

Abstract
Characterization and interpretation of disease-associated alterations of protein glycosylation are the central aims of the emerging glycoproteomics projects, which are expected to lead to more sensitive and specific diagnosis and improve therapeutic outcomes for various diseases. Here we report a new approach to identify carbohydrate-targeting serum biomarkers, termed isotopic glycosidase elution and labeling on lectin-column chromatography (IGEL). This technology is based on glycan structure-specific enrichment of glycopeptides by lectin-column chromatography and site-directed tagging of N-glycosylation sites by 18O during the elution with N-glycosidase. The combination of IGEL with 8-plex isobaric tag for relative and absolute quantitation (iTRAQ) stable isotope labeling enabled us not only to identify N-glycosylation sites effectively but also to compare glycan structures on each glycosylation site quantitatively in a single LC/MS/MS analysis. We applied this method to eight sera from lung cancer patients and controls, and finally identified 107 glycopeptides in their sera, including A2GL_Asn151, A2GL_Asn290, CD14_Asn132, CO8A_Asn417, C163A_Asn64, TIMP1_Asn30, and TSP1_Asn1049 which showed the significant change of the affinity to Concanavalin A (ConA) lectin between the lung cancer samples and the controls (p < 0.05 and more than twofold change). These screening results were further confirmed by the conventional lectin-column chromatography and immunoblot analysis using additional serum samples. Our novel methodology, which should be valuable for diverse biomarker discoveries, can provide high-throughput and quantitative profiling of glycan structure alterations.
Glycan structure variations often show highly organ-specific manners (1, 2), as well as those manners that correlate with diverse disease states, (3, 4) e.g., cancer and inflammation. Thus, the carbohydrates are currently attracting a great deal of attention as specific targets of cancer biomarkers and therapy (5). In fact, certain changes of glycan structures are already in clinical use as serum biomarkers, such as AFP-L3 (6), and glycosylation at the specific site of therapeutic antibody proved to be essential for its therapeutic effect (7). Advances in proteomic technologies and analysis have stimulated a great interest in application of MS to identify glycosylation sites (8, 9) or analyze glycan structures (10, 11) from various biological specimens, but the comprehensive techniques which allow quantitative profiling of glycan structures on each glycosylation site have not been developed.
The two major issues facing recent glycoproteomic studies are the difficulties in glycopeptide-specific enrichment tools involving lectin-column chromatography and the detection of glycopeptides in mass spectrometers. In the conventional lectin-column chromatography experiments, glycoprotein enrichment from complicated protein mixtures, such as human sera, resulted in a heavy contamination of hapten sugar, salts, and nonspecific proteins caused by protein-protein interactions of serum proteins (12). Even when the digested peptide mixture was subjected to the lectin-column chromatography, salt contamination and the eluting sugar-dependent biases of elution efficiency were inevitable. Moreover, the straightforward analysis of the eluted glycopeptides by MS was hardly possible without further deglycosylation and desalting steps.
In this study we report our new approach for the identification of carbohydrate-targeting biomarkers, termed isotopic glycosidase elution and labeling on lectin-column chromatography (IGEL),1 which is based on glycan structure-specific enrichment of glycopeptides by lectin- column chromatography and site-directed labeling of N-glycosylation sites by water-18O during the elution with N-glycosidase. We combined this method with 8-plex isotobaric tag for relative and absolute quantitation (iTRAQ) labeling for relative quantification of glycopeptides and applied them to search for carbohydrate-targeting serum biomarkers in lung cancer patient sera.
EXPERIMENTAL PROCEDURES
Serum Samples
Archived human serum samples were obtained with informed consent from 12 patients who had lung adenocarcinoma and six patients who had chronic obstructive pulmonary disease (COPD) at Hiroshima University Hospital. Serum samples as normal controls were also obtained with informed consent from six healthy volunteers who received medical examinations at Hiroshima University Hospital. Serum was collected using standard protocol from whole blood by centrifugation at 1,500 × g for 10 minutes and stored at −150°C. This study was approved by individual institutional ethical committees.
Removal of Serum Abundant Proteins
Fourteen abundant proteins in serum (albumin, immunoglobulinG [IgG], antitrypsin, IgA, transferrin, haptoglobin, fibrinogen, alpha2-macroglobulin, alpha1-acid glycoprotein, IgM, apolipoprotein Al, apolipoprotein All, complement C3, and transthyretin) were removed from 40 μL of each serum sample using 4.6 × 100 mm Multiple Affinity Removal System (MARS) LC Column - Human 14 (Agilent Technologies, Santa Clara, CA) coupled with Prominence HPLC system (Shimadzu, Tokyo, Japan). The unbound fractions were directly loaded to 4.6 × 50 mm mRP-C18 column (Agilent) and desalted with the protocols recommended by the manufacturer. The eluted proteins were vacuum-dried and subjected to trypsin digestion.
