [DOI] [PubMed] [Google Scholar]
- 145.Ishihama Y., Sato T., Tabata T., Miyamoto N., Sagane K., Nagasu T., Oda Y. (2005) Quantitative mouse brain proteomics using culture-derived isotope tags as internal standards. Nat. Biotechnol. 23, 617–621 [DOI] [PubMed] [Google Scholar]
- 146.Sihlbom C., Wilhelmsson U., Li L., Nilsson C. L., Pekny M. (2007) 14-3-3 expression in denervated hippocampus after entorhinal cortex lesion assessed by culture-derived isotope tags in quantitative proteomics. J. Proteome Res. 6, 3491–3500 [DOI] [PubMed] [Google Scholar]
- 147.Rasmussen N., Ditzel H. J. (2009) Scanning the cell surface proteome of cancer cells and identification of metastasis-associated proteins using a subtractive immunization strategy. J. Proteome Res. 8, 5048–5059 [DOI] [PubMed] [Google Scholar]
- 148.Giltnane J. M., Rimm D. L. (2004) Technology insight: Identification of biomarkers with tissue microarray technology. Nat. Clin. Pract. Oncol. 1, 104–111 [DOI] [PubMed] [Google Scholar]
- 149.Thomson T. A., Zhou C., Chu C., Knight B. (2009) Tissue microarray for routine analysis of breast biomarkers in the clinical laboratory. Am. J. Clin. Pathol. 132, 899–905 [DOI] [PubMed] [Google Scholar]
- 150.Mager S. R., Oomen M. H., Morente M. M., Ratcliffe C., Knox K., Kerr D. J., Pezzella F., Riegman P. H. (2007) Standard operating procedure for the collection of fresh frozen tissue samples. Eur. J. Cancer 43, 828–834 [DOI] [PubMed] [Google Scholar]
- 151.Wasielewski R., Hasselmann S., Rüschoff J., Fisseler-Eckhoff A., Kreipe H. (2008) Proficiency testing of immunohistochemical biomarker assays in breast cancer. Virchows Arch. 453, 537–543 [DOI] [PubMed] [Google Scholar]
- 152.Camp R. L., Chung G. G., Rimm D. L. (2002) Automated subcellular localization and quantification of protein expression in tissue microarrays. Nat. Med. 8, 1323–1327 [DOI] [PubMed] [Google Scholar]
- 153.Rubin M. A., Zerkowski M. P., Camp R. L., Kuefer R., Hofer M. D., Chinnaiyan A. M., Rimm D. L. (2004) Quantitative determination of expression of the prostate cancer protein alpha-methylacyl-CoA racemase using automated quantitative analysis (AQUA): a novel paradigm for automated and continuous biomarker measurements. Am. J. Pathol. 164, 831–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Harigopal M., Heymann J., Ghosh S., Anagnostou V., Camp R. L., Rimm D. L. (2009) Estrogen receptor co-activator (AIB1) protein expression by automated quantitative analysis (AQUA) in a breast cancer tissue microarray and association with patient outcome. Breast Cancer Res. Treat. 115, 77–85 [DOI] [PubMed] [Google Scholar]
- 155.Kozarova A., Petrinac S., Ali A., Hudson J. W. (2006) Array of informatics: applications in modern research. J. Proteome Res. 5, 1051–1059 [DOI] [PubMed] [Google Scholar]
- 156.Sanchez-Carbayo M. (2006) Antibody arrays: technical considerations and clinical applications in cancer. Clin. Chem. 52, 1651–1659 [DOI] [PubMed] [Google Scholar]
- 157.MacBeath G. (2002) Protein microarrays and proteomics. Nat. Genet. 32, (suppl.) 526–532 [DOI] [PubMed] [Google Scholar]
- 158.Gerber S. A., Rush J., Stemman O., Kirschner M. W., Gygi S. P. (2003) Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. U.S.A. 100, 6940–6945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Pratt J. M., Simpson D. M., Doherty M. K., Rivers J., Gaskell S. J., Beynon R. J. (2006) Multiplexed absolute quantification for proteomics using concatenated signature peptides encoded by QconCAT genes. Nat. Protoc. 1, 1029–1043 [DOI] [PubMed] [Google Scholar]
- 160.Mirzaei H., McBee J. K., Watts J., Aebersold R. (2008) Comparative evaluation of current peptide production platforms used in absolute quantification in proteomics. Mol. Cell. Proteomics 7, 813–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zucker S., Hymowitz M., Conner C., Zarrabi H. M., Hurewitz A. N., Matrisian L., Boyd D., Nicolson G., Montana S. (1999) Measurement of matrix metalloproteinases and tissue inhibitors of metalloproteinases in blood and tissues: clinical and experimental applications. Ann. N. Y. Acad. Sci. 878, 212–227 [DOI] [PubMed] [Google Scholar]
- 162.Anderson N. L., Anderson N. G. (2002) The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics 1, 845–867 [DOI] [PubMed] [Google Scholar]
- 163.Pardo M., García A., Antrobus R., Blanco M. J., Dwek R. A., Zitzmann N. (2007) Biomarker discovery from uveal melanoma secretomes: identification of gp100 and cathepsin D in patient serum. J. Proteome Res. 6, 2802–2811 [DOI] [PubMed] [Google Scholar]
