

Abstract
The recent elucidation of the sodium/galactose symporter structure from the Vibrio parahaemolyticus bacterium, vSGLT, has revealed a similarity in the core architecture with transporters from different gene families, including the leucine transporter (LeuT). Even though several transporters sharing this core have been structurally determined over the past few years, vSGLT is the only one crystallized in the substrate-bound inward-facing conformation so far. In this study, we report the first insight into the dynamics and coordination of the galactose (Gal) and proposed Na+ ion in vSGLT using a series of molecular dynamics simulations with a total time of about 0.1 μs. Our study reveals new residues, not observed in the crystal structure, which closely interact with the Na+ ion or the substrate for extended times, and shows that in the crystallized conformation, a Na+ ion placed at the site equivalent to Na2 in LeuT can escape into the intracellular (IC) space in the absence of external forces. We have identified the highly conserved Asp189 as a likely binding residue on the pathway of Na+ into the IC cavity. The release of Gal, on the other hand, requires the rotation of the side chain of the inner hydrophobic gate, Tyr263, without a significant change in vSGLT backbone conformation. Our simulations further show that the crystal structure represents but one accessible binding pose of Gal and Na+ among an ensemble of microstates, and that the Gal undergoes versatile alternate interactions at the binding pocket.
Secondary transporters are molecular machines that allow the translocation of molecules across the cell membrane using the electrochemical gradient of ions, often sodium, across the membrane. The energy released as these ions move down their electrochemical gradient is used to power the ‘uphill’ movement of the substrate.1,2 One of the major families of secondary transporters, the sodium solute symporters (SSS) with over 250 members, co-transports sodium with sugars, amino acids, vitamins, osmolytes and inorganic ions3 and has diverse functions including transport of sugars and urea,4 glucosensation and tumor suppression.5,6
The recently reported crystal structure of vSGLT,7 a member of the SSS family, shows an overall topology similar to several evolutionarily-distant transporters which have been crystallized in various states along the transport cycle.7–13 The first crystal obtained for this structural family belongs to the leucine transporter (LeuT), later shown to be a transporter of other amino acids as well.14 The LeuT structure has a pseudo-two-fold symmetry such that the transmembrane (TM) helices 1–5 can be structurally superimposed on helices 6–10 upon ~180° rotation around an axis through the plane of the membrane.11 Another prominent feature of this core is the break/irregularity in the first α-helix of each repeat (helices 1 and 6) about halfway across the membrane exposing backbone polar groups to interact with the substrate or Na+ ions.11 Whether it is helices 1–10 in LeuT, 2–11 in vSGLT or 3–12 in the Na+/betaine symporter, BetP,13 that form the transport core, these unique structural features observed near the substrate and/or ion binding sites seem to be a commonality among these families of transporters despite their lack of sequence similarity.15 According to the mechanism of alternate access, the transporter undergoes cooperative changes in structure during the transport cycle, which causes the crevice near the binding-pocket to alternate between intracellular (IC) and extracellular (EC) exposure. The EC-exposed binding pocket binds the substrate and Na+ ion(s). The transporter then shifts to a conformation where the binding site is inaccessible to the EC or the IC space, followed by the exposure of the binding site to the IC medium where the substrate and Na+ ion(s) are released. The inward-facing transporter can then reorient to face the EC medium again, and so on. The crystal structures obtained so far have captured transporters in various states of this transport cycle; whereas several crystals have been obtained for the outward-facing conformation with the substrate or a competitive inhibitor as in the case of LeuT, the only crystal structure resolved to date for the inward-facing conformation with the substrate bound is that of vSGLT.7
The vSGLT crystal is a parallel dimer7 where each monomer contains a galactose (Gal) molecule bound approximately midway across the membrane (Fig. 1A) in a site similar to that observed in LeuT. Gal is liganded by residues on TM helices 2, 3, 7, 8 and 11, which are equivalent to the helices 1, 2, 6, 7 and 10 in LeuT (Fig. 1B–D). These, together with helices 4 and 9, the counterparts of helices 3 and 8 in LeuT, lie at the core of the protein. As in the case of the sodium/glucose transporter SGLT1, which can transport glucose as well as Gal,16 vSGLT is not specific to Gal and can bind other sugars such as fucose and transport glucose.17,18 However, in contrast to SGLT1 which apparently co-transports two Na+ ions for every glucose molecule, in vSGLT the stoichiometry is 1: 1.18 Even though no functionally-relevant cation(s) could be observed in the vSGLT crystal structure, the Na+ binding site has been suggested to be similar to the Na2 site in LeuT.11 This is supported by structural homology between the two transporters, sequence similarity among the members of the SSS family, and mutational approaches.18
Fig. 1. The crystal structure and simulated system of vSGLT.