Digestion of Serum Proteins
The dried proteins were resuspended in 50 μL of [8 mol/L Urea, 50 mmol/L ammonium bicarbonate (Sigma, St. Louis, MO)] and reduced with 10 mmol/L tris(2-carboxyethyl)phosphine (Sigma) at 37°C for 30 minutes and alkylated in 50 mmol/Liodoacetamide (Sigma) with 50 mmol/L ammonium bicarbonate for 45 minutes in the dark at 25°C. Porcine trypsin (Promega, Madison, WI) was added for a final enzyme to protein ratio of 1:20. The digestion was conducted at 37°C for 16 hours, followed by quenching the reaction with the addition of trifluoroacetic acid at the final concentration 0.4%. The digested peptides were then desalted with Oasis HLB cartridges (Waters, Milford, MA) according to the manufacturer's instructions except for the use of binding solution (0.1% trifluoroacetic acid with 2% acetonitrile) and elution solution (0.1% trifluoroacetic acid with 70% acetonitrile). The eluted peptides were vacuum-dried and subjected to 8-plex iTRAQ labeling.
8-plex iTRAQ Labeling
The dried peptides were resuspended in 30 μL of [500 mmol/L triethylammonium bicarbonate (Sigma), 50% acetonitrile, pH 8.5] and combined with a vial of 8-plex iTRAQ reagent (Applied Biosystems, Foster City, CA) that was reconstructed with 70 μL of ethanol. After incubation for two hours with gentle shaking at ambient temperature, eight samples were mixed and vacuum-dried.
Capturing Glycopeptides and Glycosylation Site-Directed Stable Isotope Labeling in the Lectin-Column Chromatography
The iTRAQ-labeled peptides were resuspended in 500 μL of [100 mmol/L ammonium bicarbonate, 10% acetonitrile] and applied to the 1 mL polypropylene column (Qiagen, Valencia, CA) packed with 500 μL of ConA-agarose beads (Seikagaku Biobusiness Corporation, Tokyo, Japan). After 60 minutes of incubation with gentle rotation at ambient temperature, the column was washed with 1 mL of [100 mmol/L ammonium bicarbonate, 10% acetonitrile] five times and subsequently 500 μL of [100 mmol/L ammonium bicarbonate, 10% acetonitrile, 90% water (18O 98%, Wako Pure Chemical Industries, Osaka, Japan)] three times. The column was then incubated with 5 units (5 μL) of N-glycosidase F (Roche Applied Science) in 500 μL of [100 mmol/L ammonium bicarbonate, 10% acetonitrile, 90% water (18O 98%)] at 37°C for 12 hours. The eluate and additional five rinses with 500 μL of [100 mmol/M ammonium bicarbonate, 10% acetonitrile] were combined and vacuum-dried.
Strong Cation Exchange (SCX) Chromatography
The peptides were resuspended in 20 μL of [0.1% trifluoroacetic acid, 70% acetonitrile] and loaded to 0.2 × 200 mm MonoCap SCX column (GL Science, Tokyo, Japan) with the use of solvent A [10 mmol/L ammonium formate, 25% acetonitrile] and solvent B [1 mol/L ammonium formate, 25% acetonitrile] at a flow rate of 5 μL/min. During the multistep linear gradient of solvent B 0 to 15% for 15 minutes and 15 to 100% for 5 minutes, the eluted fraction was separately collected each minute, followed by vacuum-drying.
LC/MS/MS Analysis and the Peptide Identification
The fractions of SCX chromatography were individually analyzed with QSTAR Elite MS (Applied Biosystems) combined with UltiMate 3000 nano-flow HPLC system (DIONEX Corporation, Sunnyvale, CA). The peptides were separated on 75 μm × 150 mm l-Column (Chemicals Evaluation and Research Institute) using solvent A [0.1% formic acid, 2% acetonitrile] and solvent B [0.1% formic acid, 70% acetonitrile] with the multistep linear gradient of solvent B 5 to 45% for 65 minutes and 45 to 95% for 10 minutes at a flow rate 200 nL/min. QSTAR Elite mass spectrometer was used in standard MS/MS data-dependent acquisition mode. The 1 s survey MS spectra were collected (m/z 400–1800) followed by three MS/MS measurements on the most intense parent ions (30 counts/sec threshold, +2-+4 charge state, m/z 500–2000 mass range for MS/MS), using the manufacturer's “smart exit” setting (SIDA= 3.0). Previously targeted parent ions were excluded from repetitive MS/MS acquisition for 40 seconds (100 mDa mass tolerance). The reporter region was enhanced and collision energy was adjusted by the manufacturer's setting “Using iTRAQ Reagent”.
