

Abstract
Histone acetylation is a key modification that regulates chromatin accessibility. Here we show that treatment with butyrate or other histone deacetylase (HDAC) inhibitors does not induce histone hyperacetylation in metaphase-arrested HeLa cells. When compared to similarly treated interphase cells, acetylation levels are significantly decreased in all four core histones and at all individual sites examined. However, the extent of the decrease varies, ranging from only slight reduction at H3K23 and H4K12 to no acetylation at H3K27 and barely detectable acetylation at H4K16. Our results show that the bulk effect is not due to increased or butyrate-insensitive HDAC activity, though these factors may play a role with some individual sites. We conclude that the lack of histone acetylation during mitosis is primarily due to changes in histone acetyltransferases (HATs) or changes in chromatin. The effects of protein phosphatase inhibitors on histone acetylation in cell lysates suggest that the reduced ability of histones to become acetylated in mitotic cells depends on protein phosphorylation.
Keywords: Histone acetylation, mitosis, HDAC inhibitors, butyrate, metaphase chromosomes, protein phosphatases
Introduction
Histones are the most abundant proteins associated with DNA in eukaryotic chromosomes. The core histones H2A, H2B, H3 and H4 wind the DNA into bead-like nucleosome cores [1, 2], while histone H1 organizes the “string of beads” into a 30 nm chromatin fiber [3, 4]. In addition to packaging the DNA and neutralizing some of its negative charge, the histones also play roles in DNA replication, repair and transcription.
Histone function is modulated by several different post-translational modifications, one of which is the reversible acetylation of lysine residues in the N-terminal tails of the four core histones. Histone acetylation is a dynamic process involving a balance between histone acetyltransferase (HAT) and histone deacetylase (HDAC) activities, and it is important because it correlates with gene transcription or readiness for transcription [5-11].
Normally only a small percentage of core histone molecules is more than monoacetylated. However, histone acetyl groups turn over rapidly [12]. In cultured cell lines, 70% of the acetate incorporated into histones in a short pulse turns over with a half-life of about 3 min, and the rest with a half-life of 30-40 min [12]. As a result, treatment of interphase cells for a few hours with HDAC inhibitors such as sodium butyrate, valproic acid, trichostatin A (TSA), apicidin or oxamflatin leads to significant histone hyperacetylation [13-20]. Evidence indicates that butyrate and TSA are competitive inhibitors of HDACs [21-22], and based on structural similarity to butyrate or acetyllysine, apicidin [23] and valproate probably are also. The mode of inhibition by oxamflatin has not been reported.
Histone acetylation levels are lower during mitosis than during interphase in mammalian cells [24-33] and in Physarum polycephalum [34, 35]. Histone acetylation is also decreased during the later stages of meiosis in Lilium microsporocytes [36] and in mouse oocytes [37]. The reason for the decrease in histone acetylation at mitosis is not known. However, it could be related to the cessation of transcription during mitosis in higher eukaryotes (e.g., [38-41]), either as a cause or a consequence.
In the work reported here, we have explored the possible reasons for underacetylation of histones at mitosis by treating metaphase-arrested HeLa cells with HDAC inhibitors. We find that this treatment results in little or no increase in core histone acetylation. Since the effect is seen in bulk chromatin, it is not due merely to the cessation of transcription. Our results suggest that the causes may be complex, but that the phenomenon reflects reduced turnover of histone acetates in mitotic cells and decreased ability of HATs to act on histones in mitotic chromatin. In vitro experiments suggest that decreased histone acetylation at mitosis is dependent on mitosis-specific protein phosphorylation of an as-yet unknown target.
Materials and Methods
Chemicals, Media and Antibodies
Microcystin LR was dissolved at 1 mM in 50 mM Tris-Cl pH 7.0 and stored in aliquots at −20°C. Calyculin A was prepared as a 100 μM solution in methanol and stored at 2°C. Cantharidin was prepared as a 200 mM solution in N,N-dimethylformamide (DMF) and stored at 2EC. Sodium butyrate was made as a 5 M stock solution in 0.9% NaCl and 20 mM sodium phosphate and adjusted to pH 7.4. Trichostatin A (TSA), oxamflatin and apicidin were prepared as 1 mg/mL solutions in dimethylsulfoxide (DMSO) and stored at −20°C. Media and components were obtained from Gibco or Sigma. All other reagents were obtained from Sigma unless otherwise noted.
Some of the antibodies recognizing specific core histone acetylations were gifts from Dr. Hiroshi Kimura (Osaka) or Dr. Maria Vogelauer (Edinburgh). Antisera from the Turner laboratory were prepared and characterized as described by Turner and Fellows [29] and White et al. [42]. The following were used (all rabbit polyclonal antibodies unless otherwise noted): anti-H2AK5ac (Turner, R123); anti-H2BK12ac/K15ac (Turner, R209); anti-H3K9ac (Upstate, 07-352), anti-H3K18ac (Upstate, 07-354); anti-H3K23ac (Upstate, 07-355); anti-H3K27ac (H. Kimura, 309, mouse monoclonal [43]); anti-H4K8ac (Upstate, 07-328); anti-H4K8ac (Turner, R403); anti-H4K12ac (Upstate, 07-595); anti-H4K12ac (Upstate, 06-761); anti-H4K16ac (Turner, R251); and anti-pan-H4, loading control (Upstate, 05-858). Note that Upstate 07-354 has been found to react with both H3K18ac and H3K14ac (M. Vogelauer, personal communication), and R209 requires either H2BK12 or H2BK15 to be acetylated, or both.
Cell Culture and Metaphase-Arrest
All biochemical experiments used suspension cultures of either H-HeLa [44] or HeLa S3. H-HeLa cells were grown in Eagle's MEM as previously described [45]. HeLa S3 cells were grown in RPMI-1640 supplemented with penicillin/streptomycin and 10% fetal bovine serum and diluted daily to 2.0 – 2.5 H 105/mL. For metaphase arrest, cells were first synchronized with thymidine [46] and then arrested with nocodazole as described previously [45]. Mitotic indices were typically 80-95% for H-HeLa and 95-98% for HeLa S3. In no case were any differences in results observed between the two strains.
Treatment with HDAC Inhibitors; Isolation of Mitotic Chromosomes and Interphase Nuclei
Cell cultures with 2 – 4 H 105 cells/mL were typically treated with 10 mM sodium butyrate, 1.0 Φg/mL trichostatin A, 2.0 Φg/mL apicidin, or 2.0 Φg/mL oxamflatin. For most experiments, cells were placed on ice immediately at the end of the treatment period and metaphase chromosome clusters were isolated as previously described [47, 48]. Lysis Buffer (LB) consisted of 10 mM Na+-Hepes, pH 7.4, 10 mM NaCl, 5 mM MgCl2, 0.5 M sucrose and 0.1% NP40, and Resuspension Buffer (RB) had the same composition but without sucrose. The lysate was subjected to 6 strokes in a glass-glass Dounce homogenizer with a tight fitting pestle (Wheaton Glass) and the chromosome clusters were pelleted through a layer consisting of RB plus 1.2 M sucrose. Crude interphase nuclei were isolated similarly, except using 15 – 20 strokes of the Dounce homogenizer and omitting the 1.2 M sucrose layer. Lysis solutions contained 2 mM p-chloromercuriphenyl sulfonate (PCMPS) or 2 mM p-hydroxymercuribenzoate (PHMB) to block histone dephosphorylation [49] and 10 mM butyrate or 1 μg/mL TSA to prevent histone deacetylation.
For the experiment shown in Fig. 5, cells were released from a thymidine block and after 4 hrs both nocodazole and 10 mM butyrate were added. At 16 hrs after release from thymidine, cells (mitotic index 60 – 70%) were harvested. Individual metaphase chromosomes were isolated by the aqueous method of Marsden and Laemmli [50] and purified away from nuclei by differential centrifugation. For comparison, nuclei were isolated from interphase (unsynchronized) cells that had been treated with 10 mM butyrate for 12 hrs.
Fig. 5.
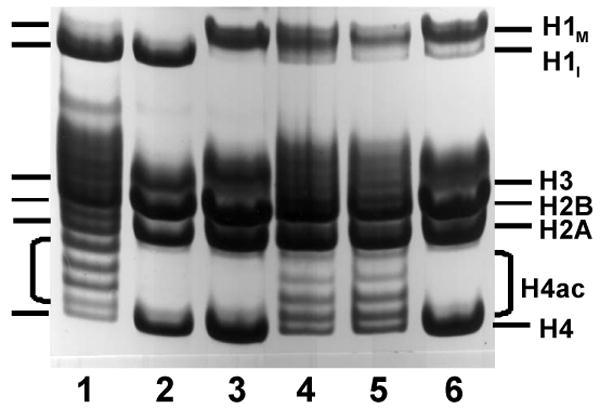
Treatment of synchronized cells with butyrate during entry into metaphase-arrest allows isolation of mitotic chromosomes containing hyperacetylated histones. Histones were extracted from the following: Lane 1, interphase nuclei isolated from butyrate-treated cells; lane 2, interphase nuclei isolated from untreated cells; lane 3, metaphase chromosomes isolated from untreated cells (note the shift in position of histone H1 due to its phosphorylation at mitosis); lanes 4 and 5, two separate preparations of metaphase chromosomes isolated from cells treated with butyrate starting in mid-S-phase and continuing until they arrested in mitosis (as in Fig. 3, lane 5); lane 6, a mixture of the samples shown in lanes 1 and 3, to give approximately the same ratio of H1M to H1I (and therefore the same amount of interphase contamination) as seen in lanes 4 and 5. Acid-extracted histones were analyzed by acid-urea polyacrylamide gel electrophoresis. Only the portion of the gel containing the histones is shown.