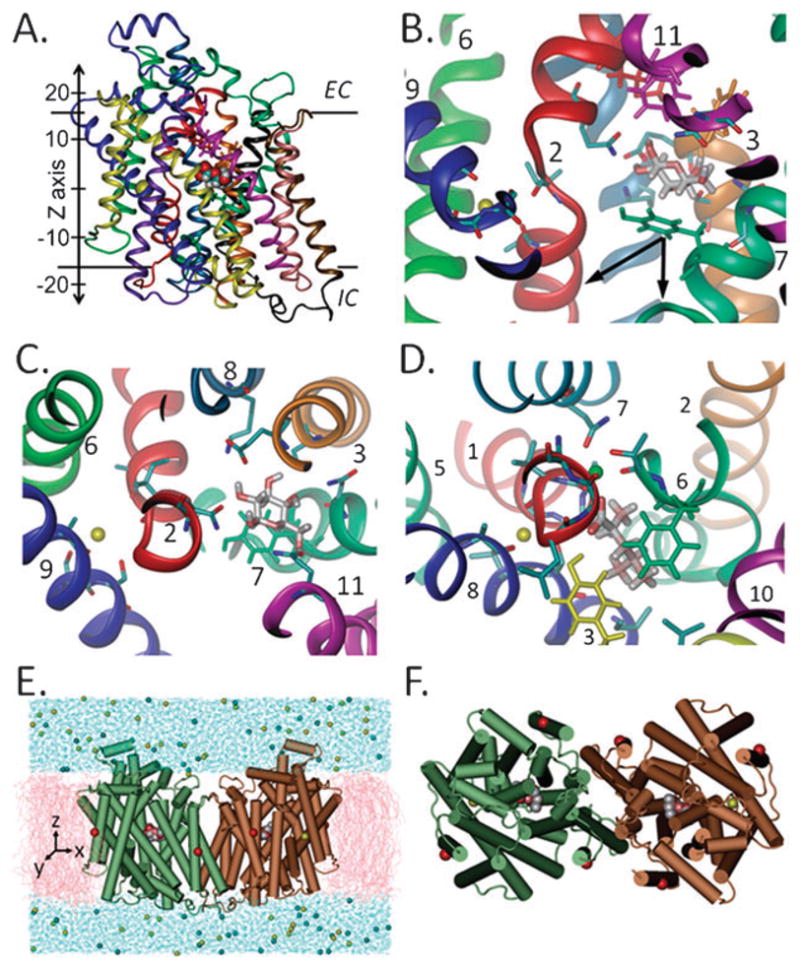
The whole monomer (A) or binding site region (B and C) is viewed here through the plane of the membrane (A and B) or from the external side (C). Panels C and D compare the monomeric binding site of SGLT (C), which contains the proposed Na+ ion (yellow sphere), and that of LeuT (D) with Na1 and Na2 (green and yellow spheres, respectively). All helices are colored similarly in A–C and as their equivalents in LeuT (D). Residues at the internal (green) and external hydrophobic gates in vSGLT (magenta, red and orange) and at the external gate in LeuT (green and yellow) are drawn in licorice. The simulated system (E and F), composed of the dimer embedded in a membrane bilayer (pink) and solvated in water (blue), is shown here (E, side-view and F, top view) with the α-carbons where harmonic constraints were applied (red spheres, see Methods). The arrows in (B) indicate the two directions of pulling in setups 4–11.
To gain insights into the collective dynamics and coordination of Gal and sodium in vSGLT, we have performed about 0.1 μs of molecular dynamics (MD) and steered MD (SMD)19,20 simulations on vSGLT in different setups (ESI† Table S1). In our SMD simulations, Gal was pulled from the binding site towards the IC space to investigate the possible pathway(s) of unbinding and conformational changes, or lack thereof, involved in this step. All simulations described here were performed on the dimer even though hSGLT1 and vSGLT function as monomers.18,21 Using the dimer was based on the obtained crystal structure and it enabled us to increase the statistical significance of our results as independent events within the two core domains were simultaneously observed for both monomers A and B.
Results
Dynamic coordination of the sodium ion
Both the identity and number of groups that coordinate the Na+ showed considerable variations during the simulations. As a measure of the temporal evolution of Na+ position and interactions, we probed the instantaneous distances of Na+ to the proposed coordinating groups (7), along with the corresponding interaction energies in both monomers (Fig. 2 and ESI† Fig. S1). Fluctuations in the distances began as soon as the constraints of the equilibration phase were released. Notably, stepwise increases or decreases were intermittently observed in the distances of the nearest oxygens from Na+, indicative of compensating switches between the ligating roles of these atoms, such that Na+ ion is coordinated almost at all times by two or more residues, in addition to water molecules (Fig. 3). The atoms located within 2.6 Å from Na+, which took successive turns in stabilizing the cation, include the hydroxyl oxygens (Oγ) of Ser364 and Ser365 on H9, and the carbonyl oxygens (O) of Ala361 on H9 and Ala62 and Ile65 on H2, and the carboxylic oxygens (Oδ1 and Oδ2) of Asp189 on H6.