Identification of Glycosylation Sites
ProteinPilot 2.0 software (Applied Biosystems) with the Paragon Algorithm was used for the identification and relative abundance quantification of proteins. Tandem MS data were searched against the human protein database from SwissProt 57.4 (20,400 sequences). The search parameters were trypsin as enzyme, carbamidomethylation at cysteines as fixed modification, and thorough IDas search method. We report only peptide identifications with Conf. > 95 and protein identifications with ProtScore >1.3, which represents > 95% statistical confidence in ProteinPilot. Particularly, we used protein identifications with more than two peptide identifications. Whereas common modifications such as oxidation of methionine and deamidation of asparagine were included automatically as variable modifications in Paragon Algorithm, we additionally entered a sentence in “ProteinPilot.DataDictionary.xml” file to define the IGEL-tag as follows;
<Mod rKey = “0”>
<Nme>User modH on Asn</Nme>
<TLC>UNH</TLC>
<Tgt>Asparagine</Tgt>
<TS>255</TS>
<Fma>Ob</Fma>
<RpF>NH</RpF>
<NLF></NLF>
<IIF></IIF>
<Chg>0</Chg>
</Mod>
The peptide identification data including iTRAQ-based quantification results was exported into the XML-formatted file from ProteinPilot software. We manually excluded peptides with “low confidence”, “no iTRAQ”, “no quant”, and “weak signal” and used peptides with the value “Used = 1” for the further quantification analysis. Furthermore, we accepted only the following modifications; iTRAQ8plex@N-term, iTRAQ8plex(K), Carbamidomethyl(C), Oxidation(M), and User modH on Asn(N). After removing the redundancy and selecting the peptides with unique amino acid sequence and modifications, peptides possessing “User modH on Asn(N)” modification (described above) were extracted, which were expected to be derived from N-glycosylated peptides. Next, the existence of N-glycosylation consensus sequence N x S/T was checked.
Glycosylation Site-Specific Quantification Analysis
The peak area of eight reporter ions from each identified peptide was used for the following quantification analysis. We converted the absolute value of the area into the relative difference from the normal levels by dividing eight reporter values by the average of the area from three healthy controls labeled with iTRAQ-113, 114, and 115 (NAvr). The ratios obtained from the sample with ConA lectin purification were described here as [(113/NAvr)ConA, …, (121/NAvr)ConA]. We further analyzed samples without ConA lectin purification to examine relative protein concentrations among eight samples, termed as [(113/NAvr)Pre, …, (121/NAvr)Pre]. These ratios for a typical protein were calculated from the average of all peptide quantification data assigned to that protein. Finally, comparative glycan structure changes among eight samples were calculated as follows.
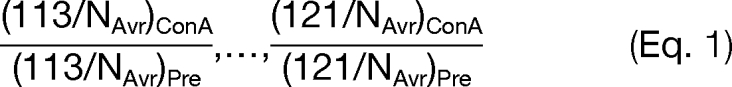 |
ConA Spin Column Purification and Immunoblot Analysis
Twenty microliters each of 24 serum samples were purified with MARS Human-14 column and mRP-C18 column as described above. The immunodepleted protein samples were resuspended with 10 μL of [8 mmol/L Urea, 50 mmol/L Tris-HCl (8.0)] thoroughly and diluted with 90 μL of the Binding/Wash Buffer (Glycoprotein Isolation Kit ConA, Thermo Scientific, Waltham, MA). The ConA-bound fractions were collected in accordance with the manufacturer's instructions, excepting three times repeated elution with 50 μL of the elution buffer (Glycoprotein Isolation Kit ConA, Thermo Scientific). The 20 μL aliquots of the eluate were run on 10% SDS-polyacrylamide gel and electroblotted onto a PVDF membrane. The blots were probed with anti-TIMP1 antibody (Abcam, Cambridge, MA). After incubation of the membrane with horseradish peroxidase-conjugated secondary antibody (GE Healthcare), the reactivity was visualized on x-ray films (Kodak) using a chemiluminescent detection kit (PerkinElmer Life Sciences).
RESULTS
Isotopic Glycosidase Elution and Labeling on Lectin-Column Chromatography (IGEL)
Our IGEL technology clearly resolved major problems for lectin-column chromatography and mass spectrometric analysis of glycosylated peptides. Since the eluted product of the IGEL method was completely deglycosylated peptides containing 18O-tagged N-glycosylation sites dissolved in volatile solution, it was directly analyzable by MS (Fig. 1A). Although 1-Da mass shift by deamidation of asparagine residues (Asn to Asp) and glutamine residues (Gln to Glu) could be detected in mass spectrometric analysis, the 3-Da increase of the asparagine residue was more specific to the IGEL process, which enabled precise identification of glycosylation sites. Furthermore, the combination with 8-plex iTRAQ labeling of peptides diminished potential experimental errors of lectin-column chromatography and allowed quantitative comparison of glycan structures on serum proteins from eight individuals. To identify carbohydrate-targeting serum markers for lung cancer, we applied this IGEL method to eight human serum samples, consisting of sera from three healthy controls, two chronic obstructive pulmonary disease (COPD) patients, and three lung cancer patients (Fig. 1B, supplemental Table 1).
Fig. 1.

Concept of IGEL technology and schematic representation of IGEL-based biomarker screening. (A) Tryptic digests from eight clinical samples were labeled with 8-plex iTRAQ tags and loaded to lectin-column chromatography. After replacement of the solvent with H218O, glycopeptides which bind to lectin- column specifically by glycan-lectin interaction were eluted with N-glycosidase. As the eluate includes 18O-tagged “peptides” without any involatile salts, it can be directly subjected to LC/MS/MS analysis and usual database search, which results in simultaneous identification of N-glycosylation sites and quantification of site-specific affinity changes to the lectin. (B) Following immuno-depletion of 14 abundant proteins, eight serum samples were processed with or without IGEL purification (termed ConA or Pre, respectively). The relative difference from the normal level (e.g., 113/NAvr) was used to compare the affinity variations to ConA lectin among eight samples (see Method for procedural details).