Light microscopy: Preparation of chromosome spreads and immunostaining
For immunofluorescence microscopy, metaphase chromosome spreads were prepared from human female lymphoblastoid cells grown in RPMI 1640 medium plus 10% fetal bovine serum. Cells in exponential growth were treated for 6 hrs with 1 mM sodium valproate, including the final 2 hrs with 0.1 μg/mL colcemid (KaryoMax, Gibco). Cells were then pelleted by centrifugation, washed twice with cold phosphate buffered saline (PBS), resuspended in 75mM KCl at 2×105 cells/ml and left at room temperature for 10 min. 200μL aliquots of the swollen cell suspension were spun onto glass slides at 1800 rpm for 10min in a Shandon Cytospin 4. Slides were then immersed for 10 min at room temperature in KCM buffer (120mM KCl, 20mM NaCl, 10mM Tris/HCl pH 8.0, 0.5mM EDTA, 0.1% Triton X-100). Immunolabelling was carried out for 1hr at 4°C as described previously [51] with antisera diluted 200-400 fold in KCM supplemented with 1-1.5% BSA (Sigma). For all labelings, the secondary antibody was FITC-conjugated goat anti-rabbit immunoglobulin (Sigma F1262) diluted 150-fold in KCM, 1% BSA. Slides were washed twice in KCM (5 min at room temperature), fixed in 4% v/v formaldehyde (10 min at room temperature), rinsed in deionised water and mounted in Vector Shield (Vector Lab) supplemented with DAPI (Sigma) at 2μg/ml.
Observation of HAT Activity in Lysates
HeLa cells were pelleted, washed twice with saline, and lysed at 1.0 H 107 cells/mL in HAT buffer consisting of 50 mM Tris-Cl pH 8.0, 50 mM KCl, 0.1 mM EDTA, 5% glycerol, 1 mM phenyl methyl sulfonylfluoride (PMSF; from a stock solution 0.5 M in 2-propanol), 10 mM sodium butyrate, 1 mM dithiothreitol, 1 mM acetyl coenzyme A and 0.2% Nonidet P40. The protein phosphatase inhibitor cantharidin [52] was used at a final concentration of 1 mM. DMF was evaporated with a gentle stream of air and the cantharidin redissolved in the appropriate volume of HAT buffer. Lysates were incubated for 2 hrs at 37°C with agitation to keep chromosomes or nuclei in suspension. Chromosomes or nuclei were then pelleted and histones extracted.
Histone Extraction, Polyacrylamide Gel Electrophoresis, and Detection
Histones were extracted from pelleted chromosomes or nuclei with 0.2 M H2SO4 as previously described [49]. For SDS-polyacrylamide gels, sample preparation and electrophoresis were carried out as described by Laemmli and Favre [53]. All gels contained 15% acrylamide and 0.4% N,N′-methylene-bis-acrylamide. For 3H-labelling experiments (e.g., Fig. 2), 16 cm long gels were used. For immunoblotting experiments (e.g., Fig. 3), 6 cm long mini-gels were used. For acid-urea gel electrophoresis [54], samples of acid-extracted histones were dried in 1.5 mL microcentrifuge tubes using a CentriVap (Labconco) and redissolved in 3 – 6 μL of sample buffer for loading. 16 cm long gels containing 15% acrylamide, 0.1% N,N′-methylene-bis-acrylamide, 2.5 M urea and 5.4% acetic acid were pre-run as described in [55] and then run at 300 V for 5 – 6 hrs until the blue component of the methyl green marker reached the bottom of the gel.
Fig. 2.
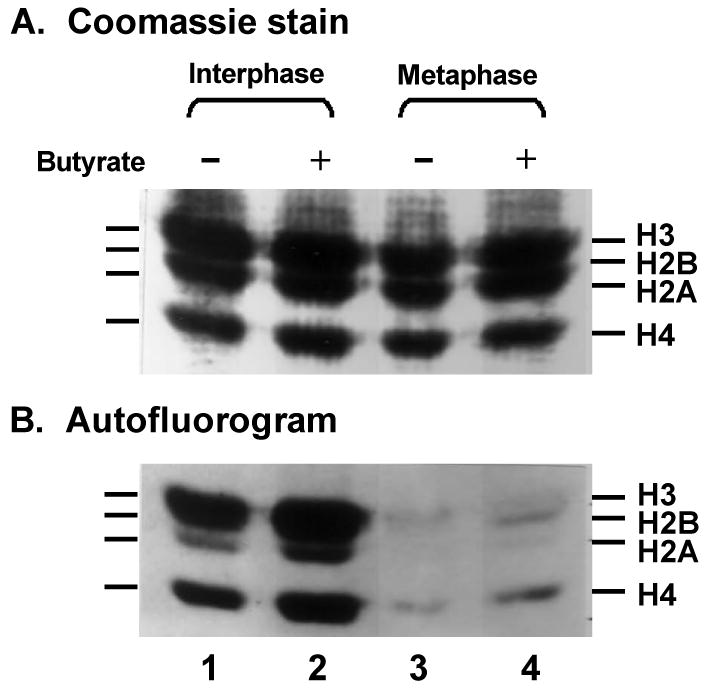
Sodium butyrate does not induce hyperacetylation of any of the four core histones in metaphase-arrested HeLa cells. Interphase and metaphase-arrested cultures were grown under identical conditions in the presence of [3H]-acetate for 1 hr, with or without the addition of 10 mM sodium butyrate. Histones were extracted and separated by SDS-polyacrylamide gel electrophoresis; (A) Coomassie blue stain; (B) autofluorography to detect incorporated [3H]-acetate. Only the portion of the gel containing the core histones is shown. Note that although H3 and H2B are not resolved from one another on this gel, it is clear that neither of them is highly labeled in metaphase cells, even in the presence of butyrate. Incorporation of [3H]-acetate into acetyl coenzyme A pools was comparable in all cultures.
Fig. 3.
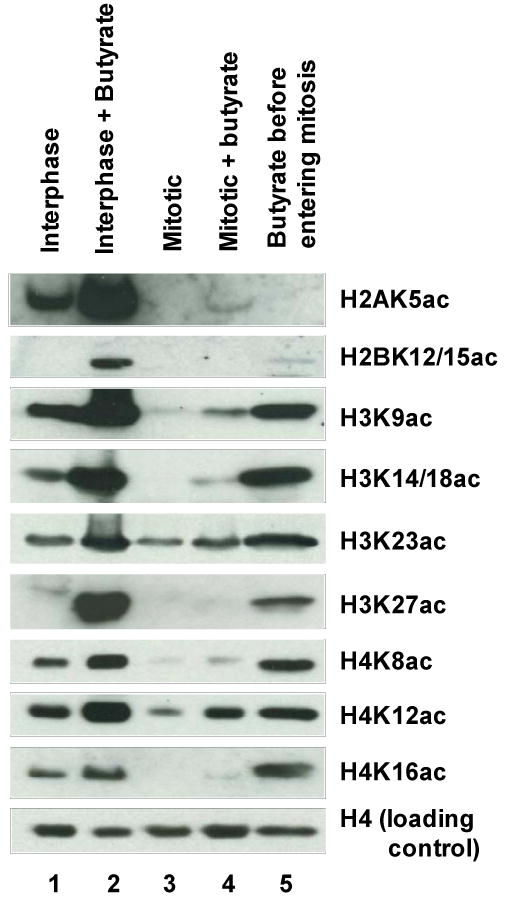
Levels of acetylation at specific sites in core histones from mitotic and interphase cells, with and without butyrate treatment. Histones were separated on SDS-polyacrylamide gels and acetylation was detected by immunoblotting with specific anti-acetylhistone antibodies. Gels run with the same samples and the same loadings and stained with Coomassie blue or probed with anti-pan histone H4 as a loading control indicated that the five samples contained essentially the same amounts of each of the four core histones. For the samples in lanes 1-4, cultures were treated for 4 hrs with or without 10 mM sodium butyrate and histones were extracted from isolated chromosome clusters or nuclei: lane 1, interphase, untreated; lane 2, interphase, treated with butyrate; lane 3, metaphase-arrested, untreated; lane 4, metaphase-arrested, treated with butyrate. The metaphase-arrested culture (lanes 3 and 4) had a mitotic index of 97% and showed no detectable interphase histone H1 (H1I) on acid-urea gels. For the sample in lane 5, a culture synchronized in mid-S-phase was treated with 10 mM butyrate for 12 hrs until most of the cells had arrested in metaphase. Individual chromosomes were then isolated and histones extracted. On an acid-urea gel, the interphase form of histone H1 (H1I) was undetectable, demonstrating that this sample was free of contamination by interphase histones.
Gels were stained with 0.1% Coomassie Brilliant Blue R250 (BioRad) in 50% methanol, 10% acetic acid and destained in 5% methanol, 10% acetic acid. For detection of 3H-labelled proteins, Coomassie-stained gels were soaked in water to remove methanol and acetic acid, soaked in Fluoro-Hance (Research Products International) for at least 30 min, and finally dried on Whatman 3MM CHR filter paper (which was first wetted in Fluoro-Hance) in a vacuum gel drier. Autofluorography was carried out using Fuji RX Medical X-ray film that had been sensitized by pre-exposure to an even, low-intensity flash of light [56].
For immunoblotting, proteins were electrophoretically transferred to Hybond ECL nitrocellulose membranes (Amersham Biosciences) in a BioRad Miniprotean apparatus. Membranes were blocked with 5% milk in phosphate-buffered saline (PBS), incubated with the primary antibody (typically 1:500 to 1:1000) in 5% milk in PBS, washed 3 times in PBS, and incubated with the appropriate HRP-conjugated secondary antibody in 3% milk in PBS. Bands were detected by chemiluminescence (ECL, GE Health Sciences).
[3H]-Acetate Labeling of Histones In Vivo
The procedure was modified from that of Cousens et al. [16]. For a given experiment, to insure that the concentrations of metaphase and interphase chromatin were equal [57], cells from 50 mL of each culture were pelleted, washed twice with cold 0.9% NaCl and lysed in 10 mL RB. A 100 ΦL aliquot of this lysate was diluted to 5 mL with 1% aqueous SDS, mixed well and its absorbance measured at 260 and 280 nm. Based on these measurements, metaphase-arrested and interphase cultures were adjusted to the same concentration of chromatin before adding [3H]-acetate.