Fig. 2. Dynamic character of sodium ion coordination and energetics.

Time evolution of the distances (top) and interaction energies (bottom) between the Na+ ion and the indicated atoms are shown for monomer A in setup 1. Results for monomer B are presented in ESI† Fig. S1.
Fig. 3. Coordination of the sodium ion at, and beyond, the binding site.

Snapshots of the Na+ ion in monomer A at 4 ns (upper left), 10 ns (upper right), 19 ns (lower left) and 25 ns (lower right) from setup 1 are viewed through the plane of the membrane. Underlined residues indicate groups within 2.6 Å.
The average distances between Na+ and these atoms during the MD trajectories exhibited clear departures from those observed in the crystal structure (ESI† Table S2). We note in particular the increase in the separation between Na+ and H9 residues A361, S364 and S365 due to the dislocation of Na+ away from its original position. Furthermore, the association with oxygens on the Asp189 side chain appeared to be energetically dominant over all other sodium-coordinating groups as may be clearly seen in Fig. 2. Another residue observed in our simulations to interact with Na+, albeit to a lesser extent, was Gln192, located at the helical turn adjoining Asp189. The average energies of interaction with Na+ ion listed in Table S2† clearly show there is a competition between helices H2 and H6, and more specifically between Ala62, Ala63 and Ala65 on H2 (complemented by Ala361 on H9) and Asp189 on H6, consistently observed in both monomers.
The interaction with the Na+ ion in the inward-facing conformation of vSGLT is not limited to the protein but includes water molecules (Fig. 3 and ESI† Fig. S2). These water molecules enter the transporter crevice and reach the Na+ binding site from the IC side. During the 30 ns simulation (setup 1), water molecules contributed on average about 40% to the total energy of interaction with the Na+ ion in both monomers (Table S2†) and this was also confirmed in setup 3. The number of water molecules optimally binding the Na+ ion (within 2.6 Å) was observed to vary between 2 and 6. The more water molecules bound, the less effective the coordination by the transporter, such that the overall interaction energy experienced by the Na+ ion was more or less maintained (see the inversions between the red and blue curves in Fig. S2†).
Sodium ions tend to leave their original binding site and bind Asp189 on H6
Sodium ions in both monomers exhibited similar dynamic features, as may be seen from the comparisons of Fig. 2, and Fig. S1 and S2†. The ability of Na+ ions to move away from their initial binding site in each monomer, their high tendency to visit alternative binding sites between H2, H6 and H8, and the strong preference for binding Asp189 were robust features reproduced in two independent runs (setups 1 and 3) and both monomers in each case. All four Na+ ions were invariably observed to leave their initial binding site at various times during the simulations. Coordination by Asp189 served as a highly preferred binding interaction (Fig. 2) in all independent runs even though Asp189 is not part of the initial binding site and requires a translation of Na+ by ~5 Å down the z-axis relative to its initial position.
In setup 3, the Na+ ion in monomer A, initially at a distance of ~13 Å from the Gal molecule in the same monomer was able to approach Gal within 3 Å in 5 ns, Na+ in monomer B moved within the 1st nanosecond to the close neighborhood of Asp189, where it remained coordinated by the carboxylic group of Asp189 in addition to Val185 backbone carbonyl for the remainder of the simulation. This tendency of Na+ to come and/or remain in close proximity of Gal was also seen in the SMD runs.
Gal remains bound but enjoys significant mobility
In contrast to Na+ which was spontaneously able to leave the binding site with no external force, Gal remained in the binding site at all times during runs 1 and 3. However, the position of Gal within the binding site was not fixed and large distance fluctuations were seen in the coordination of Gal by the groups located at the binding site,7 as shown in Fig. 4A and Fig. S3.† These fluctuations ran in parallel to changes in hydrogen bond formation patterns, exhibiting a versatility of interactions that Gal was able to form in the binding pocket (Table S3†) with enough space therein to rotate by about 90° (Fig. 4B). The hydrogen bonds seen in the crystal structure between Gal(C2–OH) and Asn69(Oε), Gal(C3–O/C2–O) and Lys294(Hζ), or Gal(C4–O) and Asn260(H–Nδ) were readily broken; whereas other hydrogen bonds, not previously observed, were formed and remained stable for extended periods during the simulations, such as those between Gal(C1–O) and Asn64(H–Nδ) and between Gal(C2–O) and Ser91(H–Oγ).
Fig. 4. Versatility of interactions of galactose with the transporter.