Because serum abundant proteins, particularly abundant glycosylated proteins, critically interfere with the association between serum minor components and lectins, the 14 most abundant serum proteins were removed from sera using the MARS Human 14 column before IGEL. In this step, 92.8% w/w of serum proteins on the average was immunodepleted (supplemental Fig. 1). The protein recovery rate of the desalting process by mRP-C18 column chromatography was 98% (supplemental Fig. 2). After tryptic digestion, peptides were labeled with 8-plex iTRAQ stable isotope tags. We optimized the peptide labeling protocol by reacting one vial of 8-plex iTRAQ reagent to 25 μg of serum peptide mixture so that 94.2% of lysine residues and N-terminal amino acids of peptides were labeled by iTRAQ (supplemental Fig. 3). The vacuum-dried iTRAQ-tagged peptides were applied to the ConA lectin-column chromatography.
Evaluation of the Accuracy and Efficiency of the IGEL Tagging
Preliminarily, we evaluated the transduction efficiency of 18O in the IGEL-purified peptides using QSTAR Elite LC/MS/MS system. The typical MS or MS/MS spectra of the identified peptide in our screening, T165LTLFNVTR173 or T521LTLFNVTR529 from the CEAM5 protein (well-known tumor marker CEA), were shown in Figure 2. The TLTLFNVTR sequence was present in the two locations in the CEAM5 protein. When PNGase F reaction was conducted in H216O as a control experiment, CEAM5 165–173 or 521–529 peptides were detected at m/z = 685.40 with doubly charged state in MS analysis (Fig. 2A). The MS/MS experiment and the subsequent database search showed an amino acid conversion of asparagine to aspartic acid in this sequence, resulting in 1-Da increase of the molecular weight (Fig. 2B), whereas a 3-Da increase of the same peptide (m/z = 686.39) was observed from the sample eluted in the presence of H218O by IGEL method (Figs. 2C and 2D). The peak of m/z 685.40 corresponding to the CEAM5 peptide with 16O-incorporated asparagine residue was hardly detectable in Figure 2C, indicating that H216O contamination in PNGase F reaction with H218O could be almost negligible for the mass spectrometric analysis. Additionally, the MS/MS spectrum acquired from the precursor ion m/z 686.39 clearly showed the 3-Da increase of the asparagine residue (Fig. 2D). The iTRAQ reporter ions detected in the MS/MS spectrum of m/z 685.40 (Fig. 2E) and m/z 686.39 (Fig. 2F) indicated that CEAM5 165–173 or 521–529 peptide was dominantly detected in one lung cancer serum, which was labeled with iTRAQ-121.These data implicated that the relative abundance of eight reporter ions was not affected by the use of elution buffer containing H218O, and the IGEL method could have a great potential for the reliable identification of glycosylation sites on even small quantity of proteins, such as serum CEA (ng/mL range). Moreover, the use of 8-plex iTRAQ tags enabled the relative quantification of glycan structures on each glycosylation site.
Fig. 2.

Representative mass spectra of carcinoembryonic antigen-related cell adhesion molecule 5 (CEAM5, well known as CEA) peptides processed with IGEL. The MS spectra of CEAM5 165–173 or 521–529 peptides eluted with glycopeptidase-F in H216O (A) or H218O (C) were shown. The MS/MS spectrum of the same peptide in each condition and identified fragment ions were shown in (B) or (D), respectively. The reporter ions of 8-plex iTRAQ tags were magnified from the spectrum (B) or (D) and indicated by arrow heads in (E) or (F), respectively. Reporters from three healthy controls and three lung cancer samples were indicated with blue or red arrow heads.