The total amount of [3H]-acetate (DuPont NEN, 25 mCi/mL in ethanol) to be used in an experiment was placed in a single flask, the solvent evaporated with a gentle stream of air, and the [3H]-acetate redissolved in warm medium. Metaphase and interphase cultures were then treated at the same time with the same amount of [3H]-acetate and incubated identically at 37EC with magnetic stirring. Incorporation was stopped by adding two volumes of ice cold 0.9% NaCl and placing the cultures on ice for 15 min. Cells were washed twice in cold 0.9% NaCl, nuclei or chromosome clusters were isolated, and finally histones were extracted with 0.2 M H2SO4.
Lysate supernatants were saved and used to compare the relative specific activity of acetyl coenzyme A in the metaphase and interphase cells by HPLC using a modification of the method of Hosokawa et al. [58]. Samples were centrifuged at 14,000 rpm for 2 min in a microcentrifuge to remove large particulates. Next, HCl was added to a final concentration of 0.2 N and the samples again centrifuged. The remaining samples were dried using a CentriVap, redissolved in 0.2 M ammonium acetate, and fractionated using a Shimadzu LC-10AS HPLC with SPD-10A UV-Visible detector, a 25 cm C-18 column (Alltech) and a flow rate of 1 mL/min. During the first 10 min, the solvent consisted of 2% acetonitrile and 98% 0.2 M ammonium acetate. During the next 15 min, the acetonitrile concentration rose linearly to 30%. Fractions were collected and 3H determined by scintillation counting. Acetyl coenzyme A (Sigma) and [3H]-acetyl coenzyme A (DuPont NEN) eluted at 20.5 min.
Histone Deacetylase Assay
The assay was modified from [16]. Substrate histones were labeled in vivo with [3H]-acetate and extracted with 0.2 M H2SO4 as described above, redissolved in 1 mM HCl, dried in aliquots using the CentriVap, and stored at !20EC. Fluorography of SDS gels showed that core histones were the only 3H-labeled proteins present.
For the assay, labeled histones (containing approximately 1000 cpm/Φg) were dissolved at 1 mg/mL in RB, and interphase and metaphase cells were lysed at equivalent concentrations (see above) in LB. For the reaction, prewarmed substrate histone solution (20 ΦL) and crude cell lysate (20 ΦL) were mixed and incubated at 37EC. The reaction was stopped by adding 20 ΦL carrier histones (calf thymus histone type II-AS, Sigma, dissolved at 1 mg/mL in 0.1 M acetic acid and 0.4 N HCl) and 40 ΦL of a saturated solution of Reinicke salt (ammonium tetrathiocyanodiammonochromate). Samples were mixed well, placed on ice for at least 30 min and then centrifuged for 5 min at full speed in a microcentrifuge to pellet the 3H-labeled histones. The [3H]-acetate released by deacetylase action was determined in 75 ΦL of the supernatant by scintillation counting.
Typically the amount of [3H]-acetate released was followed as a function of time. As a control, a baseline was determined with LB in place of lysate (i.e., with no deacetylase present), and in this case every time point contained less than 100 cpm. With lysate, the amount of [3H]-acetate released (above the 100 cpm background) was linear from 0 to 9 mins of incubation (Fig. 7A). For a given time point, the amount of 3H released was proportional to the lysate concentration up to 2 H 107 cells/mL (Fig. 7B).
Fig. 7.

Assay of histone deacetylase activity in lysates of interphase and metaphase-arrested HeLa cells. The assay is based on release of 3H from exogenous acid-extracted histones that were labeled in vivo with [3H]-acetate (see Materials and Methods). (A) A lysate of 2.0 H 107 cells/mL was tested for deacetylase activity over a time course of 9 mins. The release of 3H is linear over time. (B) Various concentrations of lysate, up to 2.0 H 107 cells/mL, were tested for deacetylase activity at one time point (5 min). The amount of [3H]-acetate released is proportional to the lysate concentration. (C), (D) Comparison of HDAC activity in lysates (of equivalent concentration) of (C) interphase and (D) metaphase-arrested HeLa cells. 10 separate deacetylase assays were run using interphase lysates and 8 using metaphase-arrested lysates. Error bars indicating two standard deviations from the mean are shown for each time point. Note that the deacetylase activity is virtually identical for metaphase ( — ) and interphase ( — ) lysates. (E), (F) Effects of protein phosphatase inhibitors and butyrate on deacetylase activity in HeLa lysates: (E) interphase lysates; (F) metaphase lysates. Cells were lysed either without added inhibitors ( — ), with 50 mM sodium butyrate (∼—∼) to inhibit HDACs, or with protein phosphatase inhibitors: 1 μM microcystin-LR ( — ) and 100 nM calyculin A ( — ).
Results
HDAC Inhibitors do not induce hyperacetylation of histone H4 in metaphase-arrested HeLa cells
Treatment of interphase mammalian cells with histone deacetylase (HDAC) inhibitors is known to induce hyperacetylation of core histones (e.g., [13-19]). In order to determine whether the same is true of mitotic cells, HeLa cultures were synchronized in S-phase by treatment with 2.5 mM thymidine for 20-24 hrs, released from the thymidine block, and treated 4 hrs later with 0.25 μg/mL nocodazole to arrest in metaphase [45,46]. At 16 hrs after release from thymidine, the metaphase-arrested cells were treated with 10 mM sodium butyrate. For comparison, interphase (unsynchronized) cultures were treated in parallel. At various times after treatment, samples were immediately placed on ice and chromosomes or nuclei were isolated [47,48]. Histones were extracted with acid and H4 acetylation was analyzed by acid urea gel electrophoresis. This method separates proteins partly on the basis of charge and is able to resolve histone H4 species containing zero, one, two, three or four acetylated residues (e.g., [13-15]).
The results of one such experiment are shown in Fig. 1A and B. With interphase cells (Fig. 1B), histone H4 becomes highly acetylated after only 1 hr. By contrast, metaphase-arrested cells (Fig. 1A) show little or no increase in H4 acetylation even after 5 hrs of treatment Note that in Fig. 1A, histone H1 is almost entirely in the highly phosphorylated (and lower mobility) form characteristic of mitosis (H1M). This confirms that the culture was arrested to a high mitotic index.
Fig. 1.

Lack of hyperacetylation of histone H4 following treatment of metaphase-arrested HeLa cells with HDAC inhibitors. (A), (B) Time course of treatment of (A) metaphase-arrested and (B) interphase (unsynchronized) HeLa cell cultures with 10 mM sodium butyrate. Histones were extracted from isolated chromosomes or nuclei at the indicated times after treatment, separated by acid-urea polyacrylamide gel electrophoresis [54] and stained with Coomassie blue. Note that H4 is highly acetylated in interphase cells after one or more hours with butyrate, but no increase in H4 acetylation is seen in metaphase-arrested cells even after five hours of treatment. (C), (D) Effects of other HDAC inhibitors on histone H4 acetylation. Aliquots of (C) metaphase-arrested and (D) interphase (unsynchronized) cultures were treated in parallel for 4 hrs with 1.0 Φg/mL trichostatin A (lanes 1), 2.0 Φg/mL apicidin (lanes 2), 2.0 Φg/mL oxamflatin (lanes 3), or 10 mM sodium butyrate (lanes 4), or incubated for 4 hrs with no treatment (Ctrl). Histones were extracted and analyzed as in (A) and (B). All four inhibitors induce extensive acetylation of histone H4 in interphase but not in metaphase-arrested cells. The positions of histone H4 and its acetylated forms, as well as mitotic (phosphorylated) and interphase histone H1 (H1M and H1I, respectively) are indicated at the right. The abundances of H1M and H1I verify that the cells were indeed predominantly mitotic in (A) and (C) and interphase in (B) and (D). Only the portions of the gels containing the histones are shown.
Other HDAC inhibitors give essentially the same results as butyrate. The acid-urea gel shown in Fig. 1C and D compares histones of metaphase-arrested cells and interphase cells that have been incubated for 4 hrs with 1.0 μg/mL Trichostatin A (lanes 1), 2.0 μg/mL apicidin (lanes 2), 2.0 μg/mL oxamflatin (lanes 3), or 10 mM sodium butyrate (lanes 4), or without any HDAC inhibitor (Ctrl). With interphase cells (Fig. 1D), comparison with the control (Ctrl) shows that in all cases histone H4 becomes hyperacetylated. In metaphase-arrested cells (Fig. 1C) on the other hand, there is little or no increase in histone H4 acetylation with any of the four HDAC inhibitors. Note again in Fig. 1C that histone H1 is mainly in its mitotic, highly phosphorylated form (H1M).
Lack of hyperacetylation after treatment of metaphase-arrested cells with butyrate is also observed with histones H2A, H2B and H3
The results shown in Fig. 1 demonstrate that treatment with HDAC inhibitors leads to hyperacetylation of histone H4 in interphase HeLa cells but not in metaphase-arrested cells. To test whether the same is true of H2A, H2B and H3, metaphase-arrested and interphase cultures were treated simultaneously with sodium butyrate and [3H]-acetate for 1 hour. Separate portions of the same cultures were incubated with [3H]-acetate in the same way but without butyrate. Histones were then extracted and separated by SDS-polyacrylamide gel electrophoresis. Total protein was visualized by Coomassie blue staining (Fig. 2A) and [3H]-acetate-labeled proteins were observed by autofluorography (Fig. 2B).