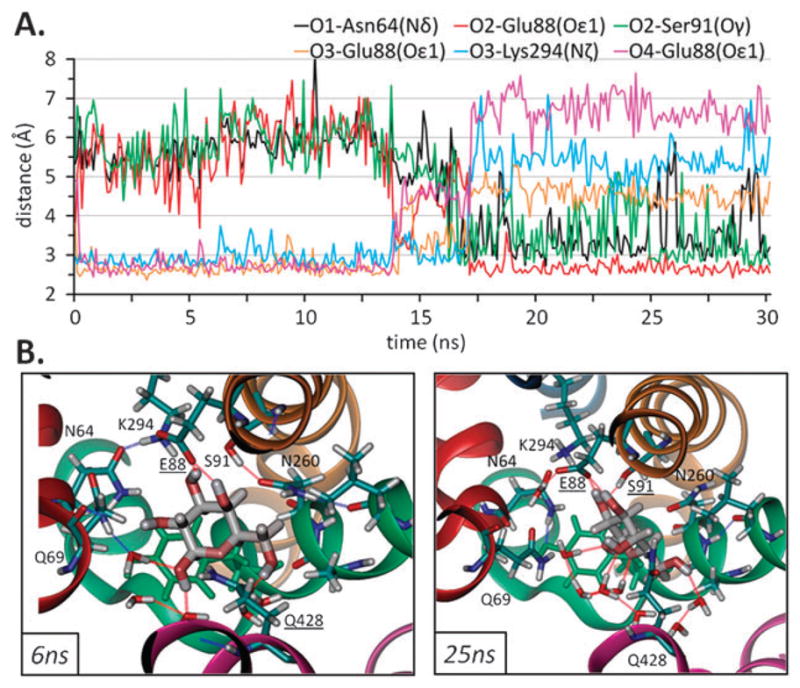
(A) Preferential interactions between Gal and monomer B in setup 1 (distances between non-H atoms are shown here). (B) Two snapshots of galactose in the binding pocket viewed from the EC side are shown here are for monomer B at 6 ns (left) and 25 ns (right) from setup 1. Residues within 3.5 Å from Gal are shown here together with residues (underlined) forming a H-bond (dashed lines). Different coloring styles for Gal (charge), the transporter residues (CPK) and Tyr263 (green) are used for clarity. Coloring of helices is as in Fig. 1.
Fig. 4 illustrates the conformational switch observed for Gal. Similar to the distance profiles observed for atoms near Na+, a number of polar groups appear to successively take turns in coordinating the Gal at all times (see the portion below 3 Å in panel A) and the same behavior was observed for both monomers (Fig. S3†). However, as in the case of LeuT, no significant interaction between Gal and Na+ ion, located at the Na2 position in LeuT, was observed in either monomer, provided the Na+ ion was within the confines of the initial binding site (Table S3†).
Overall, two charged residues Glu88 on H3 and Lys294 on H8 exhibit the strongest interactions that stabilize the Gal at its original binding site (Table S3.B†). These attractive interactions presumably prevented the translocation of Gal away from its binding site while allowing for rotational motions. Glu88 had a more pronounced effect (an average interaction of −28 kcal mol−1) due to the simultaneous formation of two H-bonds with Gal hydroxyls during a significant portion of the runs (Fig. 4). Other hydrogen bonds that were formed, albeit not as frequent and persistent as those with these two residues, are those with the side-chains of Asn64, Asn260 and Gln428. Occasional hydrogen bonds with Gln69, Ser91, Trp264 and the backbone of Ala259 also contributed, to a weaker extent, to Gal stabilization whereas Glu68 interacted with Gal unfavorably (Table S3.B†).
Access of water molecules to the Gal binding site
The side-chains of Met73, Tyr87 and Phe424 were observed to serve as the EC gate, continually preventing access of water molecules into the binding site (Fig. 1B). On the other hand, despite the presence and occasional obstruction of the aromatic side-group of Tyr263, water from the IC side was able to reach and form hydrogen bonds with Gal (Fig. 4). In fact, the total energy of interaction of Gal with the surroundings was on average ~−80 kcal mol−1, about 25% of which were contributed by water molecules (setups 1 and 3) (Fig. S4 and Table S3.B†).
Unbinding of galactose using CV-SMD
An important step in the transport cycle is the dissociation of the substrate from the binding site in the inward-facing conformation. The bulky side-chain of Tyr263 on H7, with the plane of its aromatic ring lying generally parallel to the ring of Gal located ~4 Å “above” it, would be a first barrier that inhibits downward movement of Gal. Over the course of 30 ns of free/unbiased MD, no dissociation of Gal from its binding site was observed. In order to accelerate this process, we performed constant-velocity SMD (CV-SMD) at 2 and 4 Å ns−1 using a soft-spring of 7.14 kcal/(mol Å2). Pulling forces were exerted to the mass center of Gal towards the cytoplasm along the negative z-axis (setups 4–8) or at an angle of~60° to the z-axis (9–11) (Fig. 1B). The latter direction might allow the extraction of Gal from its binding site with less force, and hence less energy, since the Gal molecule would not be pulled directly against the aromatic ring of Tyr263. The latter direction was also chosen to enable the passage through the IC water-filled cavity with apparently minimal obstruction by the protein residues.