Quantitative Profiling of Lung Cancer Specific Glycosylations by IGEL
We applied this method to the wide screening for the cancer-associated glycan structure alterations in lung cancer patients' sera. When we undertake the quantification analysis of glycosylations, it is important to be aware of the fact that the quantification analysis using ConA-purified peptides can be reflected both by their affinity changes to the ConA lectin column and by the difference of their protein concentrations. To eliminate the effect of protein concentrations and accurately quantify the glycan structure alterations, we simultaneously performed LC/MS/MS analyses of iTRAQ-tagged peptides without ConA lectin-column purification (termed pre-ConA purified sample), as well as the same sample after ConA lectin- column purification (termed ConA-purified sample) (Fig. 1B). For the identification of glycopeptides from ConA-purified sample, 10,709 peptides were automatically identified with ProteinPilot software from 61,216 MS/MS analyses. By removing the redundancy and peptides with unreliable identification or very weak signals under the criteria described under “Experimental Procedures,” we identified 127 unique peptides including 119 peptides possessing 18O-incorporated asparagine residues (termed IGEL-tagged asparagine residues). The enrichment yield of glycopeptides using theConA-IGEL procedure was 93.7%. Although ProteinPilot had great advantages in that the selection of variable modifications was completely automated and the peptide quantification were performed simultaneously with the peptide identification, the search results by ProteinPilot included a large number of improper modifications, such as dehydration of serine + phosphorylation of threonine + dethiomethylation of methionine on a peptide. Therefore, we excluded the unreliable modification data using the criteria described under “Experimental Procedures.” Among the 119 peptides, all of the IGEL-tagged asparagine residues on 107 peptides coincided with the N-glycosylation consensus sequences “N x S/T (x is one of any amino-acids),” while the other 12 peptides had at least one IGEL-tagged asparagine residue that did not match the consensus sequences above (supplemental Table 2). Finally, 101 unique N-glycosylation sites were found on 119 glycopeptides, which were assigned to 56 serum proteins (supplemental Table 3). However, LC/MS/MS analysis on pre-ConA purified samples identified 274 serum proteins (supplemental Table 4), and 50 proteins among them were commonly observed in the list of the glycopeptides identified from ConA-purified samples, which were corresponding to 97 glycopeptides out of 119 glycopeptides that IGEL identified confidently from only ConA-purified samples. Although six proteins which were identified from only ConA-purified samples were not able to be applied to the quantification analysis because of the lack of the protein concentration information from pre-ConA lectin purified samples, they included a serum minor component integrin alpha-1 (> 3.4 ng/mL) (13) (supplemental Table 5). These findings indicated significant improvement of the detection limit of mass spectrometric analysis by the enrichment of IGEL method.
Next we quantified site-directed variations of the glycan structures on each of these 97 glycopeptides. For relative quantification using 8-plex iTRAQ tags, the area of the reporter ions derived from eight samples (m/z = 113, 114, 115, 116, 117, 118, 119, and 121) was divided by the averaged peak area (NAvr) of three controls (m/z = 113, 114, and 115), and the comparative affinity variations to ConA lectin among eight samples were calculated as described under “Experimental Procedures” (supplemental Table 6). The distributions of the relative changes of the 97 identified glycopeptides among eight serum samples were plotted in Figure 3A. For instance, glycopeptides corresponding to CEAM5 165–173 and 521–529 showed 46.7-fold affinity change to ConA lectin in lung cancer patients' sera. To identify glycosylation sites on which the glycan structures varied in lung cancer-specific manner, we extracted peptides satisfying the following two criteria: (1) the affinity to ConA lectin showed more than twofold change in all three lung cancer patients' sera, compared with the normal level (NAvr), and (2) showing the p value in the Student's t test between the control group (113, 114, 115, 116, and 117) and the lung cancer group (118, 119, and 121) was less than 0.05. This selection identified eight glycopeptides as candidates for serum glycan biomarkers of lung cancer (Fig. 3B and Table 1). Among them, six peptides showed more than twofold higher affinity to ConA lectin in all lung cancer sera compared with normal levels, whereas two peptides showed less than 0.5-fold lower affinity to ConA lectin in all lung cancer sera. We selected four glycopeptides, Monocyte differentiation antigen CD14 (CD14) 132–139, Scavenger receptor cysteine-rich type 1 protein M130 (C163A) 59–71, Metalloproteinase inhibitor 1 (TIMP1) 23–37 and Thrombospondin-1 (TSP1) 1047–1059, as potential carbohydrate-targeting tumor markers whose glycan structure changes were specific to three lung cancer patients' sera. The representative MS/MS spectra corresponding to the four candidates were shown in Figure 4A–4D. We found drastic changes in the relative intensities of each reporter ion between the samples before ConA lectin-column purification (Pre-ConA) and after purification (ConA-purified) for all of four peptides. Here, the relative difference of the reporter ion intensities in Pre-ConA samples indicates the difference of the identified protein concentration. By subtracting the protein concentration ratios (left side panels in Fig. 4) from the peptide quantification results of ConA-purified samples (right side panels in Fig. 4), accurate glycosylation status changes could be assessed. Thus, lung cancer[n dash]associated enhancement of the affinity to ConA lectin was observed for three glycopeptides CD14 132–139, C163A 59–71, and TIMP1 23–37, whereas significant attenuation was observed for TSP1 1047–1059 glycopeptide.
Fig. 3.

IGEL-based screening of lung cancer-associated glycan structure alterations. A, Relative affinity variations to ConA lectin for 97 glycopeptides were plotted. The bold line, log2(ratio) = 0, indicates the average of three normal controls (NAvr) labeled with iTRAQ-113, 114, and 115. Detailed information of peptide sequences and ratios were available in supplemental Table 6. Two COPD samples and three lung cancer samples were labeled with iTRAQ-116, 117 and iTRAQ-118, 119, 121, respectively. B, 8 glycopeptides whose affinity to ConA lectin showed more than twofold or less than 0.5-fold change in all three lung cancer patient samples compared with normal level (NAvr) and p < 0.05 in the t test between the control group (113 to 117) and the lung cancer group (118, 119, and 121) were extracted from A and displayed in the bar charts. Accession number and the sequence of each peptide were shown. Identified N-glycosylation sites were indicated with asterisks. TSP1 1047–1059 peptide was observed at charge state +3 and +4.