It is clear from Fig. 2B that much less [3H]-acetate is incorporated into histones in metaphase-arrested cells than in interphase cells. In interphase cells in the absence of butyrate (lane 1), labeling of histone H2A is detectable and labeling of H3 and H4 is quite strong. In the presence of butyrate, even more [3H]-acetate is incorporated into histones H2A, H3 and H4 in interphase cells (lane 2). In metaphase-arrested cells, on the other hand, labeling of histones is only barely detectable in the absence of butyrate (lane 3) and it is only slightly increased in the presence of butyrate (lane 4). (The small amount of labeling of H3 and H4 seen in Fig. 2B lanes 3 and 4 may be due in part to interphase contamination.) Note that histone H2B is not resolved from H3 on this gel, so we cannot say whether or not it is acetylated in interphase cells based on this data. However, H2B has been reported to become highly acetylated in butyrate-treated interphase HeLa cells [59, 25] and it is clearly not acetylated in butyrate-treated metaphase-arrested cells (Fig. 2B, lane 4). Lack of H2B acetylation at K12 and K15 in butyrate-treated metaphase cells is confirmed in Fig. 3 (see below).
These results are not due to differences in the amount of protein loaded or differences in the efficiency of [3H]-acetate incorporation into cellular acetyl coenzyme A pools. Coomassie blue staining demonstrates that the four samples contained equivalent amounts of each histone (Fig. 2A). Analysis of cell lysate supernatants by HPLC revealed essentially the same amount of 3H-label in acetyl coenzyme A in metaphase-arrested cells and interphase cells (data not shown).
We conclude from the results in Figs. 1 and 2 that when metaphase-arrested cells are treated with butyrate none of the core histones become highly acetylated.
Butyrate treatment of metaphase-arrested cells fails to induce hyperacetylation at several specific acetylation sites in the core histones
We next asked whether the lack of histone acetylation following butyrate treatment of metaphase-arrested cells is a characteristic of all acetylation sites, or only some of them. Histone extracts were prepared from four HeLa cell cultures: (1) interphase cells (unsynchronized); (2) interphase cells as in (1) but treated 4 hrs with 10 mM butyrate; (3) metaphase-arrested cells (mitotic index 97%); and (4) metaphase arrested cells as in (3) but treated 4 hrs with 10 mM butyrate. The histones were separated on SDS-polyacrylamide gels and acetylation was detected by immunoblotting using antibodies recognizing specific acetylated lysine residues. Antibody recognizing bulk histone H4 was used as a loading control (bottom panel).
The results of this analysis are shown in the first four lanes of Fig. 3. Nine different anti-acetylhistone antibodies were used, and in all cases the level of acetylation is clearly decreased in butyrate-treated mitotic cells (lane 4) compared to butyrate-treated interphase cells (lane 2). However, it is also clear that the extent of the decrease differs for the various acetylation sites. For example, butyrate-treated mitotic cells show significant amounts of acetylation at H4K12 and H3K23, though still much less than in the butyrate-treated interphase cells. On the other hand, little or no acetylation is detected in butyrate-treated mitotic cells with anti-H2BK12ac/K15ac, anti-H4K16ac or anti-H3K27ac antibodies (Fig. 3, lane 4).
The lack of detectable H2BK12ac/K15ac and H3K27ac in butyrate-treated metaphase-arrested cells, together with the strong signals at those sites in butyrate-treated interphase cells, suggests that the metaphase-arrested culture was not detectably contaminated with interphase cells. It follows that the small amount of acetylation seen in butyrate-treated metaphase-arrested cells at H3K9, H3K14/K18, H3K23, H4K8 and H4K12 (Fig. 3, lane 4) cannot simply be attributed to interphase contamination. This is significant because the amount of acetylation at H3K9, H3K14/K18, H3K23, H4K8 and H4K12 is clearly greater in butyrate-treated metaphase cells than in untreated metaphase cells (compare Fig. 3, lanes 3 and 4). Since the increased acetylation at these sites is not due to interphase contamination, we conclude that some small amount of acetylation must be occurring at these sites in metaphase-arrested cells, though of course much less than in interphase cells. Thus, not all HATs are completely inhibited or blocked from acetylating histones in mitotic cells.
Immunofluorescence microscopy detects no change in distribution of acetylated H4 across metaphase chromosomes following HDAC inhibitor treatment
The biochemical analyses of bulk histones in Figs. 1 – 3 cannot detect changes in the distribution or level of histone acetylation that are limited to particular chromosome regions or that affect only specific chromatin types. To determine whether large scale regional changes occur following treatment with HDAC inhibitors, we used immunofluorescence microscopy to study the distribution of acetylated H4 across metaphase chromosomes. Human female lymphoblastoid cells were used because they are diploid, have an inactive X chromosome (an example of facultative heterochromatin) which is substantially underacetylated relative to other chromosomes, and provide metaphase chromosome spreads suitable for immunolabelling and karyotypic analysis. Cells were treated with the HDAC inhibitor valproic acid (VPA, [20]), a short chain fatty acid equivalent to butyrate but better tolerated by these cells. As shown in Figure 4, exposure to VPA for 6h caused no measurable increase in H4 acetylation along the chromosome arms, nor did it change the underacetylation of the constitutive heterochromatin at centromeres (arrowheads) or the facultative heterochromatin of the inactive X chromosome (arrows). Although we cannot eliminate the possibility of subtle changes below the level of resolution of the light microscope, it seems that VPA causes no detectable overall changes in either the level or chromosomal distribution of H4 acetylation.
Fig. 4.

Immunolabeling of acetylated histone H4 on human metaphase chromosomes with or without treatment with the HDAC inhibitor valproic acid (VPA).
Chromosome spreads from human female lymphoblastoid cells were immunolabeled with antibodies to H4K8ac (green) and counterstained with DAPI (pseudocolored red). VPA-treated cells were exposed to 1 mM sodium valproate for 6 hrs, including 2 hrs in colcemid prior to harvesting and chromosome preparation. This treatment induces hyperacetylation of bulk histones comparable to that shown in Fig.1B (E. Terrenoire and B.M. Turner, unpublished results). Untreated cells were exposed to colcemid only. The inactive X chromosome (white arrow) is essentially unlabeled in both treated and untreated cells, as are blocks of centric heterochromatin (white arrowheads).
Decreased ability of H3 and H4 to become acetylated following treatment with butyrate is not due to butyrate-insensitive HDAC activity
The results presented in Figs. 1 – 3 could conceivably be explained by the presence of butyrate-insensitive HDACs or by increased HDAC activity during mitosis. In these scenarios, lack of histone hyperacetylation in metaphase-arrested cells would be due to significant residual HDAC activity, even in the presence of the usual HDAC inhibitors.
To test the hypothesis that butyrate-insensitive HDACs are present during mitosis, synchronized HeLa cells were treated with 10 mM sodium butyrate for 12 hours starting in mid-S-phase and continuing through G2-phase and into metaphase-arrest. With this procedure, mitotic indices of only 60 – 70% are obtained. Therefore, to minimize interphase contamination, individual mitotic chromosomes were isolated before extracting histones.
The hypothesis predicts that such chromosomes should not contain hyperacetylated histones, because any acetyl groups added during S- or G2-phase would be removed by butyrate-insensitive deacetylases after the cells were arrested in mitosis. Contrary to this prediction, histone H4 was found to be highly acetylated in these mitotic chromosomes (Fig. 5, lanes 4 and 5). Controls in Fig. 5 include histones from interphase cells treated with butyrate (lane 1), untreated interphase cells (lane 2), and untreated mitotic cells (lane 3). The relative amounts of multiply acetylated H4 species appear to be slightly less in lanes 4 and 5 than in lane 1, perhaps because the histones had less time to become acetylated (i.e., only until the cells entered mitosis).
The presence of some H1I (the unphosphorylated interphase form of histone H1) in Fig. 5, lanes 4 and 5 raises the possibility of interphase contamination. However, the amount of H1I is relatively minor, suggesting that there is too little interphase contamination to account for the extensive acetylation of H4 seen in these samples. To test this possibility further, histones from metaphase-arrested cells that were not treated with butyrate (Fig. 5, lane 3) were mixed with histones from butyrate-treated interphase cells (Fig. 5, lane 1) in proportions to mimic the amount of H1I seen in Fig. 5, lanes 4 and 5. In such a sample, acetylated H4 is not detectable on acid-urea gels (Fig. 5, lane 6).
To further test the hypothesis that butyrate-insensitive HDACs exist in mitotic cells, samples like those in Fig. 5 lanes 4 and 5 were analyzed by immunoblotting using antibodies specific for various acetylation sites in core histones. The results (Fig. 3, lane 5) show significant acetylation at all sites in H3 and H4. On the other hand, there appears to be little or no acetylation of H2AK5 or H2BK12/K15 in this sample. Evidently those sites were either (a) acetylated when the cells entered metaphase arrest, but then deacetylated, or (b) not acetylated at the time the cells entered metaphase arrest. The first possibility would suggest that during mitosis HDACs specific for those sites are present that either have a high level of activity or are insensitive to butyrate. The second possibility, which seems more likely, would suggest that those sites are not acetylated during G2-phase.
To summarize, hyperacetylated histones are indeed found in mitotic chromosomes when the cells have been treated with butyrate during late S- and G2-phase. We conclude that, at least in the case of histones H3 and H4, the lack of acetylation in metaphase-arrested cells treated with HDAC inhibitors (Figs. 1 – 3) cannot be attributed to butyrate-insensitive HDACs active during mitosis.
Inability of histones to become hyperacetylated in metaphase-arrested cells is related to mitotic protein phosphorylation
As a first step toward eventually testing whether HATs are down-regulated during mitosis or acetylation sites are inaccessible in mitotic chromosomes, we used a simple in vitro system in which cells are lysed in the presence of acetyl coenzyme A and butyrate in a buffer favorable for HAT activity. After incubation for 2 hrs at 37°C, histones are extracted and analyzed on acid-urea gels (Fig. 6).
Fig. 6.

Acetylation of histones in crude lysates of HeLa cells. Lanes 1 – 7: Interphase (I) or metaphase-arrested (M) cells were lysed and incubated in HAT buffer as described in Materials and Methods, with (+) or without (-) 10 mM butyrate, 1 mM acetyl coenzyme A and 1 mM cantharidin. Histones were extracted and analyzed on acid-urea gels. Lanes 8 – 9: Histones extracted from interphase cells treated in vivo with (+) or without (-) 10 mM butyrate. Only the portions of the gels containing the histones are shown.