The applied force peaked at around the same range (800–1000 pN; see Fig. 5) upon pulling along the z-axis compared with that at 60° to it (Fig. S5†). However, Gal moved about twice the distance down the z-axis in the former case (as expected from cos(60°) = 1/2); no detectable rise in the conformational energy of the side-chain or backbone of Tyr263 was observed prior to, during or after the exit of the Gal from its binding pocket during all the simulations (setups 4–11); and no significant difference in the overall translocation path and associated force-displacement profiles were observed by the application of two different pulling velocities (Fig. 5). Based on these observations, we focus here on the results from a representative run (setup 6). Fig. 6 illustrates snapshots from this run, corresponding to the positions indicated by the arrows in Fig. 5.
Fig. 5. Force-displacement profiles from CV-SMD runs.

Results are shown for two different pulling velocities, 2 and 4 Å ns−1, corresponding to setups 6 and 7, respectively. The force and displacement profiles are closely superimposable upon rescaling the time elapsed in run 6 (see the upper and lower abscissa) to enable a direct comparison of the Gal displacement and experienced resistance in the two runs. The arrows show the time (and position along the z-axis) of the snapshots shown in Fig. 6.
Fig. 6. Gal leaving the binding site.

Snapshots of Gal at~2.0, 2.7, 3.6, 4.0, 5.0 and 5.8 ns (A–F, respectively), taken from setup 6 and viewed all from the same perspective, through the plane of the membrane. Only residues within 3.5 Å of Gal are shown and colored according to the helices they are located on. Panel A is representative of the initial bound state, panel B is a high energy state where the Gal is dislodged from its binding site and obstructed by Y263, the rotation of which (panel C) enables the passage though. Close interaction with N267 is observed after that (panel C–F). Y269 appears to present a second barrier to translocation (panels E and F) before release to the cytoplasm (not shown).
Rotation of Tyr263 side-chain is required for the translocation of Gal to the cytoplasm
In the five simulations where Gal was pulled along the z-axis (setups 4–8), the largest resistance to displacement was observed at the dissociation of Gal from its initial binding pose, where it was closely coordinated by several polar and charged amino acids (see Fig. 6A). Once released from the strong electrostatic interactions with the residues therein, a lateral displacement away from H8 despite steering along the z-axis was observed (panel B), succeeded by rotameric transition of the side-chain of Tyr263 to allow the movement of Gal towards the IC cavity. Interactions with other H7 residues (Y262, N267, Q268, Y269 and R273) were observed at succeeding phases of translocation inviting attention to the role of these polar and charged residues in modulating the translocation process. A second obstacle to translocation was Y269 side chain, which may be viewed as a second (outer) IC gate, in addition to the inner gate (Y263). Notably, in four out of five independent simulations the unbinding pathway was mostly similar; Gal crossed the Tyr263 side-chain along the side of H4, 9 and 11, in addition to H7.
Discussion
Our data (summarized in Table 1) show that the obtained crystal structure does not represent a tightly bound state of Na+ or a fixed geometry of Gal in the inward-facing conformation of the transporter. This is not due to instability in the structure of the transporter, as shown by the RMSD values obtained but rather mostly due to the nature of the binding pocket itself in this conformation. In addition, no decrease in backbone stability was observed in the absence of Gal and Na+, in contrast to that seen with Leu/Na+ in LeuT.22
Table 1.
Summary of the results from present simulations of galactose and sodium motions in the crystallized inward-facing open state of vSGLT
| Observations | Sodium | Galactose |
|---|---|---|
| Stability in the binding site | Unstable, can leave the binding site completely | Stable, can rotate within, but not leave, the binding site |
| Interactions within the binding site | Versatile, alternating | Versatile, alternating |
| Total interaction energy with the monomera | ~110 kcal mol−1 | ~60 kcal mol−1 |
| Amino acids exhibiting strongest interaction with the liganda | Asp189 | Glu88 |
| Accessibility to watera (% of total interaction energy) | ~40% | ~25% |
| Key amino acids on intracellular exit routeb | Exit facilitated by Asp189 | May include interactions with N267, S372, Y269 and R273 |
See Table S2.B for Na+ and Table S3.B for Gal.
See Fig. 2, 3 and ESI Fig. S1 for Na+ and Fig. 6 for Gal.