Fig. 4.

Representative MS/MS spectra of candidate biomarker glycopeptides. The whole view of MS/MS spectra, extracted area of reporter ions, fragment ion identification Tables in ProteinPilot and peptide identification information are shown for CD14 (A), C163A (B), TIMP1 (C), and TSP1 (D). The left or right side panels show peptide identification data from before or after ConA lectin-column purified samples (Pre-ConA or ConA-purified, respectively). Reporter ions corresponding to three healthy controls, two COPD samples and three lung cancer samples were indicated with blue, green, and red arrow heads.
Table I. List of 8 glycopeptides showing more than 2-fold affinity change to ConA lectin in lung cancer patients' sera.
| Accessions | m/z (z) | aConf. | bSequence | Modifications | c113/Navr | c114/Navr | c115/Navr | c116/Navr | c117/Navr | c118/Navr | c119/Navr | c121/Navr |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P02750|A2GL_HUMAN | 722.12 (+3) | 99 | K143LPPGLLAN151*FTLLR156 | iTRAQ8plex@N-term | 0.487 | 0.977 | 1.771 | 0.430 | 1.818 | 4.298 | 8.601 | 3.339 |
| iTRAQ8plex(K)@1 | ||||||||||||
| User modH on Asn(N)@9 | ||||||||||||
| P02750|A2GL_HUMAN | 653.32 (+2) | 98 | M286FSQN290*DTR293 | iTRAQ8plex@N-term | 0.588 | 0.996 | 1.686 | 0.530 | 2.433 | 6.324 | 6.919 | 2.442 |
| User modH on Asn(N)@5 | ||||||||||||
| P08571|CD14_HUMAN | 599.32 (+2) | 98 | N132*VSWATGR139 | iTRAQ8plex@N-term | 0.964 | 1.200 | 1.227 | 1.338 | 1.611 | 2.396 | 2.305 | 2.358 |
| User modH on Asn(N)@1 | ||||||||||||
| P07357|CO8A_HUMAN | 547.61 (+3) | 99 | G405GSSGWSGGLAQN417*R418 | iTRAQ8plex@N-term | 2.173 | 1.101 | 0.544 | 1.903 | 1.996 | 2.523 | 2.470 | 3.033 |
| User modH on Asn(N)@13 | ||||||||||||
| Q86VB7|C163A_HUMAN | 762.89 (+2) | 99 | A59PGWAN64*SSAGSGR71 | iTRAQ8plex@N-term | 1.503 | 1.276 | 0.861 | 0.722 | 1.903 | 3.862 | 2.412 | 4.575 |
| User modH on Asn(N)@6 | ||||||||||||
| P01033|TIMP1_HUMAN | 687.37 (+3) | 99 | F23VGTPEVN30*QTTLYQR37 | iTRAQ8plex@N-term | 0.601 | 1.492 | 1.316 | 1.614 | 1.626 | 2.695 | 2.238 | 9.126 |
| User modH on Asn(N)@8 | ||||||||||||
| P07996|TSP1_HUMAN | 558.64 (+3) | 99 | V1047VN1049*STTGPGEHLR1059 | iTRAQ8plex@N-term | 1.318 | 1.214 | 0.982 | 1.623 | 1.225 | 0.440 | 0.287 | 0.339 |
| User modH on Asn(N)@3 | ||||||||||||
| P07996|TSP1_HUMAN | 495.28 (+4) | 98 | V1047VN1049*STTGPGEHLR1059 | iTRAQ8plex@N-term | 1.088 | 1.517 | 0.904 | 0.748 | 1.113 | 0.260 | 0.449 | 0.226 |
| User modH on Asn(N)@3 | ||||||||||||
| iTRAQ8plex(T)@5 |
a The Confidence values in ProteinPilot software were shown. We set the peptide identification threshold as Conf. > 95.
b The identified N-glycosylation sites were indicated by asterisks and their amino acid numbers were also shown.
c Fold change values of the affinity to ConA lectin were calculated as relative differences from the average of three normal samples (NAvr). The ratio of more than 2-fold or less than 0.5-fold change was highlighted in red or blue, respectively.