Following this procedure, hyperacetylation of histone H4 is seen both in lysates of interphase cells (Fig. 6, lane 1) and in lysates of metaphase-arrested cells (Fig. 6, lane 2). However, when cantharidin, an inhibitor of Protein Phosphatases (PPases) 1 and 2A [52], is included during lysis of the metaphase-arrested cells, histone H4 does not become acetylated (Fig. 6, lane 3; note the accumulation of non-acetylated H4, the lowest band). This suggests that the inability of histone H4 to become acetylated following treatment of metaphase-arrested cells with butyrate depends on phosphoproteins in the mitotic cells that become dephosphorylated following cell lysis. It is well known that histone H1, histone H3 and other proteins are dephosphorylated following lysis of metaphase-arrested cells [60, 49], most likely by Protein Phosphatase 1[48]. H1 dephosphorylation (shift from the H1M to H1I position) is clearly seen in Fig. 6, lane 2, but cantharidin has prevented it in lane 3. Note that this does not necessarily imply that the lack of histone H4 acetylation is due to H1 phosphorylation, but that remains one possibility.
This result is not due to fortuitous inhibition of HATs by cantharidin, because histone H4 gets highly acetylated in lysates of interphase cells whether or not cantharidin is present (Fig. 6, lanes 4 and 5). We conclude that the repression of histone acetylation during mitosis depends on phosphorylation of as-yet-unidentified proteins that are dephosphorylated following cell lysis, but whose dephosphorylation is prevented by cantharidin.
Lack of histone acetylation in mitosis is not due to increased HDAC activity in metaphase-arrested cells
As discussed above, the results in Figs. 3 and 5 show (at least for histones H3 and H4) that butyrate-insensitive histone deacetylases are not present in metaphase-arrested HeLa cells at significant levels. The same results also suggest that decreased histone acetylation during butyrate treatment of these cells is not simply due to elevated (but still butyrate-sensitive) HDAC activity. We have tested this second possibility further by comparing HDAC activity in lysates. Lysates of interphase and mitotic cells were prepared so as to give equivalent concentrations of chromatin (see Materials and Methods). Relative deacetylase activity was determined as the amount of 3H released at various time points, using as a substrate exogenous acid-extracted histones that had been labeled in vivo with [3H]-acetate. Note that for a given lysate concentration, the amount of [3H]-acetate released is linear with respect to time (Fig. 7A) and for a given time point it is linear with respect to lysate concentration (Fig. 7B).
Fig. 7C and D indicate that HDAC activity is the same in metaphase-arrested and interphase cell lysates. However, as shown in the previous section and Fig. 6, if differences in interphase and mitotic HDAC activity existed in vivo, they would likely be due to mitosis-specific protein phosphorylation. Such differences could be erased in vitro as a result of protein dephosphorylation following cell lysis. To examine this possibility, histone deacetylase activity was assayed in lysates of metaphase-arrested and interphase cells prepared in the presence of calyculin A and microcystin LR. These compounds, like cantharidin, are inhibitors of Protein Phosphatases 1 and 2A [61, 62].
The results of this experiment are shown in Fig. 7E and F. Curiously, HDAC activity in interphase lysates is slightly lower in the presence of phosphatase inhibitors, suggesting that HDACs may be downregulated by phosphorylation during interphase (Fig. 7E). Nevertheless, it is clear that in mitotic lysates, the rate of deacetylation is unaffected by the presence of calyculin A or microcystin LR (Fig. 7F), contrary to what one would expect if HDACs were upregulated by phosphorylation during mitosis. From this result and the results in Fig. 6 we conclude that the phenomenon we observe is not primarily due to increased HDAC activity.
Discussion
Decreased acetylation of histones at mitosis in response to HDAC inhibitors
We have shown that the ability of histones to become acetylated following treatment of HeLa cells with HDAC inhibitors is significantly diminished during mitosis. Multiacetylated histone H4 isoforms are induced in interphase cells, but not in metaphase-arrested cells, by exposure to sodium butyrate [13], trichostatin A [17], apicidin [18] or oxamflatin [19]. The effect is seen with all four core histones and to varying degrees at all individual acetylation sites that have been examined. Our results suggest further that this phenomenon depends on mitosis-specific protein phosphorylation. Histone hyperacetylation occurs in lysates of metaphase-arrested cells when histones and other proteins are allowed to become dephosphorylated by endogenous phosphatases, but it does not occur when protein dephosphorylation is blocked by inhibitors of Protein Phosphatases 1 and 2A [48].
Transcription, histone acetylation and mitosis
Histone acetylation is closely connected with gene transcription [63-66] and transcription is shut off during mitosis (with some exceptions; e.g., [67]). The decreased ability of histones to become acetylated during mitosis is probably not a result of switching off transcription because it occurs in the bulk of the chromatin, not just in the small portion that was transcriptionally active during the previous interphase. However, decreased histone acetylation could be part of the cause of the cessation of transcription at mitosis.
The shut-off of transcription by all three RNA polymerases at mitosis involves phosphorylation of basal transcription factors by Cdk1/cyclin B [68-71]. This may provide the full explanation, particularly in the case of RNA Pol I [71]. However, there may be other mechanisms as well [72, 73] and the repression of core histone acetylation could be one of them. A striking feature of our data is that in butyrate-treated metaphase-arrested cells, acetylation at H4K16 is only barely detectable. H4K16 acetylation is essential for decompacting 30 nm fibers and maintaining a structure that permits DNA transcription [74, 4].
Possible explanations for the decreased ability of histones to become acetylated during mitosis
Since histone acetylation involves a balance between HDACs and HATs, two general hypotheses could explain the effect we have observed: (1) HDAC activity could be increased during mitosis, and/or become insensitive to HDAC inhibitors; and (2) the ability of HATs to acetylate histones in chromatin could be decreased during mitosis, either as a result of changes to the HATs themselves or changes in the chromatin substrate.
Based on our experiments, hypothesis (1) can be eliminated for most acetylation sites. Bulk HDAC activity is not substantially different in interphase and metaphase lysates and is equally sensitive to butyrate. In addition, mitotic chromosomes containing highly acetylated histones can be isolated by treating cells with butyrate during late S- and G2-phase. The latter result rules out the presence of butyrate-insensitive HDACs during mitosis, since histone acetyl groups added during S- or G2-phase would be removed by such HDACs when the cells reached metaphase arrest. Note that the isolation of individual mitotic chromosomes in this experiment confirms the observation [75] that histone hyperacetylation does not completely block mitotic chromosome condensation. However, hyperacetylation does lead to chromatin bridges and the failure of chromatid segregation at anaphase [75]. This phenotype is similar to what is seen in the absence of condensin [76], suggesting that histone deacetylation may be important for stabilizing condensed mitotic chromosomes.
This leaves hypothesis (2) which states that HATs are less able to acetylate histones during mitosis, either due to changes in the HATs themselves or changes in the chromatin substrate. This could be the result of down-regulation of HAT activity, displacement of HATs from chromosomes, changes in chromatin structure that make acetylation sites inaccessible to HATs, or a combination of these mechanisms. Note that the same mechanisms need not be operative for all HATs or for all sites.
Factors that could influence HAT activity through mitosis
In principle HAT activity could be down-regulated during mitosis by phosphorylation of HATs themselves or associated proteins. This seems unlikely to be the primary mechanism, however. Modulation of HAT activity by phosphorylation has been reported (e.g., [77-84]), but only Tip60 is known to be phosphorylated specifically at mitosis, and in this case the modification is stimulatory [85]. Kruhlak et al. [31] reported that HATs remain enzymatically active during mitosis, but they only assayed total HAT activity so it is still possible that some HATs may be down-regulated. One possible candidate for such a mode of control is hMOF, which is responsible for H4K16 acetylation [86-88]. Our results show that H4K16 acetylation is only barely detectable when metaphase-arrested cells are treated with butyrate, so perhaps hMOF is switched off.
Alternatively, decreased HAT activity on histones during mitosis could be due to displacement of HATs from the chromosomes, as proposed by Kruhlak et al. [31]. Failure of HATs to bind to mitotic chromosomes could be a consequence of changes in chromatin structure or a result of modification (e.g., phosphorylation) of the HATs themselves.
A final possibility is that histone acetylation sites could become inaccessible to HATs during mitosis due to structural changes in the chromatin. Gross chromosome condensation probably does not play a major role since condensed mitotic chromatin is known to be accessible to transcription factors and other proteins in vivo [89]. However, inaccessibility of acetylation sites could result from stabilization of 30 nm fibers or new interactions within the chromatin fiber. Histone phosphorylation could be involved by altering contacts between different nucleosomes or between histones and DNA.
One attractive hypothesis is that specific unacetylated lysine residues in the core histone tails may interact electrostatically with phosphate groups in histones H1 and H3 during mitosis, thereby stabilizing 30 nm chromatin fibers or condensed chromosomes. Alternatively, 30 nm chromatin fibers may be stabilized during mitosis by interaction between unacetylated H4K16 and an “acidic patch” on a neighboring nucleosome [66, 4]. In either case, accessibility of the relevant lysine residues would normally be restricted during mitosis. Furthermore, extensive acetylation of those sites would tend to disrupt the interactions and destabilize the condensed chromosomes, as observed by Cimini et al. [75].
Control of acetylation at specific lysine residues
In mammalian cells, acetylation of lysines in the N-terminal tails of histones H3 and H4 progresses in a non-random order from the mono-acetylated to the tetra-acetylated isoforms. For example, in mono-acetylated H4, K16 is the predominant acetylated residue. K16, K8 and K12 are acetylated in the di- and tri-acetylated isoforms while K5 is used only in the most highly acetylated isoforms [90, 59]. A similar situation occurs for H3 with K14 being acetylated first, followed by K23, K18 and K9 [90, 59]. Work on H4 acetylation in Tetrahymena [91] and cuttlefish [92] has led to similar conclusions.