The Na+ ion in this state is not optimally coordinated by the carbonyl groups of Ala62 and Ile65 on H2, Ala361 on H9 and the hydroxyl groups of Ser364 and Ser365 on H9. In fact, a Na+ ion placed in this site, equivalent to Na2 in LeuT, would be unable to maintain any constant position or fixed coordination with the interacting groups in the transporter. Furthermore, the Na+ ion is constantly and significantly ligated by several water molecules, even within the confines of the initial binding site. The marked mobility of Na+ and the ability to leave this site altogether help explain why neither Na+ nor other cations could be observed therein upon the elucidation of the crystal structure.7
The side-chain of Gln192, located on H6 is within H-bonding distance from the hydroxyl of Ser364 and is often seen to form a H-bond with it. Also, its side-chain interaction energy with sodium is not less than that of Ser364 or Ser365. Approximately one helical turn below Gln192 lies Asp189. In most cases, sodium was observed to leave the proposed binding-site into the cavity approximately 5 Å below towards the cytoplasm to be bound by the carboxyl group(s) of Asp189. Interestingly, both residues are fully conserved in hSGLT1, NIS and PutP indicating that they may play a common role among the members of the SSS in the coordination and release of the Na+ ion from its binding site into the IC space.
Galactose is sandwiched between two hydrophobic gates and is not completely shielded from the IC medium by Tyr263 or other residues in the binding pocket. In fact, the binding pocket itself is sufficiently large to accommodate several water molecules simultaneously resulting in water binding to Gal comprising ~ 25% of the total coordination energy. In LeuT, no accessibility of the bound Leu or Na+ to EC water, or ability of the Na+ ion to leave the binding site was reported. In addition, Leu is tightly bound to LeuT.22,23 This is due to the shape of the binding site and the conformation in LeuT which allow better geometric coordination and more occlusion from the aqueous cavities in their binding sites. Another prominent difference is probably the nature of the substrate itself since Gal has five hydroxyl groups all capable of forming hydrogen bonds whereas Leu has two oppositely charged moieties that limit its possible interactions with the transporter and strengthen the binding. Thus, Gal in the crystallized inward-facing “occluded” state of vSGLT is much less occluded from the internal aqueous medium than the Leu/Na+ from the external medium in the outward-facing occluded state of LeuT.
Our simulations do not necessarily reflect all possible interactions between Gal and the transporter in this conformation since longer simulations may reveal additional ones. Our simulations do, however, show that more than one conformation of Gal in the binding pocket is possible and this can help explain the ability of vSGLT, and probably SGLT1, to bind and transport several types of monosaccharides.17,18
In the SMD simulations, the RMSD of the backbone of the whole monomer or that of H7 which contains the inner hydrophobic gate were not detectably affected by the pulling of the Gal from its binding site indicating that no significant conformational changes are needed for the substrate to leave the binding site in this state. After disrupting the strong interactions between the transporter and Gal at the binding site, rotation of the side chain of Tyr263 appears to be necessary to provide an exit route for the substrate. The ability of Gal to rotate in the binding pocket and for water molecules to enter therein may accelerate the dissociation step from the binding site in this conformation. Since the turnover rate of galactose per vSGLT molecule has been estimated to be ~0.4 s−1,18 conformational changes in the structure of the symporter relevant to the transport cycle, with or without the bound galactose/Na+, cannot be expected to take place within the timescale of MD simulations as in this study. Therefore, even if Gal can leave the binding pocket with relatively low force, this does not rule out additional changes in the conformations of both the protein and the substrate which may take place increasing the exposure of Gal to the IC cavity even more.
In contrast to the simulations where Gal was pulled along the z-axis, those at an angle of 60° did not provide a route with lower energy barrier to exit the binding site. The observed pathway, at least as far as crossing Tyr263 goes, therefore seems more likely. Nevertheless, other pathways for Gal to leave the transporter cannot be excluded and determination of the most probable pathway, with the least energy required, to completely exit the IC cavity of the transporter will require pulling Gal at variable angles. The exerted pulling forces are comparable in magnitude to those (ca. 200–2,000 pN) adopted in comparable SMD simulations.22 Even though pulling galactose at lower velocities may turn out to require smaller, and thus more physiologically relevant, forces, accelerating the dissociation step as in our simulations is required in order to visualize an exit pathway within a reasonable timescale.
The crystal structure of an amino acid, polyamine and organocation transporter (ApcT) has been recently obtained and its structure shows similar overall architecture of the transport core as vSGLT and LeuT. The ApcT structure has also been obtained in the inward-facing conformation.8 However, the ApcT is a proton-coupled transporter, with a lysine seen in the position equivalent to Na2 in LeuT, that has been crystallized in the apo-form.8 Thus, vSGLT is the optimal system available so far for the study of the dynamics of the substrate and Na+ in the inward-facing conformation of this large structural family.
Note added in proof
A paper by Li and Tajkhorshid24 has been recently published also showing that Na+ placed in the putative sodium binding site in vSGLT can escape into the intracellular medium, facilitated by Asp189 and with no external force applied.