- 164.Whelan S. A., Lu M., He J., Yan W., Saxton R. E., Faull K. F., Whitelegge J. P., Chang H. R. (2009) Mass spectrometry (LC-MS/MS) site-mapping of N-glycosylated membrane proteins for breast cancer biomarkers. J. Proteome Res. 8, 4151–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gross M., Top I., Laux I., Katz J., Curran J., Tindell C., Agus D. (2007) Beta-2-microglobulin is an androgen-regulated secreted protein elevated in serum of patients with advanced prostate cancer. Clin. Cancer Res. 13, 1979–1986 [DOI] [PubMed] [Google Scholar]
- 166.Souder C., Leitzel K., Ali S. M., Demers L., Evans D. B., Chaudri-Ross H. A., Hackl W., Hamer P., Carney W., Lipton A. (2006) Serum epidermal growth factor receptor/HER-2 predicts poor survival in patients with metastatic breast cancer. Cancer 107, 2337–2345 [DOI] [PubMed] [Google Scholar]
- 167.Sandri M. T., Johansson H. A., Zorzino L., Salvatici M., Passerini R., Maisonneuve P., Rocca A., Peruzzotti G., Colleoni M. (2007) Serum EGFR and serum HER-2/neu are useful predictive and prognostic markers in metastatic breast cancer patients treated with metronomic chemotherapy. Cancer 110, 509–517 [DOI] [PubMed] [Google Scholar]
- 168.Witzel I., Thomssen C., Krenkel S., Wilczak W., Bubenheim M., Pantel K., Neumann R., Jänicke F., Müller V. (2006) Clinical utility of determination of HER-2/neu and EGFR fragments in serum of patients with metastatic breast cancer. Int. J. Biol. Markers 21, 131–140 [DOI] [PubMed] [Google Scholar]
- 169.Ali S. M., Carney W. P., Esteva F. J., Fornier M., Harris L., Köstler W. J., Lotz J. P., Luftner D., Pichon M. F., Lipton A. (2008) Serum HER-2/neu and relative resistance to trastuzumab-based therapy in patients with metastatic breast cancer. Cancer 113, 1294–1301 [DOI] [PubMed] [Google Scholar]
- 170.Mayer S., zur Hausen A., Watermann D. O., Stamm S., Jäger M., Gitsch G., Stickeler E. (2008) Increased soluble CD44 concentrations are associated with larger tumor size and lymph node metastasis in breast cancer patients. J. Cancer Res. Clin. Oncol. 134, 1229–1235 [DOI] [PubMed] [Google Scholar]
- 171.Lim K. T., Miyazaki K., Kimura N., Izawa M., Kannagi R. (2008) Clinical application of functional glycoproteomics—dissection of glycotopes carried by soluble CD44 variants in sera of patients with cancers. Proteomics 8, 3263–3273 [DOI] [PubMed] [Google Scholar]
- 172.Callesen A. K., Vach W., Jørgensen P. E., Cold S., Tan Q., Depont Christensen R., Mogensen O., Kruse T. A., Jensen O. N., Madsen J. S. (2008) Combined experimental and statistical strategy for mass spectrometry based serum protein profiling for diagnosis of breast cancer: a case-control study. J. Proteome Res. 7, 1419–1426 [DOI] [PubMed] [Google Scholar]
- 173.Becker S., Cazares L. H., Watson P., Lynch H., Semmes O. J., Drake R. R., Laronga C. (2004) Surfaced-enhanced laser desorption/ionization time-of-flight (SELDI-TOF) differentiation of serum protein profiles of BRCA-1 and sporadic breast cancer. Ann. Surg. Oncol. 11, 907–914 [DOI] [PubMed] [Google Scholar]
- 174.Vlahou A., Laronga C., Wilson L., Gregory B., Fournier K., McGaughey D., Perry R. R., Wright G. L., Jr., Semmes O. J. (2003) A novel approach toward development of a rapid blood test for breast cancer. Clin. Breast Cancer 4, 203–209 [DOI] [PubMed] [Google Scholar]
- 175.Knoop A. S., Bentzen S. M., Nielsen M. M., Rasmussen B. B., Rose C. (2001) Value of epidermal growth factor receptor, HER2, p53, and steroid receptors in predicting the efficacy of tamoxifen in high-risk postmenopausal breast cancer patients. J. Clin. Oncol. 19, 3376–3384 [DOI] [PubMed] [Google Scholar]
- 176.Early Breast Cancer Trialists' Collaborative Group (1998) Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet 351, 1451–1467 [PubMed] [Google Scholar]
- 177.Early Breast Cancer Trialists' Collaborative Group (2005) Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet 365, 1687–1717 [DOI] [PubMed] [Google Scholar]
- 178.Umar A., Kang H., Timmermans A. M., Look M. P., Meijer-van Gelder M. E., den Bakker M. A., Jaitly N., Martens J. W., Luider T. M., Foekens J. A., Pasa-Toliæ L. (2009) Identification of a putative protein profile associated with tamoxifen therapy resistance in breast cancer. Mol. Cell. Proteomics 8, 1278–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]