In bulk histones from untreated cells, the non-acetylated and mono-acetylated isoforms predominate, with the more highly acetylated forms being rare. However, treatment with HDAC inhibitors leads to rapid hyperacetylation and accumulation of more highly acetylated isoforms, showing that there is rapid and continuing acetate turnover at all lysines.
It is likely that multiple HATs and HDACs are involved in setting the level of acetylation at each site. The enzymes involved may also vary, either from one region of the genome to another or throughout the cell cycle. Recent work on the properties of candidate enzymes is beginning to provide clues about how lysine-specific acetylation might be regulated. In general, HATs show more specificity than HDACs, which tend to show only limited preference for particular lysines or histones [93-96]. For example, the acetyltransferase MOF shows a strong preference for H4K16. Cells in which MOF is knocked down are depleted of acetylated H4, suggesting that MOF has a dominant role in controlling bulk H4 acetylation [97]. However, the specificity of HATs also depends on both the chromatin context in which they operate and on the proteins that associate with them [98]. Thus, the HAT Rtt109 acetylates H3K14 and H3K23 in the presence of the histone chaperone protein Vps75 but acetylates H3K56 in the presence of the chaperone Asf1 [95, 99, 100].
We have shown here that turnover of H3 and H4 acetates is reduced (that is, the ability to generate hyperacetylated isoforms in the presence of HDAC inhibitors is diminished) in metaphase. We have also shown that the relative levels of acetylation of individual lysines are altered in metaphase cells. Our results are consistent with previous work showing that H4K5 and H4K12 are more frequently acetylated in the mono-acetylated isoform in metaphase than in interphase HeLa cells [29]. Altered patterns of lysine acetylation may be a consequence of differences in turnover. For example, we show here that relative to other sites, H3K23ac is much more commonly acetylated in metaphase than in interphase. During interphase, H3K23 is normally acetylated only in the di-, tri- and tetra-acetylated isoforms, but in butyrate-treated cells it predominates in the mono-acetylated isoform [59]. It may be that more rapid acetate turnover at H3K23 causes it to become the dominant site of acetylation after butyrate inhibition of HDAC activity. In the absence of HDAC inhibitors, a reduction in HDAC activity as cells progress into metaphase could exert the same effect. Reduced activity could be due to depletion or modification of the enzymes, but it could also be simply due to their dissociation from chromatin, in which case decreased HDAC activity would not be detected in assays of whole cell extracts.
In summary, it is likely that progression from non-acetylated to hyperacetylated histones requires the concerted action of multiple HATs, with a variety of site specificities, moderated by less specific HDACs. If the activity levels of individual enzymes (or their locations on chromatin) change as cells enter mitosis, then this will be reflected in changes in both the ability to generate highly acetylated histone isoforms and the extent to which individual sites are acetylated. While acetylation at individual sites would continue, albeit in a restricted way, progression to more highly acetylated forms would not, largely because the depleted, or relocated, HATs would rarely act on the same histone tail. Overall, this decrease in more highly acetylated forms of the core histones may then help to stabilize the characteristic condensed structure of mitotic chromosomes.
Acknowledgments
We are grateful to Dr. Maria Vogelauer and Dr. Hiroshi Kimura for providing acetylhistone-specific antibodies. This work was supported by the University of Wisconsin-Oshkosh Faculty Development Board and by grants R15 GM39915 and R15 GM46040 (to J. R. Paulson) and grant R01 CA69008 (to W. C. Earnshaw and S. H. Kaufmann) from the National Institutes of Health, U.S. Public Health Service. The work was also supported by grants to B.M.Turner from Cancer Research UK (programme grant C1015/A9077) and the European Union (FP6, “Epigenome” NoE 2003-9).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Δ resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryotic chromosome. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- 3.Thoma F, Koller T, Klug A. Involvement of histone H1 in the organization of the nucleosome and of the salt-dependent superstructures of chromatin. J Cell Biol. 1979;83:403–427. doi: 10.1083/jcb.83.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robinson PJ, An W, Routh A, Martino F, Chapman L, Roeder RG, Rhodes D. 30 nm chromatin fibre decompaction requires both H4-K16 acetylation and linker histone eviction. J Mol Biol. 2008;381:816–825. doi: 10.1016/j.jmb.2008.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 6.Brown CE, Lechner T, Howe L, Workman JL. The many HATs of transcription coactivators. Trends Biochem Sci. 2000;25:15–19. doi: 10.1016/s0968-0004(99)01516-9. [DOI] [PubMed] [Google Scholar]
- 7.Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64:435–459. doi: 10.1128/mmbr.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 9.Eberharter A, Becker PB. Histone acetylation: a switch between repressive and permissive chromatin. EMBO Rep. 2002;3:224–229. doi: 10.1093/embo-reports/kvf053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 11.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 12.Jackson V, Shires A, Chalkley R, Granner DK. Studies on highly metabolically active acetylation and phosphorylation of histones. J Biol Chem. 1975;250:4856–4863. [PubMed] [Google Scholar]
- 13.Riggs MG, Whittaker RG, Neumann JR, Ingram VM. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature. 1977;268:462–464. doi: 10.1038/268462a0. [DOI] [PubMed] [Google Scholar]
- 14.Vidali G, Boffa LC, Bradbury EM, Allfrey VG. Butyrate suppression of histone deacetylation leads to accumulation of multiacetylated forms of histones H3 and H4 and increased DNase I sensitivity of the associated DNA sequences. Proc Natl Acad Sci USA. 1978;75:2239–2243. doi: 10.1073/pnas.75.5.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sealy L, Chalkley R. The effect of sodium butyrate on histone modification. Cell. 1978;14:115–121. doi: 10.1016/0092-8674(78)90306-9. [DOI] [PubMed] [Google Scholar]
- 16.Cousens LS, Gallwitz D, Alberts BM. Different accessibilities in chromatin to histone acetylase. J Biol Chem. 1979;254:1716–1723. [PubMed] [Google Scholar]
- 17.Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- 18.Darkin-Rattray SJ, Gurnett AM, Myers RW, Dulski PM, Crumley TM, Allocco JJ, Cannova C, Meinke PT, Colletti SL, Bednarek MA, Singh SB, Goetz MA, Dombrowski AW, Polishook JD, Schmatz DM. Apicidin: a novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc Natl Acad Sci USA. 1996;93:13143–13147. doi: 10.1073/pnas.93.23.13143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim YB, Lee KH, Sugita K, Yoshida M, Horinouchi S. Oxamflatin is a novel antitumor compound that inhibits mammalian histone deacetylase. Oncogene. 1999;18:2461–2470. doi: 10.1038/sj.onc.1202564. [DOI] [PubMed] [Google Scholar]
- 20.Pfiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer and teratogen. J Biol Chem. 2001;276:36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- 21.Schultz BE, Misialek S, Wu J, Tang J, Conn MT, Tahilramani R, Wong L. Kinetics and comparative reactivity of human class I and class IIb histone deacetylases. Biochemistry. 2004;43:11083–11091. doi: 10.1021/bi0494471. [DOI] [PubMed] [Google Scholar]
- 22.Sekhavat A, Sun JM, Davie JR. Competitive inhibition of histone deacetylases activity by trichostatin A and butyrate. Biochem Cell Biol. 2007;85:751–758. doi: 10.1139/o07-145. [DOI] [PubMed] [Google Scholar]
- 23.Meinke PT, Colletti SL, Doss G, Myers RW, Gurnett AM, Dulski PM, Darkin-Rattray SJ, Allocco JJ, Galuska S, Schmatz DM, Wyvratt MJ, Fisher MH. Synthesis of apicidin-derived quinolone derivatives: parasite-selective histone deacetylase inhibitors and antiproliferative agents. J Med Chem. 2000;43:4919–4922. doi: 10.1021/jm0001976. [DOI] [PubMed] [Google Scholar]
- 24.D'Anna JA, Tobey RA, Barham SS, Gurley LR. A reduction in the degree of H4 acetylation during mitosis in Chinese hamster cells. Biochem Biophys Res Commun. 1977;77:187–194. doi: 10.1016/s0006-291x(77)80181-2. [DOI] [PubMed] [Google Scholar]
- 25.D'Anna JA, Tobey RA, Gurley LR. Concentration-dependent effects of sodium butyrate in Chinese hamster cells: cell-cycle progression, inner-histone acetylation, histone H1 dephosphorylation, and induction of an H1-like protein. Biochemistry. 1980;19:2656–2671. doi: 10.1021/bi00553a019. [DOI] [PubMed] [Google Scholar]