Methods
System preparation
The ~3.0 Å resolution crystal structure of vSGLT7 (accession code 3DH4) was obtained from the Protein Data Bank.25 The monomeric unit contains residues 1 to 19, 47 to 178 and 185 to 543. Residues 1–19 were eliminated from the structure. Residues 179 to 184, located in the IC loop between H5 and H6, were constructed using Sybyl 8.0 (Tripos, Inc., St. Louis, MO) by first introducing a stretch of six glycines to complete the backbone and then mutating each residue to its equivalent in vSGLT. Minimization was performed after each step. The erbium(III) ions were removed and the final monomeric structure contained residues 47 to 543 in addition to one Gal and one Na+ ion. Missing atoms from side-chains and hydrogen atoms were constructed using the psfgen plugin of VMD26 employing the CHARMM32 topology for proteins, lipids and sugars.27,28 The dimer was then placed in a POPE membrane bilayer of 150 × 115 Å. Lipid molecules overlapping with the protein were removed and the system was solvated in 150 mM NaCl. Several steps of minimization and MD simulations (total of ~400 ps) followed to ensure that the protein was optimally placed in the membrane with almost no water in between the protein and lipid. A 1 ns equilibration step was finally performed with the protein backbone + ligand + Na+ ions constrained. Minimization of the solvated vSGLT dimer placed in the membrane yielded almost the same backbone structure as in the crystal. The equilibrated system was then subjected to the various simulations described in Table S1.†
Simulation details
All simulations in this study were performed with the NAMD 2.7b129 using CHARMM32 force field for proteins, lipids and sugars28,30 with the CMAP corrections.27 All simulations were carried out under periodic boundary conditions in NPT at 310 K with a time step of 1 fs. Particle Mesh Ewald algorithm was used for calculating long-range electrostatic interactions. Constant temperature was achieved by employing Langevin dynamics with a damping constant of 2/ps. The Langevin Piston method was employed to maintain a constant pressure of 1 atm with a piston period of 100 fs and a piston decay of 50 fs. A cut-off distance of 10 Å was used for VdW interactions with a switching function of 8 Å.
Coordinates for analysis were saved every 10 ps in MD simulations (setups 1–3) and 2 ps in SMD simulations (setups 4–11). Setups 1 and 2 were performed with the constraint-free dimer whereas setups 3–11 included the dimer with harmonic-constraints on the α-carbon atoms of residues 171, 462 and 534 (on helices 5, 12 and 14, respectively). These positions were chosen for being located at approximately the middle of the helices and on outer helices that they were less likely to inhibit changes in the transport core upon pulling the galactose.
Data analysis
Analysis of the crystal structure and performed simulations was carried out using VMD 1.8.626 and the related Tcl-scripts. Molecular images and energy and RMSD calculations were made using the NAMD-energy plugin of VMD.26
Supplementary Material
Acknowledgments
Partial support from NIH grant 1R01GM086238-01 is gratefully acknowledged by IB. We thank Ahmet Bakan for his assistance with the VMD/NAMD software.
Footnotes
Electronic supplementary information (ESI) available: Supplementary figures and tables. See DOI: 10.1039/b927492h
Contributor Information
Elia Zomot, Email: enz1@pitt.edu.
Ivet Bahar, Email: bahar@pitt.edu.
References
- 1.Kanner BI. Bioenergetics of neurotransmitter transport. Biochim Biophys Acta. 1983;726:293–316. doi: 10.1016/0304-4173(83)90013-7. [DOI] [PubMed] [Google Scholar]
- 2.Kanner BI, Schuldiner S. Mechanism of transport and storage of neurotransmitters. Crit Rev Biochem Mol Biol. 1987;22:1–38. doi: 10.3109/10409238709082546. [DOI] [PubMed] [Google Scholar]
- 3.Wright EM, Loo DD, Hirayama BA, Turk E. Surprising versatility of Na+-glucose cotransporters: SLC5. Physiology (Bethesda) 2004;19:370–376. doi: 10.1152/physiol.00026.2004. [DOI] [PubMed] [Google Scholar]
- 4.Leung DW, Loo DD, Hirayama BA, Zeuthen T, Wright EM. Urea transport by cotransporters. J Physiol. 2000;528(2):251–257. doi: 10.1111/j.1469-7793.2000.00251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coady MJ, Chang MH, Charron FM, Plata C, Wallendor B, Sah JF, Markowitz SD, Romero MF, Lapointe JY. The human tumour suppressor gene SLC5A8 expresses a Na+-mono-carboxylate cotransporter. J Physiol. 