Validation for the Lung Cancer-Associated Aberrant Glycosylation on TIMP1
Because TIMP1 appeared to undergo remarkably upregulated modification of high-mannose[n dash]type oligosaccharides in the sera of advanced stage lung cancer patients (Fig. 3B), it warranted further examination in the increased number of human sera using a different experimental procedure. Therefore, sera from age- and sex-matched group of healthy volunteers (n = 6), COPD patients (n = 6), or lung cancer patients (stage-I or -II: n = 6, stage-IV: n = 6) were resolved by 1D gel electrophoresis, and the amount of TIMP1 in the sera was determined by immunoblotting, as described above. As Figure 5A shows, there was a minor difference of TIMP1 level detected in the immune-depleted sera between the control sera and the lung cancer patients' sera. However, TIMP1 was specifically enriched in the lung cancer patients' sera by ConA lectin column (Fig. 5B). Indeed, TIMP1 signals were detectable in one of the COPD samples, two of the stage-I or -II lung cancer samples, and all of the stage-IV lung cancer samples. Importantly, we included the same eight samples used in the iTRAQ-based screening (indicated by the number 113 to 121 in Fig. 5) for this validation study and found that the quantitative tendency of ConA-affinity among these eight samples was absolutely matched with the result of iTRAQ quantification (Fig. 3B). Both the IGEL-iTRAQ screening result and this validation experiment showed drastically enhanced affinity of TIMP1 to ConA lectin on the iTRAQ 121-labeled lung cancer sample. These integrated results strongly suggested that TIMP1 might undergo aberrant high-mannose[n dash]type glycosylations in advanced stage lung cancer patients' sera. Although the sample scale was still small, the results were consistent and highlight the potential of this methodology and marker.
Fig. 5.

Immunoblot analysis of TIMP1 levels in lung cancer patients' sera before or after ConA lectin-column chromatography. (A) Serum levels of TIMP1 were analyzed by immunoblot analysis using anti-TIMP1 antibody. 20 μg each of the immunodepleted serum samples were prepared from six each of healthy volunteers (Normal), COPD patients, stage-I/II lung cancer patients, or stage-IV lung cancer patients and used for the blotting. Samples used in the IGEL-iTRAQ screening experiments were indicated by the iTRAQ reporter numbers (113 to 121). (B) Immunoblot of TIMP1 in the ConA-bound fractions prepared from the same sample set in (A). The entire ConA-bound fraction from 75 μg of total protein was loaded for each lane. Samples used in the IGEL-iTRAQ screening experiments were indicated by the iTRAQ reporter numbers (113 to 121).
DISCUSSION
Although modern MS instruments allow the measurements of unseparated mixtures at high sensitivity (14), glycoprotein or glycopeptide enrichment is still indispensable to achieving detection and identification of glycosylated components in low and precious amounts of biological materials. We have only few ways to enrich glycoproteins or glycopeptides so far, such as lectin affinity, chemicals covalently bound to specific glycan structures (15, 16), and oligosaccharide-recognizing antibodies (17). However, because of their specificity to a wide variety of substrates and their simple availability, lectins are most frequently used for the purpose of the enrichment of glycans and carbohydrate biomarker discovery. Our IGEL methodology can contribute a great deal to the simplification of these complicated experimental processes, improvement of quantitative reproducibility, and enhancement of throughput for the lectin-based glycopeptide enrichment. As we identified sub-μg/mL serum proteins from the IGEL-purified sample (shown in supplemental Table 5), this method diminished a loss of scarce serum components and brought about increasing sensitivity. Regarding the glycopeptidase-based elution from the lectin column, highly specific release of glycopeptides was achieved (93.7%) with little elution of nonspecifically bound peptides, and the elution efficiency depended on neither the glycan structures nor the number of sugar chains on a peptide. Moreover, it is critical to maintain the reproducibility of lectin-column chromatography because lectin-glycan interactions are relatively weak (Kd = 10−4–10−7 mol/L) compared with antigen-antibody interactions (Kd = 10−8–10−12 mol/L) and the purification efficiency can vary by washing or elution conditions. The combination of the simple procedure of IGEL and the use of 8-plex iTRAQ tags could minimize quantitative errors caused by these technical biases. Actually, our validation experiment using conventional lectin-column chromatography and immunoblot analysis confirmed the quantitative reliability and reproducibility of IGEL-iTRAQ technology. These facts further emphasize the potential of IGEL technology, whose advantages in enrichment efficiency, throughput, consistency, and variety could contribute not only to proteomics studies but to diverse kinds of glycobiological studies and maybe even clinical practices.
ConA lectin strongly recognizes “core oligosaccharide” structure including Man (α1–6) [Man (α1–3)] Man components, which widely expresses in the N-linked sugar chains (18). It is also known that the existence of bisecting GlcNAc structure Man (α1–6) [Man (α1–3)] [GlcNAc (β1–4)] Man significantly reduces its affinity to ConA. Therefore, concerning for sugar chains on the seven glycosylation sites identified in our screening (Fig. 3), it would be conceivable that the frequency of N-glycosylation itself or addition of bisecting GlcNAc structure was significantly altered in lung cancer patients' sera. The same strategy can be expanded to various combinations of lectins and eluting glycosidases, which could achieve more comprehensive analysis of glycan structure alterations. Our preliminary experiments successfully allowed quantification and identification of serum glycopeptides using Lens culinaris agglutinin (LCA) column and N-glycosidase F elution. In addition, the higher throughput is also achievable by increasing the sample scale to 8 × n sets with a universal control in each set, which could provide some potential for high-throughput quantitative analysis for glycan structures.