- 26.D'Anna JA, Gurley LR, Tobey RA. Extent of histone modifications and H10 content during cell cycle progression in the presence of butyrate. Exp Cell Res. 1983;147:407–417. doi: 10.1016/0014-4827(83)90222-7. [DOI] [PubMed] [Google Scholar]
- 27.Moore M, Jackson V, Sealy L, Chalkley R. Comparative studies on highly metabolically active histone acetylation. Biochim Biophys Acta. 1979;561:248–260. doi: 10.1016/0005-2787(79)90508-2. [DOI] [PubMed] [Google Scholar]
- 28.Turner BM. Acetylation and deacetylation of histone H4 continue through metaphase with depletion of more acetylated isoforms and altered site usage. Exp Cell Res. 1989;182:206–214. doi: 10.1016/0014-4827(89)90292-9. [DOI] [PubMed] [Google Scholar]
- 29.Turner BM, Fellows G. Specific antibodies reveal ordered and cell-cycle-related use of histone H4 acetylation sites in mammalian cells. Eur J Biochem. 1989;179:131–139. doi: 10.1111/j.1432-1033.1989.tb14530.x. [DOI] [PubMed] [Google Scholar]
- 30.Jeppesen P, Mitchell A, Turner B, Perry P. Antibodies to defined histone epitopes reveal variations in chromatin conformation and underacetylation of centric heterochromatin in human metaphase chromosomes. Chromosoma. 1992;101:322–332. doi: 10.1007/BF00346011. [DOI] [PubMed] [Google Scholar]
- 31.Kruhlak MJ, Hendzel MJ, Fischle W, Bertos NR, Hameed S, Yan XJ, Verdin E, Bazett-Jones DP. Regulation of global acetylation in mitosis through loss of histone acetyltransferases and deacetylases from chromatin. J Biol Chem. 2001;276:38307–38319. doi: 10.1074/jbc.M100290200. [DOI] [PubMed] [Google Scholar]
- 32.McManus KJ, Hendzel MJ. The relationship between histone H3 phosphorylation and acetylation throughout the mammalian cell cycle. Biochem Cell Biol. 2006;84:640–657. doi: 10.1139/o06-086. [DOI] [PubMed] [Google Scholar]
- 33.Bonenfant D, Towbin H, Coulot M, Schindler P, Mueller DR, van Oostrom J. Analysis of dynamic changes in post-translational modifications of human histones during cell cycle by mass spectrometry. Mol Cell Proteomics. 2007;6:1917–1932. doi: 10.1074/mcp.M700070-MCP200. [DOI] [PubMed] [Google Scholar]
- 34.Chahal SS, Matthews HR, Bradbury EM. Acetylation of histone H4 and its role in chromatin structure and function. Nature. 1980;287:76–79. doi: 10.1038/287076a0. [DOI] [PubMed] [Google Scholar]
- 35.Waterborg JH, Matthews HR. Patterns of histone acetylation in the cell cycle of Physarum polycephalum. Biochemistry. 1983;22:1489–1496. doi: 10.1021/bi00275a025. [DOI] [PubMed] [Google Scholar]
- 36.Nadler KD. Histone acetylation during meiosis in Lilium microsporocytes. Exp Cell Res. 1976;101:283–292. doi: 10.1016/0014-4827(76)90379-7. [DOI] [PubMed] [Google Scholar]
- 37.Kim JM, Liu H, Tazaki M, Nagata M, Aoki F. Changes in histone acetylation during mouse oocyte meiosis. J Cell Biol. 2003;162:37–46. doi: 10.1083/jcb.200303047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor JH. Nucleic acid synthesis in relation to the cell division cycle. Ann NY Acad Sci. 1960;90:409–421. doi: 10.1111/j.1749-6632.1960.tb23259.x. [DOI] [PubMed] [Google Scholar]
- 39.Prescott DM, Bender MA. Synthesis of RNA and protein during mitosis in mammalian tissue culture cells. Exp Cell Res. 1962;26:260–268. doi: 10.1016/0014-4827(62)90176-3. [DOI] [PubMed] [Google Scholar]
- 40.Baserga R. A study of nucleic acid synthesis in ascites tumor cells by two-emulsion autoradiography. J Cell Biol. 1962;12:633–637. doi: 10.1083/jcb.12.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Terasima T, Tomach LJ. Growth and nucleic acid synthesis in synchronously dividing populations of HeLa cells. Exp Cell Res. 1963;30:344–362. doi: 10.1016/0014-4827(63)90306-9. [DOI] [PubMed] [Google Scholar]
- 42.White DA, Belyaev ND, Turner BM. Preparation of site-specific antibodies to acetylated histones. Methods. 1999;19:417–424. doi: 10.1006/meth.1999.0878. [DOI] [PubMed] [Google Scholar]
- 43.Kimura H, Hayashi-Takanaka Y, Goto Y, Takizawa N, Nozaki N. The organization of histone H3 modifications as revealed by a panel of specific monoclonal antibodies. Cell Struct Funct. 2008;33:61–73. doi: 10.1247/csf.07035. [DOI] [PubMed] [Google Scholar]
- 44.Medappa KC, McLean C, Rueckert RR. On the structure of rhinovirus 1A. Virology. 1971;44:259–270. doi: 10.1016/0042-6822(71)90258-3. [DOI] [PubMed] [Google Scholar]
- 45.Paulson JR, Ciesielski WA, Schram BR, Mesner PW. Okadaic acid induces dephosphorylation of histone H1 in metaphase-arrested HeLa cells. J Cell Sci. 1994;107:267–273. doi: 10.1242/jcs.107.1.267. [DOI] [PubMed] [Google Scholar]
- 46.Xeros N. Deoxyriboside control and synchronization of mitosis. Nature. 1962;194:682–683. doi: 10.1038/194682a0. [DOI] [PubMed] [Google Scholar]
- 47.Paulson JR. Isolation of chromosome clusters from metaphase-arrested HeLa cells. Chromosoma. 1982;85:571–581. doi: 10.1007/BF00327351. [DOI] [PubMed] [Google Scholar]
- 48.Paulson JR, Patzlaff JS, Vallis AJ. Evidence that the endogenous histone H1 phosphatase in HeLa mitotic chromosomes is protein phosphatase 1, not protein phosphatase 2A. J Cell Sci. 1996;109:1437–1447. doi: 10.1242/jcs.109.6.1437. [DOI] [PubMed] [Google Scholar]
- 49.Paulson JR. Sulfhydryl reagents prevent dephosphorylation and proteolysis of histones in isolated HeLa metaphase chromosomes. Eur J Biochem. 1980;111:89–197. doi: 10.1111/j.1432-1033.1980.tb06092.x. [DOI] [PubMed] [Google Scholar]
- 50.Marsden MP, Laemmli UK. Metaphase chromosome structure: evidence for a radial loop model. Cell. 1979;17:849–858. doi: 10.1016/0092-8674(79)90325-8. [DOI] [PubMed] [Google Scholar]
- 51.Keohane AM, O'Neill LP, Belyaev ND, Lavender JS, Turner BM. X-inactivation and histone H4 acetylation in embryonic stem cells. Devel Biol. 1996;180:618–630. doi: 10.1006/dbio.1996.0333. [DOI] [PubMed] [Google Scholar]
- 52.Honkanen RE. Cantharidin, another natural toxin that inhibits the activity of serine/threonine protein phosphatases types 1 and 2A. FEBS Lett. 1993;330:283–286. doi: 10.1016/0014-5793(93)80889-3. [DOI] [PubMed] [Google Scholar]
- 53.Laemmli UK, Favre M. Maturation of the head of bacteriophage T4. I. DNA packaging events. J Mol Biol. 1973;80:575–599. doi: 10.1016/0022-2836(73)90198-8. [DOI] [PubMed] [Google Scholar]
- 54.Panyim S, Chalkley R. High resolution acrylamide gel electrophoresis of histones. Arch Biochem Biophys. 1969;130:337–346. doi: 10.1016/0003-9861(69)90042-3. [DOI] [PubMed] [Google Scholar]
- 55.Paulson JR, Higley LL. Acid-urea polyacrylamide slab gel electrophoresis of proteins: preventing distortion of gel wells during preelectrophoresis. Anal Biochem. 1999;268:157–159. doi: 10.1006/abio.1998.3026. [DOI] [PubMed] [Google Scholar]
- 56.Bonner WM, Laskey RA. A film detection method for tritium-labelled proteins and nucleic acids in polyacrylamide gels. Eur J Biochem. 1974;46:83–88. doi: 10.1111/j.1432-1033.1974.tb03599.x. [DOI] [PubMed] [Google Scholar]
- 57.Rao J, Otto WR. Fluorimetric DNA assay for cell growth estimation. Anal Biochem. 1992;207:186–192. doi: 10.1016/0003-2697(92)90521-8. [DOI] [PubMed] [Google Scholar]
- 58.Hosokawa Y, Shimomura Y, Harris RA, Ozawa T. Determination of short-chain acyl-coenzyme A esters by high-performance liquid chromatography. Anal Biochem. 1986;153:45–49. doi: 10.1016/0003-2697(86)90058-8. [DOI] [PubMed] [Google Scholar]
- 59.Thorne AW, Kmiciek D, Mitchelson K, Sautiere P, Crane-Robinson C. Patterns of histone acetylation. Eur J Biochem. 1990;193:701–713. doi: 10.1111/j.1432-1033.1990.tb19390.x. [DOI] [PubMed] [Google Scholar]
- 60.D'Anna JA, Gurley LR, Deaven LL. Dephosphorylation of histones H1 and H3 during the isolation of metaphase chromosomes. Nucleic Acids Res. 1978;5:3195–3207. doi: 10.1093/nar/5.9.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ishihara H, Martin BL, Brautigan DL, Karaki H, Ozaki H, Kato Y, Fusetani N, Watabe S, Hashimoto K, Uemura D, Hartshorne DJ. Calyculin A and okadaic acid: inhibitors of protein phosphatase activity. Biochem Biophys Res Commun. 1989;159:871–877. doi: 10.1016/0006-291x(89)92189-x. [DOI] [PubMed] [Google Scholar]
- 62.MacKintosh C, Beattie KA, Klumpp S, Cohen P, Codd GA. Cyanobacterial microcystin-LR is a potent and specific inhibitor of protein phosphatases 1 and 2A from both mammals and higher plants. FEBS Lett. 1990;264:187–192. doi: 10.1016/0014-5793(90)80245-e. [DOI] [PubMed] [Google Scholar]