2004;557:719–731. doi: 10.1113/jphysiol.2004.063859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li H, Myero L, Smiraglia D, Romero MF, Pretlow TP, Kasturi L, Lutterbaugh J, Rerko RM, Casey G, Issa JP, Willis J, Willson JKV, Plass C, Markowitz SD. SLC5A8 a sodium transporter is a tumor suppressor gene silenced by methylation in human colon aberrant crypt foci and cancers. Proc Natl Acad Sci U S A. 2003;100:8412–8417. doi: 10.1073/pnas.1430846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Faham S, Watanabe A, Besserer GM, Cascio D, Specht A, Hirayama BA, Wright EM, Abramson J. The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na+/sugar symport. Science. 2008;321:810–814. doi: 10.1126/science.1160406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shaffer PL, Goehring A, Shankaranarayanan A, Gouaux E. Structure and mechanism of a Na+-independent amino acid transporter. Science. 2009;325:1010–1014. doi: 10.1126/science.1176088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh SK, Piscitelli CL, Yamashita A, Gouaux E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science. 2008;322:1655–1661. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weyand S, Shimamura T, Yajima S, Suzuki S, Mirza O, Krusong K, Carpenter EP, Rutherford NG, Hadden JM, O’Reilly J, Ma P, Saidijam M, Patching SG, Hope RJ, Norbertczak HT, Roach PCJ, Iwata S, Henderson PJF, Cameron AD. Structure and molecular mechanism of a nucleobase-cation-symport-1 family transporter. Science. 2008;322:709–713. doi: 10.1126/science.1164440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl–dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 12.Gao X, Lu F, Zhou L, Dang S, Sun L, Li X, Wang J, Shi Y. Structure and mechanism of an amino acid antiporter. Science. 2009;324:1565–1568. doi: 10.1126/science.1173654. [DOI] [PubMed] [Google Scholar]
- 13.Ressl S, Terwisscha van Scheltinga AC, Vonrhein C, Ott V, Ziegler C. Molecular basis of transport and regulation in the Na(+)/betaine symporter BetP. Nature. 2009;458:47–52. doi: 10.1038/nature07819. [DOI] [PubMed] [Google Scholar]
- 14.Singh SK, Piscitelli CL, Yamashita A, Gouaux E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science. 2008;322:1655–1661. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krishnamurthy H, Piscitelli CL, Gouaux E. Unlocking the molecular secrets of sodium-coupled transporters. Nature. 2009;459:347–355. doi: 10.1038/nature08143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ikeda TS, Hwang ES, Coady MJ, Hirayama BA, Hediger MA, Wright EM. Characterization of a Na+/glucose cotransporter cloned from rabbit small intestine. J Membr Biol. 1989;110:87–95. doi: 10.1007/BF01870995. [DOI] [PubMed] [Google Scholar]
- 17.Xie Z, Turk E, Wright EM. Characterization of the Vibrio parahaemolyticus Na+/Glucose cotransporter. A bacterial member of the sodium/glucose transporter (SGLT) family. J Biol Chem. 2000;275:25959–25964. doi: 10.1074/jbc.M002687200. [DOI] [PubMed] [Google Scholar]
- 18.Turk E, Kim O, le CJ, Whitelegge JP, Eskandari S, Lam JT, Kreman M, Zampighi G, Faull KF, Wright EM. Molecular characterization of Vibrio parahaemolyticus vSGLT: a model for sodium-coupled sugar cotransporters. J Biol Chem. 2000;275:25711–25716. doi: 10.1074/jbc.M003127200. [DOI] [PubMed] [Google Scholar]
- 19.Izrailev S, Stepaniants S, Balsera M, Oono Y, Schulten K. Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys J. 1997;72:1568–1581. doi: 10.1016/S0006-3495(97)78804-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kosztin D, Izrailev S, Schulten K. Unbinding of retinoic acid from its receptor studied by steered molecular dynamics. Biophys J. 1999;76:188–197. doi: 10.1016/S0006-3495(99)77188-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eskandari S, Wright EM, Kreman M, Starace DM, Zampighi GA. Structural analysis of cloned plasma membrane proteins by freeze-fracture electron microscopy. Proc Natl Acad Sci U S A. 1998;95:11235–11240. doi: 10.1073/pnas.95.19.11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Celik L, Schiott B, Tajkhorshid E. Substrate binding and formation of an occluded state in the leucine transporter. Biophys J. 2008;94:1600–1612. doi: 10.1529/biophysj.107.117580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caplan DA, Subbotina JO, Noskov SY. Molecularmechanism of ion–ion and ion–substrate coupling in the Na+-dependent leucine transporter LeuT. Biophys J. 2008;95:4613–4621. doi: 10.1529/biophysj.108.139741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li J, Tajkhorshid E. Ion-releasing state of a secondary membrane transporter. Biophys J. 2009;97:L29–L31. doi: 10.1016/j.bpj.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 27.Mackerell AD, Jr, Feig M, Brooks CL., III Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 28.Mackerell AD., Jr Empirical force fields for biological macromolecules: overview and issues. J Comput Chem. 2004;25:1584–1604. doi: 10.1002/jcc.20082. [DOI] [PubMed] [Google Scholar]
- 29.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brooks BR, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.