According to our data and previous studies, the advantages of 8-plex iTRAQ labeling system were clear, however, we should note that this system appeared to affect the number of peptide identifications negatively. Our 2D-LC/MS/MS experiment of pre-ConA column purification samples identified 274 proteins, while our previous experiment using the same conditions except for the use of 8-plex iTRAQ labeling identified more than 700 serum proteins (data not shown). This diminished number of identified proteins might be caused by the expanded scale of peptide concentrations after mixing eight samples together, by which considerable numbers of serum minor components should be obscured. Although the introduction of such multiplexed isotope labels can bring in quantitative reliability and high throughput, much heavier fractionation of peptides might be needed to improve its analytical dynamic range.
Furthermore, to pursue the clinical application of glycosylation-targeted disease biomarkers, we should consider that we have only few techniques so far to validate the glycan structure changes accurately. In particular, there is no established method by which glycan structure variations on a specific glycosylation site can be validated quickly, sensitively, and quantitatively for hundreds of samples. We here show that our sophisticated screening technique could contribute to the discovery of diverse disease-related glycan alterations, but new validation technologies are urgently required so that the glycoproteomic profiling results could lead to clinical applications as diagnostic or prognostic biomarkers in the future.
Acknowledgments
We thank Ms. Mari Kikuchi and Ms. Miki Yamato at Applied Biosystems Japan for their technical assistances.
* This study was supported in part by the grants from Toppan Printing Co., Ltd. and Shimadzu Corporation.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- IGEL
- isotopic glycosidase elution and labeling on lectin-column chromatography
- iTRAQ
- isobaric tag for relative and absolute quantitation
- COPD
- chronic obstructive pulmonary disease
- Ig
- immunoglobulin
- SCX
- strong cation exchange.
REFERENCES
- 1.Wollscheid B., Bausch-Fluck D., Henderson C., O'Brien R., Bibel M., Schiess R., Aebersold R., Watts J. D. (2009) Nat. Biotechnol. 27, 378–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown S. M., Smith D. M., Alt N., Thorpe S. R., Baynes J. W. (2005) Ann. N.Y. Acad. Sci. 1043, 817–823 [DOI] [PubMed] [Google Scholar]
- 3.Tissot B., North S. J., Ceroni A., Pang P. C., Panico M., Rosati F., Capone A., Haslam S. M., Dell A., Morris H. R. (2009) FEBS Lett. 583, 1728–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bos P. D., Zhang X. H., Nadal C., Shu W., Gomis R. R., Nguyen D. X., Minn A. J., van de Vijver M. J., Gerald W. L., Foekens J. A., Massague J. (2009) Nature 459, 1005–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawasaki N., Itoh S., Hashii N., Takakura D., Qin Y., Huang X., Yamaguchi T. (2009) Biol, Pharm. Bull. 32, 796–800 [DOI] [PubMed] [Google Scholar]
- 6.Ludwig J. A., Weinstein J. N. (2005) Nat. Rev. Cancer 5, 845–856 [DOI] [PubMed] [Google Scholar]
- 7.Suzuki E., Niwa R., Saji S., Muta M., Hirose M., Iida S., Shiotsu Y., Satoh M., Shitara K., Kondo M., Toi M. (2007) Clin. Cancer Res. 13, 1875–1882 [DOI] [PubMed] [Google Scholar]
- 8.Kaji H., Saito H., Yamauchi Y., Shinkawa T., Taoka M., Hirabayashi J., Kasai K., Takahashi N., Isobe T. (2003) Nat. Biotechnol. 21, 667–672 [DOI] [PubMed] [Google Scholar]
- 9.Kaji H., Yamauchi Y., Takahashi N., Isobe T. (2006) Nat. Protoc. 1, 3019–3027 [DOI] [PubMed] [Google Scholar]
- 10.Morelle W., Faid V., Chirat F., Michalski J. C. (2009) Methods Mol. Biol. 534, 5–21 [DOI] [PubMed] [Google Scholar]
- 11.Li B., An H. J., Hedrick J. L., Lebrilla C. B. (2009) Methods Mol. Biol. 534, 133–145 [DOI] [PubMed] [Google Scholar]
- 12.Jin S., Cheng Y., Reid S., Li M., Wang B. (2009) Med. Res. Rev. 30, 171–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bank I., Weiss P., Doolman R., Book M., Sela B. A. (1999) J. Lab. Clin. Med. 134, 599–604 [DOI] [PubMed] [Google Scholar]
- 14.Anderson N. L., Anderson N. G., Pearson T. W., Borchers C. H., Paulovich A. G., Patterson S. D., Gillette M., Aebersold R., Carr S. A. (2009) Mol. Cell. Proteomics 8, 883–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian Y., Zhou Y., Elliott S., Aebersold R., Zhang H. (2007) Nat. Protoc. 2, 334–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gontarev S., Shmanai V., Frey S. K., Kvach M., Schweigert F. J. (2007) Rapid Commun. Mass. Spectrom. 21, 1–6 [DOI] [PubMed] [Google Scholar]
- 17.Heimburg-Molinaro J., Rittenhouse-Olson K. (2009) Methods Mol. Biol. 534, 341–357 [DOI] [PubMed] [Google Scholar]
- 18.Ohyama Y., Kasai K., Nomoto H., Inoue Y. (1985) J. Biol. Chem. 260, 6882–6887 [PubMed] [Google Scholar]