- 63.Turner BM. Histone acetylation and an epigenetic code. Bioessays. 2000;22:836–845. doi: 10.1002/1521-1878(200009)22:9<836::AID-BIES9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 64.Strahl BC, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 65.Henikoff S. Histone modifications: combinatorial complexity or cumulative simplicity? Proc Natl Acad Sci USA. 2005;102:5308–5309. doi: 10.1073/pnas.0501853102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 67.Sciortino S, Gurtner A, Manni I, Fontemaggi G, Dey A, Sacchi A, Ozato K, Piaggio G. The cyclin B1 gene is actively transcribed during mitosis in HeLa cells. EMBO Rep. 2001;2:1018–1023. doi: 10.1093/embo-reports/kve223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leresche A, Wolf VJ, Gottesfeld JM. Repression of RNA polymerase II and III transcription during M phase of the cell cycle. Exp Cell Res. 1996;29:282–288. doi: 10.1006/excr.1996.0373. [DOI] [PubMed] [Google Scholar]
- 69.Gottesfeld JM, Forbes DJ. Mitotic repression of the transcriptional machinery. Trends Biochem Sci. 1997;22:197–202. doi: 10.1016/s0968-0004(97)01045-1. [DOI] [PubMed] [Google Scholar]
- 70.Heix J, Vente A, Voit R, Budde A, Michaelidis TM, Grummt T. Mitotic silencing of human rRNA synthesis: inactivation of the promoter selectivity factor SL1 by cdc2/cyclin B-mediated phosphorylation. EMBO J. 1998;17:7373–7381. doi: 10.1093/emboj/17.24.7373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kuhn A, Vente A, Dorée M, Grummt I. Mitotic phosphorylation of the TBP-containing factor SL1 represses ribosomal gene transcription. J Mol Biol. 1998;284:1–5. doi: 10.1006/jmbi.1998.2164. [DOI] [PubMed] [Google Scholar]
- 72.Dynlacht BD. Regulation of transcription by proteins that control the cell cycle. Nature. 1997;389:149–152. doi: 10.1038/38225. [DOI] [PubMed] [Google Scholar]
- 73.Long JJ, Leresche A, Kriwacki RW, Gottesfeld JM. Repression of TFIIH transcriptional activity and TFIIH-associated cdk7 kinase activity at Mitosis. Mol Cell Biol. 1998;18:1467–1576. doi: 10.1128/mcb.18.3.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shogren-Knaak M, Peterson CL. Switching on chromatin: mechanistic role of histone H4-K16 acetylation. Cell Cycle. 2006;5:1361–1365. doi: 10.4161/cc.5.13.2891. [DOI] [PubMed] [Google Scholar]
- 75.Cimini D, Mattiuzzo M, Torosantucci L, Degrassi F. Histone hyperacetylation in mitosis prevents sister chromatid separation and produces chromosome segregation defects. Mol Biol Cell. 2003;14:3821–3833. doi: 10.1091/mbc.E03-01-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vagnarelli P, Hudson DF, Ribeiro SA, Trinkle-Mulcahy L, Spence JM, Lai F, Farr CJ, Lamond AL, Earnshaw WC. Condensin and Repo-man PP1 cooperate in the regulation of chromosome architecture during mitosis. Nat Cell Biol. 2006;8:1133–1142. doi: 10.1038/ncb1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Barlev NA, Poltoratsky V, Owen-Hughes T, Ying C, Liu L, Workman JL, Berger SL. Repression of GCN5 histone acetyltransferase activity via bromodomain-mediated binding and phosphorylation by the Ku-DNA-dependent protein kinase complex. Mol Cell Biol. 1998;18:1349–1358. doi: 10.1128/mcb.18.3.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ait-Si-Ali S, Ramirez S, Barre FX, Dkhissi F, Magnaghi-Jaulin L, Girault JA, Robin P, Knibiehler M, Pritchard LL, Ducommun B, Trouche D, Harel-Belian A. Histone acetyltransferase activity of CBP is controlled by cyclin-dependent kinases and oncoprotein E1A. Nature. 1998;396:184–186. doi: 10.1038/24190. [DOI] [PubMed] [Google Scholar]
- 79.Ait-Si-Ali S, Carlisi D, Ramirez S, Upegui-Gonzalez LC, Duquet A, Robin P, Rudkin B, Harel-Belian A, Trouche D. Phosphorylation by p44 MAP kinase/ERK1 stimulates CBP histone acetyltransferase activity in vitro. Biochem Biophys Res Commun. 1999;262:157–162. doi: 10.1006/bbrc.1999.1132. [DOI] [PubMed] [Google Scholar]
- 80.Kumar BRP, Swaminathan V, Banerjee S, Kundu TK. p300-mediated acetylation of human transcriptional coactivator PC4 is inhibited by phosphorylation. J Biol Chem. 2001;276:16804–16809. doi: 10.1074/jbc.M100934200. [DOI] [PubMed] [Google Scholar]
- 81.Yuan LW, Soh JW, Weinstein IB. Inhibition of histone acetyltransferase function of p300 by PKCδ. Biochim Biophys Acta. 2002;1592:205–211. doi: 10.1016/s0167-4889(02)00327-0. [DOI] [PubMed] [Google Scholar]
- 82.Kovacs KA, Steinmann M, Magistretti PJ, Halfon O, Cardinaux JR. CCAAT/enhancer-binding protein family members recruit the coactivator CREB-binding protein and trigger its phosphorylation. J Biol Chem. 2003;278:36959–36965. doi: 10.1074/jbc.M303147200. [DOI] [PubMed] [Google Scholar]
- 83.Huang WC, Chen CC. Akt phosphorylation of p300 at Ser-1834 is essential for its histone acetyltransferase and transcriptional activity. Mol Cell Biol. 2005;25:6592–6602. doi: 10.1128/MCB.25.15.6592-6602.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen YJ, Wang YN, Chang WC. ERK2-mediated C-terminal serine phosphorylation of p300 is vital to the regulation of epidermal growth factor-induced keratin 16 gene expression. J Biol Chem. 2007;282:27215–27228. doi: 10.1074/jbc.M700264200. [DOI] [PubMed] [Google Scholar]
- 85.Lemercier C, Legube G, Caron C, Louwagie M, Garin J, Trouche D, Khochbin S. Tip60 acetyltransferase activity is controlled by phosphorylation. J Biol Chem. 2003;278:4713–4718. doi: 10.1074/jbc.M211811200. [DOI] [PubMed] [Google Scholar]
- 86.Taipale M, Rea S, Richter K, Vilar A, Lichter P, Imhof A, Akhtar A. hMOF histone acetyltransferase is required for histone H4 lysine 16 acetylation in mammalian cells. Mol Cell Biol. 2005;25:6798–6810. doi: 10.1128/MCB.25.15.6798-6810.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rea S, Xouri G, Akhtar A. Males absent on the first (MOF): from flies to humans. Oncogene. 2007;26:5385–5394. doi: 10.1038/sj.onc.1210607. [DOI] [PubMed] [Google Scholar]
- 88.Thomas T, Dixon MP, Kueh AJ, Voss AK. Mof (MYST1 or KAT8) is essential for progression of embryonic development past the blastocyst stage and required for normal chromatin architecture. Mol Cell Biol. 2008;28:5093–5105. doi: 10.1128/MCB.02202-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen D, Dundr M, Wang C, Leung A, Lamond A, Misteli T, Huang S. Condensed mitotic chromatin is accessible to transcription factors and chromatin structural proteins. J Cell Biol. 2005;168:41–54. doi: 10.1083/jcb.200407182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Turner BM, O'Neill LP, Allan IM. Histone H4 acetylation in human cells. Frequency of acetylation at different sites defined by immunolabeling with site-specific antibodies. FEBS Lett. 1989;253:141–145. doi: 10.1016/0014-5793(89)80947-0. [DOI] [PubMed] [Google Scholar]
- 91.Chicoine LG, Schulman IG, Richman R, Cook RG, Allis CD. Nonrandom utilization of acetylation sites in histones isolated from Tetrahymena. Evidence for functionally distinct H4 acetylation sites. J Biol Chem. 1986;261:1071–1076. [PubMed] [Google Scholar]
- 92.Couppez M, Martin-Ponthieu A, Sautiere P. Histone H4 from cuttlefish testis is sequentially acetylated. Comparison with acetylation of calf thymus histone H4. J Biol Chem. 1987;262:2854–2860. [PubMed] [Google Scholar]
- 93.Johnson CA, White DA, Lavender JS, O'Neill LP, Turner BM. Human class I histone deacetylase complexes show enhanced catalytic activity in the presence of ATP and co-immunoprecipitate with the ATP-dependent chaperone protein Hsp70. J Biol Chem. 2002;277:9590–9597. doi: 10.1074/jbc.M107942200. [DOI] [PubMed] [Google Scholar]
- 94.Sengupta N, Seto E. Regulation of histone deacetylase activities. J Cell Biochem. 2004;93:57–67. doi: 10.1002/jcb.20179. [DOI] [PubMed] [Google Scholar]
- 95.Berndsen CE, Denu JM. Catalysis and substrate selection by histone/protein lysine acetyltransferases. Curr Opin Struct Biol. 2008;18:682–689. doi: 10.1016/j.sbi.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Marmorstein R, Trievel RC. Histone modifying enzymes: structures, mechanisms and specificities. Biochim Biophys Acta. 2009;1789:58–68. doi: 10.1016/j.bbagrm.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Smith ER, Cayrou C, Huang R, Lane WS, Cote J, Lucchesi JC. A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol Cell Biol. 2005;25:9175–9188. doi: 10.1128/MCB.25.21.9175-9188.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Berndsen CE, Selleck W, McBryant SJ, Hansen JC, Tan S, Denu JM. Nucleosome recognition by the Piccolo NuA4 histone acetyltransferase complex. Biochemistry. 2007;46:2091–2099. doi: 10.1021/bi602366n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Berndsen CE, Tsubota T, Lindner SE, Lee S, Holton JM, Kaufman PD, Keck JL, Denu JM. Molecular functions of the histone acetyltransferase chaperone complex Rtt109-Vps75. Nat Struct Mol Biol. 2008;15:948–956. doi: 10.1038/nsmb.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fillingham J, Recht J, Silva AC, Suter B, Emili A, Stagljar I, Krogan NJ, Allis CD, Keogh MC, Greenblatt JF. Chaperone control of the activity and specificity of the histone H3 acetyltransferase Rtt109. Mol Cell Biol. 2008;28:4342–4253. doi: 10.1128/MCB.00182-08. [DOI] [PMC free article] [PubMed] [Google Scholar]